
Abstract
Acute respiratory distress syndrome (ARDS) is a high-mortality syndrome that develops following an infection or trauma, leading to a dysregulated inflammatory response in the lung that can cause tissue remodeling, pulmonary dysfunction, and death [1]. ARDS is characterized by acute respiratory failure caused by an increase of fluids in the alveolar space. The breakdown of the immune response increases the permeability of the epithelial–endothelial barrier. This results in an increased filtration of protein-rich fluid from the vascular system into the alveolar spaces, with a subsequent lung edema and a decrease in the ability of gas exchange [2].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) is a high-mortality syndrome that develops following an infection or trauma, leading to a dysregulated inflammatory response in the lung that can cause tissue remodeling, pulmonary dysfunction, and death [1]. ARDS is characterized by acute respiratory failure caused by an increase of fluids in the alveolar space. The breakdown of the immune response increases the permeability of the epithelial–endothelial barrier. This results in an increased filtration of protein-rich fluid from the vascular system into the alveolar spaces, with a subsequent lung edema and a decrease in the ability of gas exchange [2].
ARDS develops as a localized lung response that could have multiple origins including pneumonia, systemic sepsis, major surgery, or multiple trauma. Patients with ARDS have an acute onset of symptoms like severe chest pain and a decrease in their pulmonary function associated to pulmonary infiltrates in the X-ray, indicative of pulmonary edema. These symptoms usually appear after the first week of the injury [3].
ARDS accounts for more than 10% of intensive care unit (ICU) admissions worldwide and has a mortality rate of about 40% [4]. ARDS results in a diminished quality of life and lung function; and survivors often have long-term neuromuscular, cognitive, and psychological symptoms. Additionally, long hospitalization, ICU, and increased use of health care services after hospital discharge have enormous socioeconomical cost [5, 6].
Despite several decades of research, there is no disease-modifying therapy for ARDS. Because the mechanisms driving lung injury are complex and diverse, pharmacological treatments often fail, suggesting that targeting a single mediator or pathway is not enough to achieve therapeutic effects. As an alternative, after their use in several preclinical models of ARDS, cell-based therapies with mesenchymal stem/stromal cells (MSCs) are promising, as they can target multiple cellular and extracellular impairments associated with ARDS simultaneously [7].
2 Pathology of ARDS
There are multiple initiation agents for ARDS, from microorganism invasion (bacteria or viral) to mechanical stressors, that result from mechanical ventilation [8]. Beyond the initial trigger, aging, like in any other lung diseases, there is an impaired capacity of the lung to recover. In several animal models, when we compared aged with young, there was an increase in the morbidity and mortality as a consequence of an altered inflammatory response [9,10,11].
Immune activation results in the release of proinflammatory cytokines and chemokines with a primary influx of neutrophils into the alveolar space, leading to the release of metalloproteinases (MMP), myeloperoxidases (MPO), collagenase, and the generation of reactive oxygen species (ROS) [12]. This activates and attracts macrophages and lymphocytes to the site of injury with a sequent release of inflammatory cytokines including IL-6, IL-8, IL-1β, and TNFα [13].
In physiological situations, anti-inflammatory mediators act to limit the inflammatory cascade and control the tissue damage. ROS serves multiple functions such as killing phagocytosed microorganism, or the removal of cell debris and signaling. However, high and persistent levels of ROS, MMPs, and MPOs cause tissue necrosis, injury, and destruction [14]. The controlling feedback mechanism seems to be impaired during the onset of sepsis, which leads to a persistent inflammatory response after the resolution of the initial insult [15].
Along with the production of proinflammatory cytokines, other factors are secreted such as endothelin-1 (EDN1), angiotensin-2 (AGT-II), and phospholipase A2 (PLA2), which increase vascular permeability [16, 17]. The disruption of epithelial–endothelial barriers leads to a disruption of the alveolar clearance and production of surfactant [18]. Edema accumulates in the alveoli though the increase permeability of epithelial and endothelial barriers and the decrease in alveolar fluid clearance. Measurements of the protein content in BAL fluid provide an estimate of the alveolar changes and could be used as an indicator of the prognosis of the patient [19]. There is an initial phase of fluid accumulation followed by a proliferation phase, which is characterized by the increase of type II alveolar cells (AEC-II), fibroblast, myofibroblast, and matrix deposition. If the inflammation persists, then there is a disorganized repair that could lead to fibrosis [20, 21].
3 MSCs in the Treatment of ARDS
MSCs constitute a great option for the treatment of ARDS because of their ability to regulate the immune response, enhance the phagocytic clearance of bacteria and secrete factors that regulate the capillary–alveolar barrier. Additionally, MSCs appeared immune-privileged with low levels of type I HLA antigens in their surface, which allows them to escape from the patient’s immune response [22, 23]. This represents an important advantage that allows the therapeutic use of allogenic MSC [24].
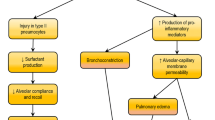
MSC could exert their effect through cell contact-dependent mechanisms and by the release of soluble factors. This chapter summarizes all the processes by which MSCs could be beneficial in the treatment of ARDS (Fig. 11.1) and the preclinical animal models of lung injury in which these have been tested.

Schematic representation of the mechanisms driving alveolar damage during ARDS and the different pathways targeted by soluble factors produced by MSCs and the consequences on lung repair
4 Mechanism of Action of MSC
Tracking MSC engraftment has been possible using MSC expressing GFP or labeled with PKH allowing the localization of the cells by fluorescent imaging [25,26,27]. There is considerable variety in the number of cells homing into the lungs. We have demonstrated an early retention in the lung of MSCs in large animal models of acute lung injury independent of the way cells were delivered [28]. However, it is well accepted that by using colocalization of surface markers, infused MSCs that localize in the lung are not differentiating into any other cell type, including alveolar or airway epithelial cells, fibroblasts, or endothelial cells. The actual knowledge suggest that most of the presence of MSCs observed in the LPS model of lung injury reflects a transient process [29] and that the protective effect seen with MSC therapy does not require MSC differentiation into any cell type [14]. Engineered MSC to overexpress important genes for the epithelial lineage like angiopoietin [30], ROR2 [31] or β-catenin [32] showed an enhanced differentiation capacity in vitro, but there was no engraftment and differentiation in in vivo models of LPS-induced acute lung injury.
Despite the initial interest in the multipotent properties of the MSCs, engraftment and differentiation in the lung [14, 33], their beneficial effect more likely derives from their capacity to be recruited by the sites of injury, interact with the host cells, and secrete soluble factors known as the secretome [34]. The MSC secretome is dynamic and could vary depending on the MSC source or the type of lung injury. It includes an extended array of bioactive molecules comprising cytokines, chemokines, growth factors, angiogenic factors, and microvesicles [2]. Table 11.1 summarizes the described molecules of the secretome and their main functions.
4.1 MSCs Reduce Endothelial and Epithelial Permeability
The integrity of the microvascular endothelium is essential to avoid the influx of protein-rich fluid from the circulation to the alveolar space. In addition to the generated edema, this permeabilization comes with inflammatory cytokines and cells which may further aggravate the ability of the endothelium to reduce edema [33]. Several MSC-secreted factors have the capacity of regulate the alveolar microvasculature reducing its permeability.
Angiopoietin-1 (Ang-1) is a ligand for endothelial Tie2 receptor activating the NF-κB pathway to prevent the formation, on AEC-II, of actin stress fibers and preserve the localization of claudin-18 [73]. Ang-1 has been shown to promote endothelial survival, reduce endothelial permeability, and inhibit leukocyte interactions by modifying cell adhesion molecules and cell junctions [36, 74, 75]. Engineered MSCs overexpressing Ang-1 further reduce protein content, albumin, and immune cells in BAL [35, 76, 77]. MSC culture in proinflammatory conditions enhances their capacity to produce Ang-1. Also, when Ang-1 was blocked with siRNA, MSCs no longer prevented epithelial permeabilization [73].
Keratinocyte growth factor (KGF), also known as fibroblast growth factor 7 (FGF7), is a critical factor for epithelial repair and stimulating epithelial cell proliferation [41]. Animal models of ALI, such as by administration of α-naphthylthiourea [37, 52] P. aeruginosa [78] or ventilator-induced lung injury [39], have shown the ability of KGF to reduce alveolar edema. Engineered MSCs overexpressing KGF improve microvascular permeability, reduce proinflammatory cytokines (IL-1β and TNFα), and increase the anti-inflammatory response (IL-10). The underlying mechanism is not completely understood but may be attributed to the promotion of AEC-II cells and the production of surfactant [41].
Hepatocyte growth factor (HGF) was found to preserve integrity of pulmonary endothelium though the inhibition of Rho GTPase, prevent the actin stress fiber formation, and preserve the gaps between endothelial cells [39, 42]. Sphingosine-1-phosphate (S1P) enhances the resistance of the endothelial barrier. This mechanism seems to be dependent of the capacity to inhibit leukocyte permeability as well as reduce levels of TNFα [47, 48]. Vascular endothelial growth factor (VEGF) has been described to reduce lung permeability, protect endothelium from apoptosis, control inflammation, and facilitate VE-cadherin recovery. Knockdown of VEGF in MSCs confirms these activities as these cells present reduced therapeutic activity on ALI models [51].
4.2 Alveolar Fluid Clearance, AFC
Alveolar fluid clearance (AFC) is the capacity of the lung epithelium to remove alveolar fluid during pulmonary edema. This process is mediated through sodium channels, aquaporin, and sodium-potassium adenosine triphosphatase Na-K-ATPase [43]. Many conditions such as high-volume mechanical ventilation, live bacteria, or proinflammatory cytokines can reduce AFC [79, 80]. The compromised capacity of AFC is used as prognostic value to determine morbidity and mortality [80]. Both AEC-I and AEC-II are involved in AFC during pulmonary edema. AEC-I has the highest permeability to water, potentially through aquaporin, which supports its role in ion transport [81,82,83]. Na-K-ATPase is expressed in both AEC-I and AEC-II and has a critical role in alveolar fluid reabsorption, where the sodium transport is followed by outflux of water in an isosmolar manner [84, 85].
Studies with intra-bronchia administration of LPS in ex-vivo perfused lungs revealed a marked decrease in AFC, and mechanisms dependent on the blood presence suggest that immune cell are required for the injurious effect of the LPS. MSCs, or their conditioned media (CM), normalized AFC in a KGF-dependent manner as siRNA for KGF reduced the therapeutic effect of the MSCs [86]. KGF increases fluid transported across the alveolar epithelium through increased trafficking of sodium transport proteins to the cell surface [87, 88]. KGF also has important functions reducing the expression of aquaporin 5 and avoiding the transdifferentiation of AEC-II toward AEC-I [38]. Another group described that KGF effects on AFC are mediated through an increased rate of epithelial repair, cell adherence, and migration [40].
Epithelial growth factor (EGF) stimulates the proliferation of epithelial cells, increases sodium channels and Na-K-APase function in in vitro alveolar epithelial cultures [54]. An in vivo rat model with aerosolized EGF showed an increase in active sodium transport, Na-K-ATPase activity, and lung fluid clearance [53]. Rat models with instilled TGF-β increase alveolar fluid clearance in a time-dependent and dose-dependent manner. This increase of alveolar clearance is driven by an increased activity of the sodium channels [55].
4.3 Immune Response Regulation
MSCs constitutively do not exert their immunomodulatory properties but instead have to be “primed” by inflammatory mediators [89]. In the context of ARDS, the acute inflammation drives this “priming” of the MSCs, activating their immunomodulatory properties [24]. MSCs were found to protect tissue damage from extraordinary inflammation by downregulate the expression of proinflammatory cytokines as IL-6, IL-8 IL-1β, IFNɣ, and TNFα and through the production of anti-inflammatory cytokines such a IL-4, IL-10, or IL-13 [71, 79, 90, 91]. Additionally, MSCs produce IL-1rN which is a cytokine that competes with IL-1β receptor binding, thus inhibiting its effects [62].
Several in vitro and in vivo studies have demonstrated that MSCs have several effects over the innate immune system. They can influence the maturation of the dendritic cells (DC) [92], increase the phagocytic capacity of monocytes [93,94,95], neutrophils [96,97,98], and modify macrophage toward immunomodulatory M2 phenotype [99, 100]. MSCs secrete prostaglandin E2 that then stimulates macrophages and monocytes to produce the anti-inflammatory cytokine IL-10 [101]. IL-10 has been reported to inhibit the rolling, adhesion, and transepithelial migration of neutrophils, thus resulting critical downstream in the MSC therapeutic effects [13, 102]. MSCs produce KGF that induces the secretion of granulocyte–macrophage colony-stimulating factor that increases alveolar macrophage phagocytosis [103]. KGF also inhibits macrophage apoptosis through a downregulation of the β-catenin pathway that increases Bcl-2 and decreases BAX and caspase-3 [104].
MSCs also can modulate the adaptive immune response. MSCs suppress B cell proliferation and terminal differentiation [105], suppress T cell proliferation [102, 106, 107], induce a switch from Th1 proinflammatory response to Th2 response, and finally increase the number of T regulatory cells [108]. MSCs secrete idoleamine 2,3-dioxygenase (IDO) upon stimulation with IFNɣ. IDO activity results in tryptophan depletion and kynurenine production that inhibits T cell proliferation [109]. TNF-stimulated gene protein 6 (KGF6) is another contributor to the immunomodulatory effects of the MSCs. In lung injury models, MSCs upregulate KGF6 that binds IL-8, blocking its function [66]. Blockage of KGF6 by siRNA completely reverses MSCs’ anti-inflammatory properties [65].
It is important to note that while MSCs secrete an extensive range of anti-inflammatory cytokines, they also have the capacity to produce several proinflammatory cytokines such as IL-6 and IL-8. The production of these cytokines has been associated with poor outcomes in ARDS patients [110, 111]; however, there is new evidence that suggests a role of these cytokines in the MSC therapeutic effect. IL-6 is usually implicated in proinflammatory responses, but apparently, it could have promiscuous functions [112,113,114]. It is not clear how IL-6 induces therapeutic effects, but its inhibition in lung injury models diminished the MSC therapeutic effect [115]. The role of IL-8 is not clear but there is evidence suggesting that IL-8 promotes the production of VEGF that promotes angiogenic effects [50].
4.4 Enhancement of Bacterial Clearance
MSCs attenuate bacterial sepsis directly by antimicrobial peptides secretion and by enhancement of macrophage phagocytosis [116]. MSCs stimulated by the presence of bacteria released β-defensin 2 (BD2) [67], LL-37 [68, 69] and lipocalin-2 [71]. BD2 is upregulated in the presence of bacteria through TLR2 and TLR4 pathways, inhibiting bacterial growth in vitro [67]. LL-37 is a cathelicidin peptide with antibacterial, antifungal, and antiviral properties [68, 117]. Lipocalin-2 is known to act on mucosal cells during pulmonary infection to regulate chemokines such as CXCL9 to reduce inflammation in front bacterial infections [70, 72, 117].
CM from stimulated MSC was found to contain high levels of antimicrobial peptides and to inhibit bacterial growth. However, when treated with inhibitors of these peptides, the effect was abolished. Animal models of lung injury show a reduced bacterial growth in the lung homogenates on the animals treated with MSCs. This effect was decreased if a neutralizing antibody against LL-37 was administered with the MSCs [92]. Similar observations were done blocking the TLR2 and TLR4 and thus the production of BD2 [67].
4.5 Transfer and Rescue of Mitochondrial Function in Target Cells
MSCs could contribute to rescue mitochondrial function in epithelial cells containing nonfunctional mitochondria [118, 119]. In vitro imaging reveals the formation of connexin-43-gap junctions between MSC and alveolar epithelial cells, allowing the transport of mitochondria to the LPS-injured epithelium. This resulted in increased ATP levels that rescued the surfactant secretion by AEC-II leading to reduce alveolar permeability and reduced mortality. An endotoxin lung injury model treated with instilled MSCs confirmed this mitochondrial transfer to the injured epithelium, leading to the restoration of its function. This rescue mechanism was abrogated when MSCs with dysfunctional mitochondria were instilled, supporting its role in restoring the lung epithelium [25]. As described in more detail in Chap. 4, another group found that the mitochondria were transferred through nanotubes, which was upregulated by the Rho-GTPase Miro1 [120].
Mitochondrial transfer to macrophages has also been described in vitro through the formation of tunneling nanotubes. Mitochondrial transfer enhanced macrophage oxidative phosphorylation and phagocytosis. The blockage of the tunneling nanotubes formation completely abolished the MSC effect on macrophages [121, 122].
5 Preclinical Models of MSCs in ARDS
5.1 Animal Models of ARDS Treated with MSCs
The therapeutic efficacy of MSCs in the treatment of lung injury has been demonstrated in numerous animal models. Most of the studies have been performed in mouse or rat models and large animal models using swine [145, 146] or sheep [147, 148]. Acute lung injury is mainly induced through infection with endotoxin or live bacteria, and via cecal ligation and puncture driving sepsis. Rat models are preferred for the study of ventilator induced lung injury. There is lot of variability in dose, timing, route of administration, and source of MSCs, but all the studies found an improvement of the injury. Table 11.2 shows the main findings in animal models of ARDS using MSCs.
Animal models have shown the potential of using MSCs as a treatment therapy for ARDS. A recent study compared the therapeutic potential and the distribution of the MSCs with different administration routes in a large animal model of acute lung injury. Endobronchial and intravenous administration of MSCs have similar rates of lung retention at 5 h post administration and recovery of arterial oxygenation in equal extent [28].
Most of the studies performed showed histological improvements upon MSC treatment with reduction of the inflammation, edema, and lung injury with reduced collagen deposition [29, 65, 69, 71, 94, 108, 123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138]. At the endothelial–epithelial level, the alveolar barrier is better maintained in the animals treated with MSCs, with lower levels of protein in the BAL [65, 123, 129, 135,136,137, 139, 140]. In vivo, MSCs display immunomodulatory properties modifying the lung immune response. At molecular levels, MSC are able to shift the cytokine profile from the pro-inflammatory status in ARDS toward a more anti-inflammatory one [124, 128, 131, 133, 135, 136, 138, 140, 141]. At the cellular level, MSCs promote a T regulatory lymphocyte response [108], increasing phagocytic activity of macrophages [69, 101, 127, 129] and reducing the neutrophil numbers [8, 125, 127, 131, 136]. All these modifications of the immune response control the inflammation and preserve the lung tissue integrity. Also, MSC perform antimicrobial properties promoting phagocytosis and bacterial killing by the secretion of LL37 peptide [69] or lipocalin [71, 94]. In addition, MSC have showed antioxidant effects in vivo, preserving plasma levels of cysteine and glutathione redox state after endotoxin administration [126, 149].
5.1.1 Development of Genetically Modified MSCs
MSCs’ capacity to migrate to the sites of inflammation makes them an attractive factor for gene-based therapy [150]. Overexpression of specific genes in MSCs can be used to enhance their therapeutic effects [25]. Most of the approaches are directed to increase their regenerative potential or their immunomodulatory capacity.
Administered MSCs overexpressing KGF improve pulmonary microvascular permeability, reducing the lung injury in the model of LPS-induced lung injury [41]. VEGF-overexpressing MSCs protect the endothelium from apoptosis, reducing the permeability and edema [51]. The expression of HGF makes MSC to protect adherent junctions, VE-cadherin of the epithelium and reduce apoptosis, preserving lung architecture [46]. β-Catenin-overexpressing MSC improves alveolar permeability, promoting the differentiation of lung precursors into AEC-II [32]. Engineered MSCs targeting the immune system have been modified to express anti-inflammatory molecules or to alter the expression of chemokines. MSCs overexpressing prostaglandin receptor [63], IL-10 [151], or IL-1rl1 [152] induce a strong shift in the cytokine profile toward an anti-inflammatory response, reducing lung inflammation and edema. Finally, MSCs with altered expression of chemokine receptors show a reduced accumulation in the lung of inflammatory cells and mediators [153].
All the previous studies described the effects of the MSC in acute lung injury when administered at the initiation of the injury, but administration of MSCs once the injury is already performed did not show significant therapeutic effects [154]. MSCs overexpressing the microRNAs let-7d (antifibrotic) or miR-154 (profibrotic) were administered 7 days after bleomycin instillation. Mice treated with let-7d expressing MSCs were found to recover quicker from the initial weight loss, while those untreated or treated with miR-154 had the lowest survival rate, although no fibrotic differences were found in the lung tissue. The effect was more immunomodulatory, altering the pattern of cytokines and the leukocyte infiltration [155].
5.1.2 Pretreatment of the MSCs to Enhance Their Potential
MSCs’ immunomodulatory effects vary depending on the immune microenvironment [156, 157]. The therapeutic effect achieved varies greatly between different studies and one possible explanation could be the resting status of the MSC. The immunosuppressive function of the MSC is enhanced by the presence of pro-inflammatory cytokines such as IFNɣ, TNFα, IL-1α, or IL-1β [158], while the presence of anti-inflammatory cytokines like IL-10 abrogates their suppressive effect and even induces the production of pro-inflammatory cytokines [159,160,161].
An early study assessing the role of the microenvironment in the treatment of acute lung injury with MSCs showed the importance of the expression of TLR4 on the MSCs. TLR4 expression of MSCs is essential for the release of prostaglandin E2 upon activation by LPS or TNFα. MSCs lacking the genes for TLR4 or their downstream mediators are unable to produce prostaglandin E2 and activate macrophages to produce IL-10 in a mouse model of acute lung injury [101].
Rojas et al. showed that MSC treatment with serum from ARDS patients containing pro-inflammatory cytokines increases their immunomodulatory function with higher production of IL-10 and IL-1β receptor agonist. Pretreated MSCs have an enhanced protective capacity, reducing lung injury, edema, and accumulation of pro-inflammatory cells and cytokines in a mouse model of acute lung injury [130].
The time point at which MSCs are administered seems to be crucial. Only MSCs injected at the time of inducing the lung injury have shown to have therapeutic effect, even though they could lose all the antifibrotic properties when administered long after the injury [154]. One of the possible mechanisms responsible for the shift in their function is the microenvironment of the injured lung. In a mouse model of irradiated lungs, TGF-β expression was incremented in the damaged lungs, and in vitro the cytokines released by the lung injured cells inhibited the differentiation of the MSC into epithelial cells [162]. A recent study of acute lung injury by acid instillation revealed that treatment with MSC worsens the acid effects, driving a fibrotic process. This process seemed to be mediated by the presence of IL-6 and fibronectin in the microenvironment, which could be driving a senescence-associated phenotype on the MSCs. The fibrotic effect was reversed when the MSC were engineered to overexpress IL-10 or HGF neutralizing both in vivo in mouse models of HCl and ventilator-induced lung injury [163].
Another factor that could influence the therapeutic effect achieved is the aging status of the MSCs. MSCs isolated from aged individuals have reduced expression of cytokine and chemokine receptors that impair their migration and activation, failing to reduce inflammation in a mouse model of acute lung injury [164].
5.1.3 Alternative Sources of MSCs Than Bone Marrow
Traditionally, MSCs obtained from bone marrow have been used in most of the preclinical and translational studies. However, there is a growing number of studies using adipose and umbilical cord blood (UC-MSC) as potentially more plentiful sources. MSCs from different sources exhibit different receptors and immunomodulatory properties, which may cause differential therapeutic effect on ARDS, but overall, animal models have shown beneficial outcomes [165,166,167,168,169,170,171].
UC-MSCs have shown higher proliferative rates and lower expression of senescence markers than BM-MSCs which could reflect a more multipotent capacity [165]. UC-MSCs have great immunomodulatory capacity in mouse models of acute lung injury, inducing a shift toward a regulatory immune response with increasing levels of IL-10 and phagocytic macrophages [60, 64]. At the regenerative level, UC-MSCs attenuate lung injury, preserving vascular permeability and protecting from apoptosis [56, 172, 173].
Adipose MSCs have the advantage of their availability and easy isolation. Adipose MSCs have shown immunomodulatory properties in animal models of acute lung injury reducing inflammation, leukocyte infiltrate and modifying the cytokine profile toward an anti-inflammatory response [59, 174,175,176]. Recently, other studies have appeared using stem cells from pulp and periodontal ligament or menstrual stem cells, showing improvement of alveolar epithelial permeability and reducing pro-inflammatory cells and cytokines in LPS-induced ARDS models [177, 178].
5.1.4 Use of Soluble Factors Generated by MSCs
Microvesicles (MV) are small circular membrane fragments that are shed from the cell surface or released from the endosomal membrane and play an important role in cell communication. This communication system has emerged early during evolution and serves as template in the further development of intercellular interaction mechanisms. MV can transfer specific genes, miRNAs, or small organelles, including mitochondria, from the MSC to the injured target cell through the connexin-43 gap junction channels.
MSC have demonstrated to have therapeutic effects in in vivo animal models but little is known about their long-term side effects, including the possibility of becoming tumorigenic. Given that the MSCs’ therapeutic effect depends on the release of soluble factors, the in vivo use of MV represents an alternative and safer approach. Analysis of the RNA of microvesicles derived from MSCs revealed mRNAs associated with transcription, proliferation, immune cell regulation, and microRNiAs as well.
Intra-tracheal instillation of MSC-derived MV reduces edema and alveolar protein levels in mouse models of acute lung injury [179]. MV also show anti-inflammatory properties reducing neutrophils and inflammatory macrophages. A partial therapeutic effect of MSC MV depends on KGF, as KGF siRNA pretreatment of MSCs partially eliminated their therapeutic benefits [180]. Further studies show that the MV-dependent activity is mediated by CD44 receptors, promoting internalization of MV into monocytes, resulting in a decreased expression of inflammatory cytokines [181].
5.2 Ex-Vivo Lung Perfusion (EVLP) Models
Ex-vivo lung perfusion (EVLP) offers a unique opportunity for in situ testing of the effects of MSCs. EVLP was originally developed as a treatment to increase the number of lungs available for transplantation. Nowadays it is not only a way to improve unacceptable lungs for transplantation, but it also represents an excellent research tool as a preclinical model for in situ testing [182]. Several groups have developed models of acute lung injury [86, 103, 183] in which the effect of MSCs was studied [86, 103, 184, 185].
A study conducted on pigs looked at the optimal route and dose for the MSCs. Intravascular delivery of MSC showed better outcomes that intratracheal administration and the optimal dose was of 5 × 106 MSC per kilogram of animal [184]. Early studies using MSCs in human lung grafts were focused on testing their capacity to restore the AFC in lungs that were unsuitable for transplantation. Intravenous administration of MSC restored AFC in injured lungs in a mechanism dependent on KGF [185].
A couple of groups have developed models of ARDS to study the disease in human lung grafts. Treatment with MSC reduces edema, improves AFC, and restores epithelial barrier permeability in ex vivo perfuse human lungs injured with E. coli endotoxin. The beneficial effect on endotoxin injured lung was almost abolished when the MSCs or their conditioned media (CM) was treated with KGF siRNA [86]. The treatment with MSC in a model of pneumonia using live bacteria restored AFC, reduced inflammation, and increased bacterial killing through increased macrophage phagocytosis. KGF was shown as one of the main factors protecting monocytes from apoptosis and increasing bacterial clearance [103]. A recent study used MSC in combination with extracorporeal membrane oxygenation (ECMO) after inoculation of E. coli endotoxin in a sheep model. The combination of the MSC and the ECMO treatment showed better histopathology changes with less inflammation [186].
The use of MSCs microvesicles has a positive result as well in the treatment of injured lung grafts. Human MSCs derived microvesicles have been used to recover lungs rejected for lung transplantation by increasing AFC and improving airway and hemodynamic parameters [187]. In another study, treatment of ex-vivo lung perfusion model of bacterial pneumonia with MSC microvesicles increased AFC, reducing protein permeability and bacterial load [188].
In summary, since the description of the protective effect of the administration of MSCs to mice with induced ARDS [29, 123], several research groups have confirmed this observation on small and large animal models and more recently in the EVLP in which ARDS is induced in human lungs. The proposed mechanisms by which MSCs can induce protection are multiple, some described in the present chapter. However, there is consensus that engraftment in the lung and differentiation into lung cells does not occurs. More recently, data generated from approved clinical trials, conducted by several academic institutions, demonstrated that the use of MSCs is safe and, in some cases, with demonstrated protection in patients with ARDS. There is still more research needed to determine the appropriate source of MSCs, route and time of administration, and the generation of modified MSCs in which the protective potential is enhanced.
References
Rawal G, Yadav S, Kumar R. Acute respiratory distress syndrome: an update and review. J Transl Int Med. 2018;6(2):74–7.
Antebi B, Mohammadipoor A, Batchinsky AI, Cancio LC. The promise of mesenchymal stem cell therapy for acute respiratory distress syndrome. J Trauma Acute Care Surg. 2018;84(1):183–91.
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L, Slutsky AS, Force ADT. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526–33.
Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353(16):1685–93.
Angus DC, Clermont G, Linde-Zwirble WT, Musthafa AA, Dremsizov TT, Lidicker J, Lave JR, NO-06 Investigators. Healthcare costs and long-term outcomes after acute respiratory distress syndrome: a phase III trial of inhaled nitric oxide. Crit Care Med. 2006;34(12):2883–90.
Walkey AJ, Summer R, Ho V, Alkana P. Acute respiratory distress syndrome: epidemiology and management approaches. Clin Epidemiol. 2012;4:159–69.
Huleihel L, Levine M, Rojas M. The potential of cell-based therapy in lung diseases. Expert Opin Biol Ther. 2013;13(10):1429–40.
Pati S, Gerber MH, Menge TD, Wataha KA, Zhao Y, Baumgartner JA, Zhao J, Letourneau PA, Huby MP, Baer LA, Salsbury JR, Kozar RA, Wade CE, Walker PA, Dash PK, Cox CS, Doursout MF, Holcomb JB. Bone marrow derived mesenchymal stem cells inhibit inflammation and preserve vascular endothelial integrity in the lungs after hemorrhagic shock. PLoS One. 2011;6(9):e25171.
Schouten LR, Schultz MJ, van Kaam AH, Juffermans NP, Bos AP, Wösten-van Asperen RM. Association between maturation and aging and pulmonary responses in animal models of lung injury: a systematic review. Anesthesiology. 2015;123(2):389–408.
Kling KM, Lopez-Rodriguez E, Pfarrer C, Mühlfeld C, Brandenberger C. Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L1–L12.
Schouten LR, Helmerhorst HJ, Wagenaar GT, Haltenhof T, Lutter R, Roelofs JJ, van Woensel JB, van Kaam AH, Bos AP, Schultz MJ, Walther T, Wösten-van Asperen RM. Age-dependent changes in the pulmonary renin-angiotensin system are associated with severity of lung injury in a model of acute lung injury in rats. Crit Care Med. 2016;44(12):e1226–35.
Martin TR, Pistorese BP, Hudson LD, Maunder RJ. The function of lung and blood neutrophils in patients with the adult respiratory distress syndrome. Implications for the pathogenesis of lung infections. Am Rev Respir Dis. 1991;144(2):254–62.
Fanelli V, Vlachou A, Ghannadian S, Simonetti U, Slutsky AS, Zhang H. Acute respiratory distress syndrome: new definition, current and future therapeutic options. J Thorac Dis. 2013;5(3):326–34.
Cardenes N, Caceres E, Romagnoli M, Rojas M. Mesenchymal stem cells: a promising therapy for the acute respiratory distress syndrome. Respiration. 2013;85(4):267–78.
Johnson CL, Soeder Y, Dahlke MH. Concise review: Mesenchymal stromal cell-based approaches for the treatment of acute respiratory distress and sepsis syndromes. Stem Cells Transl Med. 2017;6(4):1141–51.
Proudfoot AG, Hind M, Griffiths MJ. Biomarkers of acute lung injury: worth their salt? BMC Med. 2011;9:132.
Tzouvelekis A, Pneumatikos I, Bouros D. Serum biomarkers in acute respiratory distress syndrome an ailing prognosticator. Respir Res. 2005;6:62.
Berger G, Guetta J, Klorin G, Badarneh R, Braun E, Brod V, Saleh NA, Katz A, Bitterman H, Azzam ZS. Sepsis impairs alveolar epithelial function by downregulating Na-K-ATPase pump. Am J Physiol Lung Cell Mol Physiol. 2011;301(1):L23–30.
Gotts JE, Matthay MA. Mesenchymal stem cells and acute lung injury. Crit Care Clin. 2011;27(3):719–33.
Vadász I, Weiss CH, Sznajder JI. Ubiquitination and proteolysis in acute lung injury. Chest. 2012;141(3):763–71.
de Luis Cabezón N, Sánchez Castro I, Bengoetxea Uriarte UX, Rodrigo Casanova MP, García Peña JM, Aguilera Celorrio L. Acute respiratory distress syndrome: a review of the Berlin definition. Rev Esp Anestesiol Reanim. 2014;61(6):319–27.
Tu Z, Li Q, Bu H, Lin F. Mesenchymal stem cells inhibit complement activation by secreting factor H. Stem Cells Dev. 2010;19(11):1803–9.
Salazar KD, Lankford SM, Brody AR. Mesenchymal stem cells produce Wnt isoforms and TGF-beta1 that mediate proliferation and procollagen expression by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L1002–11.
Masterson C, Jerkic M, Curley GF, Laffey JG. Mesenchymal stromal cell therapies: potential and pitfalls for ARDS. Minerva Anestesiol. 2015;81(2):179–94.
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med. 2012;18(5):759–65.
Yamada M, Kubo H, Kobayashi S, Ishizawa K, Numasaki M, Ueda S, Suzuki T, Sasaki H. Bone marrow-derived progenitor cells are important for lung repair after lipopolysaccharide-induced lung injury. J Immunol. 2004;172(2):1266–72.
Rojas M, Xu J, Woods CR, Mora AL, Spears W, Roman J, Brigham KL. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol. 2005;33(2):145–52.
Cardenes N, Aranda-Valderrama P, Carney JP, Sellares Torres J, Alvarez D, Kocydirim E, Wolfram Smith JA, Ting AE, Lagazzi L, Yu Z, Mason S, Santos E, Lopresti BJ, Rojas M. Cell therapy for ARDS: efficacy of endobronchial versus intravenous administration and biodistribution of MAPCs in a large animal model. BMJ Open Respir Res. 2019;6(1):e000308.
Xu J, Woods CR, Mora AL, Joodi R, Brigham KL, Iyer S, Rojas M. Prevention of endotoxin-induced systemic response by bone marrow-derived mesenchymal stem cells in mice. Am J Physiol Lung Cell Mol Physiol. 2007;293(1):L131–41.
Xu J, Qu J, Cao L, Sai Y, Chen C, He L, Yu L. Mesenchymal stem cell-based angiopoietin-1 gene therapy for acute lung injury induced by lipopolysaccharide in mice. J Pathol. 2008;214(4):472–81.
Cai SX, Liu AR, Chen S, He HL, Chen QH, Xu JY, Pan C, Yang Y, Guo FM, Huang YZ, Liu L, Qiu HB. The orphan receptor tyrosine kinase ROR2 facilitates MSCs to repair lung injury in ARDS animal model. Cell Transplant. 2016;25(8):1561–74.
Cai SX, Liu AR, Chen S, He HL, Chen QH, Xu JY, Pan C, Yang Y, Guo FM, Huang YZ, Liu L, Qiu HB. Activation of Wnt/β-catenin signalling promotes mesenchymal stem cells to repair injured alveolar epithelium induced by lipopolysaccharide in mice. Stem Cell Res Ther. 2015;6:65.
Lee JW, Fang X, Krasnodembskaya A, Howard JP, Matthay MA. Concise review: mesenchymal stem cells for acute lung injury: role of paracrine soluble factors. Stem Cells. 2011;29(6):913–9.
Vizoso FJ, Eiro N, Cid S, Schneider J, Perez-Fernandez R. Mesenchymal stem cell Secretome: toward cell-free therapeutic strategies in regenerative medicine. Int J Mol Sci. 2017;18(9):E1852.
Papapetropoulos A, García-Cardeña G, Dengler TJ, Maisonpierre PC, Yancopoulos GD, Sessa WC. Direct actions of angiopoietin-1 on human endothelium: evidence for network stabilization, cell survival, and interaction with other angiogenic growth factors. Lab Investig. 1999;79(2):213–23.
Pizurki L, Zhou Z, Glynos K, Roussos C, Papapetropoulos A. Angiopoietin-1 inhibits endothelial permeability, neutrophil adherence and IL-8 production. Br J Pharmacol. 2003;139(2):329–36.
Guery BP, Mason CM, Dobard EP, Beaucaire G, Summer WR, Nelson S. Keratinocyte growth factor increases transalveolar sodium reabsorption in normal and injured rat lungs. Am J Respir Crit Care Med. 1997;155(5):1777–84.
Borok Z, Lubman RL, Danto SI, Zhang XL, Zabski SM, King LS, Lee DM, Agre P, Crandall ED. Keratinocyte growth factor modulates alveolar epithelial cell phenotype in vitro: expression of aquaporin 5. Am J Respir Cell Mol Biol. 1998;18(4):554–61.
Viget NB, Guery BP, Ader F, Nevière R, Alfandari S, Creuzy C, Roussel-Delvallez M, Foucher C, Mason CM, Beaucaire G, Pittet JF. Keratinocyte growth factor protects against Pseudomonas aeruginosa-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1199–209.
Atabai K, Ishigaki M, Geiser T, Ueki I, Matthay MA, Ware LB. Keratinocyte growth factor can enhance alveolar epithelial repair by nonmitogenic mechanisms. Am J Physiol Lung Cell Mol Physiol. 2002;283(1):L163–9.
Chen J, Li C, Gao X, Liang Z, Yu L, Li Y, Xiao X, Chen L. Keratinocyte growth factor gene delivery via mesenchymal stem cells protects against lipopolysaccharide-induced acute lung injury in mice. PLoS One. 2013;8(12):e83303.
Birukova AA, Alekseeva E, Mikaelyan A, Birukov KG. HGF attenuates thrombin-induced endothelial permeability by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the rho pathway. FASEB J. 2007;21(11):2776–86.
Singleton PA, Salgia R, Moreno-Vinasco L, Moitra J, Sammani S, Mirzapoiazova T, Garcia JG. CD44 regulates hepatocyte growth factor-mediated vascular integrity. Role of c-Met, Tiam1/Rac1, dynamin 2, and cortactin. J Biol Chem. 2007;282(42):30643–57.
Dong LH, Jiang YY, Liu YJ, Cui S, Xia CC, Qu C, Jiang X, Qu YQ, Chang PY, Liu F. The anti-fibrotic effects of mesenchymal stem cells on irradiated lungs via stimulating endogenous secretion of HGF and PGE2. Sci Rep. 2015;5:8713.
Seedorf G, Metoxen AJ, Rock R, Markham N, Ryan S, Vu T, Abman SH. Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1098–110.
Hu S, Li J, Xu X, Liu A, He H, Xu J, Chen Q, Liu S, Liu L, Qiu H, Yang Y. The hepatocyte growth factor-expressing character is required for mesenchymal stem cells to protect the lung injured by lipopolysaccharide in vivo. Stem Cell Res Ther. 2016;7(1):66.
Liu H, Zhang Z, Li P, Yuan X, Zheng J, Liu J, Bai C, Niu W. Regulation of S1P receptors and sphingosine kinases expression in acute pulmonary endothelial cell injury. PeerJ. 2016;4:e2712.
Ebenezer DL, Fu P, Suryadevara V, Zhao Y, Natarajan V. Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: role of S1P lyase. Adv Biol Regul. 2017;63:156–66.
Potapova IA, Gaudette GR, Brink PR, Robinson RB, Rosen MR, Cohen IS, Doronin SV. Mesenchymal stem cells support migration, extracellular matrix invasion, proliferation, and survival of endothelial cells in vitro. Stem Cells. 2007;25(7):1761–8.
Hou Y, Ryu CH, Jun JA, Kim SM, Jeong CH, Jeun SS. IL-8 enhances the angiogenic potential of human bone marrow mesenchymal stem cells by increasing vascular endothelial growth factor. Cell Biol Int. 2014;38(9):1050–9.
Yang Y, Hu S, Xu X, Li J, Liu A, Han J, Liu S, Liu L, Qiu H. The vascular endothelial growth factors-expressing character of Mesenchymal stem cells plays a positive role in treatment of acute lung injury in vivo. Mediat Inflamm. 2016;2016:2347938.
Murakami M, Nguyen LT, Zhuang ZW, Zhang ZW, Moodie KL, Carmeliet P, Stan RV, Simons M. The FGF system has a key role in regulating vascular integrity. J Clin Invest. 2008;118(10):3355–66.
Sznajder JI, Ridge KM, Yeates DB, Ilekis J, Olivera W. Epidermal growth factor increases lung liquid clearance in rat lungs. J Appl Physiol (1985). 1998;85(3):1004–10.
Danto SI, Borok Z, Zhang XL, Lopez MZ, Patel P, Crandall ED, Lubman RL. Mechanisms of EGF-induced stimulation of sodium reabsorption by alveolar epithelial cells. Am J Phys. 1998;275(1 Pt 1):C82–92.
Folkesson HG, Pittet JF, Nitenberg G, Matthay MA. Transforming growth factor-alpha increases alveolar liquid clearance in anesthetized ventilated rats. Am J Phys. 1996;271(2 Pt 1):L236–44.
Chang YS, Oh W, Choi SJ, Sung DK, Kim SY, Choi EY, Kang S, Jin HJ, Yang YS, Park WS. Human umbilical cord blood-derived mesenchymal stem cells attenuate hyperoxia-induced lung injury in neonatal rats. Cell Transplant. 2009;18(8):869–86.
Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, Chen FF, Jiang XD. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10:106.
Miyagawa I, Nakayamada S, Nakano K, Yamagata K, Sakata K, Yamaoka K, Tanaka Y. Induction of regulatory T cells and its regulation with insulin-like growth Factor/insulin-like growth Factor binding Protein-4 by human Mesenchymal stem cells. J Immunol. 2017;199(5):1616–25.
Gonzalez-Rey E, Anderson P, González MA, Rico L, Büscher D, Delgado M. Human adult stem cells derived from adipose tissue protect against experimental colitis and sepsis. Gut. 2009;58(7):929–39.
Sun J, Han ZB, Liao W, Yang SG, Yang Z, Yu J, Meng L, Wu R, Han ZC. Intrapulmonary delivery of human umbilical cord mesenchymal stem cells attenuates acute lung injury by expanding CD4+CD25+ Forkhead Boxp3 (FOXP3)+ regulatory T cells and balancing anti- and pro-inflammatory factors. Cell Physiol Biochem. 2011;27(5):587–96.
Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, Stappenbeck TS, Patterson GA, Pierce RA, Brody SL, Holtzman MJ. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. 2013;123(9):3967–82.
Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med. 2001;163(6):1384–8.
Han J, Lu X, Zou L, Xu X, Qiu H. E-Prostanoid 2 receptor overexpression promotes Mesenchymal stem cell attenuated lung injury. Hum Gene Ther. 2016;27(8):621–30.
Zhu H, Xiong Y, Xia Y, Zhang R, Tian D, Wang T, Dai J, Wang L, Yao H, Jiang H, Yang K, Liu E, Shi Y, Fu Z, Gao L, Zou L. Therapeutic effects of human umbilical cord-derived Mesenchymal stem cells in acute lung injury mice. Sci Rep. 2017;7:39889.
Danchuk S, Ylostalo JH, Hossain F, Sorge R, Ramsey A, Bonvillain RW, Lasky JA, Bunnell BA, Welsh DA, Prockop DJ, Sullivan DE. Human multipotent stromal cells attenuate lipopolysaccharide-induced acute lung injury in mice via secretion of tumor necrosis factor-α-induced protein 6. Stem Cell Res Ther. 2011;2(3):27.
Dyer DP, Thomson JM, Hermant A, Jowitt TA, Handel TM, Proudfoot AE, Day AJ, Milner CM. TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J Immunol. 2014;192(5):2177–85.
Sung DK, Chang YS, Sung SI, Yoo HS, Ahn SY, Park WS. Antibacterial effect of mesenchymal stem cells against Escherichia coli is mediated by secretion of beta- defensin- 2 via toll- like receptor 4 signalling. Cell Microbiol. 2016;18(3):424–36.
Krasnodembskaya A, Song Y, Fang X, Gupta N, Serikov V, Lee JW, Matthay MA. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem Cells. 2010;28(12):2229–38.
Devaney J, Horie S, Masterson C, Elliman S, Barry F, O'Brien T, Curley GF, O'Toole D, Laffey JG. Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax. 2015;70(7):625–35.
Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432(7019):917–21.
Gupta N, Krasnodembskaya A, Kapetanaki M, Mouded M, Tan X, Serikov V, Matthay MA. Mesenchymal stem cells enhance survival and bacterial clearance in murine Escherichia coli pneumonia. Thorax. 2012;67(6):533–9.
Guglani L, Gopal R, Rangel-Moreno J, Junecko BF, Lin Y, Berger T, Mak TW, Alcorn JF, Randall TD, Reinhart TA, Chan YR, Khader SA. Lipocalin 2 regulates inflammation during pulmonary mycobacterial infections. PLoS One. 2012;7(11):e50052.
Fang X, Neyrinck AP, Matthay MA, Lee JW. Allogeneic human mesenchymal stem cells restore epithelial protein permeability in cultured human alveolar type II cells by secretion of angiopoietin-1. J Biol Chem. 2010;285(34):26211–22.
Kwak HJ, So JN, Lee SJ, Kim I, Koh GY. Angiopoietin-1 is an apoptosis survival factor for endothelial cells. FEBS Lett. 1999;448(2-3):249–53.
Gamble JR, Drew J, Trezise L, Underwood A, Parsons M, Kasminkas L, Rudge J, Yancopoulos G, Vadas MA. Angiopoietin-1 is an antipermeability and anti-inflammatory agent in vitro and targets cell junctions. Circ Res. 2000;87(7):603–7.
Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science. 1999;286(5449):2511–4.
Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nat Med. 2000;6(4):460–3.
Mason CM, Guery BP, Summer WR, Nelson S. Keratinocyte growth factor attenuates lung leak induced by alpha-naphthylthiourea in rats. Crit Care Med. 1996;24(6):925–31.
Rogol PR. Intact epithelial barrier function is critical for resolution of alveolar edema in humans. Am Rev Respir Dis. 1991;144(2):468.
Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163(6):1376–83.
Ridge KM, Olivera WG, Saldias F, Azzam Z, Horowitz S, Rutschman DH, Dumasius V, Factor P, Sznajder JI. Alveolar type 1 cells express the alpha2 Na,K-ATPase, which contributes to lung liquid clearance. Circ Res. 2003;92(4):453–60.
Borok Z, Liebler JM, Lubman RL, Foster MJ, Zhou B, Li X, Zabski SM, Kim KJ, Crandall ED. Na transport proteins are expressed by rat alveolar epithelial type I cells. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L599–608.
Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG. Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci U S A. 2002;99(4):1966–71.
Ridge KM, Rutschman DH, Factor P, Katz AI, Bertorello AM, Sznajder JL. Differential expression of Na-K-ATPase isoforms in rat alveolar epithelial cells. Am J Phys. 1997;273(1 Pt 1):L246–55.
Schneeberger EE, McCarthy KM. Cytochemical localization of Na+-K+-ATPase in rat type II pneumocytes. J Appl Physiol (1985). 1986;60(5):1584–9.
Lee JW, Fang X, Gupta N, Serikov V, Matthay MA. Allogeneic human mesenchymal stem cells for treatment of E. coli endotoxin-induced acute lung injury in the ex vivo perfused human lung. Proc Natl Acad Sci U S A. 2009;106(38):16357–62.
Dada LA, Sznajder JI. Mechanisms of pulmonary edema clearance during acute hypoxemic respiratory failure: role of the Na, K-ATPase. Crit Care Med. 2003;31(4 Suppl):S248–52.
Planès C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277(49):47318–24.
Prasanna SJ, Gopalakrishnan D, Shankar SR, Vasandan AB. Pro-inflammatory cytokines, IFNgamma and TNFalpha, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS One. 2010;5(2):e9016.
Hayes M, Curley G, Ansari B, Laffey JG. Clinical review: stem cell therapies for acute lung injury/acute respiratory distress syndrome—hope or hype? Crit Care. 2012;16(2):205.
Liang ZX, Sun JP, Wang P, Tian Q, Yang Z, Chen LA. Bone marrow-derived mesenchymal stem cells protect rats from endotoxin-induced acute lung injury. Chin Med J. 2011;124(17):2715–22.
Yi T, Song SU. Immunomodulatory properties of mesenchymal stem cells and their therapeutic applications. Arch Pharm Res. 2012;35(2):213–21.
Barbash IM, Chouraqui P, Baron J, Feinberg MS, Etzion S, Tessone A, Miller L, Guetta E, Zipori D, Kedes LH, Kloner RA, Leor J. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: feasibility, cell migration, and body distribution. Circulation. 2003;108(7):863–8.
Krasnodembskaya A, Samarani G, Song Y, Zhuo H, Su X, Lee JW, Gupta N, Petrini M, Matthay MA. Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am J Physiol Lung Cell Mol Physiol. 2012;302(10):L1003–13.
Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815–22.
Hall SR, Tsoyi K, Ith B, Padera RF, Lederer JA, Wang Z, Liu X, Perrella MA. Mesenchymal stromal cells improve survival during sepsis in the absence of heme oxygenase-1: the importance of neutrophils. Stem Cells. 2013;31(2):397–407.
Iyer SS, Rojas M. Anti-inflammatory effects of mesenchymal stem cells: novel concept for future therapies. Expert Opin Biol Ther. 2008;8(5):569–81.
Zhou X, Dai Q, Huang X. Neutrophils in acute lung injury. Front Biosci (Landmark Ed). 2012;17:2278–83.
Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47(4):417–26.
Wakayama H, Hashimoto N, Matsushita Y, Matsubara K, Yamamoto N, Hasegawa Y, Ueda M, Yamamoto A. Factors secreted from dental pulp stem cells show multifaceted benefits for treating acute lung injury in mice. Cytotherapy. 2015;17(8):1119–29.
Nemeth K, Leelahavanichkul A, Yuen PS, Mayer B, Parmelee A, Doi K, Robey PG, Leelahavanichkul K, Koller BH, Brown JM, Hu X, Jelinek I, Star RA, Mezey E. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat Med. 2009;15(1):42–9.
Jarvinen L, Badri L, Wettlaufer S, Ohtsuka T, Standiford TJ, Toews GB, Pinsky DJ, Peters-Golden M, Lama VN. Lung resident mesenchymal stem cells isolated from human lung allografts inhibit T cell proliferation via a soluble mediator. J Immunol. 2008;181(6):4389–96.
Lee JW, Krasnodembskaya A, McKenna DH, Song Y, Abbott J, Matthay MA. Therapeutic effects of human mesenchymal stem cells in ex vivo human lungs injured with live bacteria. Am J Respir Crit Care Med. 2013;187(7):751–60.
Li B, Zhang H, Zeng M, He W, Li M, Huang X, Deng DY, Wu J. Bone marrow mesenchymal stem cells protect alveolar macrophages from lipopolysaccharide-induced apoptosis partially by inhibiting the Wnt/β-catenin pathway. Cell Biol Int. 2015;39(2):192–200.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, Uccelli A. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107(1):367–72.
Ramasamy R, Tong CK, Seow HF, Vidyadaran S, Dazzi F. The immunosuppressive effects of human bone marrow-derived mesenchymal stem cells target T cell proliferation but not its effector function. Cell Immunol. 2008;251(2):131–6.
Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005;105(5):2214–9.
Gore AV, Bible LE, Song K, Livingston DH, Mohr AM, Sifri ZC. Mesenchymal stem cells increase T-regulatory cells and improve healing following trauma and hemorrhagic shock. J Trauma Acute Care Surg. 2015;79(1):48–52; discussion 52
Meisel R, Zibert A, Laryea M, Göbel U, Däubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103(12):4619–21.
Frenzel J, Gessner C, Sandvoss T, Hammerschmidt S, Schellenberger W, Sack U, Eschrich K, Wirtz H. Outcome prediction in pneumonia induced ALI/ARDS by clinical features and peptide patterns of BALF determined by mass spectrometry. PLoS One. 2011;6(10):e25544.
Lin WC, Lin CF, Chen CL, Chen CW, Lin YS. Prediction of outcome in patients with acute respiratory distress syndrome by bronchoalveolar lavage inflammatory mediators. Exp Biol Med (Maywood). 2010;235(1):57–65.
Steensberg A, Fischer CP, Keller C, Møller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. 2003;285(2):E433–7.
Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 1994;83(1):113–8.
Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, Achong MK. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101(2):311–20.
Zhang S, Danchuk SD, Bonvillain RW, Xu B, Scruggs BA, Strong AL, Semon JA, Gimble JM, Betancourt AM, Sullivan DE, Bunnell BA. Interleukin 6 mediates the therapeutic effects of adipose-derived stromal/stem cells in lipopolysaccharide-induced acute lung injury. Stem Cells. 2014;32(6):1616–28.
Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O'Kane CM, Krasnodembskaya AD. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am J Respir Crit Care Med. 2017;196(10):1275–86.
Mutlu GM, Dumasius V, Burhop J, McShane PJ, Meng FJ, Welch L, Dumasius A, Mohebahmadi N, Thakuria G, Hardiman K, Matalon S, Hollenberg S, Factor P. Upregulation of alveolar epithelial active Na+ transport is dependent on beta2-adrenergic receptor signaling. Circ Res. 2004;94(8):1091–100.
Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A. 2006;103(5):1283–8.
Plotnikov EY, Khryapenkova TG, Vasileva AK, Marey MV, Galkina SI, Isaev NK, Sheval EV, Polyakov VY, Sukhikh GT, Zorov DB. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J Cell Mol Med. 2008;12(5A):1622–31.
Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014;33(9):994–1010.
Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, O'Kane CM, Krasnodembskaya AD. Mitochondrial transfer via Tunneling nanotubes is an important mechanism by which Mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells. 2016;34(8):2210–23.
Jackson MV, Krasnodembskaya AD. Analysis of mitochondrial transfer in direct co-cultures of human monocyte-derived macrophages (MDM) and Mesenchymal stem cells (MSC). Bio Protoc. 2017;7(9):e2255.
Gupta N, Su X, Popov B, Lee JW, Serikov V, Matthay MA. Intrapulmonary delivery of bone marrow-derived mesenchymal stem cells improves survival and attenuates endotoxin-induced acute lung injury in mice. J Immunol. 2007;179(3):1855–63.
Mei SH, Haitsma JJ, Dos Santos CC, Deng Y, Lai PF, Slutsky AS, Liles WC, Stewart DJ. Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med. 2010;182(8):1047–57.
Pedrazza L, Cunha AA, Luft C, Nunes NK, Schimitz F, Gassen RB, Breda RV, Donadio MV, de Souza Wyse AT, Pitrez PMC, Rosa JL, de Oliveira JR. Mesenchymal stem cells improves survival in LPS-induced acute lung injury acting through inhibition of NETs formation. J Cell Physiol. 2017;232(12):3552–64.
Shalaby SM, El-Shal AS, Abd-Allah SH, Selim AO, Selim SA, Gouda ZA, Abd El Motteleb DM, Zanfaly HE, El-Assar HM, Abdelazim S. Mesenchymal stromal cell injection protects against oxidative stress in Escherichia coli-induced acute lung injury in mice. Cytotherapy. 2014;16(6):764–75.
Ionescu L, Byrne RN, van Haaften T, Vadivel A, Alphonse RS, Rey-Parra GJ, Weissmann G, Hall A, Eaton F, Thébaud B. Stem cell conditioned medium improves acute lung injury in mice: in vivo evidence for stem cell paracrine action. Am J Physiol Lung Cell Mol Physiol. 2012;303(11):L967–77.
Song L, Xu J, Qu J, Sai Y, Chen C, Yu L, Li D, Guo X. A therapeutic role for mesenchymal stem cells in acute lung injury independent of hypoxia-induced mitogenic factor. J Cell Mol Med. 2012;16(2):376–85.
Maron-Gutierrez T, Silva JD, Asensi KD, Bakker-Abreu I, Shan Y, Diaz BL, Goldenberg RC, Mei SH, Stewart DJ, Morales MM, Rocco PR, Dos Santos CC. Effects of mesenchymal stem cell therapy on the time course of pulmonary remodeling depend on the etiology of lung injury in mice. Crit Care Med. 2013;41(11):e319–33.
Bustos ML, Huleihel L, Meyer EM, Donnenberg AD, Donnenberg VS, Sciurba JD, Mroz L, McVerry BJ, Ellis BM, Kaminski N, Rojas M. Activation of human mesenchymal stem cells impacts their therapeutic abilities in lung injury by increasing interleukin (IL)-10 and IL-1RN levels. Stem Cells Transl Med. 2013;2(11):884–95.
Yilmaz S, Inandiklioglu N, Yildizdas D, Subasi C, Acikalin A, Kuyucu Y, Bayram I, Topak A, Tanyeli A, Duruksu G, Karaoz E. Mesenchymal stem cell: does it work in an experimental model with acute respiratory distress syndrome? Stem Cell Rev. 2013;9(1):80–92.
Yang H, Wen Y, Bin J, Hou-You Y, Yu-Tong W. Protection of bone marrow mesenchymal stem cells from acute lung injury induced by paraquat poisoning. Clin Toxicol (Phila). 2011;49(4):298–302.
Curley GF, Hayes M, Ansari B, Shaw G, Ryan A, Barry F, O'Brien T, O'Toole D, Laffey JG. Mesenchymal stem cells enhance recovery and repair following ventilator-induced lung injury in the rat. Thorax. 2012;67(6):496–501.
Chimenti L, Luque T, Bonsignore MR, Ramírez J, Navajas D, Farré R. Pre-treatment with mesenchymal stem cells reduces ventilator-induced lung injury. Eur Respir J. 2012;40(4):939–48.
Curley GF, Ansari B, Hayes M, Devaney J, Masterson C, Ryan A, Barry F, O'Brien T, Toole DO, Laffey JG. Effects of intratracheal mesenchymal stromal cell therapy during recovery and resolution after ventilator-induced lung injury. Anesthesiology. 2013;118(4):924–32.
Hayes M, Masterson C, Devaney J, Barry F, Elliman S, O'Brien T, O'Toole D, Curley GF, Laffey JG. Therapeutic efficacy of human mesenchymal stromal cells in the repair of established ventilator-induced lung injury in the rat. Anesthesiology. 2015;122(2):363–73.
Hayes M, Curley GF, Masterson C, Devaney J, O'Toole D, Laffey JG. Mesenchymal stromal cells are more effective than the MSC secretome in diminishing injury and enhancing recovery following ventilator-induced lung injury. Intensive Care Med Exp. 2015;3(1):29.
Li J, Li D, Liu X, Tang S, Wei F. Human umbilical cord mesenchymal stem cells reduce systemic inflammation and attenuate LPS-induced acute lung injury in rats. J Inflamm (Lond). 2012;9(1):33.
Maron-Gutierrez T, Silva JD, Cruz FF, Alegria S, Xisto DG, Assis EF, Castro-Faria-Neto HC, Dos Santos CC, Morales MM, Rocco PR. Insult-dependent effect of bone marrow cell therapy on inflammatory response in a murine model of extrapulmonary acute respiratory distress syndrome. Stem Cell Res Ther. 2013;4(5):123.
Qin ZH, Xu JF, Qu JM, Zhang J, Sai Y, Chen CM, Wu L, Yu L. Intrapleural delivery of MSCs attenuates acute lung injury by paracrine/endocrine mechanism. J Cell Mol Med. 2012;16(11):2745–53.
Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4(9):e269.
Zhao Y, Yang C, Wang H, Li H, Du J, Gu W, Jiang J. Therapeutic effects of bone marrow-derived mesenchymal stem cells on pulmonary impact injury complicated with endotoxemia in rats. Int Immunopharmacol. 2013;15(2):246–53.
Yang H, Wen Y, Hou-you Y, Yu-tong W, Chuan-ming L, Jian X, Lu H. Combined treatment with bone marrow mesenchymal stem cells and methylprednisolone in paraquat-induced acute lung injury. BMC Emerg Med. 2013;13(Suppl 1):S5.
Gotts JE, Abbott J, Matthay MA. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am J Physiol Lung Cell Mol Physiol. 2014;307(5):L395–406.
Rojas M, Parker RE, Thorn N, Corredor C, Iyer SS, Bueno M, Mroz L, Cardenes N, Mora AL, Stecenko AA, Brigham KL. Infusion of freshly isolated autologous bone marrow derived mononuclear cells prevents endotoxin-induced lung injury in an ex-vivo perfused swine model. Stem Cell Res Ther. 2013;4(2):26.
Moodley Y, Sturm M, Shaw K, Shimbori C, Tan DB, Kolb M, Graham R. Human mesenchymal stem cells attenuate early damage in a ventilated pig model of acute lung injury. Stem Cell Res. 2016;17(1):25–31.
Rojas M, Cardenes N, Kocyildirim E, Tedrow JR, Caceres E, Deans R, Ting A, Bermudez C. Human adult bone marrow-derived stem cells decrease severity of lipopolysaccharide-induced acute respiratory distress syndrome in sheep. Stem Cell Res Ther. 2014;5(2):42.
Asmussen S, Ito H, Traber DL, Lee JW, Cox RA, Hawkins HK, McAuley DF, McKenna DH, Traber LD, Zhuo H, Wilson J, Herndon DN, Prough DS, Liu KD, Matthay MA, Enkhbaatar P. Human mesenchymal stem cells reduce the severity of acute lung injury in a sheep model of bacterial pneumonia. Thorax. 2014;69(9):819–25.
Iyer SS, Torres-Gonzalez E, Neujahr DC, Kwon M, Brigham KL, Jones DP, Mora AL, Rojas M. Effect of bone marrow-derived mesenchymal stem cells on endotoxin-induced oxidation of plasma cysteine and glutathione in mice. Stem Cells Int. 2010;2010:868076.
Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res. 2014;92:10–8.
Wang C, Lv D, Zhang X, Ni ZA, Sun X, Zhu C. Interleukin-10-overexpressing Mesenchymal stromal cells induce a series of regulatory effects in the inflammatory system and promote the survival of endotoxin-induced acute lung injury in mice model. DNA Cell Biol. 2018;37(1):53–61.
Martinez-Gonzalez I, Roca O, Masclans JR, Moreno R, Salcedo MT, Baekelandt V, Cruz MJ, Rello J, Aran JM. Human mesenchymal stem cells overexpressing the IL-33 antagonist soluble IL-1 receptor-like-1 attenuate endotoxin-induced acute lung injury. Am J Respir Cell Mol Biol. 2013;49(4):552–62.
Saito S, Nakayama T, Hashimoto N, Miyata Y, Egashira K, Nakao N, Nishiwaki S, Hasegawa M, Hasegawa Y, Naoe T. Mesenchymal stem cells stably transduced with a dominant-negative inhibitor of CCL2 greatly attenuate bleomycin-induced lung damage. Am J Pathol. 2011;179(3):1088–94.
Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci U S A. 2003;100(14):8407–11.
Huleihel L, Sellares J, Cardenes N, Alvarez D, Faner R, Sakamoto K, Yu G, Kapetanaki MG, Kaminski N, Rojas M. Modified mesenchymal stem cells using miRNA transduction alter lung injury in a bleomycin model. Am J Physiol Lung Cell Mol Physiol. 2017;313(1):L92–L103.
Djouad F, Fritz V, Apparailly F, Louis-Plence P, Bony C, Sany J, Jorgensen C, Noël D. Reversal of the immunosuppressive properties of mesenchymal stem cells by tumor necrosis factor alpha in collagen-induced arthritis. Arthritis Rheum. 2005;52(5):1595–603.
Li W, Ren G, Huang Y, Su J, Han Y, Li J, Chen X, Cao K, Chen Q, Shou P, Zhang L, Yuan ZR, Roberts AI, Shi S, Le AD, Shi Y. Mesenchymal stem cells: a double-edged sword in regulating immune responses. Cell Death Differ. 2012;19(9):1505–13.
Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI, Zhao RC, Shi Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2(2):141–50.
Renner P, Eggenhofer E, Rosenauer A, Popp FC, Steinmann JF, Slowik P, Geissler EK, Piso P, Schlitt HJ, Dahlke MH. Mesenchymal stem cells require a sufficient, ongoing immune response to exert their immunosuppressive function. Transplant Proc. 2009;41(6):2607–11.
Romieu-Mourez R, François M, Boivin MN, Bouchentouf M, Spaner DE, Galipeau J. Cytokine modulation of TLR expression and activation in mesenchymal stromal cells leads to a proinflammatory phenotype. J Immunol. 2009;182(12):7963–73.
Ren G, Su J, Zhang L, Zhao X, Ling W, L'huillie A, Zhang J, Lu Y, Roberts AI, Ji W, Zhang H, Rabson AB, Shi Y. Species variation in the mechanisms of mesenchymal stem cell-mediated immunosuppression. Stem Cells. 2009;27(8):1954–62.
Yan X, Liu Y, Han Q, Jia M, Liao L, Qi M, Zhao RC. Injured microenvironment directly guides the differentiation of engrafted Flk-1(+) mesenchymal stem cell in lung. Exp Hematol. 2007;35(9):1466–75.
Islam D, Huang Y, Fanelli V, Delsedime L, Wu S, Khang J, Han B, Grassi A, Li M, Xu Y, Luo A, Wu J, Liu X, McKillop M, Medin J, Qiu H, Zhong N, Liu M, Laffey J, Li Y, Zhang H. Identification and modulation of microenvironment is crucial for effective MSC therapy in acute lung injury. Am J Respir Crit Care Med. 2019;199(10):1214–24.
Bustos ML, Huleihel L, Kapetanaki MG, Lino-Cardenas CL, Mroz L, Ellis BM, McVerry BJ, Richards TJ, Kaminski N, Cerdenes N, Mora AL, Rojas M. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am J Respir Crit Care Med. 2014;189(7):787–98.
Jin HJ, Bae YK, Kim M, Kwon SJ, Jeon HB, Choi SJ, Kim SW, Yang YS, Oh W, Chang JW. Comparative analysis of human mesenchymal stem cells from bone marrow, adipose tissue, and umbilical cord blood as sources of cell therapy. Int J Mol Sci. 2013;14(9):17986–8001.
Kern S, Eichler H, Stoeve J, Klüter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. 2006;24(5):1294–301.
Wang Q, Yang Q, Wang Z, Tong H, Ma L, Zhang Y, Shan F, Meng Y, Yuan Z. Comparative analysis of human mesenchymal stem cells from fetal-bone marrow, adipose tissue, and Warton's jelly as sources of cell immunomodulatory therapy. Hum Vaccin Immunother. 2016;12(1):85–96.
Lotfy A, Salama M, Zahran F, Jones E, Badawy A, Sobh M. Characterization of mesenchymal stem cells derived from rat bone marrow and adipose tissue: a comparative study. Int J Stem Cells. 2014;7(2):135–42.
Li CY, Wu XY, Tong JB, Yang XX, Zhao JL, Zheng QF, Zhao GB, Ma ZJ. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res Ther. 2015;6:55.
Lalu MM, Moher D, Marshall J, Fergusson D, Mei SH, Macleod M, Griffin G, Turgeon AF, Rudnicki M, Fishman J, Avey MT, Skidmore B, Grimshaw JM, Stewart DJ, Singh K, McIntyre L, Canadian Critical Care Translational Biology Group. Efficacy and safety of mesenchymal stromal cells in preclinical models of acute lung injury: a systematic review protocol. Syst Rev. 2014;3:48.
McIntyre LA, Moher D, Fergusson DA, Sullivan KJ, Mei SH, Lalu M, Marshall J, Mcleod M, Griffin G, Grimshaw J, Turgeon A, Avey MT, Rudnicki MA, Jazi M, Fishman J, Stewart DJ, Canadian Critical Care Translational Biology Group. Efficacy of Mesenchymal stromal cell therapy for acute lung injury in preclinical animal models: a systematic review. PLoS One. 2016;11(1):e0147170.
Kim ES, Chang YS, Choi SJ, Kim JK, Yoo HS, Ahn SY, Sung DK, Kim SY, Park YR, Park WS. Intratracheal transplantation of human umbilical cord blood-derived mesenchymal stem cells attenuates Escherichia coli-induced acute lung injury in mice. Respir Res. 2011;12:108.
Zhang Z, Li W, Heng Z, Zheng J, Li P, Yuan X, Niu W, Bai C, Liu H. Combination therapy of human umbilical cord mesenchymal stem cells and FTY720 attenuates acute lung injury induced by lipopolysaccharide in a murine model. Oncotarget. 2017;8(44):77407–14.
Shin S, Kim Y, Jeong S, Hong S, Kim I, Lee W, Choi S. The therapeutic effect of human adult stem cells derived from adipose tissue in endotoxemic rat model. Int J Med Sci. 2013;10(1):8–18.
Zhang S, Danchuk SD, Imhof KM, Semon JA, Scruggs BA, Bonvillain RW, Strong AL, Gimble JM, Betancourt AM, Sullivan DE, Bunnell BA. Comparison of the therapeutic effects of human and mouse adipose-derived stem cells in a murine model of lipopolysaccharide-induced acute lung injury. Stem Cell Res Ther. 2013;4(1):13.
Lu H, Cook T, Poirier C, Merfeld-Clauss S, Petrache I, March KL, Bogatcheva NV. Pulmonary retention of adipose stromal cells following intravenous delivery is markedly altered in the presence of ARDS. Cell Transplant. 2016;25(9):1635–43.
Hakki SS, Kayis SA, Hakki EE, Bozkurt SB, Duruksu G, Unal ZS, Turaç G, Karaoz E. Comparison of mesenchymal stem cells isolated from pulp and periodontal ligament. J Periodontol. 2015;86(2):283–91.
Xiang B, Chen L, Wang X, Zhao Y, Wang Y, Xiang C. Transplantation of menstrual blood-derived Mesenchymal stem cells promotes the repair of LPS-induced acute lung injury. Int J Mol Sci. 2017;18(4):E689.
Park J, Kim S, Lim H, Liu A, Hu S, Lee J, Zhuo H, Hao Q, Matthay MA, Lee JW. Therapeutic effects of human mesenchymal stem cell microvesicles in an ex vivo perfused human lung injured with severe E. coli pneumonia. Thorax. 2019;74(1):43–50.
Zhu YG, Feng XM, Abbott J, Fang XH, Hao Q, Monsel A, Qu JM, Matthay MA, Lee JW. Human mesenchymal stem cell microvesicles for treatment of Escherichia coli endotoxin-induced acute lung injury in mice. Stem Cells. 2014;32(1):116–25.
Monsel A, Zhu YG, Gennai S, Hao Q, Hu S, Rouby JJ, Rosenzwajg M, Matthay MA, Lee JW. Therapeutic effects of human Mesenchymal stem cell-derived microvesicles in severe pneumonia in mice. Am J Respir Crit Care Med. 2015;192(3):324–36.
D'Cunha HC, Rojas M. Ex vivo lung perfusion: past, present, and future. ASAIO J. 2018;64(2):135–9.
Weathington NM, Alvarez D, Sembrat J, Radder J, Cardenes N, Noda K, Gong Q, Wong H, Kolls J, D'Cunha J, Mallampalli RK, Chen BB, Rojas M. Ex vivo lung perfusion as a human platform for preclinical small molecule testing. JCI Insight. 2018;3(19):95515.
Mordant P, Nakajima D, Kalaf R, Iskender I, Maahs L, Behrens P, Coutinho R, Iyer RK, Davies JE, Cypel M, Liu M, Waddell TK, Keshavjee S. Mesenchymal stem cell treatment is associated with decreased perfusate concentration of interleukin-8 during ex vivo perfusion of donor lungs after 18-hour preservation. J Heart Lung Transplant. 2016;35(10):1245–54.
McAuley DF, Curley GF, Hamid UI, Laffey JG, Abbott J, McKenna DH, Fang X, Matthay MA, Lee JW. Clinical grade allogeneic human mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):L809–15.
Kocyildirim E, Cardenes N, Ting A, Caceres E, BermUdez C, Rojas M. The use of GMP-produced bone marrow-derived stem cells in combination with extracorporeal membrane oxygenation in ARDS: an animal model. ASAIO J. 2017;63(3):324–32.
Gennai S, Monsel A, Hao Q, Park J, Matthay MA, Lee JW. Microvesicles derived from human Mesenchymal stem cells restore alveolar fluid clearance in human lungs rejected for transplantation. Am J Transplant. 2015;15(9):2404–12.
Park J, Kim S, Lim H, Liu A, Hu S, Lee J, Zhuo H, Hao Q, Matthay MA, Lee JW. Therapeutic effects of human mesenchymal stem cell microvesicles in an ex vivo perfused human lung injured with severe. Thorax. 2019;74(1):43–50.
Acknowledgments
The authors want to thank Nayra Cardenes for her help drafting the figure and to Jordan Bullock for the editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cruz, T., Rojas, M. (2019). Preclinical Evidence for the Role of Stem/Stromal Cells in Targeting ARDS. In: Burgess, J., Heijink, I. (eds) Stem Cell-Based Therapy for Lung Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-29403-8_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-29403-8_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-29402-1
Online ISBN: 978-3-030-29403-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)