
Abstract
Normal human retinal pigmented epithelium (RPE) is a monolayer of cuboidal cells in which the cytoplasm is filled with melanin pigment granules. The basal surface of an RPE cell faces the Bruch’s membrane, a five-layer structure lying between the retina and choroid. The apical surface borders the subretinal space, the apical microvilli interdigitate with the photoreceptor outer segments. The special apical-basal polarized nature of the RPE is required for the proper development of the photoreceptors and choroid. This polarization allows for multiple functions of the RPE cells. RPE cells also contain lipofuscin granules, which increase in number and size throughout life due to outer segment phagocytosis.
The total number of RPE cells has been reported to vary between 3.6 and 6.1 million in a healthy human eye, and cells are mainly regular hexagons. Both aging and certain retinal disorders are associated with not only a decline in the relative number of hexagonal RPE cells and an increased variability in sidedness (pleomorphism), but also with an increase in the variability of cell area (polymegethism). Fundamental differences have been shown in RPE cell density and morphometry across topographical location.
RPE dysfunction can play either a primary role or a secondary role through interactions with the neurosensory retina, Bruch’s membrane, and choriocapillary network in different diseases such as age-related macular degeneration, proliferative retinopathies, central and peripheral retinal dystrophies, mitochondrial diseases, and tumors of the RPE.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Retinal pigment epithelium (RPE) is a single layer of hexagonal/cuboidal epithelial cells which separates the neuroretina from the underlying choroid. These cells are arranged in a mosaic-like pattern. Regular hexagon cell pattern is thought to be the most stable and energetically the most favorable cell arrangement [1,2,3]. Embryologically, RPE is a neuroepithelial derivative. In the early development, RPE differentiates from the neuroectoderm of the optic vesicle and later becomes highly specialized [4]. The apical membrane faces the photoreceptor outer segments and interphotoreceptor matrix [5], the RPE basal lamina is in close contact with the inner collagenous layer of the Bruch’s membrane. This special polarized structure is required for the proper development of the photoreceptors and choroid and this allows for multiple functions of the RPE cells such as adsorbing scattered light to improve spatial resolution, recycling visual pigments to ensure light sensitivity of photoreceptors, and transporting nutrients and metabolites between the choriocapillaris and the neurosensory retina [6]. The impact of RPE in the regulation of eye growth has also been implicated by transmitting signals between the retina and the adjacent choroid and sclera [7]. Tight junctions between neighboring RPE cells constitute the outer blood-retinal barrier, i.e. barrier between the subretinal space and the choriocapillaris. The highly dynamic cytoskeletal elements of the plasma membrane and cytoplasm provide membrane motility, intracellular transport and mechanical strength of the cells. RPE cells show topographic differences, and they also vary within one particular region based on their melanin pigment granule content [3].
Improperly functioning RPE cells can have primary or secondary contribution to several disease processes including dry and wet age-related macular degeneration (AMD), proliferative vitreoretinopathy (PVR), central and peripheral retinal dystrophies, mitochondrial diseases, and RPE neoplasias. Progressive RPE dysfunction and cell loss may lead to a secondary degeneration of photoreceptor rods and cones due to the close relationship between the RPE and photoreceptor cells and due to the loss of barrier functions.
Normal Histology
Normal human RPE is a monolayer of cuboidal cells in which the cytoplasm is filled with melanin pigment granules (also known as melanosomes). The thickness of this layer varies according to the location and age, RPE cells are about 14 μm in height in the macular region whereas toward the periphery they become significantly shorter [8]. Numerous microvilli of the apical membrane extend to the photoreceptor outer segments. The morphology of the polarized RPE layer is visible by transmission electron microscopy (TEM), in which tight junctions, apical microvilli, basal infoldings, shed photoreceptor outer segment disks, melanin pigments, lipofuscin and melanolipofuscin granules can be observed (Fig. 1.1). Melanins are complex pigment structures in the skin, retina and uveal tract which are synthesized within melanosomes. There are two types of cells in the eye which produce melanin: (1) melanocytes in the uveal tract and (2) neuroepithelial cells, such as pigment epithelial cells in the iris, ciliary body and retina. There are substantial differences in the phenotype of melanosomes between the skin, uveal tract and retina. The RPE melanosomes are more elongated or bullet shaped whereas skin and choroidal melanosomes are more spherical [9, 10]. The basal surface of an RPE cell faces the Bruch’s membrane, a five-layer complex structure with multiple functions which lies between the retina and choroid. The RPE basement membrane (basal lamina) is the innermost layer of the Bruch’s membrane with an average thickness of 0.15 μm in young individuals [11]. The structure of this basement membrane is not unique, it consists of type IV collagens, fibronectin, laminin, heparan, chondroitin and dermatan sulfate [12]. The basal plasma membrane of the RPE cells is separated from the basement membrane and it contains several infoldings in order to increase its membrane surface for needed ion transport. Anteriorly there are less infoldings, and they are more pronounced on the posterior pole except adjacent to the optic nerve head where they tend to be less marked. Desmosome and hemidesmosome-like structures are present in the basal plasma membrane resulting in a tight connection between the RPE and Bruch’s membrane [13]. In the lateral surface, zonula occludens, zonula adherens, gap junctions, and occasional desmosomes are present to maintain paracellular permeability, cellular integrity, and cell-cell contact. Zonula occludens or tight junctions are present in the apical half of the cell and are responsible for maintaining the outer blood-retinal barrier. Na,K-ATPase function has been shown to be involved in the normal tight junction structure and permeability in human RPE cells [14]. Zonula adherens or adherens junctions are associated with a rich network of actin filaments. They have multiple functions by supporting cellular motility but also by providing mechanical strength in response to various intracellular signals [15]. The apical microvilli provide increased surface area and play role in special functions, such as phagocytosis of the shed photoreceptor outer segment disks, movements of melanin granules due to light exposure and adhesion to the neurosensory retina. The apical surface faces the subretinal space, the microvilli connect and interdigitate with the photoreceptor outer segments. Light exposure causes the pigment granules migrate into the microvilli in order to decrease the light amount that reaches the rods and cones [16,17,18,19,20]. On the contrary, in scotopic conditions the pigments migrate back to the cell body to allow more light to reach the photoreceptors [16,17,18,19,20].

Normal anatomy of an RPE cell in relation to the surrounding structures. Transmission electron microscopy photo of an RPE cell in a 75-year old Caucasian male. Melanosome (M), shed photoreceptor outer segment disc (sPR), lipofuscin granules (L), melanolipofuscin (ML), phagosome (P), nucleus (N) are seen (3600×)
RPE cells also contain lipofuscin granules, which increase in amount throughout life due to outer segment phagocytosis. The RPE cells are known to show large degrees of heterogeneity in melanin and lipofuscin granule content [3]. It has been proposed that higher melanin content of RPE cells might be protective against the formation of lipofuscin granules [21, 22]. One of the major metabolic roles of RPE cells is to degrade phagocytosed photoreceptor outer segment disk membranes, which are very densely packed structures. Young and Bok observed that approximately 10% of the outer segment disks are generated each day [23], and the same fraction of outer segment dics are phagocytosed by the RPE daily. This is on a circadian rhythm, with rod outer segments consumed at daybreak, and cone outer segments at dusk. Thus, it takes about 10 days for an outer segment disc to traverse from its proximal site of biosynthesis to the distal tip of the outer segment, where it is consumed by the RPE.
Qualitative, morphological analysis of the RPE cells has traditionally been performed during routine histopathologic examination in vitro (Fig. 1.2). More recently, non-invasive imaging modalities are capable of automatic segmentation of the human RPE layer in vivo [24,25,26,27]. Polarization-sensitive optical coherence tomography (OCT) reveals RPE cell bodies specifically in optical biopsy of the retina [27]. Adaptive optics scanning laser ophthalmoscope combined with lipofuscin autofluorescence, dual wavelength imaging and registration is an applicable tool to measure and quantify RPE cell morphometry in vivo [28].

Normal histological appearance of the human RPE in relation to the neurosensory retinal layers and Bruch’s membrane (hematoxylin and eosin, 100×) (a). En face (flat mount) light micrograph of the hexagonal RPE cells (hematoxylin & eosin, 250×) (b). Toluidine blue stain of the normal RPE layer highlighting the intracellular pigment granules (150×) (c). Transmission electron microscopy of the RPE cells with normal cell organelles (1900×) (d)
Cell Morphometry
Pleomorphism, Polymegathism
The total number of RPE cells has been reported to vary between 3.6 and 6.1 million in a healthy human eye [29, 30]. Qualitatively, both aging and degenerative retinal disorders are associated with not only a decline of the relative number of regular hexagonal RPE cells and an increase in the variability of the shape of the RPE cell and increased variability in the number of sides of a cell (pleomorphism), but also with an increase in the variability of cell area (polymegethism).
Hexagonal cells are most frequently (>50–60%) located in the fovea, and the proportion of six-sided RPE cells decreases from the fovea towards the peripheral retina [28, 31]. Cell areas are more variable in older eyes (>80 year old) than in younger ones (≤51 year old) [31]. A previous study confirmed a predominance of hexagonal cells only in the younger adult fovea, where almost 60% of cells had six neighbors [31]. However, RPE cells show continuous remodeling and rearrangement during lifetime, which is thought to reflect a propensity of RPE cells to remain tightly attached to each other so that the outer blood retina barrier remains intact at all costs.
An inter-RPE cell variability (mosaicism) has been described in animal and human studies affecting different cell properties both in macroscopic and molecular level [3]. The pigment granule content varies highly between RPE cells and the expression level of certain proteins. This heterogeneity is thought be due to both genetic and epigenetic processes and contribute to the functional diversity of the RPE layer [3].
Spatial Distribution, Topography
RPE cells extend from the edge of the optic disc to the ora serrata. Anterior to the ora serrata the RPE continues to the pars plana pigment epithelium of the ciliary body. Certain fundamental differences have been shown between RPE cell density (Fig. 1.3) and morphometry in distinct topographical locations (Fig. 1.4) and in different age groups [32]. Cell density at the posterior pole was about four times greater than at the far periphery with inverse correlation to cell area (Fig. 1.3) [32]. The literature shows large ranges of RPE cell density with high variability across age, retinal location and between individuals [3, 28, 30, 33,34,35,36,37]. The RPE cell density has been shown to decrease from the fovea (4220 cells/mm2) to the midperiphery (3002 cells/mm2) and to the peripheral retina (1600 cells/mm2) in normal human eyes [30]. Another investigation described the highest density of 7500 RPE cells/mm2 at the fovea in both younger (≤51 year old) and older subjects (>80 year old) [31]. RPE cell density decreases gradually towards the equator (it measures approximately 5000 cells/mm2 at the edge of the macula) [31]. Also foveal RPE cells are found to be significantly smaller in size than the peripheral cells [2, 31, 38].

RPE cell density (a) and area (b) from the optic nerve head (ONH) to the far peripheral retina. (With permission of the authors: Bhatia SK, Rashid A, Chrenek MA, Zhang Q, Bruce BB, Klein M, et al. Analysis of RPE morphometry in human eyes. Mol Vis. 2016;22:898–916)

Differences in RPE morphometry in the macula, mid-periphery and far-periphery demonstrated by image analysis techniques. (With permission of the authors: Bhatia SK, Rashid A, Chrenek MA, Zhang Q, Bruce BB, Klein M, et al. Analysis of RPE morphometry in human eyes. Mol Vis. 2016;22:898–916)
There is a physiologic variation in RPE cell shape between different locations, i.e. cells are flat and wider anteriorly, whereas they are elongated and narrow posterior to the equator. Salzmann reported the height of RPE cells to be 11–14 μm in the macular region and 8 μm in the other parts of the fundus [8]. The cells also contain more melanin pigment granules within the macular area.
Pathology
Extensive research of the cellular and subcellular pathogenesis of retinal disorders has identified RPE dysfunction as having either a primary role or a secondary role. RPE cells might interact with the neurosensory retina, Bruch’s membrane, and choriocapillary network in different diseases such as age-related macular degeneration (AMD), proliferative retinopathies, central and peripheral retinal dystrophies, mitochondrial diseases and tumors of the RPE.
Age-Related Changes
Normal human RPE cells are mitotically quiescent. Age-related decline of RPE density and the corresponding morphology changes (Fig. 1.5) have been extensively studied [30, 31, 34, 38,39,40,41]. In a previous investigation, the RPE cell density in the fovea decreased significantly (p < 0.001) by about 0.3% per year with increasing age [30]. Such a continuous age-related decline of RPE cell count is commonly found in the literature but the quantification of the magnitude of the decline is not well understood [31, 34, 38,39,40,41]. With age, the number of hexagonal RPE cells decreases in the fovea and parafoveal areas [31]. Ach et al. indicated that the density of five-sided RPE cells increased significantly in the fovea, whereas in the parafovea the proportion of five and seven-sided cells tends to dominate as a result of an age-related remodeling [31].

RPE cell morphology of a “young” individual (37-year-old; a, c, and e) and an “old” individual (75-year-old; b, d, and f). (With permission of the authors: Bhatia SK, Rashid A, Chrenek MA, Zhang Q, Bruce BB, Klein M, et al. Analysis of RPE morphometry in human eyes. Mol Vis. 2016;22:898–916.) Note the increase in the bluish autofluorescence in the “old” sample in comparing the right and left images. This signal is thought to reflect an increase in autofluorescent lipofuscin with age
Lipofuscin granules are known to localize close to the RPE cell borders when low lipofuscin granule density is present which is expected for young, healthy individuals [42]. RPE cells are responsible for the phagocytosis of diurnally shed photoreceptor outer segments [43]. Lipofuscin arises in the RPE from incomplete digestion of the outer-segment fragments within the lysosomes [44]. Lipofuscin accumulation in RPE cells has been proved to lead to a range of retinal disorders (Fig. 1.6).

Photoreceptor outer segment (PR-OS) degradation and lipofuscin formation in the RPE cells
The major fluorophores of lipofuscin are the bis-retinoid, N-retinylidene-N-retinylethanolamine (A2E), closely related A2E phosphatidyl esters, and retinyl dimers [45]. In clinical setting, fundus autofluorescence (FAF) is informative for monitoring RPE health and metabolism by enabling detection of autofluorescence (AF) attributable to lipofuscin and melanolipofuscin, long-lasting intracellular residual bodies rich in bisretinoid derivatives of vitamin A. The loss of fundus autofluorescence (detected as black patches in FAF) thus represents the loss of RPE cells that contain these highly fluorescent molecules (Fig. 1.7). In 2005, eight phenotypic FAF patterns have been defined to be associated with non-exudative AMD, such as normal, minimal change, focal increase, patchy, linear, lacelike, reticular and speckled pattern [46]. Diffuse and banded phenotypes are associated with a higher risk for disease progression [47].

Infrared reflectance (a) and spectral domain optical coherence tomography (b) image of the fovea in geographic RPE atrophy. Note the junction (arrow) between the normal RPE layer and the atrophic lesion (b). On fundus autofluorescence image (c), the hypo-autofluorescent areas correspond to RPE atrophy and they are surrounded by diffuse reticular hyper-autofluorescence in the junctional zone, which represent areas of current RPE cell dysfunction. This is a 76-year-old female patient with 20/25 best corrected visual acuity due to foveal sparing of geographic atrophy
Degenerations
Biometry-Related Changes
RPE has been shown to play a critical role in ocular growth regulation by controlling ion/fluid transport and signal transduction between the retina, choroid and sclera [7]. A profound decrease in the RPE cell density was reported in the equatorial to retro-equatorial region in association with longer axial length and a lower degree of decline at the midpoint between equator/posterior pole [41]. RPE cell density at the posterior pole was not significantly associated with axial length in a previous study [41].
Age-Related Macular Degeneration
With aging RPE cells gradually decrease in density and enlarge in size associated with intracellular lipofuscin accumulation [48]. Lipofuscin was found to be photocytotoxic to RPE cells in a wavelength dependent manner (390–550 nm), it reduced significantly the RPE cell viability by at least 41% 2 days after 390–550 nm exposure when compared to lipofuscin-free cells [49]. Its accumulation is associated with photooxidation, RPE cell damage, and inflammation. These alterations with age-related changes of the Bruch’s membrane (calcification and fragmentation) may lead to age-related macular degeneration [50]. However, the heterogeneous clinical picture of RPE dysfunction could be explained by the susceptibility of RPE cells in vivo due to the balance between the amount of lipofuscin granules and the antioxidant potential of the cell [49]. Moreover, RPE cell pattern has been shown to have a much reduced regularity in AMD [40].
AMD starts with the formation of basal laminar deposits and drusen and hypertrophy and loss of the RPE cells (Fig. 1.8). In more advance stages, these processes may lead to geographic atrophy (Fig. 1.7) and choroidal neovascularization. It has also been proposed that RPE cells might undergo transdifferentiation [51, 52]. Drusen are deposits of amorphous extracellular material lying between the RPE and the inner collagenous zone of Bruch’s membrane [53]. The transformation theory [54], deposition theory [55] and the vascular theory [56] have been proposed to explain the exact origin of drusen. Hard drusen may progress into atrophic AMD, whereas soft drusen may precede choroidal neovascularization. Softening of drusen is associated with accumulation of membranous debris external to the RPE basement membrane which may arise from the photoreceptors [57]. Large drusen are associated with diffuse thickening of Bruch’s membrane with basal linear deposit, whereas confluent soft drusen resemble an exudative RPE detachment [57]. Basal laminar deposit is a diffuse accumulation of hyalinized material between the RPE basal plasma membrane and its basement membrane. It is composed of widely spaced type IV collagen and other fibrillar materials [58]. They can be either the cause or consequence of compromised RPE cell functions [59]. Multiple studies concluded that there is a significant risk of progression to exudative AMD if there is a large drusen size, drusen confluence or focal macular hyperpigmentation on initial presentation [57]. Spraul et al. postulated that choroidal neovascular membranes represent a nonspecific wound-repair mechanism since their composition (i.e. RPE cells, vascular endothelial cells, fibrocytes, macrophages, photoreceptors, erythrocytes, lymphocytes, myofibroblasts, collagen, fibrin and basal laminar deposit) is not characteristic of a specific disease process [59].

Light microscopy of hard drusen (hematoxylin and eosin, 160×) (a), soft drusen (periodic acid-Schiff, 160×) (b), large drusen (hematoxylin and eosin, 160×) (c) and confluent drusen (hematoxylin and eosin, 160×) (d). (Reproduced with permission from Spraul CW, Grossniklaus HE. Characteristics of drusen and Bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol. 1997;115:267–273)
Retinal Dystrophies
Retinal dystrophy refers to a heterogeneous group of inherited central and peripheral retinal diseases associated with various mutations in the human retina affecting the retinal pigment epithelium and the photoreceptor layer [60]. These mutations may impact a number of molecular cascades by interfering with intra- and intercellular interactions in different layers of the retina. Those processes may compromise the integrity of the RPE plasma membranes (bestrophinopathies, e.g. Best vitelliform macular dystrophy) [61] and/or photoreceptor membranes (pattern dystrophies, e.g. butterfly-shaped pattern dystrophy) [62]. Histopathologically, pattern dystrophies are associated with accumulation of excessive lipofuscin in the RPE, whereas in later stages these changes are replaced by extensive atrophic depigmented areas of the RPE and degeneration of photoreceptor cells [62,63,64]. In retinitis pigmentosa, the initial degenerative changes occur in the rod photoreceptors followed by secondary changes in the cones, RPE, retinal glia and ganglion cells [65]. The RPE cells migrate to perivascular sites in the inner retina after extensive photoreceptor damage, producing the characteristic bone-spicule appearance due to the black melanin granules surrounding the branching retinal vessels [65].
Proliferative Retinopathies
Epiretinal membrane (ERM) formation can be occurring secondary to a number of pathological changes on the vitreoretinal interface. Snead et al. classified ERMs into three distinct types: (1) simple ERMs, which contain only internal limiting membrane (ILM) and laminocytes; (2) PVR/tissue repair membranes, which contain ILM, laminocytes and spindle-shaped RPE cells with extracellular stroma; (3) neovascular ERMs, which are devoid of ILM and contain vessels and hyaline stroma [66].
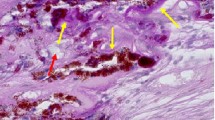
Under physiologic conditions, RPE cells are mitotically quiescent and virtually stationary. In various disease processes such as proliferative vitreoretinopathy, retinitis pigmentosa, or choroidal neovascularization, the RPE cells detach from the basement membrane and exhibit migratory potential [65, 67, 68]. The term PVR refers to various conditions characterized by cell proliferation and matrix deposition within the retina (Fig. 1.9). Essentially in PVR cases, the ERM is usually complicated by the presence of RPE cells [66]. Besides RPE cells, occasional macrophages, fibrocytes and lymphocytes can also be present with various amount of collagen indicating a tissue repair process [66].

Light microscopy of subretinal PVR showing fibrocellular tissue composed of RPE (pigmented cells), collagen, fibrocytes and macrophages (hematoxylin and eosin, 25×). This is a 10-year-old highly myopic male with a history of prematurity and long standing retinal detachment. During pars plana vitrectomy, a “napkin ring” band of PVR was removed from the subretinal space through a small retinotomy site
Mitochondrial Diseases
RPE cells are metabolically highly active and they produce reactive oxygen species (ROS) by a variety of pathways during aerobic metabolism [69]. The major site of ROS production is within mitochondria, thus mitochondrial DNA (mtDNA) is thought to be more susceptible to damage by ROS than nuclear DNA. mtDNA damage is a good biomarker of oxidative stress [70]. On the other hand, the RPE cells along with retinal ganglion cells are particularly susceptible to oxidative damage [71]. Retinal pigmentary changes and optic atrophy both are common ophthalmic manifestations of mitochondrial disorders [72]. Histopathological studies showed a primary RPE degeneration followed by secondary changes of the photoreceptors and choriocapillaris, and proved abnormally enlarged mitochondria in the RPE cells [73]. The same ROS-mediated mtDNA damage and RPE cell death mechanisms are thought to play key roles in the pathogenesis of AMD in susceptible eyes [70].
RPE Tumors
The retinal pigment epithelium often undergoes reactive hyperplasia secondary to intraocular inflammation or trauma (Fig. 1.10). Although less common, it can produce a variety of tumors and related lesions [74]. The main tumors of the RPE include congenital hypertrophy of the RPE (CHRPE), congenital simple (solitary or grouped) hamartoma, combined hamartoma, adenoma, and adenocarcinoma.

Light microscopy of RPE hyperplasia showing multiple layers of RPE cells (hematoxylin and eosin left 25×, right 100×)
Histopathologically, both solitary and grouped CHRPE lesions consist of a single layer of hypertrophied RPE cells (Fig. 1.11) which are densely packed with round macromelanosomes [75]. A hypopigmented halo is often seen around the lesion representing taller RPE cells with less and smaller melanosomes. The Bruch’s membrane is thickened, but the neurosensory retina remains unaffected [76]. Multilayered (hyperplastic) lesions with hypertrophic RPE cells are often referred to as hamartomas, i.e. abnormal amount of mature tissue in the normal anatomic location [75]. There is a distinct autosomal dominant disorder when the RPE hamartomas are associated with familial adenomatous polyposis. This disease entity was recently termed as “RPE hamartomas associated with familial adenomatous polyposis” by Shields and Shields to avoid the confusion because neither simple nor grouped hamartomas possess a higher risk of colon carcinoma [77]. Combined hamartoma of the retina and RPE has been described in juxtapapillary location when the RPE proliferation extended into the optic disc [78]. Proliferation of glial elements on the retinal surface and intraretinally was also observed [79]. The RPE cells proliferate in the neurosensory retinal layers as sheets and cords [76].

Light microscopy of RPE hypertrophy indicating a single layer of enlarged RPE cells in volume. The left figure shows RPE hypertrophy associated with the dark part of a congenital hypertrophy of the RPE (CHRPE) lesion and the right shows depigmented RPE in the lacunae in CHRPE (hematoxylin and eosin 100×)
Adenoma of the RPE usually shows tubules and cords of proliferating RPE cells, separated by basement membrane and fibrous stroma histopathologically [80]. They usually arise from the peripheral retina but can occasionally be observed juxtapapillary masquerading melanocytoma of the optic disc or peripapillary uveal melanoma [81]. Adenocarcinoma of the RPE is composed of cords and tubular proliferations of cells with pleomorphic, hyperchromatic nuclei and variable amounts of cytoplasm (Fig. 1.12). There can be focal desmoplastic reaction to the tumor and bone formation (osseous metaplasia) in some areas. RPE adenocarcinoma can be highly invasive with extensive intraocular invasion, including lens, choroidal, scleral and optic nerve invasion, extension through emissary canals to the epibulbar surface with conjunctival and orbital invasion. However, tumors of the RPE seem to have no potential to metastasize [82]. Malignant transformation of a preexisting CHRPE into an adenocarcinoma has also been reported [83].

Light microscopy of RPE adenocarcinoma. The tumor is composed of tubular and chord-like proliferations of cells with associated osseous metaplasia (a, b). The tumor has also invaded the optic nerve (c, d) (hematoxylin and eosin 5×, 25×, 5×, 100×, respectively)
Pigmented RPE lesions can clinically simulate choroidal melanoma, thus they often referred to as pseudomelanoma [84]. Most commonly, CHRPE may appear similar to choroidal melanoma on routine clinical examination. However, differentiating between a benign RPE hypertrophy and malignant choroidal melanoma is crucial. CHRPE is typically a sharply demarcated dark pigmented lesion and usually flat or slightly elevated. A pigmented or non-pigmented halo can often be seen surrounding the lesion and intralesional lacunae are present in 43% of cases [85]. The diagnosis of RPE tumors is usually made on the basis of ophthalmoscopic findings and diagnostic imaging.
References
Yamashita M, Gotoh M. Impact behavior of honeycomb structures with various cell specifications—numerical simulation and experiment. Int J Impact Eng. 2005;32:618–30.
Streeten BW. Development of the human retinal pigment epithelium and the posterior segment. Arch Ophthalmol. 1969;81:383–94.
Burke JM, Hjelmeland LM. Mosaicism of the retinal pigment epithelium: seeing the small picture. Mol Interv. 2005;5:241–9.
Fuhrmann S, Zou C, Levine EM. Retinal pigment epithelium development, plasticity, and tissue homeostasis. Exp Eye Res. 2014;123:141–50.
Röhlich P. The interphotoreceptor matrix: electron microscopic and histochemical observations on the vertebrate retina. Exp Eye Res. 1970;10:80–6.
Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–81.
Rymer J, Wildsoet CF. The role of the retinal pigment epithelium in eye growth regulation and myopia: a review. Vis Neurosci. 2005;22:251–61.
Salzmann M. Anatomie und Histologie des menschlichen Augapfels. Leipzig: F. Deuticke; 1912.
Liu Y, Hong L, Wakamatsu K, Ito S, Adhyaru BB, Cheng CY, et al. Comparisons of the structural and chemical properties of melanosomes isolated from retinal pigment epithelium, iris and choroid of newborn and mature bovine eyes. Photochem Photobiol. 2005;81:510–6.
Nakagawa H, Imokawa G. Characterization of melanogenesis in normal human epidermal melanocytes by chemical and ultrastructural analysis. Pigment Cell Res. 1996;9:175–8.
Guymer R, Bird A. Bruch’s membrane, drusen, and age-related macular degeneration. In: Marmor M, Wolfensberger T, editors. The retinal pigment epithelium. New York: Oxford University Press; 1998. p. 693–705.
Booij JC, Baas DC, Beisekeeva J, Gorgels TG, Bergen AA. The dynamic nature of Bruch’s membrane. Prog Retin Eye Res. 2010;29:1–18.
Miki H, Bellhorn MB, Henkind P. Specializations of the retinochoroidal juncture. Invest Ophthalmol. 1975;14:701–7.
Rajasekaran SA, Hu J, Gopal J, Gallemore R, Ryazantsev S, Bok D, Rajasekaran AK, et al. Am J Physiol Cell Physiol. 2003;284:C1497–507.
Pollard TD, Cooper JA. Actin and actin-binding proteins. A critical evaluation of mechanisms and functions. Annu Rev Biochem. 1986;55:987–1035.
Bruenner U, Burnside B. Pigment granule migration in isolated cells of the teleost retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1986;27:1634–43.
Burnside B, Adler R, O’Connor P. Retinomotor pigment migration in the teleost retinal pigment epithelium. I. Roles for actin and microtubules in pigment granule transport and cone movement. Invest Ophthalmol Vis Sci. 1983;24:1–15.
Mondragón R, Frixione E. Retinomotor movements in the frog retinal pigment epithelium: dependence of pigment migration on Na+ and Ca2+. Exp Eye Res. 1989;48:589–603.
Lythgoe JN, Shand J. Endogenous circadian retinomotor movements in the neon tetra (Paracheirodon innesi). Invest Ophthalmol Vis Sci. 1983;24:1203–10.
Zhang QX, Lu RW, Messinger JD, Curcio CA, Guarcello V, Yao XC. In vivo optical coherence tomography of light-driven melanosome translocation in retinal pigment epithelium. Sci Rep. 2013;3:2644.
Sundelin SP, Nilsson SE, Brunk UT. Lipofuscin-formation in cultured retinal pigment epithelial cells is related to their melanin content. Free Radic Biol Med. 2001;30:74–81.
Schraermeyer U, Heimann K. Current understanding on the role of retinal pigment epithelium and its pigmentation. Pigment Cell Res. 1999;12:219–36.
Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403.
Rangel-Fonseca P, Gómez-Vieyra A, Malacara-Hernández D, Wilson MC, Williams DR, Rossi EA. Automated segmentation of retinal pigment epithelium cells in fluorescence adaptive optics images. J Opt Soc Am A Opt Image Sci Vis. 2013;30:2595–604.
Rossi EA, Rangel-Fonseca P, Parkins K, Fischer W, Latchney LR, Folwell MA, et al. In vivo imaging of retinal pigment epithelium cells in age related macular degeneration. Biomed Opt Express. 2013;4:2527–39.
Rossi EA, Chung M, Dubra A, Hunter JJ, Merigan WH, Williams DR. Imaging retinal mosaics in the living eye. Eye (Lond). 2011;25:301–8.
Schütze C, Wedl M, Baumann B, Pircher M, Hitzenberger CK, Schmidt-Erfurth U. Progression of retinal pigment epithelial atrophy in antiangiogenic therapy of neo-vascular age-related macular degeneration. Am J Ophthalmol. 2015;159:1100–4e1.
Morgan JIW, Dubra A, Wolfe R, Merigan WH, Williams DR. In vivo autofluorescence imaging of the human and macaque retinal pigment epithelial cell mosaic. Invest Ophthalmol Vis Sci. 2009;50:1350–9.
Hogan MJ, Alvaraso JA, Weddell JE. Chapter 9. In: Hogan MJ, Alvaraso JA, Weddell JE, editors. Histology of the human eye. Philadelphia: W. B. Saunders; 1971. p. 393–522.
Panda-Jonas S, Jonas JB, Jakobczyk-Zmija M. Retinal pigment epithelial cell count, distribution, and correlations in normal human eyes. Am J Ophthalmol. 1996;121:181–9.
Ach T, Huisingh C, McGwin G Jr, Messinger JD, Zhang T, Bentley MJ, et al. Quantitative autofluorescence and cell density maps of the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2014;55:4832–41.
Bhatia SK, Rashid A, Chrenek MA, Zhang Q, Bruce BB, Klein M, et al. Analysis of RPE morphometry in human eyes. Mol Vis. 2016;22:898–916.
Gao H, Hollyfield JG. Aging of the human retina: differential loss of neurons and retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1992;33:1–17.
Watzke RC, Soldevilla JD, Trune DR. Morphometric analysis of human retinal pigment epithelium: correlation with age and location. Curr Eye Res. 1993;12:133–42.
Harman AM, Fleming PA, Hoskins RV, Moore SR. Development and aging of cell topography in the human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1997;38:2016–26.
Del Priore LV, Kuo Y-H, Tazel TH. Age-related changes in human RPE cell density and apoptosis proportion in situ. Invest Ophthalmol Vis Sci. 2002;43:3312–8.
Leung IY, Sandstrom MM, Zucker CL, Neuringer M, Snodderly DM. Nutritional manipulation of primate retinas, II: effects of age, n-3 fatty acids, lutein, and zeaxanthin on retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2004;45:3244–56.
Ts’o MO, Friedman E. The retinal pigment epithelium. 3. Growth and development. Arch Ophthalmol. 1968;80:214–6.
Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci. 1989;30:1691–9.
Rashid A, Bhatia SK, Mazzitello KI, Chrenek MA, Zhang Q, Boatright JH, et al. RPE cell and sheet properties in normal and diseased eyes. Adv Exp Med Biol. 2016;854:757–63.
Jonas JB, Ohno-Matsui K, Holbach L, Panda-Jonas S. Retinal pigment epithelium cell density in relationship to axial length in human eyes. Acta Ophthalmol. 2017;95:e22–8.
Cabral L, Unger W, Boulton M, Lightfoot R, McKechnie N, Grierson I, et al. Regional distribution of lysosomal enzymes in the canine retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1990;31:670–6.
Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–95.
Kennedy CJ, Rakoczy PE, Constable IJ. Lipofuscin of the retinal pigment epithelium: a review. Eye (Lond). 1995;9:763–71.
Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–6.
Bindewald A, Bird AC, Dandekar SS, Dolar-Szczasny J, Dreyhaupt J, Fitzke FW, et al. Classification of fundus autofluorescence patterns in early age-related macular disease. Invest Ophthalmol Vis Sci. 2005;46:3309–14.
Holz FG, Bindewald-Wittich A, Fleckenstein M, Dreyhaupt J, Scholl HP, Schmitz-Valckenberg S. Progression of geographic atrophy and impact of fundus autofluorescence patterns in age-related macular degeneration. Am J Ophthalmol. 2007;143:463–72.
Hogan MJ. Role of the retinal pigment epithelium in macular disease. Trans Am Acad Ophthalmol Otolaryngol. 1972;76:64–80.
Davies S, Elliott MH, Floor E, Truscott TG, Zareba M, Sarna T, et al. Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells. Free Radic Biol Med. 2001;31:256–65.
Spraul CW, Grossniklaus HE. Characteristics of Drusen and Bruch’s membrane in postmortem eyes with age-related macular degeneration. Arch Ophthalmol. 1997;115:267–73.
Grisanti S, Guidry C. Transdifferentiation of retinal pigment epithelial cells from epithelial to mesenchymal phenotype. Invest Ophthalmol Vis Sci. 1995;36:391–405.
Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37:855–68.
Farkas TG, Syvlester V, Archer D. The ultrastructure of drusen. Am J Ophthalmol. 1971;71:1196–205.
Donders FC. Beitrage zur pathologischen Anatomie des Auges. Arch Ophthalmol. 1854;1:106.
Muller H. Untersuchungen uber die gladuates des Auges, insbesondere die Glaslamelle der Choroidea und ihr senilen Veranderungen. Arch Ophthalmol. 1856;2:1.
Friedman E, Smith T, Kuwabara T. Senile choroidal vascular patterns and drusen. Arch Ophthalmol. 1963;69:114.
Abdelsalam A, Del Priore L, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis, natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999;44:1–29.
Löffler KU, Lee WR. Basal linear deposit in the human macula. Graefes Arch Clin Exp Ophthalmol. 1986;224:493–501.
Spraul CW, Lang GE, Grossniklaus HE, Lang GK. Histologic and morphometric analysis of the choroid, Bruch’s membrane, and retinal pigment epithelium in postmortem eyes with age-related macular degeneration and histologic examination of surgically excised choroidal neovascular membranes. Surv Ophthalmol. 1999;44:S10–32.
Broadgate S, Yu J, Downes SM, Halford S. Unravelling the genetics of inherited retinal dystrophies: past, present and future. Prog Retin Eye Res. 2017;59:53–96. https://doi.org/10.1016/j.preteyeres.2017.03.003.
Guziewicz KE, Sinha D, Gómez NM, Zorych K, Dutrow EV, Dhingra A, et al. Bestrophinopathy: an RPE-photoreceptor interface disease. Prog Retin Eye Res. 2017;58:70–88.
Zhang K, Garibaldi DC, Li Y, Green WR, Zack DJ. Butterfly-shaped pattern dystrophy: a genetic, clinical, and histopathological report. Ophthalmic Mol Genet. 2002;120:485–90.
Gass JMD. Stereoscopic atlas of macular disease. Philadelphia: Elsevier; 1997.
Birnbach CD, Jarvelainen M, Possin DE, Milam AH. Histopathology and immunocytochemistry of the neurosensory retina in fundus flavimaculatus. Ophthalmology. 1994;101:1211–9.
Li ZY, Possin DE, Milam AH. Histopathology of bone spicule pigmentation in retinitis pigmentosa. Ophthalmology. 1995;102:805–16.
Snead DR, James S, Snead MP. Pathological changes in the vitreoretinal junction 1: epiretinal membrane formation. Eye (Lond). 2008;22:1310–7.
Machemer R, Laqua H. Pigment epithelial proliferation in retinal detachment (massive periretinal proliferation). Am J Ophthalmol. 1975;80:1–23.
Miller H, Miller B, Ryan SJ. The role of retinal pigment epithelium in the involution of subretinal neovascularization. Invest Ophthalmol Vis Sci. 1986;27:1644–52.
Jin GF, Hurst JS, Godley BF. Rod outer segments mediate mitochondrial DNA damage and apoptosis in human retinal pigment epithelium. Curr Eye Res. 2001;23:11–9.
Liang FQ, Godley BF. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: a possible mechanism for RPE aging and age-related macular degeneration. Exp Eye Res. 2003;76:397–403.
Newman NJ. Mitochondrial diseases and the eye. Ophthalmol Clin North Am. 1992;5:405–24.
Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299–334.
McKechnie NM, King M, Lee WR. Retinal pathology in Kearns-Sayre syndrome. Br J Ophthalmol. 1985;69:63–75.
Shields JA, Shields CL. Tumors and related lesions of the pigment epithelium. In: Shields JA, Shields CL, editors. Atlas of intraocular tumors. Philadelphia: Lippincott, Williams and Wilkins; 1999. p. 287–307.
Lloyd WC 3rd, Eagle RC Jr, Shields JA, Kwa DM, Arbizo VV. Congenital hypertrophy of the retinal pigment epithelium. Electron microscopic and morphometric observations. Ophthalmology. 1990;97:1052–60.
Meyer CH, Gerding H. Congenital hypertrophy of the retinal pigment epithelium. In: Ryan SJ, editor. Retina. 5th ed. St. Louis: Elsevier; 2013. p. 2209–13.
Shields JA, Shields CL. Tumors and related lesions of the pigmented epithelium. Asia Pac J Ophthalmol (Phila). 2017;6:215–23.
Theobald GD, Floyd G, Kirk HQ. Hyperplasia of the retinal pigment epithelium simulating a neoplasm: report of two cases. Am J Ophthalmol. 1958;45(4 Pt 2):235–40.
Vogel MH, Zimmerman LE, Gass JD. Proliferation of the juxtapapillary retinal pigment epithelium simulating malignant melanoma. Doc Ophthalmol. 1969;26:461–81.
Shields JA, Eagle RC Jr, Dutton J, Ehya H, Shields CL. Adenocarcinoma of the retinal pigment epithelium: clinicopathologic correlation with paradoxical immunohistochemical findings. JAMA Ophthalmol. 2014;132:1249–52.
Shields JA, Melki T, Shields CL, Eagle RC Jr, Singh AD. Epipapillary adenoma of retinal pigment epithelium. Retina. 2001;21:76–8.
Shields JA, Shields CL. Tumors and related lesions of the pigment epithelium. In: Shields JA, Shields CL, editors. Intraocular tumors: an atlas and textbook. 2nd ed. Philadelphia: Lippincott, Williams & Wilkins; 2008. p. 432–83.
Shields JA, Eagle RC Jr, Shields CL, Brown GC, Lally SE. Malignant transformation of congenital hypertrophy of the retinal pigment epithelium. Ophthalmology. 2009;116:2213–6.
Shields CL, Manalac J, Das C, Ferguson K, Shields JA. Choroidal melanoma: clinical features, classification, and top 10 pseudomelanomas. Curr Opin Ophthalmol. 2014;25:177–85.
Shields CL, Mashayekhi A, Ho T, Cater J, Shields JA. Solitary congenital hypertrophy of the retinal pigment epithelium: clinical features and frequency of enlargement in 330 patients. Ophthalmology. 2003;110:1968–76.
Acknowledgements
The authors thank Nancy L’Hernault for the assistance in transmission electron microscopy. We are grateful for support provided by NIH R01EY021592, R01EY016470, P30EY006360, and an unrestricted grant to the Department of Ophthalmology at Emory University from Research to Prevent Blindness, Inc.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Szalai, E., Nickerson, J.M., Grossniklaus, H.E. (2020). RPE Histopathology and Morphometry. In: Klettner, A., Dithmar, S. (eds) Retinal Pigment Epithelium in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-28384-1_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-28384-1_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28383-4
Online ISBN: 978-3-030-28384-1
eBook Packages: MedicineMedicine (R0)