
Abstract
Understanding of the pathogenesis of inflammatory bowel disease (IBD) continues to evolve. Current evidence suggests that IBD results from an inappropriate inflammatory response to intestinal microbes in a genetically susceptible host. Dysbiosis, that is, unfavorable alteration of the composition and function of gastrointestinal (GI) microbiota or chronic pathogen infection, may be the mechanisms microbiota contribute to the pathogenesis of IBD. Several microbes have been associated with IBD, and among them, bacteria such as Clostridium species, gram-negatives, fecal microbes, and Mycobacterium avium subspecies remain exciting candidate pathogens but yet undiscovered other bacteria, viruses, and fungi also likely contribute significantly. Current therapeutic strategies targeting microbes that may be beneficial include probiotics and possibly fecal microbiota transplantation while dietary changes, prebiotics, and antibiotics have proven to be unhelpful. Newly discovered techniques focusing on molecular analysis of gut bacteria flora in combination with high genomic approaches are likely to further the insights into the role that microbes play in the development of IBD.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Inflammatory bowel disease
- Gastrointestinal microbiota
- Dysbiosis
- Gastrointestinal
- Pathogenesis
- Genetics
- NOD2
- Environment
- Diet
- Epidemiology
- Enterobacteriaceae
- Therapy
- Probiotics
- Fecal microbiota transplant
Introduction and Background
It has been well said that the human body is nothing but a vessel to carry microbes. One of the places the microbes are abundant and contribute in a number of ways is the gastrointestinal system. The host–microbe interactions in the gastrointestinal tract are bidirectional, complex, and not well understood. These host–microbe interactions in the gastrointestinal (GI) tract and especially the intestines and colon can be mutually beneficial or have adverse effects that contribute to inflammation.
On the one hand, intestinal microbial colonization is essential to nutrition, energy metabolism, and proper “conditioning” of the gastrointestinal and peripheral immune systems. On the other hand, the GI lumen can contain microbiota and microbial-derived factors that may promote inflammatory bowel disease (IBD) in the context of an underlying genetic immune defect.
The intestinal microbiota is acquired at birth but changes rapidly during the first year of life; thereafter there are only minor changes throughout childhood. In adults, each person’s unique population of fecal microbiota is fairly stable over time, but fluctuations occur in response to environmental and developmental factors and in disease.
Intestinal microbiota and the ability of the host to recognize and respond to this microbiota are important in the generation and optimal function of intestinal antimicrobial proteins, epithelial cells, immune cells, cytokines, and immunoglobulin A (IgA). The host immune system, various host conditions (obesity) and environmental factors (diet, antibiotic exposure) can influence intestinal microbial communities. The microbial community alterations can, in turn, modulate intestinal inflammatory outcomes. In patients with IBD, alterations in both the diversity and density of bacteria, in specific bacteria directly associated with the mucosa, and in the functions of the bacteria present (oxidative stress) have been described, further suggesting a significant role of intestinal microbiota in the pathogenesis of IBD.
This chapter strives to shed light on the current understanding of this complex interaction and how it contributes to the pathogenesis of inflammatory bowel disease.
Epidemiology
In North America, incidence rates of IBD range from 2.2 to 19.2 cases per 100,000 person-years for ulcerative colitis (UC) and 3.1 to 20.2 cases per 100,000 person-years for Crohn’s disease (CD) [1, 2]. Microbes play a significant role in its development as suggested by the correlation between specific microorganisms and IBD and the association between acute gastroenteritis and IBD. A case control study suggested that the risk of IBD was significantly increased in patients with a prior episode of acute gastroenteritis (OR 1.4; 95% CI 1.2–1.7) [3]. A separate population-based cohort study of patients with documented Salmonella or Campylobacter gastroenteritis also showed an increased risk of IBD, when compared with a matched control group (1.2% vs. 0.5%, HR 2.9, 95% CI 2.2–3.9) with the highest risk being in the first year after the gastroenteritis episode [4].
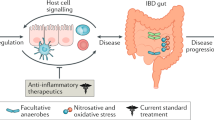
Pathogenesis of IBD (Fig. 37.1)
Inflammatory bowel disease (IBD) is an immune-mediated chronic intestinal condition. It represents heterogeneous disorders affecting the gastrointestinal tract. Two major types of IBD are ulcerative colitis (UC) and Crohn’s disease (CD) . Ulcerative colitis is a chronic inflammatory disease limited to the mucosal layer of the colon. It nearly always involves the rectum and usually extends in a proximal and continuous fashion to involve other portions of the colon. Crohn’s disease is a chronic inflammatory disease characterized by transmural inflammation and by skip lesions. Accumulating evidence suggests that inflammatory bowel disease results from an inappropriate inflammatory response to intestinal microbes in a genetically susceptible host [5].

Pathogenesis of IBD: a multifactorial concept
The human gastrointestinal tract harbors a hundred trillion different microbial organisms, including bacteria, viruses, fungi, and protozoa; this constitutes the microbiota also referred to as microbial flora [6]. Resident bacteria outnumber human somatic and germ cells tenfold and represent a combined microbial genome well in excess of the human genome and thus microbiota has the metabolic activity of a virtual organ within an organ [7]. A healthy gut microbial flora is very important for gastrointestinal functions like development of the immune system, development of host defenses, and supply of nutrients and energy. The fetal gastrointestinal tract is sterile, and colonization begins immediately after birth, which is influenced by diet, medications, and hygiene levels [7]. The establishment of a stable gut microbiota generally accompanies two big transitions in infancy. The first transition occurs soon after birth, during lactation, and results in dominance of the gut microbiota by Bifidobacterium. The second transition occurs during the weaning period, with the introduction of solid foods and continuation of breast milk feeding, and results in the establishment of an adult-type complex microbiome dominated by the phyla Bacteroidetes and Firmicutes [8].
At present, the exact etiology of IBD is unclear. However, it is believed that disruption of the immune system and/or imbalanced interactions with microbiota leads to the development of chronic intestinal inflammation and the potential addition of environmental factors triggers genetically susceptible hosts [9]. In the following section, we will be talking briefly about the different known etiologies/pathogenesis for IBD at this time.
Environmental and Genetic Factors
Important environmental risk factors for the pathogenesis of IBD include smoking, diet, antibiotics, and oral contraceptives pills (OCPs) [10, 11]. Multiple theories have been proposed to explain the unknown environmental exposures that may interact with the immune system and result in an abnormal inflammatory response to intestinal microflora. The most predominant theory is the hygiene hypothesis. This hypothesis proposes that the rising frequency of immunologic disorders can be attributed to a lack of childhood exposure to enteric pathogens. Improved sanitation and hygiene, along with decreased exposure to enteric organisms during early childhood, may lead to a greater susceptibility to develop an inappropriate immunologic response upon exposure to new antigens (e.g., a gastrointestinal infection) later in life [10].
Since the discovery of nucleotide-binding oligomerization domain-containing 2 (NOD2) , more than 160 IBD-associated gene loci have been identified. NOD2’s involvement in microbial sensing , innate and adaptive immune activation, plus its role in autophagy, the gut epithelial barrier, and shaping the gut microbiota suggest that it is a versatile protein with many roles in IBD pathogenesis. These genetic associations highlight the importance of gene–microbe–environment interactions in IBD pathogenesis. Smoking is a major environmental risk factor , with evidence for gene–microbe interactions in its contribution to disease. It has been independently demonstrated that NOD2 −/− mice have altered gut microbiota composition, and that cigarette smoke can alter NOD2 expression and function in intestinal epithelial cells [11].
Similarly, diet is another environmental risk factor for the development of inflammatory bowel disease. Studies have shown how dietary habits result in compositional changes to the microbiota and could theoretically lead to inflammation in genetically susceptible individuals. A report has shown increased dietary intake of n-6 polyunsaturated fatty acids and animal protein together with a change toward a more westernized diet increases the risk of IBD. On the other hand, surveys and case control studies have shown that a range of other dietary factors, such as refined sugar, fast foods, margarine, and dairy products increase the risk of IBD, while vegetables, fruits, fish, and dietary fiber appear to have a protective effect [11, 12].
Gut Microbiota in IBD
The gut microbiota forms a natural defense barrier and provides numerous protective, structural, and metabolic effects to the host [7, 8] (Fig. 37.2). Commensal microbes are source for nutrients and energy, like production of short-chain fatty acids (SCFAs) and vitamin synthesis. They also help in the development of immune systems like IgA production and regulation of T-helper cell. Last but not least, microbiota is also involved in the host defense, like in the production of bacteriocins and lactic acid, which act as antimicrobial factors [7, 8].

Physiological role of gut microbiota
The majority of commensal bacteria consist of gram-negative bacteria, such as Bacteroidetes, and gram-positive bacteria, such as Firmicutes [8, 9]. Gut bacteria such as Bifidobacterium can help in the production and supply of vitamins such as vitamin K and the water-soluble B vitamins [11]. The phyla Firmicutes and Bacteroidetes produce SCFAs from indigestible carbohydrates through collaboration with species specialized in oligosaccharide fermentation (e.g., Bifidobacteria). SCFAs are major anions in the colon, mainly as acetate, propionate, and butyrate. Butyrate is a primary energy source for colonic epithelial cells. Butyrate is consumed mainly by the colonic epithelium, and acetate and propionate will then become available systemically. The levels of SCFAs are significantly decreased in IBD, which may be a key factor that compromises intestinal and immune homeostasis [8]. Gut microbiota also play a very vital role in the growth of the host’s immune system. A literature search has shown how one of the bacteria Candidatus Arthromitus, also known as segmental filamentous bacteria (SFB), alone promotes the maturation of the mucosal immune system [8, 13].
Any unfavorable alteration of the composition and function of the gut microbiota is known as dysbiosis, which alters the host–microbiota interaction and the host immune system [8]. Lately, many studies have been able to identify intestinal dysbiosis if present in patients with IBD, even though it still remains largely unknown whether the severity of gut dysbiosis is the cause of, or the response to, the severity of the disease [14, 15]. Studies summarizing the gut microbiota in patients with IBD compared with controls have consistently shown changes in microbiota composition as well as reduction in overall biodiversity [11]. IBD is associated with an increased abundance of Enterobacteriaceae, including Escherichia coli and Fusobacterium. It is also associated with an increased abundance of Fusobacteriaceae, Pasteurellaceae, and Bifidobacteriaceae [11, 14]. These pathogenic bacteria have the ability to adhere to the intestinal epithelium, which in turn alters the diversity and composition of commensal microbes causing intestinal inflammation [8].
Role of Microbes
Several bacteria have been associated with the pathogenesis of IBD, including Mycobacterium avium paratuberculosis , enterotoxigenic Bacteroides fragilis , adherent/invasive Escherichia coli, Campylobacter jejuni , Listeria monocytogenes , Chlamydia sp., Aeromonas hydrophila , Salmonella typhi, and Clostridium difficile (C. diff) [16,17,18].
Clostridium difficile
Clostridium difficile (C. diff) is a gram-positive, spore-forming, toxin-producing, anaerobic bacteria. Pathogenically, it primarily affects the colon, leading to either asymptomatic carriage or clinical disease that may range in severity from mild diarrhea to fatal pseudomembranous colitis, accounting for up to 50–75% of antibiotic-associated colitis [19, 20]. C. diff infections (CDI) are known to occur more frequently in patients with IBD, many of whom may not have had previous exposure to any antibiotics. Studies also show higher rates of asymptomatic carriage of C. diff of 8.2% (9.4% in patients with UC and 6.9% in patients with CD), versus 1% in healthy volunteers [21].
Toxins A and B (also known as TcdA and TcdB) are exotoxins that are thought to be major virulence factors of C. difficile. At least four functionally distinct domains of toxins A and B have been identified including the N-terminal region, enzymatically active glucosyltransferase domain, a cysteine protease domain, a hydrophobic segment, and C-terminal region with repetitive oligopeptide repeats (CROPs) . Toxin A or B binds to the cell surface receptor with its C-terminus, followed by clathrin-mediated endocytosis of the toxin–receptor complex. A decrease in the pH inside the endosomal compartment leads to conformational changes within the toxins, permitting pore formation and translocation of the glucosyltransferase and protease domains into the cytoplasm. After activation, the cysteine protease domain autocatalytically cleaves the toxin to release the glucosyltransferase region, which inactivates the Rho family of proteins through glucosylation. Rho family of proteins is involved in intracellular signaling, and inactivation of Rho proteins in turn leads to disruption of the cell cytoskeleton and cell death. The inactivation of these proteins and cell death impairs the integrity of the membrane cytoskeleton and the barrier function of epithelial cells [22].
C. diff toxins have also been implicated in triggering a number of innate immune response pathways including NF-kB and MAP kinase. Exposure to toxin A leads to secretion on IL-8 by intestinal epithelial cells [23]. Studies suggest several other inflammatory cytokines are released in response to Toxin A and B including interleukin 18 (IL-18), IL-1beta, IL-6, and tumor necrosis factor (TNF-a). This alteration in the immune system with CDI may act as a trigger to IBD. A reduction in bile acids that occurs in IBD promotes C. diff growth and spore germination [22].
Helicobacter pylori
Helicobacter pylori (H. pylori) is a gram-negative, spiral-shaped pathogenic bacterium that has been associated with chronic gastritis, peptic ulcer disease, and gastric malignancies [24]. Helicobacter species can be subdivided into two major lineages, the gastric Helicobacter species such as H. pylori and the enterohepatic (nongastric) Helicobacter species, which predominantly colonize the intestine and the hepatobiliary system. They have been linked to chronic liver and intestinal diseases [24]. In studies related to IBD, epidemiological evidence suggests negative correlations between the incidence of H. pylori and IBD [25].
A possible mechanism for the protective role of H. pylori in the development of IBD may be an alteration of the host immunologic response away from the pro-inflammatory T-helper (Th1/Th17) response toward an increased T-regulatory cell immune response , by increasing the expression of forkhead box P3 (FOXP3) [26, 27].
Listeria
Listeria monocytogenes (L. monocytogenes) is an intracellular gram-positive bacteria identified in 0.8–3.4% of the asymptomatic population on stool analysis [28]. It has been associated with infections in the central nervous system and bacteremia in patients with immunodeficiency, children, elderly, pregnant females, as well as healthy people [29]. Several studies have investigated the affinity between IBD and L. monocytogenes. It is presumed since patients with IBD receive immunosuppressive drugs, the defensive barrier in the gut is compromised, thereby making patients vulnerable to the colonization of L. monocytogenes [30].
Mycobacterium avium Complex and Paratuberculosis
Mycobacterium avium subspecies paratuberculosis (MAP) is an obligate pathogenic organism, linked to Johne’s disease in cattle. Dalziel in 1913 first postulated the hypothesis of a link between Crohn’s disease (CD) and MAP even before CD was described as he noted the similarities with Johne’s disease, an intestinal disorder of cattle, and IBD in humans [31]. MAP causes chronic granulomatous ileitis (Johne’s disease) in cattle and sheep, similar to some pathological features seen with CD. Olsen et al. showed an increased presence of MAP-reactive T cells that were extremely reactive to MAP and produced the pro-inflammatory cytokines Interferon-γ (INFγ) and Interleukin-17 (IL17) [32].
CD patients commonly have circulating antibodies namely anti-Saccharomyces cerevisiae antibody (ASCA) [33]. The epitope for this antibody is a mannan with a specific mannose alpha of 1–3 mannose (Man alpha1–3Man) terminal disaccharide. MAP is a possible source for the ASCA mannan epitope, and probably releases a mannose-containing glycoconjugate that inhibits killing of phagocytosed E. coli by macrophages, thus causing an indirect pathogenic mechanism for CD [34]. Nucleotide-binding oligomerization domain-containing protein 2, also known as caspase recruitment domain-containing protein 15 (NOD2/CARD15) receptors mutations, is known to be associated with increased rates of intracellular survival of the bacteria, eventually causing infection, due to abnormal development of Peyer’s patches [35, 36]. Mutations in Nramp1 (natural-resistance associated macrophage protein 1) have been associated with an increased risk of mycobacterial infections, and polymorphisms in the Nramp1 promoter have been identified in patients with IBD [37]. It has also been hypothesized that even though MAP infects a large population, it only becomes pathogenic in individuals who are genetically susceptible due to an underlying genetic deficiency causing dysfunctional IFNγ activity [38].
E. coli
E. coli is a facultative gram-negative aerobe that has been found to be the numerically most dominant bacteria in the gut microbiota [39]. The association of E. coli with IBD was first suggested in 1978 when Tabaqchali et al. noted high titers of antibodies against E. coli O-antigens in patients with IBD [40]. Escherichia coli (E. coli) strains are classified into the following categories: A, B1, B2, C, D, E, and F phylogenetic groups. Group A and B1 are mainly commensal strains, while B2 and D groups are mainly pathogenic strains. Patients with IBD compared to the healthy individuals have been reported to have increased amounts of B2 and D strains of E. coli [41,42,43]. E. coli isolates from patients with IBD show increased adherence to gastrointestinal epithelial cells and are called adhesive and invasive E. coli (AIEC). AIEC have been shown to promote release of the pro-inflammatory cytokine IL-8 [44]. AIEC strains also result in increased expression of cell adhesion molecules; such as carcinoembryonic antigen related cell adhesion molecule 6 (CEACAM6 ) to promote bacterial colonization in the intestinal mucosa [45]. Studies have shown that certain genes promoting virulence are overrepresented in AIEC relative to nonpathogenic E. coli, namely, k1 and kpsm2 (effective in capsule synthesis), FynA, yersiniabactin, chu operon (utilized in iron metabolism), and ibeA gene (involved in invasion) [46, 47].
Campylobacter concisus
Campylobacter concisus (C. concisus) is a gram-negative, fastidious aerobic bacterium that normally colonizes the human oral cavity [48]; however, few studies found a significantly higher intestinal prevalence of C. concisus in patients with CD [49,50,51]. Strains of C. concisus have been shown to express a zonula occludens toxin (Zot) acquired likely through a CON-Phi2 prophage (a mechanism similar to Vibrio cholerae toxin), which enhances permeability of the epithelial cells [52]. Studies have shown an increased prevalence of Zot gene in C. concisus strains isolated from IBD patients [53].
Chlamydia pneumoniae
Chlamydia pneumoniae is a gram-negative rod suggested to have a higher prevalence in IBD patients. Mutations in the NOD2/CARD15 receptor have been studied as possible pathogenic mechanism for this bacterium. This mutation can cause activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-KB) pathway, react with basic myelin protein in the immune system, and potentially trigger an autoimmune response [54, 55].
Mycoplasma pneumoniae
Mycoplasmas are small bacteria without a rigid cell wall, existing as either commensals or pathogens. Mycoplasmas are thought of as organisms of ubiquitous distribution with the potential to cause inflammatory diseases. M. pneumoniae DNA was detected at a significantly higher rate in intestinal biopsies from patients with CD, suggesting some role in the pathogenesis of IBD [56]. Possible pathogenic mechanisms associated with Mycoplasma include increased production of various regulatory cytokines (IL-2, IL-4) and inflammatory cytokines (IL-6, IL-8) [57] as well as increased oxidative stress due to increased production of hydrogen peroxidase and superoxide radicals resulting in damage to epithelial cells [58].
Fecal Bacteria
Several studies have suggested an alteration in the relationship of commensal bacteria in the intestine and host immune system as a possible pathogenic mechanism in IBD [59, 60]. 95% of the bacteria in stool samples from healthy adults belong to the Bacteroides, Clostridium coccoides, and Clostridium leptum subgroup [61]; however, IBD patients have lower numbers of these organisms, including bifidobacteria but, in particular, the C. leptum subgroup. Studies have found a strong association between ileal CD and lack of Faecalibacterium prausnitzii. F. prausnitzii has anti-inflammatory effects on Caco-2 cells, as metabolites secreted by this bacterium blocks NF-kappa B activation and IL-8, 12 and IFN-gamma secretion, and promotes secretion of anti-inflammatory IL-10 [62] (Table 37.1).
Animal Models of Intestinal Inflammation
Several types of models for colitis are available including genetically driven, chemically induced or immune mediated , all of which are represented in mouse models of colitis. Some of the examples of genetically driven models available are interleukin-10 (IL-10) deficient mouse, resulting in uncontrolled inflammation in the gut [63, 64], the Mdr1a −/−mouse, deficient in P-glycoprotein 170, resulting in increased gut permeability, microbial translocation and colitis development [65], the SAMP1/YitFc mouse, which develops spontaneous ileitis [66], and the TRUC mouse, where deficiency in both T-bet and Rag2 causes increased TNFα responses [67]. Models that are developed by immune activation include the T cell transfer model, where naive T cells are transferred into a lymphopenic recipient mouse, that may result in wasting disease, prevented by cotransfer of regulatory T cells [68, 69] and anti-CD3ε monoclonal antibody model, in which T cell activation results in intestinal mucosal damage and a cytokine storm [70, 71]. Citrobacter rodentium or rotavirus can cause intestinal inflammation, and is used as a model to study the inflammatory response [72,73,74,75]. Models with chemically induced colitis include administration of compounds that cause epithelial damage, for example, dextran sulfate sodium (DSS) , piroxicam or 2,4,6-trinitrobenzene sulfonic acid.
Management
Options Targeting Microbes
A variety of therapeutic options have emerged to target microbes in IBD. These include probiotics, prebiotics, antibiotics, fecal microbiota transplantation (FMT), and dietary changes.
Probiotics are live microorganism preparations thought to promote human health; they have been studied in IBD with the theory that they might work to improve the balance in the gastrointestinal microbiota, thereby reducing intestinal inflammation. The mechanism by which this could occur may be due to less colonization resistance, better intestinal barrier function, signal transduction alteration, and metabolic effects [76]. Different preparations of probiotics have been used in studies. VSL#3, a combination of eight different lactic acid bacteria, has been used in numerous studies [77,78,79,80,81]. Other formulations include Nissle 1917 (a nonpathogenic E. coli strain), Lactobacillus GG, Bifidobacteria-fermented milk, and Bifidobacterium longum/Synergy [82,83,84,85,86,87,88,89], among others. Additionally, probiotic therapy has been studied in pouchitis, a known possible complication after surgical reconstruction in IBD patients [90].
Gut microbiota is affected by dietary intake. A similar concept to probiotics in the treatment of IBD is the use of prebiotics, which have been suggested to provide a metabolic substrate to promote the proliferation of beneficial microbes [91].
Fecal microbiota transplantation is another potential therapeutic option to alter gut dysbiosis in IBD. In FMT, fecal flora from a healthy person is transplanted to the diseased. This therapy has been shown to be efficacious in resolution of C. difficile-associated diarrhea and is now used in clinical practice as a treatment option [92]. If IBD is the result of dysbiosis, it is thought that FMT from a healthy donor may be able to restore symbiosis, similar to the outcomes found in C. difficile infection resolution.
Antibiotics have widely been used to treat acute and chronic pouchitis and fistulizing disease [93]. Ciprofloxacin and/or metronidazole are used in perianal CD in conjunction with other medications such as biologics [94]. Azithromycin and rifaximin have also been used to treat mild to moderate luminal CD. The theory behind the use of antibiotics is to eradicate potentially pathologic gut microbes. Use of antibiotics has been with caution, however, due to the concern of creating an imbalance in commensal organisms such as the case with the potential for development of pathogenic C. difficile colitis. A systematic review and meta-analysis of 22 studies showed significantly higher rates of CDIs in patients with CD involving their colon [95]. There was also an association with CDIs in those who used antibiotics within the last 30 days and in those using biologic medications.
Evidence, Efficacy, and Prognosis
Lactobacillus casei , a probiotic, has been shown in a study to downregulate pro-inflammatory cytokines in Crohn’s disease thereby antagonizing the inflammatory effects of host E. coli [96]. Several studies have explored this concept in mouse models. An example is C. rodentium-induced colitis in mice, which was lessened by the use of Saccharomyces boulardii, a probiotic [97]. Another example is the use of probiotics to reduce gut inflammation by means of modulating growth factors to promote epithelial restoration [98].
Several studies using VSL#3 in UC patients have shown it to be efficacious in both induction of remission as well as use in maintenance therapy [77,78,79,80,81]. Prevention of recurrence of chronic, relapsing pouchitis with the use of VSL#3 was shown in two double-blinded placebo-controlled trials [77, 78]. When VSL#3 was added to standard therapy (mesalazine and steroids) in 29 pediatric patients with UC, remission rates at 4 weeks were 92.8% compared to 36.4% in placebo [81]. When they looked at recurrence rates 1 year later, the VSL#3 group was 36.4% vs. 73.3% in the placebo. A study in patients with UC who underwent ileal-pouch-anal-anastomosis involved 4 weeks of daily lactobacilli and bifidobacteria administration to 51 UC patients. Stools samples showed an increase in the number of these bacteria during intervention as well as a decrease in involuntary defecation, leakage, abdominal cramps, fecal number, consistency, and mucus, and urge to evacuate stools [90]. Lactobacillus, Bifidobacterium, and Saccharomyces boulardii in combination, as well as Nissle 1917, were found to induce and maintain remission in moderately severe UC [99].
One of the first studies using FMT dates back to 1989, when an author of the paper, with UC, received FMT and reported disease-free remission [100]. In 2012, a systematic review showed 13 out of 18 patients with UC who achieved remission with FMT [101]. There is some conflicting evidence in other studies; two longitudinally prospective studies of FMT therapy in UC patients did not achieve clinical remission [102, 103]. There was suggestion in these studies that in most patients, the gut microbiota was indeed altered, albeit temporarily, and so may require repeat FMT therapy at periodic intervals to sustain disease remission. Additionally, there may be specific organisms which confer worse success with FMT when overrepresented in the gut flora as compared to other organisms [102]. This might be an important factor in determining which individual is more likely to respond to FMT. A more promising study of 10 UC pediatric patients showed a 79% clinical response rate to FMF therapy within 1 week [104]. A more recent meta-analysis and systematic review of 29 studies and 524 FMT-treated IBD patients has shown a concerning alternative outcome—IBD flaring after FMT [105]. The rates of IBD worsening were higher with lower GI FMT delivery as opposed to upper GI delivery, suggesting a possible site variance (16.5% vs. 5.6%). The rates of worsening were considered to be marginal (4.6%) and it was questioned as to whether it was due to other etiologies.
Prebiotic data is limited, but as of a 2014 review, had yet to show any effect as a therapeutic option in IBD [91].
It appears that the data have been conflicting regarding the use of antibiotics in CD and UC. A meta-analysis made the conclusion that antibiotics were superior to placebo in inducing remission [91]. Another had a similar encouraging conclusion that antibiotic therapy induced remission and prevented relapse in IBD patients [106]. As stated previously, however, the risk of developing a CDI is increased in those with IBD and increased in those IBD patients who used antibiotics within the last 30 days [95].
As stated by Aleksandar et al. in their work on the microbiome in IBD, there is no certain diet in IBD patients which has been shown to cause, prevent or treat this disease [107]. However, Enterobacteriaceae have been shown to be increased in IBD, and one study showed, in long-term strict vegan diets, a significant decrease in Enterobacteriaceae, as well as Bacteroides spp. and Bifidobacterium spp. [108].
In review of the variety of therapeutic options to target microbes in IBD, it appears that some have shown more promise than others and all require more data to be able to draw more solid conclusions. A significant amount of work seems to be focused on the use of probiotics and more is emerging for the use of fecal microbiota transplantation as well. Probiotic data is encouraging while FMT data is conflicting. At this time, it does not seem that any particular diet is definitely efficacious nor are prebiotics. These findings are summarized in Table 37.2.
Discussion and Future Direction
As highlighted in detail above, gut microbiota are an exciting target to study and are likely to reap rich rewards in furthering our understanding of initiation and perpetuation of chronic IBD. However, there is a complex relationship between the gut immune system and the microbiota. Accumulating evidence also suggests that the dynamic balance between microbes, particularly commensal flora, and host defensive responses at the mucosal frontier has a pivotal role in the initiation and pathogenesis of chronic IBD. However, it remains unclear whether the dysbiosis observed in IBD is a cause or a consequence of intestinal inflammation. It still remains unclear how dysbiosis regulates the gut immune system. Hence, further research into this relationship is not only likely to give us better insight into it but is almost necessary to improve outcomes of chronic IBD.
It is also important to note that the etiopathogenesis of chronic IBD is complex, multifactorial, and a combination of genetic predisposition and environmental exposures, and the gut microbiota play significant roles. Among genetic factors, NOD2 has remained the strongest genetic risk factor associated with chronic IBD development for nearly two decades, although exactly how it is related to disease onset remains elusive. Important environmental risk factors for the pathogenesis for IBD include smoking, diet, antibiotics, and OCPs. Among these, smoking has been best studied and has shown a clear association with chronic IBD. Also important is the hygiene theory, which can explain unknown environmental exposures that may interact with the immune system and result in an abnormal inflammatory response to intestinal microflora.
Several bacteria have been associated with the pathogenesis of IBD and remain the focus of research in furthering the understanding of their role in chronic IBD. The gut microbiota is composed not only of bacteria but also of viruses and fungi which also likely contribute significantly to IBD. The role of these should also be studied in detail. The role of gut microbiota is being suggested strongly by the analysis of mouse models and has revealed at least two major courses of disease: dysbiosis (involving the depletion or alteration in resident species, which can be followed by loss of colonization resistance, acute infection by bacteria that can breach the intestinal barrier, then possibly develop into chronic inflammation) and chronic pathogen infection (which can be aggravated by the presence of commensals because barrier disturbances and immunomodulation can increase immune responses to resident bacteria). The most likely pathogens remain elusive but there are a number of candidate pathogens that may have a strong contribution to the development of chronic IBD. Further efforts to identify pathogens that are commonly associated with human disease and the potential protective microbiota, depletion of which might aggravate disease, can provide clarification on these issues. The likely candidate pathogens include enterotoxigenic Bacteroides fragilis, Clostridium difficile, Helicobacter spp. and Campylobacter spp. and should be further investigated. An exciting candidate is Mycobacterium avium subspecies paratuberculosis, which has received considerable attention as a possible trigger of human IBD.
Unfortunately, our understanding of the microbial flora itself is quite incomplete. Insights into the microbial–host interrelationships are hampered by both the limited knowledge of the diversity and complexity of the microbial flora and the limitation of available tools to delineate these characteristics. Metagenomic and computational analysis of the so-called microbiome may provide a foundation to achieve an understanding of the relevant, functional diversity of the flora in the context of IBD. Understanding the distribution, dynamics, and responses to microbial flora in these disease states will probably provide insights into a number of aspects of IBD.
Since gut microbiota are such an exciting target in understanding of chronic IBD, a number of therapeutic strategies have been tried to target them. These include probiotics, prebiotics, antibiotics, fecal microbiota transplantation (FMT), and dietary changes. It is very clear that antibiotics are not efficacious and are possibly harmful in this context. Dietary changes and prebiotics also do not seem to work. Probiotics and fecal microbiota transplant remain promising. Probiotics have been shown to be efficacious and tend to be well tolerated with minimal downside. They should be considered in appropriate patients with chronic IBD. On the other hand, the role of FMT in the therapy of chronic IBD remains unclear, and various studies have provided conflicting results including raising concern for possible worsening of the disease. This may be due to the study design, the selection of population, and unclear standardization of the FMT procedure.
All of the above developments highlight the role gut microbiota plays in chronic IBD, and it is our belief that we have only scratched the surface of our understanding of this role.
Summary
The main factors playing a role in IBD are genetic, environmental, gut microbiota, and immune response. Among these, gut microbiota influences every aspect involved in causation and perpetuation of the disease with complex interactions among these factors. Yet, it remains elusive how a single agent or a short list of agents exerts a majority of this influence. The use of probiotics and fecal microbiota transplant to impact the gut microbiota looks promising but unproven. Nonetheless, gut microbiota represents a “gold mine” for both clinical and basic IBD research and this is an exciting time of discovery; breakthroughs are likely to come soon in this area.
References
Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, Benchimol EI, Panaccione R, Ghosh S, Barkema HW, Kaplan GG. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012;142(1):46.
Shivashankar R, Tremaine WJ, Harmsen WS, Loftus EV Jr. Incidence and prevalence of Crohn’s disease and ulcerative colitis in Olmsted County, Minnesota from 1970 through 2010. Clin Gastroenterol Hepatol. 2017;15(6):857.
Porter CK, Tribble DR, Aliaga PA, Halvorson HA, Riddle MS. Infectious gastroenteritis and risk of developing inflammatory bowel disease. Gastroenterology. 2008;135(3):781.
Gradel KO, Nielsen HL, Schønheyder HC, Ejlertsen T, Kristensen B, Nielsen H. Increased short- and long-term risk of inflammatory bowel disease after salmonella or campylobacter gastroenteritis. Gastroenterology. 2009;137(2):495.
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78. https://doi.org/10.1056/NEJMra0804647.
Tanaka M, Nakayama J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int. 2017;66(4):515–22.
Kim DH, Cheon JH. Pathogenesis of inflammatory bowel disease and recent advances in biologic therapies. Immune Netw. 2017;17(1):25–40.
Nishida A, Inoue R, Inatomi O, Bamba S, Naito Y, Andoh A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin J Gastroenterol. 2018;11(1):1–10.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–8.
Molodecky NA, Kaplan GG. Environmental risk factors for inflammatory bowel disease. Gastroenterol Hepatol (NY). 2010;6(5):339–46.
Sheehan D, Moran C, Shanahan F. The microbiota in inflammatory bowel disease. J Gastroenterol. 2015;50(5):495–507.
Scholz D. The role of nutrition in the etiology of inflammatory bowel disease. Curr Probl Pediatr Adolesc Health Care. 2011;41(9):248–53.
Gaboriau-Routhiau V, Rakotobe S, Lécuyer E, Mulder I, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31(4):677–89.
Lane ER, Zisman TL, Suskind DL. The microbiota in inflammatory bowel disease: current and therapeutic insights. J Inflamm Res. 2017;10:63–73.
Knights D, Lassen KG, Xavier RJ. Advances in inflammatory bowel disease pathogenesis: linking host genetics and the microbiome. Gut. 2013;62(10):1505–10.
Lidar M, Langevitz P, Shoenfeld Y. The role of infection in inflammatory bowel disease: initiation, exacerbation and protection. Isr Med Assoc J. 2009;11:558–63.
Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–21.
Burnham WR, Lennard-Jones JE, Stanford JL, Bird RG. Mycobacteria as a possible cause of inflammatory bowel disease. Lancet. 1978;2:693–6.
Monaghan T, Boswell T, Mahida YR. Recent advances in Clostridium difficile-associated disease. Gut. 2008;57:850–60.
Bartlett JG. Narrative review: the new epidemic of Clostridium difficile-associated enteric disease. Ann Intern Med. 2006;145:758–64.
Clayton EM, Rea MC, Shanahan F, Quigley EM, Kiely B, Hill C, Ross RP. The vexed relationship between Clostridium difficile and inflammatory bowel disease: an assessment of carriage in an outpatient setting among patients in remission. Am J Gastroenterol. 2009;104:1162–9.
Monaghan TM, Cockayne A, Mahida YR. Pathogenesis of Clostridium difficile infection and its potential role in inflammatory bowel disease. Inflamm Bowel Dis. 2015;21:1957–66.
Mahida YR, Makh S, Hyde S, et al. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut. 1996;38:337–47.
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–90.
Koloski NA, Bret L, Radford-Smith G. Hygiene hypothesis in inflammatory bowel disease: a critical review of the literature. World J Gastroenterol. 2008;14:165–73.
Lundgren A, Strömberg E, Sjöling A, Lindholm C, et al. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori- infected patients. Infect Immun. 2005;73:523–31.
Rad R, Brenner L, Bauer S, Schwendy S, Layland L, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131:525–37.
Watson GW, Fuller TJ, Elms J, Kluge RM. Listeria cerebritis: relapse of infection in renal transplant patients. Arch Intern Med. 1978;138:83–7.
Munoz P, Rojas L, Bunsow E, Saez E, et al. Listeriosis: an emerging public health problem especially among the elderly. J Infect. 2012;64:19–33.
Miranda-Bautista J, Padilla-Suarez C, Bouza E, Munoz P, Menchen L, Marın-Jimenez I. Listeria monocytogenes infection in inflammatory bowel dis- ease patients: case series and review of the literature. Eur J Gastroenterol Hepatol. 2014;26:1247–52.
Dalziel TK. Chronic interstitial enteritis. Br Med J. 1913;2:1068–70.
Olsen I, Tollefsen S, Aagaard C, et al. Isolation of Mycobacterium avium subspecies paratuberculosis reactive CD4 T cells from intestinal biopsies of Crohn’s disease patients. PLoS One. 2009;4:e5641.
Quinton JF, Sendid B, Reumaux D, et al. Anti-Saccharomyces cerevisiae mannan antibodies combined with antineutrophil cytoplasmic autoantibodies in inflammatory bowel disease: prevalence and diagnostic role. Gut. 1998;42:788–91.
Mpofu CM, Campbell BJ, Subramanian S, et al. Microbial mannan inhibits bacterial killing by macrophages: a possible pathogenic mechanism for Crohn’s disease. Gastroenterology. 2007;133:1487–98.
Barreau F, Meinzer U, Chareyre F, et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One. 2007;2:e523.
Elguezabal N, Chamorro S, Molina E, Garrido JM, Izeta A, Rodrigo L, et al. Lactase persistence, NOD2 status and Mycobacterium avium subsp. paratuberculosis infection associations to inflammatory bowel disease. Gut Pathog. 2012;4:6.
Kojima Y, Kinouchi Y, Takahashi S, Negoro K, Hiwatashi N, Shimosegawa T. Inflammatory bowel disease is associated with a novel promoter polymorphism of natural resistance-associated macrophage protein 1 (NRAMP1) gene. Tissue Antigens. 2001;58:379–84.
Juste RA, Elguezabal N, Pavón A, et al. Association between Mycobacterium avium subsp. paratuberculosis DNA in blood and cellular and humoral immune response in inflammatory bowel disease patients and controls. Int J Infect Dis. 2009;13:247–54.
Macfarlane S, Furrie E, Cummings JH, Macfarlane GT. Chemotaxonomic analysis of bacterial populations colonizing the rectal mucosa in patients with ulcerative colitis. Clin Infect Dis. 2004;38:1690–9.
Tabaqchali S, O’Donoghue DP, Bettelheim KA. Escherichia coli antibodies in patients with inflammatory bowel disease. Gut. 1978;19:108–13.
Johnson JR, Delavari P, Kuskowski M, Stell AL. Phylogenetic distribution of extraintestinal virulence- associated traits in Escherichia coli. J Infect Dis. 2001;183:78–88.
Petersen AM, Halkjær SI, Gluud LL. Intestinal colonization with phylogenetic group B2 Escherichia coli related to inflammatory bowel disease: a systematic review and meta-analysis. Scand J Gastroenterol. 2015;50:1199–207.
Navidinia M, Peerayeh SN, Fallah F, Bakhshi B, Sajadinia RS. Phylogenetic grouping and pathotypic comparison of urine and fecal Escherichia coli isolates from children with urinary tract infection. Braz J Microbiol. 2014;45:509–14.
Martin HM, Campbell BJ, Hart CA, et al. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93.
Barnich N, Carvalho FA, Glasser AL, Darcha C, et al. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting ileal mucosa colonization in Crohn disease. J Clin Investig. 2007;117:1566–74.
Conte MP, Longhi C, Marazzato M, Conte AL, et al. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn’s disease patients. BMC Res Notes. 2014;7:748.
Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis. 2014;20:1919–32.
Zhang L, Budiman V, Day AS, Mitchell H, Lemberg DA, Riordan SM, Grimm M, Leach ST, Ismail Y. Isolation and detection of Campylobacter concisus from saliva of healthy individuals and patients with inflammatory bowel disease. J Clin Microbiol. 2010;48:2965–7.
Zhang L, Man SM, Day AS, Leach ST, et al. Detection and isolation of Campylobacter species other than C. jejuni from children with Crohn’s disease. J Clin Microbiol. 2009;47:453–5.
Man SM, Zhang L, Day AS, Leach ST, Lemberg DA, Mitchell H. Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm Bowel Dis. 2010;16:1008–16.
Mahendran V, Riordan SM, Grimm MC, Tran TA, Major J, Kaakoush NO, Mitchell H, Zhang L. Prevalence of Campylobacter species in adult Crohn’s disease and the preferential colonization sites of Campylobacter species in the human intestine. PLoS One. 2011;6:e25417.
Zhang L, Lee H, Grimm MC, Riordan SM, Day AS, Lemberg DA. Campylobacter concisus and inflammatory bowel disease. World J Gastroenterol. 2014;20:1259–67.
Mahendran V, Tan YS, Riordan SM, Grimm MC, et al. The prevalence and polymorphisms of zonula occluden toxin gene in multiple Campylobacter concisus strains isolated from saliva of patients with inflammatory bowel disease and controls. PLoS One. 2013;8:e75525.
Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603.
Muller S, Arni S, Varga L, Balsiger B, Hersberger M, Maly F, et al. Serological and DNA-based evaluation of Chlamydia pneumoniae infection in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2006;18:889–94.
Chen W, Li D, Paulus B, Wilson I, Chadwick VS. High prevalence of Mycoplasma pneumoniae in intestinal mucosal biopsies from patients with inflammatory bowel disease and controls. Dig Dis Sci. 2001;46:2529–35.
Shibata KI, Hasebe A, Sasaki T, Watanabe T. Mycoplasma salivarium induces interleukin-6 and inter- leukin-8 in human gingival fibroblasts. FEMS Immunol Med Microbiol. 1997;19:275–83.
Baseman JB, Tully JG. Mycoplasmas: sophisticated, reemerging, and burdened by their notoriety. Emerg Infect Dis. 1997;3:21.
Willing B, Halfvarson J, Dicksved J, et al. Twin studies reveal specific imbalances in the mucosa-associated micro- biota of patients with ileal Crohn’s disease. Inflamm Bowel Dis. 2009;15:653–60.
Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12:S3–9.
Suau A, Bonnet R, Sutren M, et al. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–807.
Sokol H, Pigneur B, Watterlot L, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–6.
Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74.
Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–31.
Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–44.
Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, Chieppa M, Arseneau KO, Ley K, Cominelli F. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis. 2011;17:2566–84.
Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131:33–45.
Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–71.
Powrie F, Leach MW, Mauze S, Menon S, Caddle LB, Coffman RL. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–62.
Merger M, Viney JL, Borojevic R, Steele-Norwood D, et al. Defining the roles of perforin, Fas/FasL, and tumour necrosis factor alpha in T cell induced mucosal damage in the mouse intestine. Gut. 2002;51:155–63.
Zhou P, Streutker C, Borojevic R, Wang Y, Croitoru K. IL-10 modulates intestinal damage and epithelial cell apoptosis in T cell-mediated enteropathy. Am J Physiol Gastrointest Liver Physiol. 2004;287:G599–604.
Little LM, Shadduck JA. Pathogenesis of rotavirus infection in mice. Infect Immun. 1982;38:755–63.
Higgins LM, Frankel G, Douce G, Dougan G, MacDonald TT. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect Immun. 1999;67:3031–9.
Franco MA, Angel J, Greenberg HB. Immunity and correlates of protection for rotavirus vaccines. Vaccine. 2006;24:2718–31.
Collins JW, Keeney KM, Crepin VF, Rathinam VA, Fitzgerald KA, Finlay BB, Frankel G. Citrobacter rodentium: infection, inflammation and the microbiota. Nat Rev Microbiol. 2014;12:612–23.
Sherman PM, Ossa JC, Johnson-Henry K. Unraveling mechanisms of action of probiotics. Nutr Clin Pract. 2009;24:10–4.
Gionchetti P, Rizzello F, Venturi A, Brigidi P, et al. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: a double-blind, placebo- controlled trial. Gastroenterology. 2000;119:305–9.
Mimura T, Rizzello F, Helwig U, Poggioli G, et al. Once daily high dose probiotic therapy (VSL#3) for maintaining remission in recurrent or refractory pouchitis. Gut. 2004;53:108–14.
Tursi A, Brandimarte G, Papa A, Giglio A, et al. Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: a double-blind, randomized, placebo- controlled study. Am J Gastroenterol. 2010;105:2218–27.
Sood A, Midha V, Makharia GK, Ahuja V, et al. The probiotic preparation, VSL#3 induces remission in patients with mild-to-moderately active ulcerative colitis. Clin Gastroenterol Hepatol. 2009;7:1202–9.
Miele E, Pascarella F, Giannetti E, Quaglietta L, Baldassano RN, Staiano A. Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am J Gastroenterol. 2009;104:437–43.
Rembacken BJ, Snelling AM, Hawkey PM, Chalmers DM, Axon AT. Non-pathogenic Escherichia coli versus mesalazine for the treatment of ulcerative colitis: a randomised trial. Lancet. 1999;354:635–9.
Kruis W, Fric P, Pokrotnieks J, Lukás M, et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut. 2004;53:1617–23.
Kruis W, Schütz E, Fric P, Fixa B, Judmaier G, Stolte M. Double-blind comparison of an oral Escherichia coli preparation and mesalazine in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther. 1997;11:853–8.
Zocco MA, dal Verme LZ, Cremonini F, Piscaglia AC, et al. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment Pharmacol Ther. 2006;23:1567–74.
Bousvaros A, Guandalini S, Baldassano RN, Botelho C, et al. A randomized, double-blind trial of Lactobacillus GG versus placebo in addition to standard maintenance therapy for children with Crohn’s disease. Inflamm Bowel Dis. 2005;11:833–9.
Furrie E, Macfarlane S, Kennedy A, Cummings JH, Walsh SV, O’neil DA, Macfarlane GT. Synbiotic therapy (Bifidobacterium longum/Synergy 1) initiates resolution of inflammation in patients with active ulcerative colitis: a randomised controlled pilot trial. Gut. 2005;54:242–9.
Kato K, Mizuno S, Umesaki Y, Ishii Y, et al. Randomized placebo-controlled trial assessing the effect of bifidobacteria-fermented milk on active ulcerative colitis. Aliment Pharmacol Ther. 2004;20:1133–41.
Ishikawa H, Akedo I, Umesaki Y, Tanaka R, Imaoka A, Otani T. Randomized controlled trial of the effect of bifidobacteria-fermented milk on ulcerative colitis. J Am Coll Nutr. 2003;22:56–63.
Laake KO, Bjørneklett A, Aamodt G, Aabakken L, Jacobsen M, Bakka A, Vatn MH. Outcome of four weeks’ intervention with probiotics on symptoms and endoscopic appearance after surgical reconstruction with a J-configurated ileal-pouch-anal-anastomosis in ulcerative colitis. Scand J Gastroenterol. 2005;40(1):43–51.
Bernstein CN. Antibiotics, probiotics and prebiotics in IBD. Nestle Nutr Inst Workshop Ser. 2014;79:83–100.
van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–15.
Nitzan O, Elias M, Peretz A, Saliba W. Role of antibiotics for treatment of inflammatory bowel disease. World J Gastroenterol. 2016;22(3):1078–87.
Ledder O, Turner D. Antibiotics in IBD: still a role in the biological era? Inflamm Bowel Dis. 2018;24(8):1676–88.
Balram B, Battat R, Al-Khoury A, D’Aoust J, Afif W, Bitton A, Lakatos PL, Bessissow T. Risk factors associated with Clostridium difficile infection in inflammatory bowel disease: a systematic review and meta-analysis. J Crohns Colitis. 2019;13(1):27–38.
Llopis M, Antolin M, Carol M, Borruel N, et al. Lactobacillus casei downregulates commensals’ inflammatory signals in Crohn’s disease mucosa. Inflamm Bowel Dis. 2009;15:275–83.
Wu X, Vallance BA, Boyer L, Bergstrom KS, et al. Saccharomyces boulardii ameliorates Citrobacter rodentium-induced colitis through actions on bacterial virulence factors. Am J Physiol Gastrointest Liver Physiol. 2008;294:G295–306.
Yan F, Cao H, Cover TL, Washington MK, et al. Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through EGFR-dependent mechanism. J Clin Invest. 2011;121:2242.
Dylag K, Hubalewska-Mazgaj M, Surmiak M, Szmyd J, Brzozowski T. Probiotics in the mechanism of protection against gut inflammation and therapy of gastrointestinal disorders. Curr Pharm Des. 2014;20(7):1149–55.
Bennet JD, Brinkman M. Treatment of ulcerative colitis by implantation of normal colonic flora. Lancet. 1989;1:164.
Anderson JL, Edney RJ, Whelan K. Systematic review: faecal microbiota trans- plantation in the management of inflammatory bowel disease. Aliment Pharmacol Ther. 2012;36:503–16.
Angelberger S, Reinisch W, Makristathis A, Lichtenberger C, et al. Temporal bacterial community dynamics vary among ulcerative colitis patients after fecal microbiota transplantation. Am J Gastroenterol. 2013;108:1620–30.
Kump PK, Gröchenig HP, Lackner S, Trajanoski S, et al. Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis. 2013;19:2155–65.
Kunde S, Pham A, Bonczyk S, Crumb T, et al. Safety, tolerability, and clinical response after fecal transplantation in children and young adults with ulcerative colitis. J Pediatr Gastroenterol Nutr. 2013;56:597–601.
Qazi T, Amaratunga T, Barnes EL, Fischer M, Kassam Z, Allegretti JR. The risk of inflammatory bowel disease flares after fecal microbiota transplantation: systematic review and meta-analysis. Gut Microbes. 2017;8(6):574–88.
Khan KJ, Ullman TA, Ford AC, Abreu MT, et al. Antibiotic therapy in inflammatory bowel disease: a systemic review and meta-analysis. Am J Gastroenterol. 2011;106:661–73.
Aleksandar D, Xavier RJ, Gevers D. The microbiome in inflammatory bowel diseases: current status and the future ahead. Gastroenterology. 2014;146(6):1489–99.
Zimmer J, Hofsø D, Aasheim ET, Tanbo T, et al. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur J Clin Nutr. 2012;66:53–6.
LeBlanc JG, Laiño JE, del Valle MJ, Vannini V, et al. B-group vitamin production by lactic acid bacteria--current knowledge and potential applications. J Appl Microbiol. 2011;111(6):1297–309.
Matsuoka K, Kanai T. The gut microbiota and inflammatory bowel disease. Semin Immunopathol. 2015;37(1):47–55. https://doi.org/10.1007/s00281-014-0454-4. Epub 2014 Nov 25.
Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, Eberl G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456(7221):507–10.
Conflicts of Interest
The authors have no conflicts of interest.
There was no funding for this book chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Jatwani, S., Malhotra, B., Crout, T., Majithia, V. (2019). Microbes in the Pathogenesis of Inflammatory Bowel Disease: A Review. In: Espinoza, L. (eds) Infections and the Rheumatic Diseases. Springer, Cham. https://doi.org/10.1007/978-3-030-23311-2_37
Download citation
DOI: https://doi.org/10.1007/978-3-030-23311-2_37
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-23310-5
Online ISBN: 978-3-030-23311-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)