
Abstract
Circulating tumor cells (CTCs) provide valuable information about the molecular evolution of cancers, as they may initially respond and ultimately progress on therapy. As intact tumor cells isolated from the bloodstream, CTCs also enable assessment of heterogeneous subpopulations, and their analysis may include DNA, RNA, and protein biomarkers. New microfluidic cell isolation strategies greatly facilitate the challenge of enriching viable tumor cells from the billions of hematopoietic cells within a standard blood specimen. While counting and characterization of enriched CTCs have primarily relied on immunostaining for tumor cell-specific antigens, new RNA-based analytic platforms are providing new insight into the identity of CTCs and providing new tools for clinical applications. Single-cell RNA sequencing of CTCs reveals a high degree of heterogeneity among cancer cells from a single individual, while new digital RNA-based amplification platforms may now allow high-sensitivity and high-throughput quantitative scoring of CTCs for clinical applications. Here, we focus on transcriptomic analysis of CTCs and its relevance in understanding metastatic cancer progression and in developing diagnostic assays to monitor cancer.
Mark Kalinich and Tanya T. Kwan contributed equally to this work.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


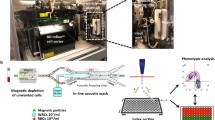
1 Introduction
The blood-borne metastasis of cancer is the leading cause of cancer-related deaths (Hanahan and Weinberg 2011). As such, CTCs provide a critical material for understanding the ability of tumor cells to intravasate into the bloodstream, either as single migratory cells or as clusters of tumor-derived fragments, survive in the circulation, and ultimately disseminate to distant organs and initiate proliferation (Poste and Fidler 1980; Allard et al. 2004; Yu et al. 2011; Stott et al. 2010; Aceto et al. 2014). While the vast majority of CTCs die in the bloodstream before giving rise to any metastatic lesion, the presence of these cells also provides a key opportunity to non-invasively sample cancer-derived materials during the evolution of the disease (Cristofanilli et al. 2004). The molecular composition of tumors evolves through the acquisition of genomic alterations and epigenetic modifications during disease progression and in response to therapeutic interventions (Easwaran et al. 2014; McGranahan and Swanton 2017). This may result in extensive tumor heterogeneity, a challenge facing the clinical management of cancer, and in the need to repeatedly adjust therapeutic regimens in response to acquired resistance mechanisms (Nardi et al. 2004). Much of our current information about cellular pathways involved in acquired drug resistance stems from repeat biopsies of tumors from patients on research protocols and from autopsy studies in patients who succumbed from extensive disease. However, the ability to repeatedly and non-invasively sample tumor cells through “liquid biopsies” may revolutionize the ability to tailor a patient’s individualized therapy to address evolving tumor characteristics. Ultimately, blood-based tumor monitoring may allow early detection of invasive cancers and enable interventions before large tumor volumes may render curative treatment impossible.
Tumor-derived components in the blood are found as whole cells (i.e., CTCs), cellular fragments (exosomes or oncosomes), or free circulating tumor DNA (ctDNA). Each of these have unique properties relevant to their isolation and enrichment from whole blood, as well as the types of molecular information that may be derived from their analysis. ctDNA is comprised of nucleosome-sized DNA fragments that are primarily informative as to the genomic composition of tumors (Wan et al. 2017). Exosomes appear to contain a partial set of tumor-derived proteins, RNA, and even some DNA (Zhang et al. 2015). Both circulating DNA and exosomes are shed by both normal and tumor tissues, and cancer-derived molecules must therefore be distinguished, using molecular tools, from those shed by normal tissues. In contrast, whole tumor cells in the circulation are extremely rare, but their enrichment is dependent upon physical or cell surface marker expression (Nagrath et al. 2007). Once isolated, however, CTCs provide the full complement of the molecular information present within individual tumor cells. Across different cancer types, CTCs are estimated to range in number from 0 to 10 cancer cells per 10 mL of whole blood, amidst 10 billion red blood cells and 10 million white blood cells (Nagrath et al. 2007). CTCs are typically defined as cells that stain for epithelial cell-specific proteins (e.g., EpCAM and cytokeratins), with the exclusion of white blood cell markers (e.g., CD45) (Cristofanilli et al. 2009). More recent studies have used lineage-associated markers (e.g., PSA for prostate cancer), rather than epithelial markers that may be less specific for a given tumor type (Miyamoto et al. 2012). In addition, epithelial-to-mesenchymal transition (EMT), a cell fate switch associated with tumor invasiveness and drug resistance complicates reliance on epithelial markers to identify CTCs (Yu et al. 2013). We have therefore preferred CTC enrichment technologies that achieve “negative depletion” of blood specimens, essentially removing hematopoietic cells, while leaving CTCs untagged and unmanipulated in the product (Ozkumur et al. 2013). Microfluidic technologies currently achieve 104 to 105 enrichment of CTCs from whole blood specimens, resulting in a cancer cell population that may be 0.1–1% pure (depending on CTC abundance within an individual specimen). Given this success in rare cancer cell enrichment from blood, the challenge remains to score and molecularly characterize these partially purified cell populations. In this review, we focus on RNA-based approaches used for interrogating CTCs (Fig. 1) and their applicability in developing diagnostic assays that can be implemented in the clinic for monitoring therapeutic responses and ultimately for early detection of cancer in high-risk individuals.
2 RNA-In Situ Hybridization Identifies Epithelial and Mesenchymal CTC Populations
Immunofluorescence staining for tumor-specific protein markers are commonly used to study CTCs enriched from patient blood. A number of technical limitations have complicated such analysis, including the relatively low signal/noise ratio evident in CTCs within the context of contaminating blood cells, which often requires the combination of highly specific antibodies with secondary fluorescent antibodies for signal amplification, together with the rigorous setting of signaling threshold, image quantitation, and automated image scanning protocols (Allard et al. 2004; Alix-Panabières and Pantel 2014). Exemplifying this challenge is the high number of blood cells that simultaneously stain positive for epithelial as well as hematopoietic markers (cytokeratin/CD45 “dual positives”), which most frequently represent non-specific antibody binding, and which may outnumber true CTCs by multiple orders of magnitude (Stott et al. 2010). In this context, novel approaches to RNA-in situ hybridization (RNA-ISH) using multiplexed oligonucleotide probes present a powerful technology, with both a high degree of sequence specificity and quantitative amplifiable signal. For instance, several studies have used RNA-ISH against multiple probes marking epithelial and mesenchymal cell states to detect CTCs enriched using microfluidic isolation, filter-based methods or Ficoll gradients (Yu et al. 2013; Payne et al. 2012; Wu et al. 2015). These studies have shown that CTCs exist not only in uniquely epithelial versus mesenchymal states, but also in more complex conditions with simultaneous coexpression of both different numbers of epithelial and mesenchymal markers. Of particular interest, longitudinal analysis of blood samples from individual breast cancer patients receiving multiple courses of therapy may show dynamic shifts, with primarily epithelial CTCs as tumors respond to new therapeutic intervention, and the emergence of mesenchymal CTCs associated with acquired resistance and clinical therapeutic failure (Yu et al. 2013). The broad application of RNA-ISH technologies in characterizing individual CTCs in this way may provide a powerful tool for characterization of tumor cell heterogeneity during the course of cancer evolution and drug resistance.
3 RNA Sequencing Identifies CTC Subpopulations and Signaling Pathways Activated in CTCs
While RNA-in situ hybridization allows for more facile probe design than antibody-based detection, it is limited in throughput and in its ability to query only a few pre-selected transcripts of interest. RNA sequencing of either bulk-enriched or single-cell populations is technically challenging but enables interrogation of the entire transcriptome. Recent advances in next-generation sequencing technologies have made it feasible to sequence thousands of single cells from tumors, producing rich datasets that can more thoroughly probe the heterogeneity of cancer (Tirosh et al. 2016). Whole transcriptomic analysis has been applied extensively to study primary and metastatic biopsies, but implementing this technology to CTCs has been complicated by the rarity of the cells and their condition after isolation using a variety of technologies (i.e., the need for unfixed cells with high-quality RNA).
The first approach to achieving CTC-specific transcriptional profiles used partially purified CTC populations subjected to single molecule (Helicos) RNA sequencing, subtracting RNA reads from matched control blood samples from that of the CTC-enriched cell populations (Yu et al. 2012). The Helicos single-molecular sequencing technology is unique in avoiding amplification of molecular templates, thereby providing highly linear measurement of transcript reads. Such subtractive strategies were best applied with CTC isolation platforms that capture cells on a fixed surface, from which single cells are not readily released but from which high-quality RNA can be isolated.

Comparison of methods to study CTC-derived RNA

CTC-ddPCR method
Indeed, early studies applying this strategy to pancreatic cancer CTCs in a mouse model, demonstrated increased non-canonical Wnt signaling in the CTC-enriched population (Yu et al. 2012), and an analysis of human melanoma CTC-enriched cell populations was noteworthy for cell motility-associated transcripts (Luo et al. 2014). However, deep sequencing of CTC-derived transcriptomes requires single-cell isolation and RNA seq, a strategy that has become possible with improving CTC enrichment technologies that allow for micromanipulation of CTCs that are unattached to a fixed surface.
For instance, the MagSweeper technology enabled single-cell RNA profiling of 87 cancer-associated genes in CTCs isolated from breast cancer patients, showing increased expression of the metastasis and EMT-associated genes (Powell et al. 2012). Using the microfluidic CTC-iChip technology, our own team has established a platform through which hematopoietic cells are antibody-tagged and depleted, leaving behind unmanipulated CTCs with 104 to 105 enrichment (Ozkumur et al. 2013). The RNA quality in untagged, unfixed CTCs is very high, and the fact that cells are delivered in suspension facilitates micromanipulation of individual CTCs. This approach was used to define comprehensive transcriptomes of single CTC collected from both mouse models and clinical specimens. In a mouse model of pancreatic cancer, CTCs were highly enriched for expression of genes encoding the extracellular matrix proteins (ECM), compared with single cells isolated simultaneously from the primary pancreatic tumor (Ting et al. 2014). This aberrant expression of ECM proteins by cancer-derived cells in circulation is of particular interest in that it suggests the ability of metastatic intermediates/precursors to direct their own microenvironmental survival signals, which are characteristically provided by stromal cells within the primary tumor.
Single-cell RNA sequencing of CTCs traveling as individual cells versus those that are part of multi-cellular CTC clusters from the blood of women with metastatic breast cancer revealed >100 genes whose expression is relatively elevated in CTC clusters (Aceto et al. 2014). Among the top genes increased in expression within CTC clusters is Plakoglobin, encoding a protein belonging to the adherence junction complex. Plakoglobin is overexpressed >200-fold within CTC clusters, and its increased expression in primary tumors is associated with poor clinical outcome. Most importantly, in mouse models of breast cancer, knockdown of Plakoglobin does not affect cell proliferation, primary tumor formation, or release of single CTCs into the bloodstream; it does, however, profoundly suppress the generation of CTC clusters in the circulation and the generation of distant metastases in the lungs. Thus, Plakoglobin appears to be a key component of the cell junctions that helps tether CTC clusters together in the bloodstream, contributing to their enhanced metastatic initiation potential (Aceto et al. 2014).
RNA sequencing analysis of single CTCs from the blood of men with metastatic prostate cancer also identified mechanisms of resistance to therapies targeting androgen receptor signaling, including non-canonical Wnt signaling through Wnt 5A (Miyamoto et al. 2015). Moreover, these studies identified a profound level of heterogeneity in castrate-resistant prostate cancer, with distinct androgen receptor (AR) gene mutations and AR splicing variants present within different cells from the same patient. Indeed, single-cell CTC analysis is poised to reveal multiple independent mechanisms of acquired drug resistance, each of which may have different kinetics as patients receive successive lines of therapy.
Taken together, transcriptomic analysis of single CTCs may provide exceptional insight into the mechanisms driving tumor progression, metastasis, and acquired drug resistance. However, the effort and cost currently associated with single-cell RNA sequencing limits this platform to discovery and research applications. For widespread and routine clinical applications, more robust and economical RNA-based quantitative assays are also available and may present shifts in diagnostic paradigms for non-invasive cancer detection and monitoring.
4 High-Throughput Diagnostic Assays Using CTC-Derived RNA Signatures
RNA-based detection of CTCs within the background of hematopoietic cells relies upon the profound differences in transcriptional profiles between these cancer cells and surrounding leukocytes. Initial attempts at RNA-based detection of CTCs relied upon RT-PCR technology, with amplification of the prostate-specific PSA transcript applied to detect prostate cancer CTCs within the mononuclear cell fraction of blood samples, and the liver-specific albumin RNA similarly used to interrogate buffy coats from patients with advanced hepatocellular carcinoma (Kar and Carr 1995; Seiden et al. 1994). Additional markers, including mRNAs for cytokeratin, melanoma-specific markers, ALDH1, telomerase, MUC1, and others, have been used to test for multiple additional cancer types (Ignatiadis et al. 2015; Gazzaniga et al. 2010; Arenberger et al. 2008; Shen et al. 2009; Pierga et al. 2007). Unfortunately, the success of these semi-quantitative RT-PCR-based analyses has been inconsistent in part because of their relatively low sensitivity and specificity in unpurified whole blood. A prevalence of 1 CTC per million leukocytes may be below the limit of detection using RT-PCR for a non-abundant transcript. Furthermore, even very low-level transcription by abundant hematopoietic cells of highly tissue-specific transcripts becomes a confounder when the tumor cells are present at such vanishingly low numbers. Indeed, higher numbers of contaminating WBCs increase the Ct values in qRT-PCR detection of identical amount of specific template, and large amount of non-specific template (equivalent of >1000 WBCs) produces SYBR Green noise independent of product amplification, interfering with the quantitative detection of the underlying signal (Pfitzner et al. 2014). The susceptibility of qRT-PCR to the inhibitory effects of large amounts of non-specific template may therefore explain the large variability and inconsistencies in reports describing CTC detection via this method. For all these reasons, we reasoned that initial enrichment of CTCs under RNA-preserving conditions, followed by quantitative digital PCR, provides a much more reliable strategy for RNA-based detection.
Droplet digital PCR (ddPCR) helps to overcome the inherent limitation posed by the presence of excess non-specific templates from contaminating cells by sequestering each individual cDNA template and PCR reagents into aqueous droplets within an oil suspension, thereby drastically increasing the effective concentration of the transcript of interest and allowing the differential expression of CTC-specific genes to be leveraged for identifying their presence (Fig. 2). Partitioning the entire cDNA sample into these droplets followed by high-cycle PCR to maximally amplify each template of interest creates a digital readout of the number of positive droplets, a measure of the prevalence of each transcript of interest (Kalinina et al. 1997). By tabulating the total number of positive and negative droplets and assuming the transcripts of interest follow a Poisson’s distribution when partitioning into droplets, the absolute number of transcripts in the sample can be imputed. ddPCR has been successfully used for detecting rare alleles in the context of free plasma DNA, where its limit of detection is at allele frequency lower than 0.01% (Vogelstein and Kinzler 1999). In RNA detection, ddPCR may be somewhat less sensitive, but it robustly detects presence of aberrant splicing variants (e.g., the androgen receptor Arv7 transcript) in RNA purified from prostate cancer CTCs (Ma et al. 2016; Parkin et al. 2017). Beyond quantifying specific cancer-associated abnormalities, ddPCR detection also offers the potential for scoring and monitoring multiple normal lineage-specific transcripts that are absent from hematopoietic cells and hence denote the presence of CTCs from a given tissue of origin. Successful application of this strategy requires extensive validation of these transcripts to ensure complete absence of signal in normal blood cells, a feat that is greatly enhanced by the initial microfluidic enrichment of CTCs and reduced abundance of contaminating leukocytes.
The normal liver expresses unique transcripts, including albumin and multiple metabolic enzymes, that are completely absent from the expression profiles of other tissues, making it an ideal proof of principle for lineage RNA-based detection of CTCs. Targeting the detection of hepatocellular carcinoma (HCC), we recently established a panel of 10 RNA markers, optimized for ddPCR amplification from CTC-iChip-enriched whole blood of patients with known liver cancer. Total cellular RNA isolated from a 0.1–1% prevalent population of HCC CTCs amidst contaminating leukocytes was subjected to whole transcriptome amplification (WTA)—a step that exponentially increases the signal from all markers and also allows a limited amount of template RNA to be interrogated simultaneously for multiple markers, an important consideration given the known heterogeneity of cancer cells. Spiking individual HCC cells into whole blood followed by microfluidic enrichment and ddPCR showed the limit of detection to be 1 cell per 5 ml of blood, with millions of transcripts of interest generated from a single-spiked cancer cell (Kalinich et al. 2017).
Critical to the successful application of any diagnostic test is the comparison between positive cases and appropriate, age-matched, and risk-matched negative controls. As expected, our HCC digital CTC assay produced negligible background signals using blood samples obtained from young healthy donors. More importantly, it was similarly negative when applied to a cohort of patients with advanced chronic cirrhosis who were at high risk for the development of HCC and were on a regular screening protocol using serial measurements of the oncofetal antigen alpha fetoprotein (AFP) and ultrasound measurements. Among patients with confirmed HCC, the sensitivity of the assay was 56% at 95% specificity when tested against a cohort of patients with chronic liver disease at high risk for developing HCC. Using this assay, we were unable to detect signal without initial CTC enrichment, pointing to the importance of both debulking normal leukocytes and applying high-sensitivity digital PCR detection.
While these results constitute a proof of principle, they also open the door toward a viable CTC-based platform for monitoring and early detection of liver cancer. HCC arises predominantly in high-risk individuals with liver cirrhosis caused by infection with hepatitis B, hepatitis C, or non-alcoholic fatty liver disease (NASH). Currently, such individuals are monitored for plasma protein AFP levels, a sensitive but non-specific biomarker, which interestingly showed poor correlation with the levels of CTC-derived AFP mRNA. Tumor secretion of AFP protein and shedding of CTCs expressing AFP and other transcripts presumably measure different aspects of tumor biology, enhancing the likelihood that combining the two assays may increase predictive value for the early detection of HCC. Indeed, in an initial cohort of 15 patients with newly diagnosed HCC, 5 were positive for both AFP protein and CTC detection, 1 was only positive for AFP protein, and 4 were only positive by CTC assay. Thus, serial monitoring using both AFP and CTC quantitation should be tested as a novel blood-based screening platform for the early detection of liver cancer in high-risk populations.
RNA-based monitoring of CTCs has broad applications beyond the measurement of tumor burden in the blood. Judicious use of biomarkers can establish indices of intracellular signaling pathways, including androgen receptor (AR) signaling in prostate cancer or estrogen receptor (ER) responsive pathways in breast cancer. For instance, in women with metastatic hormone receptor-positive breast cancer, persistence of CTC-derived transcripts indicating ER signaling despite treatment with ER-targeting therapy identifies patients likely to have rapid progression on endocrine therapy (Kwan et al. 2018). Such digital quantitation of CTC-derived RNA provides the first non-invasive blood-based pharmacodynamic measurement of ER signaling following breast cancer therapy. In addition to studies of metastatic breast cancer, in a cohort of women with early stage breast cancer, elevated CTC-derived RNA signal after initial courses of presurgical (neoadjuvant) chemotherapy was predictive of the presence of minimally residual disease at the time of surgical resection (Kwan et al. 2018). In analogous studies of prostate cancer, CTC-derived signal for the AR splicing variant AR-V7 and for the HOXB13 biomarker in men at first relapse of metastatic prostate cancer were highly correlated with abbreviated clinical response to the androgen synthesis inhibitor abiraterone. In men with localized prostate cancer, detectable CTC-derived RNA signal is correlated with extracapsular (seminal vesicle) invasion and metastasis to regional lymph nodes (Miyamoto et al. 2018). Digital quantitation of CTC-derived transcripts is also applicable in melanoma, where neural crest and carcinoembryonic antigen-associated RNAs provide robust signal of circulating cancer cells. In metastatic melanoma, serial monitoring of patients receiving immune checkpoint blockade shows a highly significant correlation between early declines in digitally quantified CTC burden and subsequent response to immunotherapy and overall survival (Hong et al. 2018). Finally, across many different types of cancer, oncogenic translocation products leading to chimeric transcripts are detectable using CTC-derived RNA analysis, leading to the appropriate application of targeted therapeutic regimens. Taken together, the convergence of high-quality enrichment of CTCs with intact RNA together with high-sensitivity RNA-based digital PCR provides new tools for the effective monitoring of cancer cells in the blood.
5 Concluding Comments
Liquid biopsies, defined as the interrogation of blood components to ascertain the properties of solid tumors, are poised to revolutionize the diagnosis and treatment of cancer. Among the multiple technologies, from circulating plasma DNA to exosomes and CTCs, each has its unique strengths and weaknesses, and each may play a greater or lesser role in a specific clinical scenario relevant to a particular tumor type. In general, ctDNA has had the benefit of ease of collection and analysis, but has been limited by the analysis of genetic variations in tumors; in contrast, CTC analyses have been limited by the technological hurdles in rare cell isolation and the biological features involved in their molecular characterization. We believe that new approaches involving automatable microfluidic negative depletion of normal blood cells to enrich for untagged and unbiased CTCs, together with RNA-based digital readouts are now poised to level the playing field, brining CTC measurements along with ctDNA into the frontline of clinical applications. These two types of liquid biopsies are highly complementary, as illustrated by the hypothetical scenario of a mutation of unknown origin identified using ctDNA, whose organ of origin may be identified by RNA-based CTC analysis. Moreover, some cancers are driven by defined genetic alterations readily identified by ctDNA, while others may be tied to epigenetic features or transcriptional changes that are invisible to DNA sequencing, but apparent by RNA-based analysis. Thus, we envision a future in which liquid biopsies with distinct capabilities may be integrated to provide a comprehensive non-invasive platform for monitoring cancer, ranging from the earliest evidence of cancer initiation or recurrence, to guiding the most effective therapeutic options for evolving cancer resistance. Finally, measuring transcriptional programs as the direct output of the genetic and epigenetic drivers promises to improve our ability to understand tumor biology and respond to its changes, opening the doors to more effective ways to diagnose, treat, and monitor cancers in the future.
References
Aceto N, Bardia A, Miyamoto DT et al (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158:1110–1122
Alix-Panabières C, Pantel K (2014) Challenges in circulating tumour cell research. Nat Rev Cancer 14:623–631
Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM (2004) Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 10:6897–6904
Arenberger P, Arenbergerova M, Vohradnikova O, Kremen J (2008) Early detection of melanoma progression by quantitative real-time RT-PCR analysis for multiple melanoma markers. Keio J Med 57:57–64
Cristofanilli M, Budd GT, Ellis MJ et al (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351:781–791
Cristofanilli M, Budd GT, Ellis MJ, et al (2009) Circulating tumor cells, disease progression, and survival in metastatic breast cancer 351:781–791. http://doi.org/10.1056/NEJMoa040766
Easwaran H, Tsai H-C, Baylin SB (2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell 54:716–727
Gazzaniga P, Gradilone A, Petracca A, Nicolazzo C, Raimondi C, Iacovelli R, Naso G, Cortesi E (2010) Molecular markers in circulating tumour cells from metastatic colorectal cancer patients. J Cell Mol Med 14:2073–2077
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Hong X, Sullivan RJ, Kalinich M et al (2018) Molecular signatures of circulating melanoma cells for monitoring early response to immune checkpoint therapy. PNAS 114:1123–1128
Ignatiadis M, Lee M, Jeffrey SS (2015) Circulating tumor cells and circulating tumor DNA: challenges and opportunities on the path to clinical utility. Clin Cancer Res 21:4786–4800
Kalinich M, Bhan I, Kwan TT et al (2017) An RNA-based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. PNAS 114:1123–1128
Kalinina O, Lebedeva I, Brown J, Silver J (1997) Nanoliter scale PCR with TaqMan detection. Nucleic Acids Res 25:1999–2004
Kar S, Carr BI (1995) Detection of liver cells in peripheral blood of patients with advanced-stage hepatocellular carcinoma. Hepatology 21:403–407
Kwan, TT, Bardia, A, Spring, LM et al (2018) A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov 8(10):1286–1299
Luo X, Mitra D, Sullivan RJ et al (2014) Isolation and molecular characterization of circulating melanoma cells. Cell Rep 7:645–653
Ma Y, Luk A, Young FP, Lynch D, Chua W, Balakrishnar B, de Souza P, Becker TM (2016) Droplet digital PCR Based Androgen Receptor Variant 7 (AR-V7) detection from prostate cancer patient blood biopsies. Int J Mol Sci 17:1264
McGranahan N, Swanton C (2017) Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168:613–628
Miyamoto DT, Lee RJ, Stott SL et al (2012) Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer. Cancer Discov 2:995–1003
Miyamoto DT, Zheng Y, Wittner BS et al (2015) RNA-Seq of single prostate CTCs implicates noncanonical Wnt signaling in antiandrogen resistance. Science 349:1351–1356
Miyamoto DT, Lee RJ, Kalinich M et al (2018) An RNA-based digital circulating tumor cell signature is predictive of drug response and early dissemination in prostate cancer. Cancer Discov CD-16-1406
Nagrath S, Sequist LV, Maheswaran S et al (2007) Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450:1235–1239
Nardi V, Azam M, Daley GQ (2004) Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol 11:35–43
Ozkumur E, Shah AM, Ciciliano JC et al (2013) Inertial focusing for tumor antigen–dependent and –independent sorting of rare circulating tumor cells. Sci Transl Med 5:179ra47–179ra47
Parkin B, Londoño-Joshi A, Kang Q, Tewari M, Rhim AD, Malek SN (2017) Ultrasensitive mutation detection identifies rare residual cells causing acute myelogenous leukemia relapse. J Clin Invest 127:3484–3495
Payne RE, Wang F, Su N, Krell J, Zebrowski A, Yagüe E, Ma X-J, Luo Y, Coombes RC (2012) Viable circulating tumour cell detection using multiplex RNA in situ hybridisation predicts progression-free survival in metastatic breast cancer patients. Br J Cancer 106:1790–1797
Pfitzner C, Schröder I, Scheungraber C, Dogan A, Runnebaum IB, Dürst M, Häfner N (2014) Digital-Direct-RT-PCR: a sensitive and specific method for quantification of CTC in patients with cervical carcinoma. Nature Publishing Group 4:3970
Pierga J-Y, Bidard F-C, Denis MG, de Cremoux P (2007) Prognostic value of peripheral blood double detection of CK19 and MUC1 mRNA positive cells detected by RT-quantitative PCR in 94 breast cancer patients with a follow up of 9 years. Mol Oncol 1:267–268
Poste G, Fidler IJ (1980) The pathogenesis of cancer metastasis. Nature 283:139–146
Powell AA, Talasaz AH, Zhang H et al (2012) Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS ONE 7:e33788
Seiden MV, Kantoff PW, Krithivas K, Propert K, Bryant M, Haltom E, Gaynes L, Kaplan I, Bubley G, DeWolf W (1994) Detection of circulating tumor cells in men with localized prostate cancer. J Clin Oncol 12:2634–2639
Shen C, Hu L, Xia L, Li Y (2009) The detection of circulating tumor cells of breast cancer patients by using multimarker (Survivin, hTERT and hMAM) quantitative real-time PCR. Clin Biochem 42:194–200
Stott SL, Lee RJ, Nagrath S et al (2010) Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med 2:25ra23–25ra23
Ting DT, Wittner BS, Ligorio M et al (2014) Single-Cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep 8:1905–1918
Tirosh I, Venteicher AS, Hebert C et al (2016) Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature Publishing Group 539:309–313
Vogelstein B, Kinzler KW (1999) Digital PCR. PNAS 96:9236–9241
Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N (2017) Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nature Publishing Group 17:223–238
Wu S, Liu S, Liu Z, Huang J, Pu X, Li J, Yang D, Deng H, Yang N, Xu J (2015) Classification of circulating tumor cells by epithelial-mesenchymal transition markers. PLoS ONE 10:e0123976
Yu M, Stott S, Toner M, Maheswaran S, Haber DA (2011) Circulating tumor cells: approaches to isolation and characterization. J Cell Biol 192:373–382
Yu M, Ting DT, Stott SL et al (2012) RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature Publishing Group 487:510–513
Yu M, Bardia A, Wittner BS et al (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339:580–584
Zhang X, Yuan X, Shi H, Wu L, Qian H, Xu W (2015) Exosomes in cancer: small particle, big player. J Hematol Oncol 8:83
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kalinich, M., Kwan, T.T., Toner, M., Haber, D.A., Maheswaran, S. (2020). Quantitative Analysis of Circulating Tumor Cells Using RNA-Based Digital Scoring. In: Schaffner, F., Merlin, JL., von Bubnoff, N. (eds) Tumor Liquid Biopsies. Recent Results in Cancer Research, vol 215. Springer, Cham. https://doi.org/10.1007/978-3-030-26439-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-26439-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-26438-3
Online ISBN: 978-3-030-26439-0
eBook Packages: MedicineMedicine (R0)