
Abstract
Fuchs endothelial corneal dystrophy (FECD), estimated to affect 4% of the US population [1], is the leading cause of endogenous corneal endothelial degeneration and the primary indication for keratoplasty worldwide [2]. FECD is a slowly progressive bilateral corneal disease that is characterized by abnormal corneal endothelial morphology, such as pleomorphism and polymegethism, decreased cell density, thickening of the Descemet membrane (DM), and formation of extracellular matrix (ECM) excrescences called guttae [3], which lead to derangements in endothelial cell function. Quality of vision is slowly compromised by corneal edema secondary to gradual failure of corneal endothelial pump action and guttae-induced visual disturbances such as decreased contrast sensitivity [4] and higher order aberrations [5]. Its pathogenesis is proposed to be an interaction between endogenous and exogenous factors [6]. The disease manifests in the early third to late fifth decade of life and has a female preponderance [7–9]. Since the first description of the disease by Ernest Fuchs more than 100 years ago [10], major advances in understanding the nature and pathogenesis of FECD have been made and are described in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Fuchs endothelial corneal dystrophy
- Corneal endothelium
- Oxidative stress
- Mitochondria
- Guttae
- Endothelial transplantation
- Descemet stripping
- DSO
Introduction
Fuchs endothelial corneal dystrophy (FECD), estimated to affect 4% of the US population [1], is the leading cause of endogenous corneal endothelial degeneration and the primary indication for keratoplasty worldwide [2]. FECD is a slowly progressive bilateral corneal disease that is characterized by abnormal corneal endothelial morphology, such as pleomorphism and polymegethism, decreased cell density, thickening of the Descemet membrane (DM), and formation of extracellular matrix (ECM) excrescences called guttae [3], which lead to derangements in endothelial cell function. Quality of vision is slowly compromised by corneal edema secondary to gradual failure of corneal endothelial pump action and guttae-induced visual disturbances such as decreased contrast sensitivity [4] and higher order aberrations [5]. Its pathogenesis is proposed to be an interaction between endogenous and exogenous factors [6]. The disease manifests in the early third to late fifth decade of life and has a female preponderance [7,8,9]. Since the first description of the disease by Ernest Fuchs more than 100 years ago [10], major advances in understanding the nature and pathogenesis of FECD have been made and are described in this chapter.
Prevalence
The prevalence of the disease varies depending on the age, origin, and ethnicity of the population. FECD affects people over 50 more frequently, from 3.3–4.1% in the Japanese population [8, 11, 12], 3.9–6.6% in the US population [13, 14], and 9.2% in residents in Reykjavik, Iceland [9]. Despite a slightly higher prevalence in Chinese Singaporean population (6.7%) [8], the prevalence of FECD appears higher in Caucasians compared to Asians. In addition, regardless of geographical distribution, females are at greater risk of developing the disease at a ratio of 2.5:1 to 3.7:1 [7,8,9, 12, 13].
Corneal Endothelial Composition and Function
Corneal Endothelial Cells
The corneal endothelium (CE), the innermost layer of the cornea, is located between the stroma and the anterior chamber of the eye and forms a monolayer of hexagonal cells derived from the neuroectoderm [15]. The corneal endothelial cells (CECs) are highly metabolically active, contain numerous intracellular mitochondria, and have cytoplasmic extensions allowing intercellular exchange [16].
The endothelial cells are arrested in the G1 phase of the cell cycle at birth [17], and thus do not proliferate, resulting in a gradual decrease in endothelial cell density during life [18]. CEC are contact-inhibited, meaning that cells stop migrating once a neighboring cell is encountered. At birth, the human CE contains approximately 5000 cells/mm [2, 19]. There is a gradual decrease in endothelial cell density [20] during life at a rate of 0.36 to 0.6% of the endothelial cells per year [ 8, 20,21,22]. In late adulthood, the normal count ranges from 2000–3000 cells/mm [2] or about 700,000 cells in total. In a normal eye, this gradual reduction of CECs has no effect on corneal transparency. In a healthy CE, the size of CECs is remarkably homogeneous (coefficient of variation of approximately 0.27–0.28) with a hexagonality percentage of 60%. The decrease in endothelial cell density leads to cellular size and shape changes; polymegethism occurs when cell size increases and pleomorphism when cell shape changes. Both result as an adaptation of the endothelial cells to their decreasing density to maintain the continuous cellular monolayer.
Barrier and Pump Function (Fig. 8.1)
The purpose of the endothelium is to maintain the constant hydration state of the cornea and, therefore, its transparency (Fig. 8.1). The CE provides stromal deturgescence both by a passive mechanism (barrier function), provided by the intercellular tight junctions [23], and an active mechanism, via Na+/K+ ATPase pumps [24], that returns solutes and water from the cornea to the anterior chamber.

Illustration of the endothelial pump function/dysfunction mechanism. Left, corneal endothelium in physiological state. The maintenance of corneal transparency is ensured by the passive endothelial barrier function (tight junctions) and by active Na+/K+ ATPase and HCO3+ pumps. Right, when the number of endothelial cells is insufficient to maintain the endothelial pump function, intrastromal edema appears, and vision decreases. Fluid infiltrates the subepithelial layer, elevating the corneal epithelium (bullae) which can break, causing pain
Its intercellular junctions prevent water from the aqueous humor from entering the stroma. As an interface between the cornea and the aqueous humor of the anterior chamber, the CE allows both trans- and paracellular transport of nutrients while providing resistance to the entry of the aqueous humor into the stroma [25].
The endothelial pump mechanism is still not completely understood. It is not strictly a mechanism of active transport of water molecules at the level of the endothelium; in fact, the movement of water through the endothelium is essentially based on the osmotic gradient created by the different pumps. The path of the water molecules is mainly paracellular, and a small amount of water moves via type 1 aquaporins (AQP1). The water is transported from the stroma to the aqueous humor at a rate of 6–8 mL/h/cm [2]. The endothelial pump mechanism is most likely based on Na+/K+ ATPase pumps located on the basolateral sides of the endothelial cells, on intracellular carbonic anhydrase, on Na+/H+ and HCO3+/Cl− exchangers, on Na+/HCO3+ and Na+/K+/Cl− cotransporters, and on an apical elimination route of bicarbonate [24].
Descemet Membrane
The Descemet membrane (DM), initially described by the French physician Jean Descemet in 1770 [26], is secreted by CECs [27] and is located between the posterior stroma and the monolayer of CECs. Like all basement membranes, the DM is a compact layer composed of extracellular matrix (ECM) biomolecules, including glycoproteins and glycosaminoglycans [28]. Its composition and ultrastructure vary with the stage of development and the presence of pathological conditions [29] with distinct layers forming over time [30]. During fetal development, the DM acquires a complex lamellar structure; it is 3 μm thick at birth and reaches 6–10 μm in adulthood [30, 31]. The anterior part of the DM is formed during the prenatal period, approximately from 12 weeks after conception and until birth [30]. From 16 weeks of gestation, striations appear to constitute the anterior banded layer (ABL), termed the fetal layer, which is composed mainly of collagen IV and III [32, 33].
At birth, the secretion of collagen VIII by CECs decreases when the cells pass from the proliferative to a nonproliferative state while that of collagen IV continues [27, 34]. The secreted ECM then loses its striated appearance and gradually becomes a nonbanded layer (PNBL), termed the neonatal layer [29]. In adulthood, the DM is composed mainly of collagen IV, VIII, and I [35]. Its porous structure allows the exchange of stroma towards the endothelium and the anterior chamber; in fact, it has pores with an average size of 38 nm that specifically serve this purpose [36]. Over time, membrane material can accumulate in certain parts of the DM; in particular, in the periphery of the adult cornea, endothelial deposits, termed Hassall-Henle bodies, can be observed. These represent posterior growths of the DM and are only visible histologically [31].
The DM is elastic [37], permeable to water, solid, and resistant to the proteolytic action of metalloproteases. As the basement membrane of endothelial cells, it participates in the maintenance of dehydration of the corneal stroma.
Pathogenesis of Fuchs Dystrophy
Overview
FECD is characterized by an accelerated, progressive decline in the number of CECs, which leads to polymegethism and pleomorphism [38]. At first, only central corneal endothelial cells are affected, but later in the disease course peripheral endothelial cells are also involved. The DM ECM homeostasis is compromised, as evidenced by the formation of guttae and the disorganization of ECM collagen fibrils [35]. Recently, evidence has established that guttae size might play a role in FECD by creating a noxious environment for the CECs [39, 40]. After a significant decrease in endothelial cell density (below 350–500 cells/mm [2]) [41], the endothelial pump is not able to ensure the deturgescence of the cornea (Fig. 8.1). This results in elevated corneal hydration levels, disruption of the collagen fiber arrangement, and loss of corneal transparency. The water present in the aqueous humor infiltrates the cornea, causing stromal edema, and eventually resulting in the formation of painful epithelial bullae [42]. The progressive corneal edema prevents light from being properly transmitted in the eye and thus compromises vision. While FECD is the most common cause of endothelial dysfunction, other causes include postsurgical, inflammatory, infectious, or traumatic.
Exogenous Factors
Smoking [43] and ultraviolet radiation [44] have been reported as exogenous factors associated with Fuchs dystrophy and are suggested to increase oxidative stress, resulting in accelerated apoptosis of corneal endothelial cells.
Endogenous Factors
Genetic Factors
The genetic basis of FECD is complex and heterogeneous, demonstrating variable expressivity and incomplete penetrance. It was not until the early 1950s that the initial reports of its heritability emerged [45,46,47]. Over the past decade, significant progress has been made in understanding the genetics of Fuchs dystrophy. Studies in families with multiple generations of members with Fuchs dystrophy have identified several chromosomal loci associated with the pathology, classified according to the affected chromosome (IC3D classification [48]): FECD1, chromosome 1; FECD2, chromosome 13; FECD3, chromosome 18; FECD4, chromosome 20; FECD5, chromosome 5; FECD6, chromosome 10; FECD7, chromosome 9; and FECD8, chromosome 15 (Fig. 8.2).

Genetics of Fuchs dystrophy
With the identification of four single-nucleotide polymorphisms (SNPs ; namely, rs17595731, rs613872, rs9954153, and rs2286812), the TCF4 gene has been associated with Fuchs dystrophy, with the SNP rs613872 being the marker for the most robust of the pathologies [49] due to the production of a CTG trinucleotide expansion sequence in the 3rd intron of TCF4 within chromosome 18q21.1 (henceforth referred to as the ‘CTG18.1’ locus) [50]. The prevalence of CTG18.1 in FECD varies depending on ethnicity [51,52,53,54,55,56] with up to 79% of FECD Caucasian patients harboring these expanded sequences [50, 51, 57]. TCF4 encodes the E2-2 transcription factor, which serves a regulatory function in the expression of multiple other genes [58]. Similar to myotonic dystrophy type 1 (DM1), which is characterized by an expanded CTG trinucleotide repeat sequence in the Dystrophia Myotonica type 1 protein kinase (DMPK) gene [59], these repeat expansion sequences negatively affect cellular function via the formation of toxic RNA foci [50], which sequesters critical transcription factors and, in turn, may cause dysfunction of the cytoskeleton and cell adhesion. Additionally, evidence has also emerged to suggest a direct correlation between repeat length at the CTG18.1 locus and FECD disease severity [53]. Furthermore, the predictive value of CTG18.1 trinucleotide repeat expansion (TNR) length on clinical progression of FECD has been characterized showing that patients harboring an expanded CTG 18.1 allele are more likely to experience clinical progression of FECD [60]. Recently, evidence has also suggest that TNR may contribute to disease pathogenesis through production of cytosolic repeat-associated non-ATG (RAN) translation products, a mechanism suggested in other repeat expansion diseases [61].
Additional genes, such as KANK4, LAMC1, and ATP1B1, have recently been linked to Fuchs dystrophy [62]. Although additional studies are still required for understanding the exact involvement of these genes in the pathophysiology of Fuchs dystrophy, it is known that KANK4 and LAMC1 play an important role in modulating intra- and extracellular interactions. Similarly, ATP1B1 is the gene that encodes the Na+/K+ ATPase pump, the central mechanism of normal endothelial function [63].
Nonsense mutations encoding the A2 chain of collagen VIII (COL8A2, p.R155Q, p.R304Q, p.R434, and p.Q455K) have been demonstrated in early Fuchs dystrophy (FECD1) [64], while mutations in the TCF8 gene (or ZEB1) have been detected in posterior polymorphic dystrophy [65] and Fuchs dystrophy [66]. At the same time, mutations in the SLC4A11 gene have been reported as causes of congenital hereditary endothelial dystrophy (CHED) [67] and were associated with Fuchs dystrophy in 2007 [68].
Although the vast majority of FECD cases appear to be associated with the trinucleotide repeat expansion on chromosome 18, further research on the genetics of Fuchs dystrophy is still required to determine how each of the associated genetic defects participate in disease pathogenesis.
Cellular Dysfunction
Oxidative Stress and Mitochondrial Dysfunction
In the eye, CECs have the second highest level of aerobic metabolism (after retinal photoreceptors) [69], which makes them highly sensitive to oxidative stress. In 2010, Jurkunas et al. demonstrated oxidative stress as an integral part of the pathophysiology of Fuchs dystrophy [70]. In this study, the authors showed that some antioxidative regulators, such as NRF2, are downregulated and that oxidative DNA damage and apoptosis are significantly higher in patients with FECD. Subsequent studies have confirmed this finding and have demonstrated that oxidative damage to mitochondrial DNA leads to mitochondrial dysfunction, cellular stress, senescence, and apoptosis [16, 44, 71,72,73].
Toxic Environment
Aberrant Secretion of ECM and Toxicity of Guttae
In addition to endothelial cell abnormalities, Fuchs dystrophy is characterized by a thickening of the DM associated with the presence of guttae in the posterior part of the cornea (Fig. 8.3). The accumulation of glycation end products [74] and certain proteins has also been described as a contributory factor in FECD. An increase in collagen IV, laminin, fibronectin, and several other components of ECM has been demonstrated in the DM of FECD patients [75].

Guttae viewed in a histological section (a) and by bright field optical microscopy (b). (a) Histological section of the DM. Guttae are the dome-shaped excrescences of the DM, varying in size and diameter. (b) Bright field optical microscopy of cornea. Left, extreme corneal periphery; right, corneal center. The guttae are present in the center and extend towards the periphery. Their size and number increase from the periphery to the center. In advanced stages of the disease, guttae become confluent
The ECM, which is present in all tissues, provides mechanical support for cells and influences differentiation, migration, activation, behavior, and cell death [76]. Thus, changes in ECM composition due to the aberrant production of ECM proteins affect the physiological activity of cells and can lead to their dysfunction. Although the genesis of guttae is still poorly understood, several studies support the hypothesis that, in FECD, the homeostasis of the ECM of the DM is compromised with disease severity related to the number and size of the guttae, which promote disorganization of ECM collagen fibrils [35]. The exact composition of the guttae is still unclear. However, the expression of transforming growth factor-beta-induced protein (TGFBIp) and clusterin was found to be increased in guttae-forming deposits [77]. A recent study from Kocaba and colleagues demonstrated that guttae cause cellular stress in CECs, promoting senescence and apoptosis [39].
Barrier and Pump Dysfunction
As endothelial cell damage progresses in FECD, the number of CECs decreases and the cells lose their tight junctions, causing loss of the barrier function and reduction in the ability to actively pump solutes from the corneal stroma due to endothelial pump dysfunction. Additionally, thickening of the DM might also negatively impact fluid exchange from the stroma to the anterior chamber. In advanced stages of the disease, the severe damage to the endothelium leads to endothelial decompensation: the capacity of the barrier and the aqueous pumps is exceeded, and intracorneal edema occurs, adversely impacting corneal transparency and visual acuity (Fig. 8.1).
Towards a Unified Theory of FECD Pathogenesis
Vicious Cycle of Fuchs Dystrophy
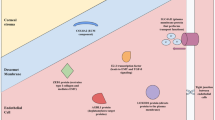
We have already discussed the role of oxidative stress, mitochondrial dysfunction, and perturbations of the ECM in FECD (Fig. 8.4). These are not the only cellular processes purported to be involved in this disease, however. Others include the unfolded protein response and epithelial-mesenchymal transition (EMT) [78]. Genetic factors and environmental stressors, such as UV exposure and hormonal influences, also contribute in yet unclear ways, to the end result of increased apoptosis and endothelial dysfunction. How these pathways interact to cause FECD is not yet known. Many questions remain. Despite genetic heterogeneity, FECD has remarkable phenotypic homogeneity, which must be accounted for by any theory of disease pathogenesis. A recent review has put forth the first unified theory of FECD pathogenesis (Fig. 8.5), seeking to tie together the disparate cellular processes and genetic mutations that have been implicated thus far and placing mitochondrial dysfunction at the core of disease pathogenesis [78].

Vicious cycle of Fuchs dystrophy. DM Descemet membrane, ECM extracellular matrix, UV ultraviolet

Working theory of FECD pathogenesis. (From Sarnicola et al. [78]. Open access). ALS-FTD amyotrophic lateral sclerosis with front-temporal dementia
Insights from Neurodegenerative Diseases
CEC are derived from the neuroectoderm via the neural crest and it has been suggested that Fuchs can be considered as a “neurodegenerative” disease [79]. Indeed, many of the same cellular mechanisms (mitochondrial dysfunction, oxidative stress, repeat expansion) implicated in FECD have been shown in classic neurodegenerative diseases including Parkinson, Alzheimer, and Huntington diseases and amyotrophic lateral sclerosis [80,81,82,83]. It is likely that FECD studies will be aided by lessons learned from these disorders. Conversely, it is likely that the study of classic neurodegenerative diseases will be enhanced by insights garnered in the study of diseased endothelium from FECD, which is routinely removed as part of surgical management.
Diagnosis of FECD
Diagnosis of FECD is mainly clinical, based on slit lamp findings, corneal pachymetry, and endothelial cell imaging. Multiple staging scales for FECD have been described. One of the most commonly used one (Krachmer classification) classifies the disease in four clinical stages [84]:
Stage 1 (Fig. 8.6) represents the initial manifestation of the disease. Nonconfluent guttae are present in the posterior part of the central cornea and are visible as dark spots by direct illumination, highlighted in retroillumination. Patients are usually asymptomatic at this stage; however, visual quality may deteriorate due to light scattering by the central guttae. Stage 2 is defined by a reduction in the number, increase in the size (polymegethism), and loss of the hexagonal shape (pleomorphism) of corneal endothelial cells, as shown by specular or confocal microscopy. Guttae become confluent centrally and extend towards the peripheral cornea. Corneal edema may be evident on exam or by measurement of corneal thickness. Early morning vision is often affected due to increased corneal edema after eye closure during the night. In stage 3, vision deterioration begins to last longer and may be present throughout the day. Corneal stromal edema increases, eventually leading to epithelial and subepithelial bullae. Pain, epiphora, and light sensitivity occur if the bullae rupture. In stage 4, prolonged and chronic corneal edema induces subepithelial fibrosis, stromal opacification , and neovascularization of the cornea. This stage is rarely seen in the developed world due to the success of modern corneal transplantation techniques.

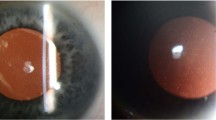
Retroillumination photo of nonconfluent central corneal guttae in FECD
Additional examinations are useful for the diagnosis of early disease and follow-up. Corneal pachymetry can be used to monitor endothelial function during the evolution of the disease. Attention should be paid to the time of the day the measurement is made since this can impact corneal deturgescence. Specular microscopy allows the observation and measurement of guttae, which appear as dark spots. Moreover, observation of decreased endothelial cell density, accompanied by polymegethism and pleomorphism, is beneficial for early diagnosis of the disease (Fig. 8.7).

Specular microscopy of a patient with early FECD
With an axial resolution of 11–18 μm, the anterior segment optical coherence tomography (AS-OCT) is potentially useful for visualizing changes in the endothelium and the DM during the progression of FECD (Fig. 8.8) [85]. The DM appears hyperreflective, thickened, irregular, and multilayered (Fig. 8.8a). Epithelial and subepithelial changes can also be observed: subepithelial bullae, thickening of the epithelium, and subepithelial fibrosis are seen in advanced cases (Fig. 8.8b).

AS-OCT of cornea from FECD patients. (a) Hyperreflectivity and thickening of the DM, subepithelial fibrosis and thickening of the epithelium. (b) Undulation of the DM and subepithelial bullae and major thickening of the epithelium in advance FECD
In vivo confocal microscopy (IVCM) allows a superior image compared to specular microscopy and is helpful for the diagnosis of the disease especially in case of corneal edema or fibrosis [86]. Guttae appear as hyporeflective zones, with a hyperreflective center that corresponds to the apices of the guttae (Fig. 8.9). IVCM can demonstrate of epithelial bullae, subepithelial fibroblast and collagen infiltration, reduction of subbasal corneal nerve density, reduced anterior keratocyte density, and fibroblastic transformation of stressed keratocytes in the stroma.

In vivo confocal microscopy on a FECD patient with advanced disease. Guttae are defined by hyporeflective zones with hyperreflective center; no endothelial cells are visible
Management of Fuchs Dystrophy
Medical management of early Fuchs dystrophy consists of topical hypertonic saline drops or ointment to shorten morning edema. Patients may use the ointment at bedtime and/or the drops upon awakening, typically one drop every 5 minutes for a total of three to four drops. This treatment does not change the course of the disease. Patients do note stinging with these agents, which reduces acceptance. A hairdryer held 5–10 inches from the cornea may also be used to facilitate corneal deturgescence. As the disease progresses, these interventions will no longer be effective. There is no convincing evidence to support the use of topical steroids in the management of Fuchs dystrophy.
Once patients are symptomatic from their FECD, surgery is the preferred treatment. Surgical management of endothelial dysfunction has undergone a revolution in the past 20 years. Interested readers who wish a historical perspective on this field are referred to two recent reviews [78, 87]. In 2019, selective endothelial keratoplasty (EK) is the most common transplantation technique, having surpassed penetrating keratoplasty in 2011. Selective endothelial transplantation techniques continue to evolve. At present, Descemet stripping endothelial keratoplasty (DSEK) is the most common technique, although Descemet membrane endothelial keratoplasty (DMEK) is increasing in popularity. These two surgeries vary in the amount of donor stroma that is transplanted, with DMEK having none and DSEK having 70–150 microns. DMEK provides more rapid visual recovery but has more complications in the immediate postoperative period. However, both DSEK and DMEK represent a real advance in the management of endothelial dysfunction, compared to penetrating keratoplasty.
Advances continue to be made. Kinoshita and colleagues have pioneered the use of cultured cells in the management of endothelial dysfunction and have recently published their initial case series [88]. Cultured cells offer the ability to treat multiple (potentially up to 100) patients from a single donor cornea. However, substantive regulatory hurdles (and insurance issues in the United States) must be surmounted before this technique will enjoy widespread acceptance.
Another exciting advance in the treatment of FECD is Descemet stripping only (DSO), independently pioneered by Colby in the United States and Moloney in Australia since 2014 [89, 90]. Multiple lines of evidence in 2012–2013 suggested that the endothelium in FECD might be capable of “self-rejuvenation.” There had been anecdotal case reports of corneal clearance after inadvertent removal of Descemet membrane or of attempted EK in which the donor tissue was unable to be placed [91, 92]. Melles presented a series of DMEK cases in which the donor tissue did not adhere to the host cornea, yet corneal clearance occurred in Fuchs dystrophy patients, but not in cases of pseudophakic bullous keratopathy [93]. Koizumi published a single case of a patient with FECD who attained corneal clearance after endothelial destruction with cryotherapy and treatment with a topical Rho kinase (ROCK) inhibitor [94]. Finally, an initial small series of deliberate Descemet removal was performed, although results were inconsistent [95].
The series by Borkar, Veldman, and Colby was the first to demonstrate success of DSO, with 10 of their 13 patients achieving corneal clearance, at an average of 12 weeks after a 4 mm deliberate Descemet stripping [89]. The first surgery in this series was done in January 2014. The patient, now 5 years out, retains a clear, compact cornea with excellent vision and an endothelial cell count of approximately 900 cells/mm [2]. Moloney published his initial series in 2017 and was the first to report the use of a topical ROCK inhibitor after DSO [90].
After a several year period where DSO was treated with skepticism, the corneal community has begun to embrace DSO as a treatment for focal Fuchs dystrophy. Subsequent studies have suggested that a “peeling” rather than a “stripping” technique may improve corneal clearance rate [96]. In an investigator-initiated randomized clinical trial of ripasudil, a ROCK inhibitor approved for treatment of glaucoma in Japan, corneal clearance occurred more rapidly and final endothelial cell count was increased in DSO patients treated with ripasudil compared with those who did not use this medication [97]. A multinational industry-sponsored study of ripasudil as an adjunct for DSO is currently in the planning stages. Finally, a recent head-to-head comparison of DMEK vs. DSO showed equivalent visual outcomes. Although DSO patients took longer to clear than DMEK patients, DSO was associated with fewer postsurgical complications [98].
Descemet stripping only is a cost-effective and time-efficient surgery. Further studies are needed to determine the optimal patient population for this nongraft treatment, but its potential is enormous.
Conclusions
Our understanding of the biological basis of FECD has improved dramatically in the last 20 years, as have our options for surgical management. Future strategies for management such as gene-editing remain as exciting possibilities. Even more appealing is the possibility of preventive strategies to address this very common corneal condition.
References
Moller HU, Sunde L. Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2013;54(1).
EBAA. Surgical use and indications for corneal transplant statistical report analysis-2016. Washington, DC: Eye Bank Association of America; 2017.
Vedana G, Villarreal G Jr, Jun AS. Fuchs endothelial corneal dystrophy: current perspectives. Clin Ophthalmol. 2016;10:321–30.
Watanabe S, Oie Y, Fujimoto H, et al. Relationship between corneal Guttae and quality of vision in patients with mild Fuchs’ endothelial corneal dystrophy. Ophthalmology. 2015;122(10):2103–9.
Wacker K, McLaren JW, Amin SR, Baratz KH, Patel SV. Corneal high-order aberrations and backscatter in Fuchs’ endothelial corneal dystrophy. Ophthalmology. 2015;122(8):1645–52.
Schmedt T, Silva MM, Ziaei A, Jurkunas U. Molecular bases of corneal endothelial dystrophies. Exp Eye Res. 2012;95(1):24–34.
Afshari NA, Pittard AB, Siddiqui A, Klintworth GK. Clinical study of Fuchs corneal endothelial dystrophy leading to penetrating keratoplasty: a 30-year experience. Arch Ophthalmol. 2006;124(6):777–80.
Kitagawa K, Kojima M, Sasaki H, et al. Prevalence of primary cornea guttata and morphology of corneal endothelium in aging Japanese and Singaporean subjects. Ophthalmic Res. 2002;34(3):135–8.
Zoega GM, Fujisawa A, Sasaki H, et al. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology. 2006;113(4):565–9.
Fuchs E. Dystrophia epithelialis cornea. Graefes Arch Clin Exp Ophthalmol. 1910;76:478–508.
Nagaki Y, Hayasaka S, Kitagawa K, Yamamoto S. Primary cornea guttata in Japanese patients with cataract: specular microscopic observations. Jpn J Ophthalmol. 1996;40(4):520–5.
Higa A, Sakai H, Sawaguchi S, et al. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: the Kumejima study. Arch Ophthalmol. 2011;129(3):332–6.
Goar EL. Dystrophy of the corneal endothelium (Cornea Guttata), with report of a histologic examination. Trans Am Ophthalmol Soc. 1933;31:48–59.
Lorenzetti DW, Uotila MH, Parikh N, Kaufman HE. Central cornea guttata. Incidence in the general population. Am J Ophthalmol. 1967;64(6):1155–8.
Katikireddy KR, Schmedt T, Price MO, Price FW, Jurkunas UV. Existence of neural crest-derived progenitor cells in normal and Fuchs endothelial dystrophy corneal endothelium. Am J Pathol. 2016;186(10):2736–50.
Benischke AS, Vasanth S, Miyai T, et al. Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy. Sci Rep. 2017;7(1):6656.
Joyce NC, Meklir B, Joyce SJ, Zieske JD. Cell cycle protein expression and proliferative status in human corneal cells. Invest Ophthalmol Vis Sci. 1996;37(4):645–55.
Joyce NC. Proliferative capacity of the corneal endothelium. Prog Retin Eye Res. 2003;22(3):359–89.
Bahn CF, Glassman RM, MacCallum DK, et al. Postnatal development of corneal endothelium. Invest Ophthalmol Vis Sci. 1986;27(1):44–51.
Bourne WM, Nelson LR, Hodge DO. Central corneal endothelial cell changes over a ten-year period. Invest Ophthalmol Vis Sci. 1997;38(3):779–82.
Tuft SJ, Coster DJ. The corneal endothelium. Eye (Lond). 1990;4(Pt 3):389–424.
Moller-Pedersen T. A comparative study of human corneal keratocyte and endothelial cell density during aging. Cornea. 1997;16(3):333–8.
Ramachandran C, Srinivas SP. Formation and disassembly of adherens and tight junctions in the corneal endothelium: regulation by actomyosin contraction. Invest Ophthalmol Vis Sci. 2010;51(4):2139–48.
Bonanno JA. Molecular mechanisms underlying the corneal endothelial pump. Exp Eye Res. 2012;95(1):2–7.
Ohrloff C, Schalnus R, Spitznas M. Quantitative control of the function of the corneal endothelium by fluorophotometry in the anterior eye segment. Klin Monatsbl Augenheilkd. 1986;189(1):24–7.
Descemet J, Demours P, Vincent P. RÈponse de M. Descemet,... ‡ la lettre de M. Demours,... insÈrÈe dans le “Journal de mÈdecine” du mois de novembre dernier. 1770.
Kabosova A, Azar DT, Bannikov GA, et al. Compositional differences between infant and adult human corneal basement membranes. Invest Ophthalmol Vis Sci. 2007;48(11):4989–99.
LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (Maywood). 2007;232(9):1121–9.
Levy SG, Moss J, Sawada H, Dopping-Hepenstal PJ, McCartney AC. The composition of wide-spaced collagen in normal and diseased Descemet's membrane. Curr Eye Res. 1996;15(1):45–52.
Murphy C, Alvarado J, Juster R. Prenatal and postnatal growth of the human Descemet's membrane. Invest Ophthalmol Vis Sci. 1984;25(12):1402–15.
Hayashi S, Osawa T, Tohyama K. Comparative observations on corneas, with special reference to Bowman's layer and Descemet’s membrane in mammals and amphibians. J Morphol. 2002;254(3):247–58.
Ljubimov AV, Burgeson RE, Butkowski RJ, Michael AF, Sun TT, Kenney MC. Human corneal basement membrane heterogeneity: topographical differences in the expression of type IV collagen and laminin isoforms. Lab Investig. 1995;72(4):461–73.
Sawada H, Konomi H, Hirosawa K. Characterization of the collagen in the hexagonal lattice of Descemet’s membrane: its relation to type VIII collagen. J Cell Biol. 1990;110(1):219–27.
Hopfer U, Fukai N, Hopfer H, et al. Targeted disruption of Col8a1 and Col8a2 genes in mice leads to anterior segment abnormalities in the eye. FASEB J. 2005;19(10):1232–44.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of Fuchs’ endothelial corneal dystrophy support that the extracellular matrix of Descemet's membrane is disordered. J Proteome Res. 2014;13(11):4659–67.
Last JA, Liliensiek SJ, Nealey PF, Murphy CJ. Determining the mechanical properties of human corneal basement membranes with atomic force microscopy. J Struct Biol. 2009;167(1):19–24.
Mohammed I, Ross A, Britton JO, Said DG, Dua HS. Elastin content and distribution in endothelial Keratoplasty tissue determines direction of scrolling. Am J Ophthalmol. 2018;194:16.
Borboli S, Colby K. Mechanisms of disease: Fuchs’ endothelial dystrophy. Ophthalmol Clin N Am. 2002;15(1):17–25.
Kocaba V, Katikireddy KR, Gipson I, Price MO, Price FW, Jurkunas UV. Association of the Gutta-induced microenvironment with corneal endothelial cell behavior and demise in Fuchs endothelial corneal dystrophy. JAMA Ophthalmol. 2018;136(8):886–92.
Rizwan M, Peh GS, Adnan K, et al. In vitro topographical model of Fuchs dystrophy for evaluation of corneal endothelial cell monolayer formation. Adv Healthc Mater. 2016;5(22):2896–910.
Agarwal A, Jacob S, Agarwal A, Agarwal S, Kumar MA. Iatrogenic descemetorhexis as a complication of phacoemulsification. J Cataract Refract Surg. 2006;32(5):895–7.
Bourne WM. Biology of the corneal endothelium in health and disease. Eye (Lond). 2003;17(8):912–8.
Zhang X, Igo RP Jr, Fondran J, et al. Association of smoking and other risk factors with Fuchs’ endothelial corneal dystrophy severity and corneal thickness. Invest Ophthalmol Vis Sci. 2013;54(8):5829–35.
Liu C, Vojnovic D, Kochevar IE, Jurkunas UV. UV-A irradiation activates Nrf2-regulated antioxidant defense and induces p53/Caspase3-dependent apoptosis in corneal endothelial cells. Invest Ophthalmol Vis Sci. 2016;57(4):2319–27.
Mortelmans L. Familial form of Fuchs’ corneal dystrophy. Ophthalmologica. 1952;123(2):88–99.
Levitt JM, Lloyd RI. Congenital and familial endothelial defects. Am J Ophthalmol. 1952;35(3):342–9.
Stocker FW. The endothelium of the cornea and its clinical implications. Trans Am Ophthalmol Soc. 1953;51:669–786.
Weiss JS, Moller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies--edition 2. Cornea. 2015;34(2):117–59.
Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuchs’s corneal dystrophy. N Engl J Med. 2010;363(11):1016–24.
Wieben ED, Aleff RA, Tosakulwong N, et al. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS One. 2012;7(11):e49083.
Luther M, Grunauer-Kloevekorn C, Weidle E, et al. TGC Repeats in Intron 2 of the TCF4 Gene have a Good Predictive Power Regarding to Fuchs Endothelial Corneal Dystrophy. Klin Monatsbl Augenheilkd. 2016;233(2):187–94.
Mootha VV, Hussain I, Cunnusamy K, et al. TCF4 triplet repeat expansion and nuclear RNA foci in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2015;56(3):2003–11.
Soliman AZ, Xing C, Radwan SH, Gong X, Mootha VV. Correlation of severity of Fuchs endothelial corneal dystrophy with triplet repeat expansion in TCF4. JAMA Ophthalmol. 2015;133(12):1386–91.
Nakano M, Okumura N, Nakagawa H, et al. Trinucleotide repeat expansion in the TCF4 gene in Fuchs’ endothelial corneal dystrophy in Japanese. Invest Ophthalmol Vis Sci. 2015;56(8):4865–9.
Vasanth S, Eghrari AO, Gapsis BC, et al. Expansion of CTG18.1 trinucleotide repeat in TCF4 is a potent driver of Fuchs’ corneal dystrophy. Invest Ophthalmol Vis Sci. 2015;56(8):4531–6.
Mootha VV, Gong X, Ku HC, Xing C. Association and familial segregation of CTG18.1 trinucleotide repeat expansion of TCF4 gene in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55(1):33–42.
Skorodumova LO, Belodedova AV, Antonova OP, et al. CTG18.1 expansion is the best classifier of late-onset Fuchs’ corneal dystrophy among 10 biomarkers in a cohort from the European Part of Russia. Invest Ophthalmol Vis Sci. 2018;59(11):4748–54.
Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ. The emerging roles of TCF4 in disease and development. Trends Mol Med. 2014;20(6):322–31.
Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302(5652):1978–80.
Soh YQ, Peh Swee Lim G, Htoon HM, et al. Trinucleotide repeat expansion length as a predictor of the clinical progression of Fuchs’ endothelial corneal dystrophy. PLoS One. 2019;14(1):e0210996.
Soragni E, Petrosyan L, Rinkoski TA, et al. Repeat-associated non-ATG (RAN) translation in Fuchs’ endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2018;59(5):1888–96.
Afshari NA, Igo RP Jr, Morris NJ, et al. Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy. Nat Commun. 2017;8:14898.
Li Y, Yang J, Li S, et al. N-myc downstream-regulated gene 2, a novel estrogen-targeted gene, is involved in the regulation of Na+/K+-ATPase. J Biol Chem. 2011;286(37):32289–99.
Biswas S, Munier FL, Yardley J, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet. 2001;10(21):2415–23.
Krafchak CM, Pawar H, Moroi SE, et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am J Hum Genet. 2005;77(5):694–708.
Riazuddin SA, Zaghloul NA, Al-Saif A, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86(1):45–53.
Vithana EN, Morgan P, Sundaresan P, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006;38(7):755–7.
Vithana EN, Morgan PE, Ramprasad V, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17(5):656–66.
Brightbill FS, McDonnell P, McGhee CNJ, Farjo AA, Serdarevic O. Corneal endothelium: structure and function in health and disease. Maryland Heights: Mosby Elsevier; 2009.
Jurkunas UV, Bitar MS, Funaki T, Azizi B. Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy. Am J Pathol. 2010;177(5):2278–89.
Halilovic A, Schmedt T, Benischke AS, et al. Menadione-induced DNA damage leads to mitochondrial dysfunction and fragmentation during rosette formation in Fuchs endothelial corneal dystrophy. Antioxid Redox Signal. 2016;24(18):1072–83.
Azizi B, Ziaei A, Fuchsluger T, Schmedt T, Chen Y, Jurkunas UV. p53-regulated increase in oxidative-stress--induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model. Invest Ophthalmol Vis Sci. 2011;52(13):9291–7.
Katikireddy KR, White TL, Miyajima T, et al. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free Radic Biol Med. 2018;116:19–30.
Wang Z, Handa JT, Green WR, Stark WJ, Weinberg RS, Jun AS. Advanced glycation end products and receptors in Fuchs’ dystrophy corneas undergoing Descemet’s stripping with endothelial keratoplasty. Ophthalmology. 2007;114(8):1453–60.
Weller JM, Zenkel M, Schlotzer-Schrehardt U, Bachmann BO, Tourtas T, Kruse FE. Extracellular matrix alterations in late-onset Fuchs' corneal dystrophy. Invest Ophthalmol Vis Sci. 2014;55(6):3700–8.
Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol. 2011;3(12).
Jurkunas UV, Bitar M, Rawe I. Colocalization of increased transforming growth factor-beta-induced protein (TGFBIp) and Clusterin in Fuchs endothelial corneal dystrophy. Invest Ophthalmol Vis Sci. 2009;50(3):1129–36.
Sarnicola C, Farooq AV, Colby K. Fuchs endothelial corneal dystrophy: update on pathogenesis and future directions. Eye Contact Lens. 2019;45(1):1–10.
Zhu AY, Eberhart CG, Jun AS. Fuchs endothelial corneal dystrophy: a neurodegenerative disorder? JAMA Ophthalmol. 2014;132(4):377–8.
Cai Q, Tammineni P. Alterations in mitochondrial quality control in Alzheimer's disease. Front Cell Neurosci. 2016;10:24.
Kumar A, Singh A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front Pharmacol. 2015;6:206.
Tobin AJ, Signer ER. Huntington’s disease: the challenge for cell biologists. Trends Cell Biol. 2000;10(12):531–6.
Lee S, Shang Y, Redmond SA, Urisman A, Tang AA, Li KH, Burlingame AL, Pak RA, Jovičić A, Gitler AD, Wang J, Gray NS, Seeley WW, Siddique T, Bigio EH, Lee VM, Trojanowski JQ, Chan JR, Huang EJ. Activation of HIPK2 promotes ER stress-mediated neurodegeneration in amyotrophic lateral sclerosis. Neuron. 2016;91(1):41–55.
Krachmer JH, Purcell JJ Jr, Young CW, Bucher KD. Corneal endothelial dystrophy. A study of 64 families. Arch Ophthalmol. 1978;96(11):2036–9.
Kaluzny BJ, Szkulmowska A, Szkulmowski M, Bajraszewski T, Kowalczyk A, Wojtkowski M. Fuchs’ endothelial dystrophy in 830-nm spectral domain optical coherence tomography. Ophthalmic Surg Lasers Imaging. 2009;40(2):198–200.
Zhang J, Patel DV. The pathophysiology of Fuchs’ endothelial dystrophy – a review of molecular and cellular insights. Exp Eye Res. 2015;130:97–105.
Jafri M, Colby K. New insights into corneal endothelial regeneration. Curr Ophthalmol Rep. 2019;7(1):37–44.
Kinoshita S, Koizumi N, Ueno M, Okumura N, Imai K, Tanaka H, et al. Injection of cultured cells with a ROCK inhibitor for bullous keratopathy. N Engl J Med. 2018;378(11):995–1003.
Borkar DS, Veldman P, Colby KA. Treatment of Fuchs endothelial dystrophy by descemet stripping without endothelial Keratoplasty. Cornea. 2016;35(10):1267–73.
Moloney G, Petsoglou C, Ball M, Kerdraon Y, Hollhumer R, Spiteri N, et al. Descemetorhexis without grafting for Fuchs endothelial dystrophy-supplementation with topical Ripasudil. Cornea. 2017;36(6):642–8.
Koenig SB. Long-term corneal clarity after spontaneous repair of an iatrogenic descemetorhexis in a patient with Fuchs dystrophy. Cornea. 2013;32(6):886–8.
Shah RD, Randleman JB, Grossniklaus HE. Spontaneous corneal clearing after Descemet's stripping without endothelial replacement. Ophthalmology. 2012;119(2):256–60.
Dirisamer M, Ham L, Dapena I, van Dijk K, Melles GR. Descemet membrane endothelial transfer: “free-floating” donor Descemet implantation as a potential alternative to “keratoplasty”. Cornea. 2012;31(2):194–7.
Koizumi N, Okumura N, Ueno M, et al. Rho-associated kinase inhibitor eye drop treatment as a possible medical treatment for Fuchs corneal dystrophy. Cornea. 2013;32(8):1167–70.
Bleyen I, Saelens IE, van Dooren BT, van Rij G. Spontaneous corneal clearing after Descemet’s stripping. Ophthalmology. 2013;120(1):215.
Davies E, Jurkunas U, Pineda R 2nd. Predictive factors for corneal clearance after Descemetorhexis without endothelial Keratoplasty. Cornea. 2018;37(2):137–40.
Macsai MS, Shiloach M. Use of topical rho kinase inhibitors in the treatment of Fuchs dystrophy after Descemet stripping only. Cornea. 2019;38(5):529–34.
Huang MJ, Kane S, Dhaliwal DK. Descemetorhexis without endothelial Keratoplasty versus DMEK for treatment of Fuchs endothelial corneal dystrophy. Cornea. 2018;37(12):1479–83.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kocaba, V., Colby, K. (2020). Fuchs Endothelial Corneal Dystrophy: Rethinking an Old Disease with Insights from the Laboratory and Clinical Practice. In: Colby, K., Dana, R. (eds) Foundations of Corneal Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-25335-6_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-25335-6_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-25334-9
Online ISBN: 978-3-030-25335-6
eBook Packages: MedicineMedicine (R0)