
Abstract
Herpes Simplex Virus-1 (HSV-1) infections in the eye often originate in the cornea before assuming a latent state in the trigeminal ganglion. During primary infection and upon injury or reactivation, HSV-1 can lead to significant corneal damage. Nanoparticles (NPs) are an emerging strategy for drug delivery to the cornea because they improve the long-term release of anti-HSV-1 drugs, such as nucleoside analogues. Acyclovir, ganciclovir, and valacyclovir have been successfully delivered using both polymer and lipid-based NPs in vitro. Solid silica dioxide NPs have been used to deliver the cathelicidin, LL-37, which prevented HSV-1 infection in corneal epithelial cells. Iron oxide nanoparticles have also been adapted to deliver an anti-HSV-1 DNA vaccine that successfully reduced corneal opacity and HSV-1 markers in a mouse model. Overall, NPs show promise as a delivery method for anti-HSV-1 strategies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


8.1 Introduction
Effective drug delivery to the cornea requires drug release into the tear fluid on the surface of the cornea to treat epithelial conditions and penetrance into the cornea proper to treat deeper tissues [1,2,3]. Ideally, the drug will accumulate in the aqueous humor located in the anterior chamber between the cornea and the lens, which acts as a reservoir for additional drug release. However, drug delivery to the cornea is still far from optimal. Although eye drops and other topical medications are commonly used, less than five percent of topically applied drugs can reach deeper ocular targets [4]. Thus, many drugs destined for the cornea, e.g., to treat infections, are still administered systemically, to allow a small percentage to reach deeper ocular targets. Nanoparticles (NPs) that can penetrate the tear film or extracellular space between cells have therefore been developed in an attempt to optimize drug delivery to the cornea. NPs developed to date include those delivering therapeutic drugs and growth factors, to those used for diagnostics.
In this chapter, we briefly review the various NP-based therapies that are being developed to treat corneal pathologies. We focus on the treatment of Herpes Simplex Virus (HSV) infections as a model disease, as this is a significant problem worldwide and is the most prevalent cause of infectious corneal blindness in both the developing and developed world.
8.2 Herpes Simplex Virus and Eye Infections
HSV serotype 1 or HSV-1 is the viral strain that causes eye infections. The global prevalence of ocular HSV-1 is estimated at 1.5 million individuals, including 40,000 new cases of monocular visual impairment annually [5]. Ocular herpes is one of the most severe forms of HSV infection. It can lead to a broad panel of ocular damage, including lid ulceration, conjunctivitis, keratitis, anterior uveitis, and rare but severe retinal disease.
The primary herpes infection is asymptomatic in approximately 94% of cases [6]. However, after a phase of replication at the primary infection site (cornea, peri-ocular, or oro-pharyngeal epithelium), the virus enters the corneal nerve endings and gains access to the trigeminal ganglion by retrograde transport. This is followed by a replication phase in this ganglion, after which the virus enters a state of latency, despite the control of the replication by the immune responses [7, 8]. Liedtke et al. found through examination of the trigeminal ganglia from 109 human cadavers at forensic post-mortems that the age-group specific prevalence of HSV neuronal latency increases from 18% in 0–20 years to 100% in persons older than 60 [9]. The virus can then periodically reactivate, particularly after a trigger like immunosuppression (e.g., medically induced, AIDS), surgery, UV or cold exposure. The risk of reactivation is correlated with the latency viral load in the trigeminal ganglion. The latent virus produces progeny that is transported to the periphery of the nerve. The virus then replicates in the cells near the nerve endings. This can lead to asymptomatic viral shedding or symptomatic recurrent infection.
The cornea is the primary site of HSV-1 infection, known as Herpes Simplex Keratitis (HSK) (Fig. 8.1).

Schematic for HSV infection and the different sites of herpes ocular infection
HSV-1 corneal infection presents in diverse forms (frequency of appearance reported in the epidemiologic study of Labetoulle et al. [10]): dendritic keratitis (56.3%), geographic keratitis (9.8%), stromal keratitis (29.5%), punctate keratitis (4%), corneal ulcer (1.8%), limbal lesion (1.1%), and other forms (2.5%) including endothelitis, central corneal oedema, paralimbal interstitial keratitis, and neuroparalytic keratitis. These infections often lead to an irreversible opacification or scarring of the cornea, with scars that result in severe visual impairment. The only treatment to recover functional vision in these eyes is corneal transplantation. However, herpetic recurrences significantly increase the risk of graft failure [5, 11]. In cases of end-stage disease or when the risk for graft failure is too high, an artificial cornea or keratoprosthesis (KPros) becomes an alternative option. This final treatment, however, is overshadowed by severe and frequent side effects, including glaucoma, retroprosthetic membrane formation, infection and dehiscence.
Latency and subsequent reactivation in response to a large number of documented but mechanistically still largely ill-defined triggers make treatment difficult. Nucleoside analogues, including acyclovir (ACV), valacyclovir (VAC) and ganciclovir (GCV), are highly effective at terminating HSV-1 DNA elongation, which inhibits viral proliferation [12]. However, because they target the viral DNA polymerase, they can only act when the virus is in its replicative form. Furthermore, their poor solubility and lack of penetrance across the cornea prevent sufficient accumulation in the anterior segment to efficiently treat ocular HSV-1 infection, which explains the use of oral formulations [13]. But oral administration remains a suboptimal method for treating a local eye infection as a high systemic concentration is needed to yield sufficient drug concentration in the cornea. However, this increases the risk of systemic side effects. Oral administration does not result in rapid accumulation of sufficient levels of nucleoside analogues for the treatment of the local infection. Systemic exposure of the virus to nucleoside analogues also potentially induces the development of resistance to the antiviral drug. Ideally, new ocular formulations of ACV that address the lack of solubility and corneal penetration would provide a more direct and effective method of treating herpes keratitis [3].
The eye is a particularly challenging field for pharmacology. Several barriers restrict the entry of drug molecules: the tear film, the cornea with its different layers, the conjunctiva, the sclera, as well as the blood-aqueous and blood-retina barriers. Topical administration (eye-drops) is the primary approach for drug delivery in ophthalmology, being used in more than 90% of cases. The topical treatment is released in the tear film and penetrates through the corneal layers to the deeper tissues [1,2,3]. Ideally, a drug penetrating the cornea will accumulate in the aqueous humor, which in turn acts as a reservoir for additional drug release and treatment of the intraocular tissues. Despite its significant barrier function (less than 5% of the total administered doses reaches the aqueous humor [4, 14]), the cornea remains the primary site of absorption for intraocular drugs. To facilitate release kinetics, nanoparticles (NPs) must have the capacity to release the drug into aqueous solutions, penetrate the corneal epithelial, stromal and endothelial layers and sustain drug release once in the aqueous humor.
We summarize below the development of encapsulated formulations of nucleoside analogues for ocular administration from the first liposomal formulations developed in the late 1980s to current developments in nanoparticle formulations designed to prevent HSV-1 infections.
8.3 History of Encapsulation of Acyclovir in the Eye
8.3.1 Liposomes
Norley et al. developed the first encapsulated form of ACV in 1986 [15, 16]. Using a liposomal formulation targeted with HSV glycoprotein D (gD), they were able to demonstrate binding to human corneal epithelial cells (HCECs) infected with HSV-1 and inhibition of replication at a concentration of 10 µg/mL of ACV.
In 1999, Fresta et al. examined the impact of phospholipid type and liposomal charge on the efficacy of ACV delivery [17]. Their results demonstrated that negatively charged dipalmitoylphosphatidylcholine-cholesterol-dimethyldioctadecyl glycerol bromide liposomes synthesized by reverse-phase evaporation were the most effective, with the highest ACV content. In rabbits, this formulation increased the ACV concentration in the aqueous humor from 2.11 ± 1.80 µg/mL with soluble ACV to 88.95 ± 12.31 µg/mL. The effect of liposomal charge was re-examined by Law et al. in 2000 [18]. Liposomes composed of phosphatidylcholine, cholesterol, stearylamine or dicetylphosphate containing ACV were synthesized with either a positive or negative charge. In rabbits, the concentration of ACV was highest in the corneal tissue and aqueous humor after the administration of positively charged liposomes. These results indicated that lipid carriers are effective at resolving the solubility issues associated with nucleoside analogues penetrating the cornea and paved the way for future lipid nanoparticle formulations.
8.3.2 Chitosan
In 1997, Genta et al. developed the first chitosan microparticle formulation of ACV [19]. The particles were 25 µm in diameter. In an in vivo test in rabbits, the microsphere (MS) formulation was able to increase the concentration of ACV in the aqueous humor in comparison to ACV alone, and it doubled the length of time that ACV was present in the aqueous humor.
8.4 Ocular Nanoparticles for Nucleoside Analogues
8.4.1 Polymer-Based Nanoparticles
8.4.1.1 Poly-(D,L-Lactic Acid)
Poly-(D,L-Lactic Acid) (PLA) NPs were developed by Giannavola et al. to test the effect of poly(ethylene glycol) (PEG) coating on ACV release [20]. NPs containing PLA of three molecular weights (LMW—16,000; MMW—109,000; HMW—209,00) were synthesized. Drug release assays demonstrated that lower molecular weight PLA resulted in greater ACV release. The LMW-PLA nanoparticles coated with PEG resulted in improved drug release over the PLA-only NPs. In normal rabbit eyes, the PEG-PLA encapsulated ACV NPs resulted in the highest concentration of ACV (424 ± 24 µg/mL/min) released into the aqueous humor. This effect was decreased to the level of PLA-only NPs with the pre-administration of the topical irritant N-acetylcysteine (221 ± 40 µg/mL/min), which suggests that the formulation may be less effective in an inflamed eye. All NP formulations had greater aqueous humor concentration than soluble ACV.
8.4.1.2 Poly-(Lactic-Co-glycolic Acid)
Jwala et al, developed poly-(lactic-co-glycolic acid) (PLGA) NPs based on ACV prodrugs [21]. PLGA is a US FDA-approved biodegradable polymer that has been used extensively in micro-capsule formulations [22]. The prodrugs were synthesized with the addition of L- and D- valine. LL-ACV and LD-ACV showed the highest degradation rate constants of log[[pro-drug concentration]] versus time in cultured cells and rabbit ocular tissues, and were used in subsequent experiments. PLGA NPs with varying LA:GA ratios (100:0, 75:25, 65:35, 50:50) were tested for encapsulation of the ACV prodrugs. LL-ACV PLGA 75:25 NPs and LD-ACV PLGA 65:35 NPs showed the greatest entrapment efficiency (EE) and drug content. In vitro, the LL-ACV NPs showed a greater controlled release time than the LD-ACV NPs. The release of both prodrugs was extended after incorporation within PLGA-PEG-PLGA thermosensitive gels.
Yang et al. applied the methodology of Jwala et al. to PLGA NPs with GCV prodrugs [23, 24]. They first developed NPs containing LL-GCV, LD-GCV, and DL-GCV with sizes ranging from 116 to 143 nm in diameter and EE of 38.7–45.3% [23]. As these formulations showed successful biphasic drug release and did not demonstrate cytotoxicity, they synthesized a fluorescein isothiocyanate (FITC) PLGA conjugate to visualize cellular uptake of the NPs into human corneal epithelial cells (HCECs) [24]. The FITC-PLGA-NPs ranged from 115 to 145 nm with zeta-potentials around −13 mV. Fluorescent microscopy demonstrated the successful uptake of the NPs by the HCECs indicating that they could successfully penetrate the corneal epithelium.
8.4.1.3 Chitosan
Calderon et al. compared chitosan microspheres (CMS) and nanoparticle (CNPs) of ACV encapsulated in chitosan [25]. Chitosan NPs crosslinked with tripolyphosphate (TPP) were prepared using ionotropic gelation. The CNPs were 240.0 ± 62.4 nm in diameter, while the CMS was 6.2 ± 0.5 µm. The CMS had a higher EE (75.46%) than the CNPs (15.73%), but both had positive zeta-potentials. Both spheres and particles released approximately 75% of the encapsulated drug over 25 h in solution for a total of 430 and 80 µg for CMPs and CNPs, respectively. Due to the comparatively lower ACV release from the CNP, only the CMS were assayed for slug mucosal irritation, which is used to simulate ocular irritation, where they showed a moderate level of irritation.
8.4.1.4 Polycaprolactone
Ramyadevi et al. developed polycaprolactone (PCL) NPs encased in a thermosensitive Pluronic F-127 and Carbopol gel [26]. The NPs were synthesized using solvent evaporation using varying amounts of PCL and concentrations of surfactant. The resulting NPs were 172–329 nm in diameter with an EE of 28–58%. Drug release from the gel varied from 7 to 32% of the encapsulated drug. The formulation PCL3 composed of 100 mg PCL and 0.5% Pluronic F-127 with a size of 201.4 nm and an EE of 64% was used to develop the thermosensitive gel formulations as it showed the greatest sustained release and no cytotoxicity. The gel formulation with the best performance was synthesized using 15% w/v Pluronic F-127 and 0.3% Carbopol 940. The gel showed a linear drug release of 4% for the first hour and 14% at 8 h, following Korsmeyer-Peppas kinetics.
8.4.1.5 Solid Prodrug Nanoparticles
Stella et al. created a novel prodrug by conjugating ACV to 1,10,2-trisnorsqualenoic acid resulting in 40-trisnorsqualenoylacyclovir (SQ-ACV) [27]. The SQ-ACV NPs were formed by spontaneous precipitation resulting in NPs ranging from 113 to 254 nm in diameter with a polydispersity index <0.1 and a negative zeta-potential. After topical administration to rabbits, the SQ-ACVNPs had a greater concentration of total ACV in the tear fluid (11 ± 2.06 mg/ml min−1 vs. 2.97 ± 0.34 mg/ml min−1) than soluble ACV. The ACV concentration was higher for the SQACV NPs at 1 h (0.294 ± 0.119 µg/mL) than soluble ACV (0.098 ± 0.051 µg/mL).
8.4.2 Lipid Nanoparticles
8.4.2.1 Nanostructured Lipid Carriers
Seyfoddin et al. compared nanostructured lipid carriers (NLC) to solid lipid nanoparticles (SLN) in 2013 [28]. SLNs were synthesized as microemulsions from varying amounts of Compritol alone, in combination with stearic acid or cithrol GMS using the hot oil-in-water method in the presence of Tween-20. The Compitrol SLNs were optimized using different types and concentrations of surfactants, resulting in a 400 mg Compritol SLN containing 40 mg ACV synthesized using 2% Tween-40 (SLN3). SLN3 was 465.86 ± 7.15 nm in size with a negative zeta-potential and a polydispersity index (PDI) of 0.530 ± 0.05. The NLCs were synthesized using 400 mg of Comptriol containing Capryol-90 and/or Lauroglycol-90 in the presence of Tween-40. The resulting NPs were 319–656 nm in size with negative zeta-potential and PDI from 0.265 to 0.752. In vitro drug release using a diffusion chamber was similar for SLN3, NLC4, and NLC5. The penetration of the NPs was measured using ex vivo bovine corneas in a Franz-type diffusion chamber. NLC5 showed the greatest ACV concentration through the cornea, followed by NLC4. Interestingly, SLN3 showed lower corneal penetration than free ACV.
8.4.2.2 Solid Lipid Nanoparticles
Valacyclovir (VAC) is a prodrug that is converted into ACV within the patient’s body. SLNs containing VAC were developed by Kumar et al. [29]. The SLNs were generated using emulsification/evaporation of stearic acid or tristearin, using poloxamer and sodium taurocholate as surfactants, in different rations of lipid:surfactant. The SLNs produced had a particle size from 202.5 to 431.7 nm. The PDI was 0.252 ± 0.06 and 0.598 ± 0.03 with all particles other than SLN-2 demonstrating a PDI > 0.5. In vitro drug release assays showed a total release of 60% of the encapsulated drug. The kinetics showed an initial burst release followed by sustained release. Ex vivo corneal penetration assays demonstrated that SLN-4 and SLN-6 had the highest corneal flux, while SLN-3, -4 and -6 had the highest corneal retention (Fig. 8.2). Irritation assays were conducted for SLN-6 using a Hen’s Egg Test Chorio Allantoic Membrane (HET-CAM) assay with 0.1 N sodium hydroxide as a positive control. SLN-6 did not result in lysis, hemorrhage or coagulation, with the same HET-CAM score as the saline control. Histopathology of SLN-6 in an ex vivo goat corneal model showed morphology similar to untreated controls.

Reproduced with permission from Kumar et al. [29]
Ex vivo corneal permeation of VAC formulations (**P ≤ 0.01; statistically very significant as analyzed by Dunnett multiple comparison test).
8.5 Experimental Ocular Nanoparticles for Delivery of Nucleoside Alternatives
More recently, cationic peptides have been developed as alternatives to nucleosides for inhibiting HSV infections. Amongst the various peptides examined are the innate cationic peptides such as cathelicidins and defensins produced by the cornea [30]. Humans produce only one cathelicidin, of which a 37-amino acid sequence was found to have anti-viral activity against HSV. This peptide, known as LL-37, is highly cationic and has been shown to have anti-HSV-1 activity. LL-37, its derivatives and other innate peptides are being proposed as new treatments and therefore also require optimal delivery to the cornea. Other cationic peptides with anti-HSV activity include the entry blocker (EB) peptide, which consists of the fibroblast growth factor 4 (FGF4) signal sequence with an additional N-terminal RRKK sequence [31].
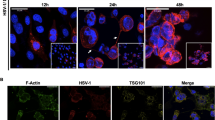
Lee et al. used silica dioxide NPs to encapsulate LL-37 and EB for delivery to corneal epithelial cells, using an immortalized line of human epithelial cell as a model line [32]. The LL-37 containing NPs were further encapsulated within a collagen- 2-methacryloyloxyethyl phosphorylcholine (collagen-MPC) hydrogel. LL-37 released from the NPs in implants blocked HSV-1 infection of HCECs more effectively than EB. Controls consisted of free LL-37 within the hydrogels, where the peptides diffused out rapidly and had little effect. The released LL-37 blocked HSV-1 by interfering with viral binding (Fig. 8.3a, c). However, in pre-infected HCECs, LL-37 delayed but did not prevent viral spreading nor clear viruses from the infected cells (Fig. 8.3b, d).

Reproduced with permission from Lee C-J et al. [32]
a The prophylactic effects of collagen-MPC hydrogels that incorporated LL-37 SiNPs, free LL-37, or nothing in HSV-1 infection (MOI ¼ 0.1). Red immunofluorescence indicated localization of HSV-1 infected cells; cell nuclei are stained with DAPI (blue). b Effects of LL-37 release on virus spreading, after inoculation with HSV-1 (MOI ¼ 0.05). c Quantification of HSV-1 titers from (a). d Quantification of HSV-1 titers corresponding (b).
8.6 Ocular Nanoparticles for Herpes Vaccine Delivery
There is currently no available vaccine against HSV infection [33]. A number of studies published from 1980s to recent times showed that vaccine development attempts were largely unsuccessful. In 2011, Hu et al. used iron oxide (Fe3O4) NPs to improve the delivery of a DNA vaccine against HSV-1 [34]. The DNA vaccine consists of a pRSC vector with cDNA for both HSV glycoprotein D (gD) and interleukin-21 (IL-21). This vector was co-administered with Fe3O4 NPs coated with glutamic acid in a mouse HSV-1 challenge model. The mice were immunized with pRSC-gD-IL-21 with and without NPs, pRSC-gD, and pRSC three times at two weeks intervals. Quantification of the vaccine efficacy was conducted by measuring secreted IgA, serum neutralizing antibody, IL-4, and IFN-γ. For all measures, DNA + NPs improved the expression of markers over the DNA vaccine alone. Three weeks after the final immunization, the mice were infected with HSV-1 and followed for 15 days. Figure 8.4 shows the clinical appearance of the cornea at follow up and histopathology of the cornea and conjunctiva. The vaccine-containing NP group had minimal corneal opacity and normal corneal and conjunctival structure, but the vaccine only or vector only groups showed corneal opacity and structural damage.

Adapted from Figures 5 and 6, Hu et al. [34]
Corneal and conjunctival response to HSV- 1 challenge after vaccination.
8.7 Outlook and Future Perspective
A wide variety of nanoparticles have been used to improve the delivery of nucleoside analogues to the cornea; however, these investigations are limited to early in vivo drug delivery and toxicology studies. There is considerable variability in the relationship between NP size and polydispersity index (PDI), zeta-potential, and entrapment efficiency (EE). While size versus PDI, and size versus EE can be loosely approximated with a linear relationship, where increased size results in increased polydispersity and EE, there is no obvious relationship between size and zeta-potential (Fig. 8.5). Chitosan is distinct from the other polymeric NPs in both zeta-potential and morphology. Where most polymeric NPs have a negative zeta-potential (Fig. 8.5) and spherical shape, the CNPs have a positive zeta-potential and red blood cell-like appearance. The SLNs have a smooth surface that is more reminiscent of a polymeric NPs, while the NLCs have highly textured surfaces. Like most polymeric NPs, the NLCs have a negative zeta-potential but their larger size and unique surfaces make their increased penetration across the cornea (in comparison to SLNs) more likely to be dependent on their hydrophobic properties than their particle size. This suggests that smaller NPs are not always superior drug carriers unless they have equally effective particle design.

Summary of nucleoside analogue nanoparticle properties. a Nanoparticle size (nm). b Size (nm) versus polydispersity index (PDI). c Nanoparticle size versus zeta-potential (mV). d Nanoparticle size versus entrapment efficiency (%). Original data obtained from cited literature in this chapter
It is unfortunate that researchers have used highly variable methodologies and units to compare NP diffusion across the cornea, making it unclear which NPs are the most effective drug delivery vehicles. The drug release studies have used a more consistent methodology, with several formulas showing a high total release, but this measure is not indicative of overall NP performance in vivo. It is also problematic that researchers developing NP formulations assume the efficacy of nucleoside analogues without conducting tests on their antiviral efficacy in vitro or in vivo, i.e., NPs are only the delivery vehicle. Resistance to ACV is a rising problem, as strains with thymidine kinase mutations have been identified that are insensitive to ACV in immunocompetent clinical patients [35] and hence, problems have arisen for patients who are prescribed prophylactic systemic doses of ACV or VAC. It is critical that NP formulations are studied in clinical strains of HSV-1, as well as reference strains, to ensure their efficacy.
The development of NPs that can deliver high doses directly to the cornea goes hand-in-hand with the development of effective anti-HSV agents. Alternatives to nucleosides, such as the innate host defense peptide LL-37, have been studied as potential anti-HSV-1 agents. NP formulations with alternative mechanisms of action will be important for treatment, as well as prophylaxis. The vaccine utilizing Fe3O4 NPs as delivery vehicles is probably the most exciting development described in this article, because they represent a novel approach for the prevention of ocular HSV-1. This study was also a comprehensive one that included an in vivo HSV-1 challenge model that demonstrated the efficacy of the DNA vaccine and Fe3O4 NP combination. Overall, NPs developed for the treatment of ocular HSV-1 remain an emerging field with a great deal of potential for drug development. The standardization of NP assessment has improved the ability to compare the properties of the NP formulations, but a standard in vivo model for corneal diffusion and efficacy would greatly improve the ability of researchers to determine if new formulations are improvements over existing NPs.
References
Agarwal P, Rupenthal ID. In vitro and ex vivo corneal penetration and absorption models. Drug Deliv Transl Res. 2016;6(6):634–47.
Reimondez-Troitino S, Csaba N, Alonso MJ, de la Fuente M. Nanotherapies for the treatment of ocular diseases. Eur J Pharm Biopharm. 2015;95(Pt B):279–93.
Sharma A, Taniguchi J. Review: Emerging strategies for antimicrobial drug delivery to the ocular surface: implications for infectious keratitis. Ocul Surf. 2017;15(4):670–9.
Gaudana R, Jwala J, Boddu SH, Mitra AK. Recent perspectives in ocular drug delivery. Pharm Res. 2009;26(5):1197–216.
Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol. 2012;57(5):448–62.
Liesegang TJ, Melton LJ III, Daly PJ, Ilstrup DM. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol. 1950;107(8):1155–9.
Kennedy DP, Clement C, Arceneaux RL, Bhattacharjee PS, Huq TS, Hill JM. Ocular herpes simplex virus type 1: is the cornea a reservoir for viral latency or a fast pit stop? Cornea. 2011;30(3):251–9.
Al-Dujaili LJ, Clerkin PP, Clement C, McFerrin HE, Bhattacharjee PS, Varnell ED, Kaufman HE, Hill JM. Ocular herpes simplex virus: how are latency, reactivation, recurrent disease and therapy interrelated? Future Microbiol. 2011;6(8):877–907.
Liedtke W, Opalka B, Zimmermann CW, Lignitz E. Age distribution of latent herpes simplex virus 1 and varicella-zoster virus genome in human nervous tissue. J Neurol Sci. 1993;116(1):6–11.
Labetoulle M, Auquier P, Conrad H, Crochard A, Daniloski M, Bouee S, El Hasnaoui A, Colin J. Incidence of herpes simplex virus keratitis in France. Ophthalmology. 2005;112(5):888–95.
Fry M, Aravena C, Yu F, Kattan J, Aldave AJ. Long-term outcomes of the Boston type I keratoprosthesis in eyes with previous herpes simplex virus keratitis. Br J Ophthalmol. 2018;102(1):48–53.
De Clercq E, Holy A. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat Rev Drug Discov. 2005;4(11):928–40.
Sanchez-Lopez E, Espina M, Doktorovova S, Souto EB, Garcia ML. Lipid nanoparticles (SLN, NLC): overcoming the anatomical and physiological barriers of the eye—Part II—Ocular drug-loaded lipid nanoparticles. Eur J Pharm Biopharm. 2017;110:58–69.
Hughes PM, Olejnik O, Chang-Lin JE, Wilson CG. Topical and systemic drug delivery to the posterior segments. Adv Drug Deliv Rev. 2005;57(14):2010–32.
Norley SG, Huang L, Rouse BT. Targeting of drug loaded immunoliposomes to herpes simplex virus infected corneal cells: an effective means of inhibiting virus replication in vitro. J Immunol. 1986;136(2):681–5.
Norley SG, Sendele D, Huang L, Rouse BT. Inhibition of herpes simplex virus replication in the mouse cornea by drug containing immunoliposomes. Invest Ophthalmol Vis Sci. 1987;28(3):591–5.
Fresta M, Panico AM, Bucolo C, Giannavola C, Puglisi G. Characterization and in-vivo ocular absorption of liposome-encapsulated acyclovir. J Pharm Pharmacol. 1999;51(5):565–76.
Law SL, Huang KJ, Chiang CH. Acyclovir-containing liposomes for potential ocular delivery. Corneal penetration and absorption. J Control Release. 2000;63(1–2):135–40.
Genta I, Conti B, Perugini P, Pavanetto F, Spadaro A, Puglisi G. Bioadhesive microspheres for ophthalmic administration of acyclovir. J Pharm Pharmacol. 1997;49(8):737–42.
Giannavola C, Bucolo C, Maltese A, Paolino D, Vandelli MA, Puglisi G, Lee VH, Fresta M. Influence of preparation conditions on acyclovir-loaded poly-d, l-lactic acid nanospheres and effect of PEG coating on ocular drug bioavailability. Pharm Res. 2003;20(4):584–90.
Jwala J, Boddu SH, Shah S, Sirimulla S, Pal D, Mitra AK. Ocular sustained release nanoparticles containing stereoisomeric dipeptide prodrugs of acyclovir. J Ocul Pharmacol Ther. 2011;27(2):163–72.
Makadia HK, Siegel SJ. Poly Lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–97.
Yang X, Shah SJ, Wang Z, Agrahari V, Pal D, Mitra AK. Nanoparticle-based topical ophthalmic formulation for sustained release of stereoisomeric dipeptide prodrugs of ganciclovir. Drug Deliv. 2016;23(7):2399–409.
Yang X, Sheng Y, Ray A, Shah SJ, Trinh HM, Pal D, Mitra AK. Uptake and bioconversion of stereoisomeric dipeptide prodrugs of ganciclovir by nanoparticulate carriers in corneal epithelial cells. Drug Deliv. 2016;23(7):2532–40.
Calderon L, Harris R, Cordoba-Diaz M, Elorza M, Elorza B, Lenoir J, Adriaens E, Remon JP, Heras A, Cordoba-Diaz D. Nano and microparticulate chitosan-based systems for antiviral topical delivery. Eur J Pharm Sci. 2013;48(1–2):216–22.
Ramyadevi D, Sandhya P. Dual sustained release delivery system for multiple route therapy of an antiviral drug. Drug Deliv. 2014;21(4):276–92.
Stella B, Arpicco S, Rocco F, Burgalassi S, Nicosia N, Tampucci S, Chetoni P, Cattel L. Nonpolymeric nanoassemblies for ocular administration of acyclovir: pharmacokinetic evaluation in rabbits. Eur J Pharm Biopharm. 2012;80(1):39–45.
Seyfoddin A, Al-Kassas R. Development of solid lipid nanoparticles and nanostructured lipid carriers for improving ocular delivery of acyclovir. Drug Dev Ind Pharm. 2013;39(4):508–19.
Kumar R, Sinha VR. Lipid nanocarrier: an efficient approach towards ocular delivery of hydrophilic drug (valacyclovir). AAPS Pharm Sci Tech. 2017;18(3):884–94.
Gordon YJ, Huang LC, Romanowski EG, Yates KA, Proske RJ, McDermott AM. Human cathelicidin (LL-37), a multifunctional peptide, is expressed by ocular surface epithelia and has potent antibacterial and antiviral activity. Curr Eye Res. 2005;30(5):385–94.
Bultmann H, Busse JS, Brandt CR. Modified FGF4 signal peptide inhibits entry of herpes simplex virus type 1. J Virol. 2001;75(6):2634–45.
Lee CJ, Buznyk O, Kuffova L, Rajendran V, Forrester JV, Phopase J, Islam MM, Skog M, Ahlqvist J, Griffith M. Cathelicidin LL-37 and HSV-1 corneal infection: peptide versus gene therapy. Transl Vis Sci Technol. 2014;3(3):4.
Johnston C, Gottlieb SL, Wald A. Status of vaccine research and development of vaccines for herpes simplex virus. Vaccine. 2016;34(26):2948–52.
Hu K, Dou J, Yu F, He X, Yuan X, Wang Y, Liu C, Gu N. An ocular mucosal administration of nanoparticles containing DNA vaccine pRSC-gD-IL-21 confers protection against mucosal challenge with herpes simplex virus type 1 in mice. Vaccine. 2011;29(7):1455–62.
Piret J, Boivin G. Antiviral resistance in herpes simplex virus and varicella-zoster virus infections: diagnosis and management. Curr Opin Infect Dis. 2016;29(6):654–62.
Acknowledgements
The authors have no conflict of interest. FS is supported by a FRQNT PhD studentship. FXG is supported by a (Berthe Fouassier) France Foundation studentship. MG holds the Caroline Durand Foundation Research Chair for Cellular Therapy of Diseases of the Eye, Université de Montréal.
Disclosure
All authors have read and approved the final version.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Simpson, F., Gueriot, FX., Brunette, I., Griffith, M. (2019). Nanoparticles for Cornea Therapeutic Applications: Treating Herpes Simplex Viral Infections. In: Alarcon, E., Ahumada, M. (eds) Nanoengineering Materials for Biomedical Uses. Springer, Cham. https://doi.org/10.1007/978-3-030-31261-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-31261-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-31260-2
Online ISBN: 978-3-030-31261-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)