
Abstract
The human being is a superorganism composed of human cells and its associated microbiota. Humans did not emerge alone along evolution but in coexistence and intricate metabolic integration with microorganisms. The microorganisms that co-evolve and co-live with humans are called the microbiota. The human gut microbiota is a dynamic taxonomically complex community that participates in several processes related to normal function of the host-microbiota superorganism, maintaining the health status. Changes to the social aspects of the Western civilization and technological developments impacted on the evolutionary host-microbes’ association. As a consequence of the disruption to this equilibrium, immunological, endocrine, metabolic and neurological alterations have arisen. Maternal diet, lifestyle, mode of delivery, administration of antibiotics to the mother during pregnancy, early nutrition (breastfeeding or formula) and treatment with antibiotics in newborns are crucial factors that affect microbiota structure. Microbiota and epigenome are involved in the reduced or increased risk to develop different microbiome-associated diseases in adult life.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
The purpose of this chapter is to analyze the emergence of diseases specific to the Western civilization and their potential relationship with the depletion of the human-associated microbiota. This evolutionary perspective takes into consideration the fact that integration of systems is a common pattern of life. The human being is a superorganism composed of human cells and organs, and its associated microbiota. Humans did not emerge alone along evolution but in coexistence and intricate metabolic integration with microorganisms. Changes to the social aspects of the Western civilization and technological developments impacted the evolutionary host-microbes’ association. The relatively recent alteration of the coexistence between host and microbiota has led to the emergence of diseases related to an over reactive immune system. A better knowledge of the human superorganism will yield approaches to restore or modulate the microbiota to treat or alleviate symptoms of these emergent diseases.
Origin of the Human-Microbes Superorganism
Maturana and Varela (Varela et al. 1974) created a new term to explain life: autopoiesis. Literally, it means self-production. It is the common trait of all living organisms. Cells and organisms produce a myriad of molecules and create a net of functionality to maintain their distinctiveness, cohesiveness, and relative autonomy. This net of components maintains cell self-production (Varela et al. 1974; Zelený 1981). According to this perspective, it is not correct to state that the organism neither adapts to the milieu nor the milieu selects changes in the organisms. The environment and the niche do not pre-exist to individuals. Organism and milieu change concomitantly. Maturana calls ‘ontogenic drift’ the process in which each organism is part of the niche where others organisms change and change it, in reciprocal building of their niches. The diversity of species is the result of reproduction and maintenance of autopoiesis and the systemic conservation of their organization in relation with other organisms or lineages (Maturana-Romesin and Mpodozis 2000).
There is an intrinsic force, inseparable of life, that causes system evolution: drastic changes result in collapse of the system or its re-organization (an evolutive step). This should not be confused with vitalism. The inevitable and inseparable propulsion of life to evolve is not extern to the organism but a characteristic that defines life. So there is nothing outside life that defines what is fit and what is not. Under this perspective, there is no selector (because it implies a teleological thinking) and natural selection is what an observer sees in the differential reproduction of two lineages of organism in different historical moments. The process behind the observed “selection” remains hidden. That is, the conservation of phenotypes in a natural drift (ontogenic and phylogenetic drift) is inseparable from autopoiesis (Maturana-Romesin and Mpodozis 2000). The properties of life imply evolution.
There are crucial mechanisms in evolution that involve the maintenance of this autopoiesis and they imply integration. Main steps of evolution are based on integration processes. A crucial mechanism is symbiogenesis. According to Margulis and Sagan (2003) symbiosis is simply the living together of organisms that are different from each other and symbiogenesis is the origin of evolutionary novelty via symbiosis (Margulis 2010; Margulis and Sagan 2003). As an example, the emergence of the eukaryotic cell involved the integration of pre-existent bacterial cells. Moreover, there is strong evidence that mitochondria and chloroplasts are evolutionary structures originated from different ancestors, with the mitochondria evolving from proto-Rickettsiales, proto-Rhizobiales, proto-alphaproteobacteria and current alphaproteobacterial species (Georgiades and Raoult 2012). The symbiogenetic theory was developed by Lynn Margulis and, independent and previously, by Ivan Wallin, Paul Poitier, Konstantin Merezhkovski and Boris Kozo-Polianski (Kozo-Polyansky and Margulis 2010; Brucker and Bordenstein 2012; Wallin 1927; Symbiosis as a Source of Evolutionary Innovation 2017).
Consistently, the nuclear structure of eukaryotes is another example of an evolutionary integration step (Bell 2001; Forterre 2006). The role of viruses in the emergence of nucleus demonstrate the importance of those microoorganisms in evolution, based on their ability to integrate into the nucleic acids, modifying genomic behaviour and regulation. This integrative process is associated with the emergence of new structures (Feschotte and Gilbert 2012; Belyi et al. 2010; Ding and Lipshitz 1994; Gifford et al. 2008; Jamain et al. 2001; Medstrand and Mager 1998; Villarreal and DeFilippis 2000). For example, virus-related genes are involved in the process of placentation in mammals (Mallet et al. 2004). Likewise, retrotransposons participate in the regulation of genes related to the histocompatibility in humans, other mammals and invertebrates (Ding and Lipshitz 1994; McDonald et al. 1997; Kidwell and Lisch 2000).
A crucial evolutionary mechanism is horizontal gene transfer (HGT). The high prevalence of HGT events in Bacteria, Archaea and Eukaryotes has resulted in the mosaicism of genomes. It is difficult to identify a single common ancestor for the gene repertoire of any organism. HGT is not only extensive and directional but also ongoing, and acquired genes are related to metabolism and biogenesis with crucial value in evolution (Deschamps et al. 2014; Fuchsman et al. 2017; Craig et al. 2009; Hotopp 2011). HGT has a central importance in evolution of microorganism and metazoan hosts. As examples, endogenous viral elements from different families (Bornaviridae, Filoviridae, Bunyaviridae, Flaviviridae, Parvoviridae, Hapadnaviridae) are part of animal genomes including primates (Holmes 2011; Katzourakis and Gifford 2010; Gilbert et al. 2010). Human genome sequencing has demonstrated the high content of bacterium- and virus-related genes including retrotransposons (Mallet et al. 2004; Hotopp 2011; Gilbert et al. 2010; Gifford et al. 2008; Mi et al. 2000). This reveals that the genomic structure is the result of a dynamic equilibrium between genetic and cellular processes. The primary structure of the DNA is the result of the continuous feedback between an organism and the rest of living beings and the environment in a net of mutual building (Belyi et al. 2010; Gifford et al. 2008; Casjens 2003; Merhej and Raoult 2012). Evolution of species including metazoan is described like a rhizome by some researchers, pointing out different and mixed origins of genomic sequences in species (Georgiades and Raoult 2011, 2012). Ramulu et al. (2012) have emphasized that many proposals have emerged replacing the tree-like pattern, that consider a the existence of a common ancestor of organisms, with more complex models such as the “reticulate evolution”, “synthesis of life”, “web of life” or “network of life” (Ramulu et al. 2012).
The mechanisms of integration and symbiopoiesis are presently at work. It is possible to understand that co-evolution of humans and microorganisms results in an intricate metabolism and homeostasis, and this is not possible to maintain if any part of this symbiosis is altered or depleted. The microorganisms that co-evolve and co-live with humans are called the microbiota. In fact, the human body contains more bacterial cells than human cells. Recently, it has been stated that human:microorganisms cells ratio is 1,3:1, but that takes into account bacterial cells (Sender et al. 2016), but not eukaryotic components of the microbiota (Parfrey et al. 2011). All organisms emerge in relation with their environment and the surrounding organisms that coevolve with them as a unity. Humans arose with the microorganisms that defined their metabolism, their systems, and their structure.
The human gut microbiota is a dynamic taxonomically complex community that participates in several processes related to normal function of the host-microbiota superorganism, maintaining the health status (Kau et al. 2011; Lederberg and McCray 2001; Salvucci 2016). These include vitamin production, digestion and utilization of carbohydrates and lipids, energy homeostasis, tryptophan metabolism regulation, integrity of intestinal barrier and angiogenesis (Arrieta et al. 2014; van der Meulen et al. 2016; Evans et al. 2013). As an example, large primates are incapable of vitamin C synthesis. As a consequence, humans have an evolved dependence on fruit and vegetables since our genes for vitamin synthesis were lost (Rook 2011).
Short-chain fatty acids (SCFAs), mainly acetic, propionic and butyric acids are products of bacterial fermentation in the intestinal tract. Cross-feeding interactions between bifidobacteria and butyrate-producing colon bacteria, such as Faecalibacterium prausnitzii (clostridial cluster IV) and Anaerostipes, Eubacterium, and Roseburia species (clostridial cluster XIVa) result in an enhancement of butyrate production. SCFAs are a source of ATP for intestinal epithelial cells (IECs) and modulate IECs and leukocytes development, survival and function through activation of G protein coupled receptors (FFAR2, FFAR3, GPR109a and Olfr78). SCFAs also modulate enzymatic activity and transcription factors including histone acetyltransferase and deacetylase, and the hypoxia-inducible factor (Corrêa-Oliveira et al. 2016; Kasubuchi et al. 2015).
The gut microbiota produces and regulates compounds with crucial local effects, but that also influence the function of distal organs and systems. In fact, metabolites generated by the gut microbiota have been shown to influence brain chemistry and behaviour independently from the autonomic nervous system, gastrointestinal neurotransmitters or inflammation through the microbiota-brain axis (Foster and McVey Neufeld 2013). This is a two-way communication pathway between the central nervous system and the intestine (Foster and McVey Neufeld 2013; Bercik et al. 2012). Butyrate has profound effects on mood and behaviour in mice (Schroeder et al. 2007). The microbial metabolism in the gut can also influence the level of neurotransmitters or hormones (Allen et al. 2013; O’Mahony et al. 2015). As examples, GABA and serotonin, related to anxiety, depression and mood states are influenced by the microbiota (Foster and McVey Neufeld 2013; Bercik et al. 2010). Inflammatory bowel diseases (IBD) are often accompanied by disorders of the nervous system such as irritability, anxiety, and depression (Hayley et al. 2016; Lee and Chua 2011). Moreover, dysbiosis can contribute to the development of psychiatric disorders in patients with intestinal symptoms (Bercik et al. 2010; Borre et al. 2014; Collins et al. 2012; Cryan and Dinan 2012; Dinan et al. 2014; Nemani et al. 2014).
The continuum human-microbiota represents a step of symbiosis along the genomic evolution. According to this perspective, the human genome would be a “hard drive” where is possible to observe integrated sequences of different origins as well as the integration processes. The human genome is consequently the result of the evolutionary history of the superorganism human-microbiota and their environmental interactions. This superorganism also includes mobile elements like plasmids, transposons, integrons, and bacteriophages that are called the ‘mobilome’ (Siefert 2009). These genetic elements constitute a genetic pool that fuel HGT and a sensitive response to environmental changes.
Main social and environmental changes along history caused variations in the human-microbes superorganism. These epidemiological transitions had and have today a profound impact on human health.
Epidemiological Transitions and Western Diseases
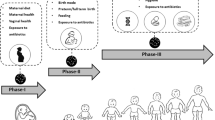
Two major epidemiological transitions resulted in dramatic changes to the human superorganism (McKeown 2009; Rook 2010). These shifts involved alterations in social behaviour and organization, which can be correlated with significant changes in the microbiota structure (Fig. 1). Two million years ago humans were nomadic hunter-gatherers (Palaeolithic period). With the establishment of populations, humans became sedentary. An agricultural revolution ensued with a profound shift in human lifestyle (Neolithic period, 10,000 years BC). In this period, new foods like cereals and dairy products appeared, and population density increased. It is reasonable to assume that dramatic changes occurred in the microbiota after the first epidemiological transition. However, this shift did not result in loss of microbial diversity because, until the modern era, more than 97% of the population still lived in rural environments (Rook 2011). However, the new lifestyle increased faecal–oral transmission and prolonged contact with animals (Rook 2012). Viruses that were acquired previous to Neolithic have been maintained, including herpes, papilloma, adeno-, parvo-, some entero-, and perhaps hepatitis B virus (Rook 2012; Van Blerkom Linda 2003; Leal Éde and Zanotto 2000). It is possible that during this period characterized by a marked increase in population density, humans started suffering from “modern” diseases such as measles, influenza, and dengue (Rook 2012; Leal Éde and Zanotto 2000). Helminths, Mycobacteria, Helicobacter pylori, Salmonella, Toxoplasma and lactobacilli were acquired during this period (Rook 2011, 2012; Cremonini and Gasbarrini 2003).

Fig. 1 Epidemiological transitions. The second epidemiological transition had great impact on human microbiota. Social and technological changes led to a depletion of microorganisms associated to human being.
The second remarkable shift in human lifestyle and dietary habits started with the Industrial Revolution less than 200 years ago, accompanied with a radical move, away from local and seasonal foods (McKeown 2009). People started to migrate into cities and the use of sanitization increased. This resulted in a diminished or delayed contact with microorganisms and viruses. With this second transition there were significant lifestyles changes. Sanitization was improved with the use of soap detergents, washed food, chlorinated water and, eventually, antibiotics. This transition was characterized by a significant decrease in orofecal transmission. Life in the city implied advances in housing with a wide use of construction materials like concrete and asphalt, and less soil material. Less contact with animals also contributed to de-worming. As a result, changes to the microbiota included decreased helminths, H. pylori, Salmonella, Toxoplasma and Mycobacterium tuberculosis (Fig. 1) (McKeown 2009; Rook 2009, 2012).
Changes in dietary habits were important factors in the alteration of the host–microbe symbiosis during epidemiological transitions since diet drives the generation of gut microbe-derived bioactive metabolites (Hunter 2008). According to recently published studies, the ancestral diet was based on high fiber intake and maintained a well-balanced microbiota. The microbiota was more structurally and functionally diverse with enhanced polysaccharide breakdown capacities. This highly diverse microbiota has been maintained in rural and remote populations from developing countries compared to urban industrialized populations (Crittenden et al. 2014; West et al. 2015; Turroni et al. 2016; Smits et al. 2017), with gut bacterial taxa or functions that may have disappeared due to cultural Westernization (Crittenden et al. 2014). Conversely, the Western diet is characterized by a high intake of animal products and sugars, the use of food preservatives, and a low intake of plant-based foods like fruits, vegetables, and whole grain cereals.
Researchers have compared microbiota of communities from Tanzania (Hadza tribe) with that from Western countries. Hunter-gatherers that live today as thousands years ago maintaining an ancestral diet, showed lower inflammation than individuals from the Western hemisphere. Hunter-gatherers have a more diverse enzymatic repertoire for utilizing carbohydrates in their microbiota than those from healthy American subjects (Schnorr et al. 2014). These studies made possible to identify organisms and functions that would have been abundant in the ancestral microbiota, but are diminished or absent from the modern city environment (Rook 2009, 2010). Specifically, the Hadza microbiota has higher functional capacity for utilization of plant carbohydrates than the microbiota of Americans (Crittenden et al. 2014). The community consume tubers and baobab year-round. Conversely, the American microbiota with less consumption of fiber-rich diet have a greater mucin-utilization capacity than Hadzas (Turroni et al. 2016; Smits et al. 2017). These data are in agreement with other studies that indicate that the microbiota of people in urbanized regions is characteristic of a diet limited in plant-derived complex carbohydrates, with many bacterial species underrepresented or missing (Kau et al. 2011; Findley et al. 2016). Fiber-rich diets fuel the gut microbiota metabolism and maintain resident bacterial populations (Thorburn et al. 2014; De Filippo et al. 2010). The depletion of microbiota has been clearly intensified by social factors like industrialization and Western diets with consequences to equilibrium and homeostasis.
As a consequence of the disruption to the host-microbiota equilibrium, immunological, endocrine, metabolic and neurological alterations have arisen (Arrieta et al. 2014; Foster and McVey Neufeld 2013; Hayley et al. 2016; Rook 2009; Paun and Danska 2015; van der Meulen et al. 2016). Human diseases like inflammatory bowel diseases (IBD), irritable bowel syndrome (IBS), cancer, diabetes, asthma, allergies, obesity and metabolic syndrome appear to be related to the depleted microbiota (Fig. 2).

Fig. 2 Microbiota depletion. Human superorganism lost part of the microbiota along the last epidemiological transition. This depletion of microbiota is related to different emergent diseases with impact on gut-brain axis, immune system, gut and systemic disorders.
Obesity and metabolic syndrome have been recognized as epidemic diseases associated with Western diets and low microbial diversity. The altered microbiota in obese subjects has lower proportion of Bacteroidetes and more Firmicutes than healthy individuals (Remely et al. 2014; Ley et al. 2005). The obese microbiota can be inherited from parents with high fat diets and it has an increased capacity to harvest energy from the diet (Ley et al. 2005; Myles et al. 2013), producing significantly higher total body fat. Concomitantly, immunological alterations include infiltration of adipose tissue by macrophages, CD8+ T cells and CD4+ T cells, and high expression of inflammatory cytokines such as IL-6, IL-17, TNF- alpha and interferon-gamma (Kau et al. 2011). Additionally, obese individuals usually have high endotoxemia caused by LPS infiltration due to an increased intestinal permeability. A recent study showed that the diversity of the gut microbiota and the degree of methylation of the FFAR3 promoter region were significantly lower in obese and type 2 diabetic individuals compared to lean individuals. FFAR3 is a G protein couple receptor involved in the development and survival of IECs (Kasubuchi et al. 2015). Prebiotics and Bifidobacterium strains lowered the uptake of LPS from the gut lumen reinforcing tight junctions of epithelial cells (Al-Sheraji et al. 2015; Fukuda et al. 2011; Meyer and Stasse-Wolthuis 2009).
The genetic background necessary to develop these disorders (intrinsic factors) can be influenced by the metabolism of the gut microbiota (extrinsic factors) (Salvucci 2013, 2016; Proal et al. 2009). For example, multiple genetic risk variants associated with the type 1 (T1D) and type 2 (T2D) diabetes have been postulated. Most T1D-associated variants are in genes controlling immunity (e.g. HLA), whereas T2D-associated genes control transcription, adipo-cytokine signals and β cell cycle regulation. Naturally, the increased incidence of T1D and T2D is not due to only to the genetic background, but the result of gene interactions with dynamic environmental risk factors, particularly impactful in early childhood (Paun and Danska 2016). In the same way, an altered gut microbiota characterized by low diversity and resilience has been associated to T1D and T2D (Paun and Danska 2016).
Crohn’s disease (CD) is a condition in which the lining of the gastrointestinal tract becomes inflamed, causing severe diarrhoea and abdominal pain (Reddy and Fried 2009). The incidence of CD has increased in Western countries. Both CD and ulcerative colitis (UC) are idiopathic pathologies of IBD. The manifestations of this disease include an aggressive cellular immune response, over expression of pro inflammatory mediators in different T lymphocyte subsets (Th1 and Th17, Th2), abnormal antigen presentation, and aberrant thymic education with a concomitant penetration of the intestinal barrier by luminal bacteria. Underlying inflammation triggers Crohn’s disease and the luminal microbiota change. This process leads to nonstandard host-microbiota interactions that can aggravate the disease. In fact, CD patients present decreased bacterial diversity and richness (Imhann et al. 2016; Khanna and Raffals 2017). Moreover, reports agree that the phyla Bacteroidetes and Proteobacteria are increased, while the phylum Firmicutes is decreased in patients with UC. Also Bifidobacterium, Roseburia, Faecalibacterium and Phascolarctobacterium are in reduced abundance compared with healthy individuals (Imhann et al. 2016; Khanna and Raffals 2017) . Recently, it was reported that there are significant differences in the gut microbiota of healthy individuals who carried a high genetic risk for IBD, including a decrease in species of Roseburia known to be butyrate producers (Imhann et al. 2016).
The remnants of ancestral integration of the microbial DNA into the human genome can be detected today. Helicobacter pylori and other potentially pathogenic microorganisms, like Toxoplasma, which possibly only infects humans accidentally, can co-habit with the host with detrimental effects (Cremonini and Gasbarrini 2003; Marini et al. 2007). Likewise, herpes viruses like varicella-zoster remain dormant in the human nervous system after an infection in the cranial nerve ganglia and autonomic ganglia. Viruses can reactivate and cause neurologic conditions (Gilden et al. 2000). Schizophrenia has been associated with gene loci that include an ancestral cytomegalovirus (Børglum et al. 2014). All of the above listed organisms leave entire copies or fragments of their genomes, which remain lodged within us for the rest of our lives interacting with our nervous system (Kramer and Bressan 2015). Parasite co-habitation can drive evolutive changes of host organisms (Vannier-Santos and Lenzi 2011). With epidemiological transitions, there was an intensification in sanitization and de-worming of humans that might have led to immunological imbalances. Parasites could act as regulators of immune system helping to control excessive inflammatory responses (Helmby 2015). For example, the induction of suppressive regulatory T cells (Tregs) is a common mechanism to regulate inflammatory effects (Paun and Danska 2015). Interestingly, activation of Tregs appears to be a feature of both microbiota colonization (Gifford et al. 2008; Jamain et al. 2001) and helminth parasite infection (Medstrand and Mager 1998). Bacteroides fragilis, Bifidobacterium infantis, Clostridium spp., and Lactobacillus spp., as well nematode parasites, such as Heligmosomoides polygyrus and Strongyloides ratti can induce or suppress regulatory Tregs (Reynolds et al. 2015). Studies in animal models have demonstrated that intestinal helminth infections can inhibit the development of intestinal inflammation (Rook 2009; Reddy and Fried 2009; Reynolds et al. 2015). Likewise, pre-clinical assays have suggested a beneficial effect of helminth infections on inflammatory bowel conditions, allergies, asthma and multiple sclerosis (Reddy and Fried 2009; Correale and Farez 2011). Specifically, treatment with embryonated Trichuris suis eggs resulted in significant disease remission in patients with UC and CD (Reddy and Fried 2007).
Microbiota restoration by fecal microbiota transplantation (FMT) is another potential emerging therapeutic. This therapy has shown to be a successfully treatment in recurrent Clostridium difficile infection (CDI) (Gianotti and Moss 2017). However, CD like other autoimmune diseases, has a more complex pathogenesis and the success of FMT can be dependant of additional factors including the donor microbial profile, inflammatory burden, and the microbial diversity of the recipient (Gianotti and Moss 2017).
We can conclude from this section that two major epidemiological transitions (establishment of nomadic hunter-gatherers and industrial revolution), with the accompanying cultural and dietary shifts led to a significant reduction in microbiota diversity, with a concomitant loss of function that could be correlated to the increased occurrence of auto immune diseases. Restoration of the gut microbiota by FMT and helminth therapies are being considered as emergent treatments.
The Infant Microbiota and the Impact of Antibiotics
The natural drift of human-microbiota superorganism and the maintenance of its autopoiesis are observed in inheritance of the microbiota. This superorganism inherited epigenetic changes that are responsible of fine tuning along their development. This epigenetic landscape is involved in the adaptation to the environment including the interaction with the microbiota at different stages of development (Indrio et al. 2017).
The placental microbiome is composed of four dominant phyla: Proteobacteria, Bacteroidetes, Actinobacteria and Firmicutes (Zheng et al. 2017). Then, the mother’s microbiota provides the newborn with a specific microbial inoculum at birth. Vaginal delivery supplies the baby with a bacterial composition resembling their mother’s vaginal microbiota. Lactobacillus, Prevotella, and Sneathia are the predominant groups. Babies delivered by Cesarean section acquire bacteria that resembles those present on the skin, like Staphylococcus, Corynebacterium, and Propionibacterium (De Filippo et al. 2010; Koleva et al. 2015; Dominguez-Bello et al. 2010, 2016). Vaginal delivery may afford the neonate immediate access at birth to microbiota that allows maximal energy harvest during the incipient hours of life (Mueller et al. 2015). In fact, studies suggest a correlation between C-section delivery and the immune system due to the essential role of the maternal microbiota on the development of the perinatal immune system. Increased risk for asthma, allergies and obesity has been reported for C-section delivered infants (Ximenez and Torres 2017). Coincidentally, babies born via C-section have a different associated microbiome at least during the first 4 weeks of age. Full-term vaginally delivered infants present higher diversity compared to C-section babies (Ximenez and Torres 2017; von Mutius 2017). They also displayed an increased faecal abundance of Firmicutes, Bacteroides and Bifidobacterium and lower abundance of Actinobacteria compared to C-section babies (Ximenez and Torres 2017; Hill et al. 2017). However, the discrepancies between groups gradually disappear and are not observed by 6 months of age.
As elaborated in Part III, Chapter “Early Gut Microbiome, a Good Start in Nutrition and Growth May Have Lifelong Lasting Consequences”, after birth, breastfeeding provides the baby with the microorganisms and immunological components to promote optimal growth. Moreover, breast milk also contains complex oligosaccharides (prebiotics) that promote the establishment of beneficial bacteria like Bifidobacterium longum (Indrio et al. 2017; Mueller et al. 2015). This is can be considered as the first example of how bacteria in food “feed” the gut microbiota.
The infant gut microbiota differ significantly from the adult microbiota. In fact its compositional structure and function are attained only after dynamic changes experienced during the first 3 years of life. Staphylococcus, Streptococcus and Enterobacteria are the first colonizers of the gut (Wampach et al. 2017; Voreades et al. 2014) . Then, these taxa are replaced by facultative anaerobic bacteria of the phyla Actinobacteria and Firmicutes (Turroni et al. 2012). At adulthood, 90% of microbes colonizing the gut are represented by only six phyla, Firmicutes, Bacteroides, Actinobacteria, Fusobacteria, Proteobacteria, and Verrucomicrobia (Arumugam et al. 2011). Other factors impact the infant gut microbiota including mom’s diet and environmental factors. The degree of impact is more relevant early in life, when the gut microbiota has not yet been fully established.
Antibiotic treatment is a factor that can perturb the microbiota early in life and could be a missing link in autoimmunity disorders (Iizumi et al. 2017). Antibiotic use in children has become widespread. In fact, children in the US are prescribed a mean of three courses of antibiotic treatment before they are 2 years of age. This represents more than the double from European countries. A recent cross-national study has shown that South Korean children had the highest rate of antimicrobial prescriptions, with 3.41 prescribed courses per child-year during the first 2 years of life. Italy and Spain had a mean of 1 and 1.6, respectively, while Norway had only 0.5 courses per child-year (Youngster et al. 2017).
Antibiotic use, administered either orally or intravenously, reduces gut microbiota diversity (Iizumi et al. 2017) (Fig. 2). Repeated exposure to antibiotics during the first year of life caused a less stable microbial community, which lasted until the third year, and a decreased diversity (Maturana-Romesin and Mpodozis 2000). Also an epidemiological study showed that children that received antibiotics in the first 6 months of life had a significantly higher risk of being overweight at 7 years old (Trasande et al. 2013). Mouse studies have demonstrated the effects of antibiotics on microbiota. For example, penicillin G, V and vancomycin have been associated with increased weight, fat mass and insulin resistance (Iizumi et al. 2017). Likewise, azithromycin significantly increased weight gain risk. Other antibiotics including meropenem, cefotaxime and ticarcillin-clavulanate (Gibson et al. 2016) administered to preterm neonates affected severely intestinal species richness. Macrolides had a similar impact in children aged 2–7 years changing composition of the gut microbiome, decreased richness, and decreased abundance of Bifidobacterium, with increased levels of Proteobacteria (Enterobacteriaceae) and Bacteroidetes (Korpela et al. 2016). These changes persisted up to 2 years after macrolide treatment and were associated with increased risk of asthma and obesity (Korpela et al. 2016).
Administration of antibiotics to the mother during pregnancy may also affect the oral microbiota of the newborn. Maternal intrapartum antibiotic treatment is a key regulator of the initial neonatal oral microbiota. The oral microbiota of the infants was more similar to the oral microbiota than to the placenta or gut microbiota of the mother. Families belonging to Proteobacteria were abundant after antibiotics exposure of the mother while Streptococcaceae, Gemellaceae and Lactobacillales dominated in unexposed neonates (Gomez-Arango et al. 2017).
Finally, antibiotics have been shown to delay the maturation of microbiota, due to reduction of Lachnospiraceae, Erysipelotrichaceae and Clostridiales. These families are very sensitive to antibiotic exposure and this causes a reduction in production of butyrate and other SCFA. These reductions impact on infant immunity, signalling epithelial cell, colonic T regulatory cells and macrophages and maturation of the gut (Ximenez and Torres 2017). Therefore, Lachnospiraceae is useful as an indicator of microbiota maturation.
There is strong evidence that maternal exposure of antibiotics and administration of antibiotics in neonates alter and delay the maturation of microbiota. These changes have consequences at immunological level and it is associated to higher risk of diseases in adult life. Early antibiotic exposure may have other long term consequences related to higher risk to inflammatory and immune diseases. Also behaviour, anxiety, blood-brain-barrier integrity and brain cytokines expression could be related to antenatal and postnatal antibiotic exposure.
Epigenetics and the Gut Microbiota
Epigenetics refers to modifications of the genome that do not alter the DNA sequence but cause mitotically and meiotically heritable changes (Morgan and Whitelaw 2008). There is a wide variety of mechanisms that reduce, activate or inactivate genes and regulatory networks influencing early cellular differentiation, and creating new phenotypic traits during pregnancy and within the neonatal period (Indrio et al. 2017; Liu 2007). A number of antenatal and postnatal factors, including diet and composition of the microbiota, contribute to epigenetic changes that have an influence on lifelong health and disease by modifying inflammatory molecular pathways and the immune response (Indrio et al. 2017).
The main epigenetic mechanism is the methylation of cytosine residues in DNA, which results in remodelling of the chromatin structure and RNA-mediated regulation. These modifications may upregulate or downregulate gene expression according to the type of change and its position. A group of enzymes catalyze DNA methylation. It consists in methylation of cytosine residues followed by guanine or adenine and the consequent suppression of gene expression (Abdolmaleky et al. 2015).
Other epigenetic mechanisms are acetylation and phosphorylation. Different amino acids of histone tails can be methylated, acetylated or phosphorylated (Alam et al. 2017). Action of acetyltransferases results in acetylation of histone residues that increases accessibility of nucleosomal DNA to transcription factors, thus increasing the expression levels of corresponding genes (Alam et al. 2017).
Epigenetic modifications have key roles in the development of human organs, especially the central nervous system during the embryo-fetal, perinatal, and later stages of life (Alam et al. 2017; Jablonka and Raz 2009). These mechanisms are widely observed in nature acting in response to environmental factors and interact in a regulatory network involving more than 1000 microRNAs. Each microRNA can target hundreds of transcripts increasing cell adaptability in a tissue-specific manner. They demonstrate how environmental factors increase genomic flexibility, being maintained through generations (Table 1) (Alam et al. 2017).
Perturbations of the perinatal microbiota by specific practices (lack of skin contact, cesarean delivery, formula feeding, antibiotics) play a role in the susceptibility to late-onset diseases like obesity, diabetes, allergies, asthma, and other autoimmune disorders potentially by developing a particular genetic repertoire and modulating the immune development through epigenetic modifications (Salvucci 2016; Indrio et al. 2017; Jablonka and Raz 2009; Bossdorf et al. 2008). Individuals inherited from parents particular epigenetic changes (acetylation, methylation or phosphorylation of genes) that influence the adaptation of the newborn and are related to metabolism and immunological status (Myles et al. 2013).
Depletion of the gut microbiota and the related inflammatory, immune and neuroendocrine manifestations have shown to be linked by epigenetic changes (Table 1) (Myles et al. 2013; Indrio et al. 2017; Morgan and Whitelaw 2008; Liu 2007; Jablonka and Raz 2009). The fetal epigenetic program is influenced by diet and the microbiota along their development. Malnutrition or overnutrition during pregnancy cause negative effects on the offspring health at childhood and adulthood (Lee 2015; Alfaradhi et al. 2016; Roberts et al. 2015). For example, in a mouse study supplementation with folic acid, vitamin B12, methionine, zinc, betaine, and choline resulted in higher rates of allergic airway inflammation due to excessive methylation of the runt-related transcription factor 3 (RUNX3), a mediator of T-lymphocyte differentiation (Håberg et al. 2009) . Zinc status can exert a fundamental influence on the epigenome. Zinc deficiency during intrauterine life and childhood could contribute to the development of chronic inflammatory diseases by aberrant methylation (Tomat et al. 2011).
Microbiota has the ability to induce epigenetic changes in the human-microbiota superorganism (Salvucci 2013). Among the mechanisms through which intestinal bacteria can influence human health, epigenetic modifications are the most important. The epigenome influences the establishment of the microbiota but also microorganisms can introduce epigenetic changes in genes relevant to immunological, metabolic, and neurological development and functions. For instance, SCFAs that regulate gene expression by DNA methylation or histone modifications are crucial metabolites of microbiota (Indrio et al. 2017; McKenzie et al. 2017).
The host-microbial interactions that characterize human superorganism start before the early postnatal period. In fact, the microbiota found in the amniotic fluid starts to modulate the foetus epigenetically since its placental life (West et al. 2015; Zheng et al. 2017; Urushiyama et al. 2017). Moreover, diet, antibiotic exposure and other environmental factors influence the microbiota composition and their epigenetic changes. All these factors contribute the higher or lesser risk to develop allergies and inflammatory diseases (Indrio et al. 2017; Cremonini and Gasbarrini 2003; Schaub et al. 2009).
It is difficult to establish a causal relationship between the diversity or prevalence of certain species in microbiota and the epigenome. Still, there are some clues related to the ratios of Firmicutes/Bacteroides and distinct DNA methylation profiles. Ratios associated with metabolic disorders have also differences in methylation in genes related to obesity, metabolism, and inflammation (Kumar et al. 2014). In obesity, there are differences in methylation of the promoter of SCD5 that encodes an stearoyl-coenzyme A (CoA) desaturase, which has a key function in the catalysis of monounsaturated fatty acids from saturated fatty acids. The promoter was more methylated in the groups of individuals with higher Firmicutes/Bacteroides ratio (Kumar et al. 2014). In IBD there are epigenetic marks that define the visceral hypersensitivity and modulate stress-induced visceral pain. Altered microbiota profiles are concomitant (Indrio et al. 2017; Jeffery et al. 2012).
Epigenetic changes like aberrant DNA methylation, histone modifications and dysregulation of micro-RNAs are linked to the pathogenesis of mental disorders. Moreover, a number of psychiatric drugs modulates features of the epigenome, for instance, tubastatin can restore the reduction in tubulin acetylation observed in Rett syndrome (Abdolmaleky et al. 2015). Valproate, lithium, lamotrigine, haloperidol, clozapine, olanzapine and risperidone alter the expression of many miRNAs (Abdolmaleky et al. 2015) . This implies that epigenetic modifications are a plausible alternative for treatment of mental disorders and, in consequence, modulation of gut microbiota can be a blank for therapies. The gut microbiota contributes to epigenetic fine-tuning confirming its role as an ontogenic missing link in mental illnesses. These changes are not only indirect effects mediated by metabolic by-products, it was observed that infection with some bacteria such as Helicobacter pylori is specifically linked to DNA methylation and may decrease expression of O6-methylguanine DNA methyltransferase (Sepulveda et al. 2010).
Maternal diet, lifestyle, mode of delivery, early nutrition and gut microbiota define an epigenome of newborn that potentially has a crucial role in the development of microbiota-related diseases. The mother (superorganism) transmits epigenetic changes to the foetus that interact with the microbiota and introducing changes at this level can be considered the missing link that could define the success of treatments like helminth therapy and FMT in the diseases related to the microbiota depletion, including mental disorders. The epigenetic heredity allows the child to be adapted to the environment that the mother has experienced. Many other antenatal and postnatal factors could distort that synchrony. The probiotic treatments and modulation of microbiota should take into account the ability of some bacteria to induce the epigenetic changes to re-establish the homeostasis. The role for microbiota-induced epigenetic modifications and their effects is an emerging research field that is in the initial stages and its development will contribute to a better understanding of the interrelationship host-microbiota superorganism.
Conclusions
Scientific and anecdotal evidence seem to suggest that overall loss of microbial diversity and the loss of specific bacterial groups associated with two historical epidemiological transitions could be potentially correlated with an over-reactive immune system and consequent increased occurrence of allergies and other autoimmune disorders (Salvucci 2013; Proal et al. 2009; Tlaskalová-Hogenová et al. 2004). The common background of immune, inflammatory or systemic imbalance point to treatments aiming to restore the gut microbiota through probiotics, prebiotics and diet are plausible treatments for these emergent diseases (van der Meulen et al. 2016).
References
Abdolmaleky, H. M., Zhou, J.-R., & Thiagalingam, S. (2015). An update on the epigenetics of psychotic diseases and autism. Epigenomics, 7(3), 427–449. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/26077430.
Alam, R., Abdolmaleky, H. M., & Zhou, J.-R. (2017). Microbiome, inflammation, epigenetic alterations, and mental diseases. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 174(6), 651–660.
Alfaradhi, M. Z., Kusinski, L. C., Fernandez-Twinn, D. S., Pantaleão, L. C., Carr, S. K., Ferland-McCollough, D., et al. (2016). Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, lean male mice offspring. Endocrinology, 157(11), 4246–4256.
Allen, S. J., Watson, J. J., Shoemark, D. K., Barua, N. U., & Patel, N. K. (2013). GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacology & Therapeutics, 138(2), 155–175.
Al-Sheraji, S. H., Amin, I., Azlan, A., Manap, M. Y., & Hassan, F. A. (2015). Effects of Bifidobacterium longum BB536 on lipid profile and histopathological changes in hypercholesterolaemic rats. Beneficial Microbes, 6(5), 661–668.
Anderson, C. M., Ralph, J. L., Johnson, L., Scheett, A., Wright, M. L., Taylor, J. Y., et al. (2015). First trimester vitamin D status and placental epigenomics in preeclampsia among Northern Plains primiparas. Life Sciences, 129, 10–15.
Arrieta, M.-C., Stiemsma, L. T., Amenyogbe, N., Brown, E. M., & Finlay, B. (2014). The intestinal microbiome in early life: Health and disease. Frontiers in Immunology, 5, 427. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4155789/.
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature, 473(7346), 174–180.
Bell, P. J. L. (2001). Viral eukaryogenesis: Was the ancestor of the nucleus a complex DNA virus? Journal of Molecular Evolution, 53(3), 251–256.
Belyi, V. A., Levine, A. J., & Skalka, A. M. (2010). Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: The parvoviridae and circoviridae are more than 40 to 50 million years old. Journal of Virology, 84(23), 12458–12462.
Bercik, P., Verdu, E. F., Foster, J. A., Macri, J., Potter, M., Huang, X., et al. (2010). Chronic gastrointestinal inflammation induces anxiety-like behavior and alters central nervous system biochemistry in mice. Gastroenterology, 139(6), 2102–2112.e1.
Bercik, P., Collins, S. M., & Verdu, E. F. (2012). Microbes and the gut-brain axis. Neurogastroenterology & Motility, 24(5), 405–413.
Børglum, A. D., Demontis, D., Grove, J., Pallesen, J., Hollegaard, M. V., Pedersen, C. B., et al. (2014). Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Molecular Psychiatry, 19(3), 325–333.
Borre, Y. E., O’Keeffe, G. W., Clarke, G., Stanton, C., Dinan, T. G., & Cryan, J. F. (2014). Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends in Molecular Medicine, 20(9), 509–518.
Bossdorf, O., Richards, C. L., & Pigliucci, M. (2008). Epigenetics for ecologists. Ecology Letters, 11(2), 106–115.
Brucker, R. M., & Bordenstein, S. R. (2012). Speciation by symbiosis. Trends in Ecology & Evolution, 27(8), 443–451.
Casjens, S. (2003). Prophages and bacterial genomics: What have we learned so far? Molecular Microbiology, 49(2), 277–300.
Chernoff, Y. O. (2001). Mutation processes at the protein level: Is Lamarck back? Mutation Research, 488(1), 39–64.
Chu, D. M., Antony, K. M., Ma, J., Prince, A. L., Showalter, L., Moller, M., et al. (2016). The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Medicine, 8(1), 77.
Collins, S. M., Surette, M., & Bercik, P. (2012). The interplay between the intestinal microbiota and the brain. Nature Reviews. Microbiology, 10(11), 735–742.
Correale, J., & Farez, M. F. (2011). The impact of parasite infections on the course of multiple sclerosis. Journal of Neuroimmunology, 233(1–2), 6–11.
Corrêa-Oliveira, R., Fachi, J. L., Vieira, A., Sato, F. T., & Vinolo, M. A. R. (2016). Regulation of immune cell function by short-chain fatty acids. Clinical & Translational Immunology, 5(4), e73.
Craig, J. P., Bekal, S., Niblack, T., Domier, L., & Lambert, K. N. (2009). Evidence for horizontally transferred genes involved in the biosynthesis of vitamin B1, B5, and B7 in Heterodera glycines. Journal of Nematology, 41(4), 281–290.
Cremonini, F., & Gasbarrini, A. (2003). Atopy, Helicobacter pylori and the hygiene hypothesis. European Journal of Gastroenterology & Hepatology, 15(6), 635–636.
Crittenden, A. N., Henry, A. G., Mabulla, A., Consolandi, C., Peano, C., Luiselli, D., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nature Communications, 5, 3654.
Cryan, J. F., & Dinan, T. G. (2012). Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nature Reviews. Neuroscience, 13(10), 701–712.
Davison, J. M., Mellott, T. J., Kovacheva, V. P., & Blusztajn, J. K. (2009). Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. The Journal of Biological Chemistry, 284(4), 1982–1989.
De Filippo, C., Cavalieri, D., Di Paola, M., Ramazzotti, M., Poullet, J. B., Massart, S., et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America, 107(33), 14691–14696.
Deschamps, P., Zivanovic, Y., Moreira, D., Rodriguez-Valera, F., & López-García, P. (2014). Pangenome evidence for extensive interdomain horizontal transfer affecting lineage core and shell genes in uncultured planktonic thaumarchaeota and euryarchaeota. Genome Biology and Evolution, 6(7), 1549–1563.
Dinan, T. G., Borre, Y. E., & Cryan, J. F. (2014). Genomics of schizophrenia: Time to consider the gut microbiome? Molecular Psychiatry, 19(12), 1252–1257.
Ding, D., & Lipshitz, H. D. (1994). Spatially regulated expression of retrovirus-like transposons during Drosophila melanogaster embryogenesis. Genetical Research, 64(03), 167–181.
Dominguez-Bello, M. G., Costello, E. K., Contreras, M., Magris, M., Hidalgo, G., Fierer, N., et al. (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proceedings of the National Academy of Sciences of the United States of America, 107(26), 11971–11975.
Dominguez-Bello, M. G., De Jesus-Laboy, K. M., Shen, N., Cox, L. M., Amir, A., Gonzalez, A., et al. (2016). Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nature Medicine, 22(3), 250–253.
Evans, J. M., Morris, L. S., & Marchesi, J. (2013). The gut microbiome: The role of a virtual organ in the endocrinology of the host. The Journal of Endocrinology, 218(8), R37–R47.
Feschotte, C., & Gilbert, C. (2012). Endogenous viruses: Insights into viral evolution and impact on host biology. Nature Reviews. Genetics, 13(4), 283–296.
Findley, K., Williams, D. R., Grice, E. A., & Bonham, V. L. (2016). Health disparities and the microbiome. Trends in Microbiology, 24(11), 847–850.
Forterre, P. (2006). The origin of viruses and their possible roles in major evolutionary transitions. Virus Research, 117(1), 5–16.
Foster, J. A., & McVey Neufeld, K.-A. (2013). Gut-brain axis: How the microbiome influences anxiety and depression. Trends in Neurosciences, 36(5), 305–312.
Fuchsman, C. A., Collins, R. E., Rocap, G., & Brazelton, W. J. (2017). Effect of the environment on horizontal gene transfer between bacteria and archaea. PeerJ, 5, e3865. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5624296/.
Fukuda, S., Toh, H., Hase, K., Oshima, K., Nakanishi, Y., Yoshimura, K., et al. (2011). Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature, 469(7331), 543–547.
Georgiades, K., & Raoult, D. (2011). The rhizome of Reclinomonas americana, Homo sapiens, Pediculus humanus and Saccharomyces cerevisiae mitochondria. Biology Direct, 6, 55.
Georgiades, K., & Raoult, D. (2012). How microbiology helps define the rhizome of life. Frontiers in Cellular and Infection Microbiology, 2, 60.
Gianotti, R. J., & Moss, A. C. (2017). Fecal microbiota transplantation: From Clostridium difficile to inflammatory bowel disease. Gastroenterología y Hepatología, 13(4), 209–213.
Gibson, M. K., Wang, B., Ahmadi, S., Burnham, C.-A. D., Tarr, P. I., Warner, B. B., et al. (2016). Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nature Microbiology, 1, 16024.
Gifford, R. J., Katzourakis, A., Tristem, M., Pybus, O. G., Winters, M., & Shafer, R. W. (2008). A transitional endogenous lentivirus from the genome of a basal primate and implications for lentivirus evolution. Proceedings of the National Academy of Sciences of the United States of America, 105(51), 20362–20367.
Gilbert, S. F., McDonald, E., Boyle, N., Buttino, N., Gyi, L., Mai, M., et al. (2010). Symbiosis as a source of selectable epigenetic variation: Taking the heat for the big guy. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 365(1540), 671–678.
Gilden, D. H., Kleinschmidt-DeMasters, B. K., LaGuardia, J. J., Mahalingam, R., & Cohrs, R. J. (2000). Neurologic complications of the reactivation of Varicella–Zoster Virus. The New England Journal of Medicine, 342(9), 635–645.
Gomez-Arango, L. F., Barrett, H. L., McIntyre, H. D., Callaway, L. K., Morrison, M., & Dekker, N. M. (2017). Antibiotic treatment at delivery shapes the initial oral microbiome in neonates. Scientific Reports, 7, 43481.
Håberg, S. E., London, S. J., Stigum, H., Nafstad, P., & Nystad, W. (2009). Folic acid supplements in pregnancy and early childhood respiratory health. Archives of Disease in Childhood, 94(3), 180–184.
Hayley, S., Audet, M.-C., & Anisman, H. (2016). Inflammation and the microbiome: Implications for depressive disorders. Current Opinion in Pharmacology, 29, 42–46.
Helmby, H. (2015). Human helminth therapy to treat inflammatory disorders—Where do we stand? BMC Immunology, 16, 12. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4374592/.
Hernández-Valero, M. A., Rother, J., Gorlov, I., Frazier, M., & Gorlova, O. Y. (2013). Interplay between polymorphisms and methylation in the H19/IGF2 gene region may contribute to obesity in Mexican-American children. Journal of Developmental Origins of Health and Disease, 4(6), 499–506.
Hill, C. J., Lynch, D. B., Murphy, K., Ulaszewska, M., Jeffery, I. B., O’Shea, C. A., et al. (2017). Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome, 5(1), 4.
Hollingsworth, J. W., Maruoka, S., Boon, K., Garantziotis, S., Li, Z., Tomfohr, J., et al. (2008). In utero supplementation with methyl donors enhances allergic airway disease in mice. The Journal of Clinical Investigation, 118(10), 3462–3469.
Holmes, E. C. (2011). The evolution of endogenous viral elements. Cell Host & Microbe, 10(4), 368–377.
Hotopp, J. C. (2011). Horizontal gene transfer between bacteria and animals. Trends in Genetics, 27(4), 157–163.
Hunter, P. (2008). We are what we eat. The link between diet, evolution and non-genetic inheritance. EMBO Reports, 9(5), 413–415.
Iizumi, T., Battaglia, T., Ruiz, V., & Perez Perez, G. I. (2017). Gut microbiome and antibiotics. Archives of Medical Research, 48(8), 727–734. Retrieved from https://www.sciencedirect.com/science/article/pii/S0188440917302333.
Imhann, F., Vila, A. V., Bonder, M. J., Fu, J., Gevers, D., Visschedijk, M. C., et al. (2016). Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut, 67(1), 108–119.
Indrio, F., Martini, S., Francavilla, R., Corvaglia, L., Cristofori, F., Mastrolia, S. A., et al. (2017). Epigenetic matters: The link between early nutrition, microbiome, and long-term health development. Frontiers in Pediatrics, 5, 178. Retrieved from http://journal.frontiersin.org/article/10.3389/fped.2017.00178/full?&utm_source=Email_to_rerev_&utm_medium=Email&utm_content=T1_11.5e4_reviewer&utm_campaign=Email_publication&journalName=Frontiers_in_Pediatrics&id=286554.
Jablonka, E., & Raz, G. (2009). Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. The Quarterly Review of Biology, 84(2), 131–176.
Jamain, S., Girondot, M., Leroy, P., Clergue, M., Quach, H., Fellous, M., et al. (2001). Transduction of the human gene FAM8A1 by endogenous retrovirus during primate evolution. Genomics, 78(1–2), 38–45.
Jeffery, I. B., O’Toole, P. W., Öhman, L., Claesson, M. J., Deane, J., Quigley, E. M. M., et al. (2012). An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut, 61(7), 997–1006.
Jensen Pena, C., Monk, C., & Champagne, F. A. (2012). Epigenetic effects of prenatal stress on 11beta-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One, 7(6), e39791.
Kaati, G., Bygren, L. O., & Edvinsson, S. (2002). Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. European Journal of Human Genetics, 10(11), 682–688.
Kasubuchi, M., Hasegawa, S., Hiramatsu, T., Ichimura, A., & Kimura, I. (2015). Dietary gut microbial metabolites, short-chain fatty acids, and host metabolic regulation. Nutrients, 7(4), 2839–2849.
Katzourakis, A., & Gifford, R. J. (2010). Endogenous viral elements in animal genomes. PLoS Genetics, 6(11), e1001191.
Kau, A. L., Ahern, P. P., Griffin, N. W., Goodman, A. L., & Gordon, J. I. (2011). Human nutrition, the gut microbiome and the immune system. Nature, 474(7351), 327–336.
Khanna, S., & Raffals, L. E. (2017). The microbiome in Crohn’s disease: Role in pathogenesis and role of microbiome replacement therapies. Gastroenterology Clinics of North America, 46(3), 481–492.
Khot, V., Chavan-Gautam, P., & Joshi, S. (2015). Proposing interactions between maternal phospholipids and the one carbon cycle: A novel mechanism influencing the risk for cardiovascular diseases in the offspring in later life. Life Sciences, 129, 16–21.
Khot, V. V., Chavan-Gautam, P., Mehendale, S., & Joshi, S. R. (2017). Variable methylation potential in preterm placenta: Implication for epigenetic programming of the offspring. Reproductive sciences (Thousand Oaks, Calif.), 24(6), 891–901.
Kidwell, M. G., & Lisch, D. R. (2000). Transposable elements and host genome evolution. Trends in Ecology & Evolution, 15(3), 95–99.
Koleva, P. T., Bridgman, S. L., & Kozyrskyj, A. L. (2015). The infant gut microbiome: Evidence for obesity risk and dietary intervention. Nutrients, 7(4), 2237–2260.
Korpela, K., Salonen, A., Virta, L. J., Kekkonen, R. A., Forslund, K., Bork, P., et al. (2016). Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nature Communications, 7, 10410.
Kozo-Polyansky, B. M., & Margulis, L. (2010). Symbiogenesis: A new principle of evolution (198p). Cambridge, MA: Paperbackshop UK Import.
Kramer, P., & Bressan, P. (2015). Humans as superorganisms: How microbes, viruses, imprinted genes, and other selfish entities shape our behavior. Perspectives on Psychological Science, 10(4), 464–481.
Kumar, H., Lund, R., Laiho, A., Lundelin, K., Ley, R. E., Isolauri, E., et al. (2014). Gut microbiota as an epigenetic regulator: Pilot study based on whole-genome methylation analysis. mBio, 5(6), e02113–e02114.
Langley-Evans, S. C. (2000). Critical differences between two low protein diet protocols in the programming of hypertension in the rat. International Journal of Food Sciences and Nutrition, 51(1), 11–17.
Leal Éde, S., & Zanotto, P. M. (2000). Viral diseases and human evolution. Memórias do Instituto Oswaldo Cruz, 95, 193–200.
Lederberg, J., & McCray, A. T. (2001). 'Ome sweet 'omics—A genealogical treasury of words. The Scientist., 15(7), 8.
Lee, H.-S. (2015). Impact of Maternal Diet on the Epigenome during In Utero Life and the Developmental Programming of Diseases in Childhood and Adulthood. Nutrients, 7(11), 9492–9507.
Lee, Y. Y., & Chua, A. S. B. (2011). Influence of Gut Microbes on the Brain-Gut Axis (Gut 2011;60:307-317). Journal of Neurogastroenterology and Motility, 17(4), 427–429.
Ley, R. E., Backhed, F., Turnbaugh, P., Lozupone, C. A., Knight, R. D., & Gordon, J. I. (2005). Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America, 102(31), 11070–11075.
Liu, Y. (2007). Like father like son. A fresh review of the inheritance of acquired characteristics. EMBO Reports, 8(9), 798–803.
Mallet, F., Bouton, O., Prudhomme, S., Cheynet, V., Oriol, G., Bonnaud, B., et al. (2004). The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proceedings of the National Academy of Sciences of the United States of America, 101(6), 1731–1736.
Margulis, L. (2010). Symbiogenesis. A new principle of evolution rediscovery of Boris Mikhaylovich Kozo-Polyansky (1890–1957). Paleontological Journal, 44(12), 1525–1539.
Margulis, L., & Sagan, D. (2003). Acquiring genomes: A theory of the origin of species (1st ed., 256p). Princeton, NJ: Basic Books.
Marini, E., Maldonado-Contreras, A. L., Cabras, S., Hidalgo, G., Buffa, R., Marin, A., et al. (2007). Helicobacter pylori and intestinal parasites are not detrimental to the nutritional status of Amerindians. The American Journal of Tropical Medicine and Hygiene, 76(3), 534–540.
Maturana-Romesin, H., & Mpodozis, J. (2000). The origin of species by means of natural drift. Revista Chilena de Historia Natural, 73(2), 261–310. Retrieved from https://www.biodiversitylibrary.org/part/116245.
McDonald, J. F., Matyunina, L. V., Wilson, S., Jordan, I. K., Bowen, N. J., & Miller, W. J. (1997). LTR retrotransposons and the evolution of eukaryotic enhancers. Genetica, 100(1–3), 3–13.
McKenzie, C., Tan, J., Macia, L., & Mackay, C. R. (2017). The nutrition-gut microbiome-physiology axis and allergic diseases. Immunological Reviews, 278(1), 277–295.
McKeown, R. E. (2009). The epidemiologic transition: Changing patterns of mortality and population dynamics. American Journal of Lifestyle Medicine, 3(1 Suppl), 19S–26S.
Medstrand, P., & Mager, D. L. (1998). Human-specific integrations of the HERV-K endogenous retrovirus family. Journal of Virology, 72(12), 9782–9787.
Merhej, V., & Raoult, D. (2012). Rhizome of life, catastrophes, sequence exchanges, gene creations, and giant viruses: How microbial genomics challenges Darwin. Frontiers in Cellular and Infection Microbiology, 2, 113. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3428605/.
Meyer, D., & Stasse-Wolthuis, M. (2009). The bifidogenic effect of inulin and oligofructose and its consequences for gut health. European Journal of Clinical Nutrition, 63(11), 1277–1289.
Mi, S., Lee, X., Li, X., Veldman, G. M., Finnerty, H., Racie, L., et al. (2000). Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature, 403(6771), 785–789.
Molinier, J., Ries, G., Zipfel, C., & Hohn, B. (2006). Transgeneration memory of stress in plants. Nature, 442(7106), 1046–1049.
Morgan, D. K., & Whitelaw, E. (2008). The case for transgenerational epigenetic inheritance in humans. Mammalian Genome, 19(6), 394–397.
Mueller, N. T., Bakacs, E., Combellick, J., Grigoryan, Z., & Dominguez-Bello, M. G. (2015). The infant microbiome development: Mom matters. Trends in Molecular Medicine, 21(2), 109–117.
Myles, I. A., Fontecilla, N. M., Janelsins, B. M., Vithayathil, P. J., Segre, J. A., & Datta, S. K. (2013). Parental dietary fat intake alters offspring microbiome and immunity. Journal of Immunology (Baltimore, MD: 1950), 191(6), 3200–3209.
Nemani, K., Hosseini Ghomi, R., McCormick, B., & Fan, X. (2014). Schizophrenia and the gut-brain axis. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 56C, 155–160.
O’Mahony, S. M., Clarke, G., Borre, Y. E., Dinan, T. G., & Cryan, J. F. (2015). Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behavioural Brain Research, 277, 32–48.
Parfrey, L. W., Walters, W. A., & Knight, R. (2011). Microbial eukaryotes in the human microbiome: ecology, evolution, and future directions. Frontiers in Microbiology, 2, 153. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3135866/.
Park, J. H., Stoffers, D. A., Nicholls, R. D., & Simmons, R. A. (2008). Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. The Journal of Clinical Investigation, 118(6), 2316–2324.
Paun, A., & Danska, J. S. (2015). Immuno-ecology: How the microbiome regulates tolerance and autoimmunity. Current Opinion in Immunology, 37, 34–39.
Paun, A., & Danska, J. S. (2016). Modulation of type 1 and type 2 diabetes risk by the intestinal microbiome. Pediatric Diabetes, 17(7), 469–477.
Proal, A. D., Albert, P. J., & Marshall, T. (2009). Autoimmune disease in the era of the metagenome. Autoimmunity Reviews, 8(8), 677–681.
Ramulu, H. G., Raoult, D., & Pontarotti, P. (2012). The rhizome of life: What about metazoa? Frontiers in Cellular and Infection Microbiology, 2, 50.
Reddy, A., & Fried, B. (2007). The use of Trichuris suis and other helminth therapies to treat Crohn’s disease. Parasitology Research, 100(5), 921–927.
Reddy, A., & Fried, B. (2009). An update on the use of helminths to treat Crohn’s and other autoimmunune diseases. Parasitology Research, 104(2), 217–221.
Remely, M., Aumueller, E., Jahn, D., Hippe, B., Brath, H., & Haslberger, A. G. (2014). Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Beneficial Microbes, 5(1), 33–43.
Reynolds, L. A., Finlay, B. B., & Maizels, R. M. (2015). Cohabitation in the intestine: Interactions among helminth parasites, bacterial microbiota, and host immunity. Journal of Immunology (Baltimore, MD: 1950), 195(9), 4059–4066.
Roberts, V. H. J., Frias, A. E., & Grove, K. L. (2015). Impact of maternal obesity on fetal programming of cardiovascular disease. Physiology, 30(3), 224–231.
Rook, G. A. (2009). Review series on helminths, immune modulation and the hygiene hypothesis: The broader implications of the hygiene hypothesis. Immunology, 126(1), 3–11.
Rook, G. A. (2010). 99th dahlem conference on infection, inflammation and chronic inflammatory disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clinical and Experimental Immunology, 160(1), 70–79.
Rook, G. A. W. (2011). Hygiene and other early childhood influences on the subsequent function of the immune system. Digestive Diseases (Basel, Switzerland), 29(2), 144–153.
Rook, G. A. W. (2012). Hygiene hypothesis and autoimmune diseases. Clinical Reviews in Allergy and Immunology, 42(1), 5–15.
Ruden, D. M., & Lu, X. (2008). Hsp90 affecting chromatin remodeling might explain transgenerational epigenetic inheritance in Drosophila. Current Genomics, 9(7), 500–508.
Salvucci, E. (2013). Crohn’s disease within the hologenome paradigm. In Crohn’s disease: Classification, diagnosis and treatment options (pp. 19–32). New York: Nova Publishers.
Salvucci, E. (2016). Microbiome, holobiont and the net of life. Critical Reviews in Microbiology, 42(3), 485–494.
Schaub, B., Liu, J., Höppler, S., Schleich, I., Huehn, J., Olek, S., et al. (2009). Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. Journal of Allergy and Clinical Immunology, 123(4), 774–782.e5.
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nature Communications, 5, 3654. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3996546/.
Schroeder, F. A., Lin, C. L., Crusio, W. E., & Akbarian, S. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biological Psychiatry, 62(1), 55–64.
Sender, R., Fuchs, S., & Milo, R. (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biology, 14(8), e1002533.
Sepulveda, A. R., Yao, Y., Yan, W., Park, D. I., Kim, J. J., Gooding, W., et al. (2010). CpG methylation and reduced expression of O6-methylguanine DNA methyltransferase is associated with Helicobacter pylori infection. Gastroenterology, 138(5), 1836–1844.e4.
Shorter, J., & Lindquist, S. (2006). Destruction or potentiation of different prions catalyzed by similar Hsp104 remodeling activities. Molecular Cell, 23(3), 425–438.
Siefert, J. L. (2009). Defining the mobilome. Methods in Molecular Biology, 532, 13–27.
Smits, S. A., Leach, J., Sonnenburg, E. D., Gonzalez, C. G., Lichtman, J. S., Reid, G., et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science, 357(6353), 802–806.
Strakovsky, R. S., Zhang, X., Zhou, D., & Pan, Y.-X. (2014). The regulation of hepatic Pon1 by a maternal high-fat diet is gender specific and may occur through promoter histone modifications in neonatal rats. The Journal of Nutritional Biochemistry, 25(2), 170–176.
Suter, M. A., Chen, A., Burdine, M. S., Choudhury, M., Harris, R. A., Lane, R. H., et al. (2012). A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. The FASEB Journal, 26(12), 5106–5114.
Symbiosis as a Source of Evolutionary Innovation. MIT Press. 2017. Retrieved from https://mitpress.mit.edu/books/symbiosis-source-evolutionary-innovation
Thorburn, A. N., Macia, L., & Mackay, C. R. (2014). Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity, 40(6), 833–842.
Tlaskalová-Hogenová, H., Stepánková, R., Hudcovic, T., Tucková, L., Cukrowska, B., Lodinová-Zádníková, R., et al. (2004). Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunology Letters, 93(2–3), 97–108.
Tomat, A. L., Costa, M., de Los, Á., & Arranz, C. T. (2011). Zinc restriction during different periods of life: Influence in renal and cardiovascular diseases. Nutrition (Burbank, Los Angeles County, Calif.), 27(4), 392–398.
Tosh, D. N., Fu, Q., Callaway, C. W., McKnight, R. A., McMillen, I. C., Ross, M. G., et al. (2010). Epigenetics of programmed obesity: Alteration in IUGR rat hepatic IGF1 mRNA expression and histone structure in rapid vs. delayed postnatal catch-up growth. American Journal of Physiology-Gastrointestinal and Liver Physiology, 299(5), G1023–G1029.
Trasande, L., Blustein, J., Liu, M., Corwin, E., Cox, L. M., & Blaser, M. J. (2013). Infant antibiotic exposures and early-life body mass. International Journal of Obesity, 37(1), 16.
Turroni, F., Peano, C., Pass, D. A., Foroni, E., Severgnini, M., Claesson, M. J., et al. (2012). Diversity of bifidobacteria within the infant gut microbiota. PLoS One, 7(5), e36957.
Turroni, S., Fiori, J., Rampelli, S., Schnorr, S. L., Consolandi, C., Barone, M., et al. (2016). Fecal metabolome of the Hadza hunter-gatherers: A host-microbiome integrative view. Scientific Reports, 6, 32826.
Urushiyama, D., Suda, W., Ohnishi, E., Araki, R., Kiyoshima, C., Kurakazu, M., et al. (2017). Microbiome profile of the amniotic fluid as a predictive biomarker of perinatal outcome. Scientific Reports, 7(1), 12171.
Van Blerkom Linda, M. (2003). Role of viruses in human evolution. American Journal of Physical Anthropology, 122(37), 14–46.
van der Meulen, T. A., Harmsen, H., Bootsma, H., Spijkervet, F., Kroese, F., & Vissink, A. (2016). The microbiome-systemic diseases connection. Oral Diseases, 22(8), 719–734.
Vannier-Santos, M. A., & Lenzi, H. L. (2011). Parasites or cohabitants: Cruel omnipresent usurpers or creative “eminences grises”? Journal of Parasitology Research, 2011, 1–19.
Varela, F. G., Maturana, H. R., & Uribe, R. (1974). Autopoiesis: The organization of living systems, its characterization and a model. Currents in Modern Biology, 5(4), 187–196.
Villarreal, L. P., & DeFilippis, V. R. (2000). A hypothesis for DNA viruses as the origin of eukaryotic replication proteins. Journal of Virology, 74(15), 7079–7084.
von Mutius, E. (2017). The shape of the microbiome in early life. Nature Medicine, 23(3), 274–275.
Voreades, N., Kozil, A., & Weir, T. L. (2014). Diet and the development of the human intestinal microbiome. Frontiers in Microbiology, 5, 494. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4170138/.
Wallin, I. E. (1927). Symbionticism and the origin of species (208p). Baltimore: Williams & Wilkins Company. Retrieved from http://archive.org/details/symbionticismori00wall
Wampach, L., Heintz-Buschart, A., Hogan, A., Muller, E. E. L., Narayanasamy, S., Laczny, C. C., et al. (2017). Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Frontiers in Microbiology, 8, 738. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5411419/.
West, C. E., Jenmalm, M. C., & Prescott, S. L. (2015). The gut microbiota and its role in the development of allergic disease: A wider perspective. Clinical & Experimental Allergy, 45(1), 43–53.
Ximenez, C., & Torres, J. (2017). Development of the microbiota in infants and its role in maturation of the gut mucosa and the immune system. Arch Med Res, 48(8), 666–680. Retrieved from https://www.sciencedirect.com/science/article/pii/S0188440917302369.
Youngster, I., Avorn, J., Belleudi, V., Cantarutti, A., Díez-Domingo, J., Kirchmayer, U., et al. (2017). Antibiotic use in children—A cross-national analysis of 6 countries. The Journal of Pediatrics, 182(Supplement C), 239–244.e1.
Zelený, M. (1981). Autopoiesis, a theory of living organizations (342p). New York: North Holland.
Zheng, J., Xiao, X., Zhang, Q., Mao, L., Yu, M., Xu, J., et al. (2017). The placental microbiota is altered among subjects with gestational diabetes mellitus: A pilot study. Frontiers in Physiology, 8, 675.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Salvucci, E. (2019). The Disappearing Microbiota: Diseases of the Western Civilization. In: Azcarate-Peril, M., Arnold, R., Bruno-Bárcena, J. (eds) How Fermented Foods Feed a Healthy Gut Microbiota. Springer, Cham. https://doi.org/10.1007/978-3-030-28737-5_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-28737-5_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28736-8
Online ISBN: 978-3-030-28737-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)