
Abstract
Heat shock protein 90 (HSP-90) has been identified in many disease processes including cancer, neurodegeneration, autoimmune diseases, and cancers. Great effort has been expended in the development of specific inhibitors of the N-terminal and C-terminal domains. Inhibitors of post-translational modification have also been developed. Herein, we explore the available inhibitors and those in development, discuss the relevant disease processes, and examine the pitfalls and promises of targeting HSP-90 for therapeutic intervention.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
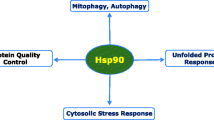
Heat-shock protein 90 (HSP-90) regulates the stability, activation , and degradation of a diverse array of proteins associated with growth, proliferation, and survival (Burlison et al. 2006; Neckers and Ivy 2003; Schopf et al. 2017; Schwock et al. 2008). Thus, it is core to regulation of protein stability and protein-degradation pathways and modulating transcription factors, signaling transduction networks, and kinases (Schopf et al. 2017). It facilitates the survival of cells during stress response and exhibits a pronounced anti-apoptotic and stabilization effect. Thus, HSP-90 has been associated with development and progression of a wide range of pathological conditions including cancers, diabetes, Gaucher disease, neurodegenerative diseases, and autoimmune dysfunction (Hoter et al. 2018; Kasperkiewicz et al. 2011; Lackie et al. 2017; Luo et al. 2010; Rice et al. 2008; Russo et al. 2006; Trepel et al. 2010; Tukaj et al. 2015; Whitesell and Linquist 2005; Yang et al. 2013).
1.1 Cancer
A commonality in many human cancers is the overexpression of HSP-90; the drastic two-to-three-fold induction of HSP90 seen in several cancers results in increase stabilization of client proteins (Barrott and Haystead 2013). The pronounced increase of HSP-90 in stress conditions can reach up to 6% of total protein (Prodromou 2016; Taipale et al. 2010). As consequence of increase HSP-90, the stabilization of its client protein results in the protects mutated or up-regulated oncoproteins. The aberrant protection of pro-survival and proliferation-related proteins such as telomerases, B-Raf, Akt, p53, VEGFR, HIF1α, HER-2, tyrosine kinases, steroid hormone receptors contribute to tumorigenesis, metastasis, and invasiveness (Banerji 2009; Beliakoff and Whitesell 2004; Hoter et al. 2018; Jhaveri and Modi 2012; Whitesell and Linquist 2005). As HSP90 acts as a regulator of HSF-1, the major hub of HSF transcriptional expression, HSP90 production causes dysregulation of HSF-1 transcriptional activity which leads to alterations in chaperone expression (Duerfeldt and Blagg 2010).
1.2 Neurodegenerative Diseases
HSP-90 works in concert with other chaperone machinery to refold misfolded proteins to prevent toxic accumulation. However, as in the case in various cancers, the stabilization of HSP-90 client proteins can have deleterious consequences outside of the context of normal physiological conditions. The stabilization of proteins associated with diseases results in manifestation of various neurodegenerative disease (Lackie et al. 2017; Luo et al. 2010). The HSP-90 client protein stabilization is a major facilitator for the accumulation of intrinsically disordered proteins (Karagoz et al. 2014; Luo et al. 2010). HSP-90 interacts with and stabilizes Tau, (Dickey et al. 2007; Hoter et al. 2018; Karagoz et al. 2014) a microtubule associated protein that mediates axonal transport. Tau hyperphosphorylation and aggregation is a classical hallmark of Alzheimer’s disease; the accumulation is also associated with other neurodegenerative disease such as progressive supranuclear palsy and Pick’s disease (Gong and Igbal 2008; Guo et al. 2017; Lee et al. 2001; Shelton et al. 2017). The HSP-90/Tau interface is associated with the neurodegenerative pathologic state and is well explained by enhanced stabilization of hyperphosphorylated Tau, which exacerbates aberrant neural activity seen in tauopathies (Shelton et al. 2017).
The aggregation of intrinsically disordered factors is also associated with Huntington’s disease and Parkinson’s disease (Lackie et al. 2017; Luo et al. 2010). HSP-90 interacts with Huntington protein (HTT) and leucine-rich repeat kinase 2 (Baldo et al. 2012; Wang et al. 2008). Interestingly, as HSP-90 is an established regulator of HSF-1, evidence suggests that, through the repression of HSP-90, other molecular chaperones systems such as HSP-70 can be enhanced and can facilitate increase neuroprotective function in otherwise pathogenic conditions (Luo et al. 2010).
1.3 Gaucher Disease
Gaucher disease is a rare autosomal recessive lysosomal disorder driven by genetic mutations in GBA gene encoding the lysosomal enzyme glucocerebrosidase (GCase); the mutations result in metabolic dysfunction and wide-spread organ dysfunction due to effects of the drastic accumulation of GCase substrate (Brady et al. 1966; Hruska et al. 2008; Stirnemann et al. 2017; Yang et al. 2013). The diminished amount of GCase results in the toxic accumulation of the GCase substrate, glucosylceramide (Stirnemann et al. 2017). GBA mutations cause protein misfolding and diminished protein instability resulting in increased retention of GCase in the endoplasmic reticulum (Ron and Horowitz 2005; Stirnemann et al. 2017; Yang et al. 2013). The intrinsic changes in the conformation of mutant GCase results in premature degradation and increased GCase turnover (Yang et al. 2013). HSP-90 is critical for targeting misfolded GCase for proteasomal degradation and directly interacts with GCase to direct the misfolded GCase to cellular ER and proteasomal degradation pathway (Yang et al. 2013). The increase degradation of GCase results in enhanced disease severity. This paradigm is in contrast with other diseases associated with HSP-90, as the interaction does not enhance the accumulation of its client protein but rather directs the client protein for degradation.
1.4 Diabetes and Associated Complications
Several of the key players mentioned above constitute a regulatory pathway for insulin sensitivity. Transcription of HSP-70 is regulated by HSF-1, which in turn is activated by inhibition of HSP-90 (Lee et al. 2013). Further, inhibition of HSP-90 leads to inhibition of JNK1 and thus improved insulin signaling; in the mouse model in this study, HSP-90 inhibition reversed hyperglycemia (Lee et al. 2013). In another study of diabetic mice, inhibition of HSP-90 with 17-dimethylaminoethylamino-17-demethoxygeldanamycin (DMAG) lessened renal damage and atherosclerosis incurred by hyperglycemia and hyperlipidemia as evidenced by decrease in albuminuria, renal lesions, and proinflammatory genes (Lazaro et al. 2015). Further study of diabetic atheroprotection with DMAG in a diabetic mouse model found HSP-90 inhibition to be protective by induction of nuclear factor erythroid-derived 2-like (Nrf2) (Lazaro et al. 2017). Hypercoagulability in diabetes was found to be dependent on a glucose-regulated interaction between HSP-90α and annexin II, which promotes the generation of plasmin (Lei et al. 2004).
1.5 Autoimmune Disease
HSP-90 machinery is involved in adaptive and innate immune responses via mediating the activation of immune cells (Srivastava 2002; Taipale et al. 2010). It is instrumental in the function of natural killer cells, macrophages, lymphocytes, dendritic cell maturation, neutrophils (Kasperkiewicz et al. 2011; Srivastava 2002). The chaperone has been implicated in inflammation, antigen presentation, and immune cell activation (Srivastava 2002; Taipale et al. 2010). Client proteins of HSP-90 include inflammation regulating kinases IKK and JAK (Madrigal-Matute et al. 2010; Prodromou 2016; Zhang and Burrows 2004). These kinases modulate transcriptional regulators STAT and NF-kB which in turn dictate the expression of many pro-inflammation factors (Madrigal-Matute et al. 2010; Prodromou 2016). Thus, mounting evidence has demonstrated the importance of HSP-90 in regulating pro-inflammation responses and immune cell leading to the hypothesis that HSP-90 plays a critical function in auto-immune disease (DeBoer et al. 1970; Madrigal-Matute et al. 2010; Ron and Horowitz 2005; Stebbins et al. 1997). In support of this hypothesis, studies have implicated HSP90 in autoimmune diseases such as systemic lupus erythematosus, rheumatoid arthritis, allergic rhinitis, and other autoimmune diseases such as bullous skin diseases (Kasperkiewicz et al. 2011; Rice et al. 2008; Russo et al. 2006; Srivastava 2002).
2 Targeting HSP-90
2.1 Modulating HSP90 Function by Perturbation of PTMs
The post-translational modifications of HSP-90 alter the chaperone dynamics and perturbs the interaction with co-chaperones, substrates, and can influence enzyme activity (Jackson 2012; Kekapure et al. 2009; Scroggins et al. 2007). Acetylation of HSP-90 at the middle domain results in a marked decrease in its function by impeding the ability to interact with co-chaperones and client proteins, changing the dynamic conformation cycles (Aoyagi and Archer 2005; Kovacs et al. 2005; Mollapour and Neckers 2012; Scroggins et al. 2007). Targeting acetylation presents an avenue to modulate the activity of HSP-90. Reversible protein acetylation regulates a wide range of biochemical processes involving HSP-90 (Kovacs et al. 2005; Yu et al. 2002). The inhibition of histone deacetylase induces hyperacetylation HSP-90; the acetylated form of the chaperone has decreased affinity for ATP and target proteins (Bali et al. 2004). HDAC, while traditionally defined by their role in deacetylation of histones, have been found to act on a larger range of substrates including HSP-90 (Bali et al. 2004; Fiskus et al. 2007). Additionally, HDAC have been found to influence drug resistance to chemotherapeutics and diverse HSP90 inhibitors (Chai et al. 2017; Wang et al. n.d.). Interestingly, HDAC proteins such as HDAC6 also regulate the interactions with HSP90 and HSF master regulator, thereby affecting the transcriptional network of other HSP systems (Boyault et al. 2007; de Zoeten et al. 2011; Prodromou 2016).
Inhibiting deacetylation through HDAC inhibitors (HDACI) presents a promising avenue in which HSP90 chaperone cycling and function can be impeded. Ultimately, HDAC influences the stability of a plethora of downstream targets of HSP-90. HDACI have anti-tumorigenic properties correlating with diminished accumulation of HSP-90 target proteins related to pro-survival and pro-growth (Bali et al. 2004; Ding et al. 2017; Park et al. 2008). The inhibition of HSP-90 chaperone function by HDACIs results in degradation of oncoproteins such as AKT, FLT-3, BCR-ABL, RAF-1, VEGFR1, VEGFR2, JAK2, RASGRP1 and CRAF (Bali et al. 2004; Ding et al. 2017; Park et al. 2008). The resulting degradation of the oncoproteins by HSP-90 stabilization leads to dramatic changes in cell cycle control and proliferation. Modulating HDAC6 and HSP90, through HDACI has been studied in the context of ameliorating autoimmunity by affecting T-regulatory cells (Chiosis et al. 2001). Additionally, NF-κB function is impaired by HDACI inhibition of HDAC6. It is thought that the increase in the acetylation of HSP90 results in reduced stability and degradation of IKK. The reduced stability of IKK in turn causes aberrant NF-κB function (Kovacs et al. 2005; Regna et al. 2015; Trepel et al. 2010). Thus, the HDAC/HSP-90 interface presents a promising target to impede autoimmunity. While initial findings in autoimmunity have shown that targeting HDAC6 show some diminished HSP90 function, further studies are needed (Regna et al. 2015).
HDACI can also promote the stability of HSP90 proteins. In the case of Gaucher’s disease, the production of mutated GCase results reduced of accumulation of the enzyme through HSP-90-directed degradation (Wang et al. 2008). HDACI, such as LB-205 and SAHA, results in increase acetylated form of HSP-90 and impairs the binding of HSP-90 to GCase (Yang et al. 2013). The increase in the accumulation of the mutated GCase in part increases the functional activity of the GCase thereby limiting toxic accumulation of the GCase substrate, glucosylceramide (Yang et al. 2013).
2.2 Targeting HSP-90 C-terminus
While traditional targeting of HSP-90 for therapeutics have predominantly developed to target the N-terminal domain (NTD) of the protein, novel approaches act to impede the C-terminal function of HSP-90. Inhibition of HSP-90 activity through the NTD perturbs the repressive effect of HSP-90 on HSF-1 which subsequently activates heat shock response. The activation of the heat shock response is thought to facilitate resistance which dampens the effect of HSP90 inhibitors (Yang et al. 2013). Therefore, a major driving force of targeting C-terminus is that prior trials with N-terminal targeting small molecules result in rapid development of resistance to the inhibitory molecules (Donnelly and Blagg 2008; Solárová et al. 2015).
The C-terminal inhibitors are subdivided into two categories; inhibitors that directly target the C-terminus and inhibitors that disrupt the binding of HSP90 to co-chaperones at the C-terminus (Koay et al. 2016). Similar to the NTD, the CTD contains a nucleotide binding site, however, lacks ATPase activity (Schopf et al. 2017). While the nucleotide binding site differs in terms of binding affinity and binding specificity, selective targeting of the nucleotide binding site has shown promising applications in inhibiting chaperone function (Donnelly and Blagg 2008; Solárová et al. 2015). Coumarin-based antibiotics were among the first inhibitors found to target the CTD (Solárová et al. 2015). Initially, this class of small molecules were found to inhibit the ATPase activity of ATP-gyrases; further biochemical classification has shown weak affinity towards the nucleotide binding site of CTD (Burlison et al. 2006; Donnelly and Blagg 2008; Solárová et al. 2015). The binding of novobiocin, coumarin-antibiotic, indeed affects the stability of HSP-90 client proteins and prompted the development of synthetic derivatives of the substrate. These nucleotide binding inhibitors induce conformational changes thought to impede and release protein-interaction by disrupting the dimerization of HSP-90 (Allan et al. 2006; Gormley et al. 1996; Solárová et al. 2015).
Novobiocin exerted anti-tumorigenic properties towards certain cancer lines; however, it lacked the efficacy that N-terminal inhibitors showed. Development of synthetic novobiocin derivatives sought to amend the poor efficacy by improving HSP90 inhibition. KU174, and KU675 analogues of novobiocin have shown strong anti-proliferation activity towards prostate cancer lines (Eskew et al. 2011; Liu et al. 2015; Solárová et al. 2015). Other novobiocin analogues developed have shown potential neuroprotective properties and provide an avenue in which HSP-90 inhibitors can be studied in the context of neurodegeneration (Donnelly and Blagg 2008).
Additional molecules have been tested and developed to target the nucleotide binding site of the CTD. These small molecules include the recent discovery of dihydropyrimidinone and analogs of bisphenol A such as NSC145366 as novel classes of CTD binding compounds (Goode et al. 2017; Terracciano et al. 2018). While, their therapeutic potential has not been fully explored, the continual development of CTD inhibiting agents provides an avenue in which HSP-90 can be inhibited without the potential drawback of driving drug resistance. Current research is focused on the neuroprotective properties of dihydropyridine derivatives and may be candidate therapeutic molecule for Alzheimer’s disease (Roe et al. 2018). Recently, a novel C-terminal targeting hexapeptide, amioxyrone, was found to bind to specifically target CTD and inhibit dimerization (Bhatia et al. 2018). The targeting of CTD results in the reduced stabilization, downregulation, and degradation of HSP-90 client oncoproteins without the induction of the heat shock response (Bhatia et al. 2018). The hexapeptide showed effectiveness in leukemic cell lines and leukemia stem cells which demonstrated a novel approach in targeting chronic myeloid leukemia (Bhatia et al. 2018).
The alternative strategy of affecting the C-terminal function is by utilizing small molecules that disrupt the binding of HSP90 to co-chaperones. The C-terminal domain of HSP-90 possess MEEVD residue that regulates the interactions with TPR domain containing co-chaperones (Buchner 1999; Wandinger et al. 2008). TPR co-chaperones have tremendous importance in HSP-90 regulation (Schopf et al. 2017). These chaperones modulate the conformation of HSP-90 and interactions with co-chaperones; therefore the HSP90 chaperone machinery is affected (Schopf et al. 2017). While no natural inhibitors have been discovered to target the MEEVD region, exploration of the TPR-domain binding interface has led to the development of synthetic molecules to target the HSP-90-TPR binding interface (Sidera and Patsavoudi 2014). Recently, C-terminal modulators including modified variants of SM molecules: SM122, SM145, SM253, and SM258, have been developed to interact with Hsp90 and block the binding of TRR-domain containing co-chaperones (Koay et al. 2016). These SM molecules disrupt TRR-containing proteins, FKBP52 and HOP (Koay et al. 2016).
2.3 Targeting HSP90 N-Terminus
The inhibition of HSP-90 at the N-terminus can be divided into geldanamycin/ geldanamycin derivatives and purine-based inhibitors. The classic targeting of HSP-90 began with the natural analogs geldanamycin, herbimycin, and macbecin (DeBoer et al. 1970). Out of these three, geldanamycin was the most potent inhibitor due to its ability to more effectively bind to the NTD of HSP-90 and prevent ATP binding to the pocket; it also functioned to inhibit HSP-90 dimerization with heat shock factor 1 (HSF-1) which lead to heat shock response through transcriptional activation of factors such as HSP27, HSP40, HSP70, and HSP90 (Zou et al. 1998). The carbamate group of geldanamycin represents one of its core interacting domains with HSP-90 as it may form a hydrogen bonding network within the pocket and elimination of which abolished geldanamycin function (Stebbins et al. 1997).
The major weaknesses of geldanamycin, however, is its low solubility, difficulty in crossing the blood-brain barrier, and most important of all, its induction of the heat shock response from inhibiting HSP-90. This response is comprised of the cells upregulating transcription of heat-shock proteins to properly compensate for the disruption of protein folding (Sittler et al. 2001). To account for the problems associated with geldanamycin, a semi-synthetic derivative of geldanamycin called tanespimycin (17-AAG) was created that improved the ADME activity while decreasing the toxicity and heat shock response generated by geldanamycin (Goetz et al. 2003). As a result, 17-AAG provided a stronger candidate for HSP-90 inhibition. Despite the improvements made by 17-AAG, induction of the heat shock response and resulting toxicity lead to the computer screening of multiple compounds targeting the NTD to develop new compounds that limited the harmful effects of the previous generation of NTD- targeting inhibitors (Table 10.1).
Purine-based inhibitors were a completely synthetic class of HSP-90 inhibitors designed to target the ATP-binding pocket of the NTD created from the complete crystal structure of HSP90 ATP/ADP complex (Prodromou et al. 1997). These inhibitors have increased potency so that side effects associated with the geldanamycin analogs would be minimized at therapeutic doses. Through screening processes, many derivatives of these purine-based inhibitors were created, the first of which was PU3. This derivative mimicked the binding of ATP in the NTD pocket in its closed conformation (Chiosis et al. 2001). The discovery of PU3 opened the door for a HSP90 NTD inhibitors that could potentially be brought through clinical trials due to the decreased toxicity.
Currently, couple of these purine and purine-like inhibitors are undergoing clinical trials. BIIB021 is a member of the purine inhibitors being used to treat chronic lymphocytic leukemia and in a combination trial to treat HER2 (+) metastatic breast cancer (Table 10.1). Frequent grade 3 and 4 toxicities are associated with its use in chronic lymphocytic leukemia such as fatigue and hyponatremia while diarrhea, partial seizure, and nausea have been associated with its use in metastatic breast cancer (Elfiky et al. 2008). Subsequently, this lead to the use of BIIB021 use in gastrointestinal stromal tumor treatment refractory to imatinib and sunitinib where it was well-tolerated and showed metabolic changes in the patients that primarily lead to the stabilization of the tumors (Dickson et al. 2012).
Another major compound of interest is PU-H71, another purine class inhibitor shown in preclinical studies to be effective against breast cancer, hepatocellular carcinoma, and Bcl-6-dependenet diffuse B-cell lymphoma cell lines. Human studies focused on patients with advanced refractory cancers revealed that PU-H71 was well-tolerated, but its discontinuation of supply did not allow a strong therapeutic index to be determined; Evidence suggested stability of disease as the average response (Speranza et al. 2018).
3 Conclusions
Herein, we have examined the role of HSP-90 in several disease entities and the routes that have been examined for therapeutic development. Historically, the C-terminal domain and PTM inhibitors have shown the most promise. Future directions will likely focus on combining these inhibitory steps and perhaps developing conjugated inhibitors to bolster delivery and efficacy.
Abbreviations
- 17-AAG:
-
Tanespimycin
- ADME:
-
absorption, distribution, metabolism, excretion
- AKT:
-
protein kinase B
- b-RAF:
-
B-Raf proto-oncogene
- c-RAF:
-
RAF proto-oncogene serine/threonine-protein kinase
- CTD:
-
C-terminal domain
- DMAG:
-
17-dimethylaminoethylamino-17-demethoxygeldanamycin
- ER:
-
endoplasmic reticulum
- FKBP:
-
FK506 binding protein
- FLT:
-
Fms-like tyrosine kinase
- GBase:
-
glucocerebrosidase
- HDAC:
-
histone deacetylase
- HDACI:
-
HDAC inhibitor
- HER:
-
human epidermal growth factor receptor
- HIF:
-
hypoxia inducible factor
- HOP:
-
HSP70-HSP90 organizaing protein
- HSF:
-
heat shock factor
- HSP:
-
heat shock protein
- HTT:
-
huntington protein
- IKK:
-
IkB kinase
- JAK:
-
Janus kinase
- JNK:
-
c-Jun N-terminal kinases
- NF-kB:
-
nuclear factor kappa light-chain enhancer of activated B cells
- Nrf:
-
nuclear factor erythroid 2-related factor
- NTD:
-
N-terminal domain
- RAF:
-
rapidly accelerated fibrosarcoma
- RASGRP:
-
RAS guanyl-releasing protein
- SAHA:
-
suberoyl anilide hydroxamic acid
- STAT:
-
signal transducer and activators of transcription
- TPR:
-
tetraotricopeptide repeat
- VEGFR:
-
vascular endothelial growth factor receptor
References
Allan RK, Mok D, Ward BK et al (2006) Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J Biol Chem 281:7161–7171
Aoyagi S, Archer TK (2005) Modulating molecular chaperone Hsp90 functions through reversible acetylation. Trends Cell Biol 15(11):565–567
Baldo B, Weiss A, Bibel M et al (2012) A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biol Chem 287:1406–1414
Bali P, George P, Cohen P et al (2004) Superior activity of the combination of histone deacetylase inhibitor LAQ824 and the FLT-3 kinase inhibitor PKC412 against human AML cells with mutant FLT-3. Clin Cancer Res 10:4991–4997
Banerji U (2009) Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 15:9–14
Barrott JJ, Haystead TA (2013) HSP90, an unlikely ally in the war on cancer. FEBS J 280(6):1381–1396
Beliakoff J, Whitesell L (2004) Hsp90: an emerging target for breast cancer therapy. Anti-Cancer Drugs 15:651–662
Bhatia S, Diedrich D, Frieg B et al (2018) Targeting HSP90 dimerization via the C terminus is effective in imatinib-resistant CML and lacks the heat shock response. Blood 132(3):307–320
Boyault C, Zhang Y, Fritah S et al (2007) HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21:2172–2218
Brady RO, Kanfer JN, Bradley RM et al (1966) Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J Clin Invest 45:1112–1115
Buchner J (1999) Hsp90 & co. – a holding for folding. Trends Biochem Sci 24:136–141
Burlison J, Neckers L, Smith AB et al (2006) Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of Hsp90. JACS 128(48):15529–15536
Chai RC, Vieusseux JL, Lang BJ et al (2017) Histone deacteylase activity mediates acquired resistance towards structurally diverse hsp90 inhibitors. Mol Oncol 11(5):567–583
Chiosis G, Timaul MN, Lucas B et al (2001) Small molecule designed to bind to the adenine nucleotide pocket of HSP-90 causes Her2 degradation and the growth arrest and differentiation of breast cancer cells. Chem Biol 8(3):289–299
DeBoer C, Meulman PA, Wnuk RJ et al (1970) Geldanamycin, a new antibiotic. J Antibiot (Tokyo) 23:442–447
Dickey CA, Kamal A, Lundgren K et al (2007) The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 117:648–658
Dickson MA, Okuno SH, Keohan ML et al (2012) Phase II study of the HSP90-inhibitor BIIB021 in gastrointestinal stromal tumors. Ann Oncol: Off J Eur Soc Med Oncol 24(1):252–257
Ding H, Peterson KL, Correia C et al (2017) Histone deacetylase inhibitors interrupt HSP90-CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia 31(7):1593–1602
Donnelly A, Blagg B (2008) Novobiocin and additional inhibitors of Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem 15:2702–2717
Duerfeldt A, Blagg B (2010) Hsp90 inhibition: elimination of shock and stress. Bioorg Med Chem Lett 20(17):4983–4987
Elfiky A, Saif MW, Beeram M et al (2008) BIIB021, an oral, synthetic non-ansamycin Hsp90 inhibitor: phase I experience. J Clin Oncol 26:2503
Eskew JD, Sadikot T, Morales P et al (2011) Development and characterization of a novel C-terminal inhibitor of Hsp90 in androgen dependent and independent prostate cancer cells. BMC Cancer 11:468
Fiskus W, Ren Y, Mohapatra A et al (2007) Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-alpha levels and transcriptional activity: a result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin Cancer Res 13:4882–4890
Goetz MP, Toft DO, Ames MM et al (2003) The HSP90 chaperone complex as a novel target for cancer therapy. Ann Oncol 14(8):1169–1176
Gong C, Igbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15:2321–2328
Goode KM, Petrov DP, Vickman RE et al (2017) Targeting the Hsp90 C-terminal domain to induce allosteric inhibition and selective client downregulation. Biochim Biophys Acta 1861:1992–2006
Gormley N, Orphanides G, Meyers A et al (1996) The interaction of coumarin antibiotics with dragments of DNA gyrase B. Biochemistry 3:5083–5092
Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol (Berl) 133:665–704
Hoter A, El-Sabban M, Naim H (2018) The HSP90 family: structure, regulation, function, and implications in health and disease. Int J Mol Sci 19:1–33
Hruska K, LaMarca M, Scott C et al (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29:567–583
Jackson SE (2012) Hsp90: structure and function. In: Jackson S (ed) Molecular chaperones, Topics in current chemistry, vol 328. Springer, Berlin
Jhaveri K, Modi S (2012) HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv Pharmacol 65:471–517
Karagoz G, Duarte A, Akoury E et al (2014) Hsp90-tau complex reveals molecular basis for specificity in chaperone action. Cell 156:963–974
Kasperkiewicz M, Müller R, Manz R et al (2011) Heat-shockprotein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood 117(23):6135–6142
Kekapure V, Dannenberg A, Subbaramaiah K (2009) HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J Biol Chem 284:7436–7445
Koay YC, Wahyudi H, McAlpine SR (2016) Reinventing HSP90 inhibitors: blocking C-terminal binding events to HSP90 by using dimerized inhibitors. Chem Eur J 22:18572–11858
Kovacs J, Murphy P, Gaillard S et al (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18:601–607
Lackie R, Maciejewski A, Ostapchenko V et al (2017) The HSP70/HSP90 chaperone machinery in neurodegenerative diseases. Front Neurosci 11:1–23
Lazaro I, Oguiza A, Recio C et al (2015) Targeting HSP90 ameliorates nephropathy and atherosclerosis through suppression of NF-κB and STAT signaling pathways in diabetic mice. Diabetes 64(10):3600–3613
Lazaro I, Oguiza A, Recio C et al (2017) Interplay between HSP90 and Nrf2 pathways in diabetes-associated atherosclerosis. Clin Investig Arterioscler 29(2):51–59
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Lee JH, Gao J, Kosinski PA, Elliman SJ et al (2013) Heat shock protein 90 (HSP90) inhibitors activate the heat shock factor 1 (HSF1) stress response pathway and improve glucose regulation in diabetic mice. Biochem Biophys Res Commun 430(3):1109–1113
Lei H, Romeo G, Kazlauskas A (2004) Heat shock protein-90α-dependent translocation of annexin II to the surface of endothelial cells modulates plasmin activity in the diabetic rat aorta. Circ Res 94:902–909
Liu W, Vielhauer GA, Holzbeierlein JM et al (2015) KU675, a concomitant heat-shock protein inhibitor of Hsp90 and Hsc70 that manifests isoform selectivity for Hsp90α in prostate cancer cells. Mol Pharmacol 88(1):121–130
Luo W, Sun W, Taldone T et al (2010) Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener 5:1–8
Madrigal-Matute J, Lopez-Franco O, Blanco-Colio LM et al (2010) Heat shock protein 90 inhibitors attenuate inflammatory responses in atherosclerosis. Cardiovasc Res 86:330–337
Mollapour M, Neckers L (2012) Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim Biophys Acta 1823:648–655
Neckers L, Ivy SP (2003) Heat shock protein 90. Curr Opin Oncol 15:419–424
Park J, Kim S, Choi M, Lee J et al (2008) Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem Biophys Res Commun 368:318–322
Prodromou C (2016) Mechanisms of Hsp90 regulation. Biochem J 473:2439–2452
Prodromou C, Rose SM, O’Brien R et al (1997) Identification and structural characterization of the ATP/ADP-binding site in the HSP-90 molecular chaperone. Cell 90(1):65–75
Regna N, Vieson M, Gojmerac A et al (2015) HDAC expression and activity is upregulated in diseased lupus-prone mice. Int Immunopharmacol 29:494–503
Rice JW, Veal JM, Fadden RP et al (2008) Small molecule inhibitors of Hsp90 potently affect inflammatory disease pathways and exhibit activity in models of rheumatoid arthritis. Arthritis Rheum 58(12):3765–3775
Roe M, Wahab B, Torok Z et al (2018) Dihydropyridines allosterically modulate Hsp90 providing a novel mechanism for heat shock protein co-induction and neuroprotection. Front Mol Biosci 5(51):1–14
Ron I, Horowitz M (2005) ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum Mol Genet 14(16):2387–2398
Russo CD, Polak PE, Mercado PR et al (2006) The heat-shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin suppresses glial inflammatory responses and ameliorates experimental autoimmune encephalomyelitis. J Neurochem 99(5):1351–1362
Schopf F, Biebl M, Buchner J (2017) The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18:345–360
Schwock J, Pham NA, Cao MP et al (2008) Efficacy of Hsp90 inhibition for induction of apoptosis and inhibition of growth in cervical carcinoma cells in vitro and in vivo. Cancer Chemother Pharmacol 61:669–681
Scroggins BT, Robzyk K, Wang D et al (2007) An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol Cell 25:151–159
Shelton L, Koren J, Blair L (2017) Imbalances in the Hsp90 chaperone machinery: implications for Tauopathies. Front Neurosci 11:1–12
Sidera K, Patsavoudi E (2014) HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov 9:1–20
Sittler A, Lurz R, Lueder G et al (2001) Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum Mol Genet 10(12):1307–1315
Solárová Z, Mojžiš J, Solár P (2015) Hsp90 inhibitor as a sensitizer of cancer cells to different therapies (review). Int J Oncol 46:907–926
Speranza G, Anderson L, Chen AP et al (2018) First-in-human study of the epichaperome inhibitor PU-H71: clinical results and metabolic profile. Investig New Drugs 36(2):230–239
Srivastava P (2002) Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol 2(3):185–194
Stebbins CE, Russo AA, Schneider C et al (1997) Crystal structure of an HSP90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 18(2):239–250
Stirnemann J, Belmatoug N, Camou F et al (2017) A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18(2):441
Taipale M, Jarosz DF, Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11:515–528
Terracciano S, Russo A, Chini MG et al (2018) Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Sci Rep 8:1709
Trepel J, Mollapour M, Giaccone G et al (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10:537–549
Tukaj S, Zillikens D, Kasperkiewicz M (2015) Heat shock protein 90: a pathophysiological factor and novel treatment target in autoimmune bullous skin diseases. Exp Dermatol 24:567–571
Wandinger S, Richter K, Buchner J (2008) The Hsp90 chaperone machinery. J Biol Chem 283(27):18473–18477
Wang L, Xie C, Greggio E et al (2008) The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2J. Neurosciences 28:3384–3391
Wang Z, Hu P, Tang F et al (n.d.) HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett 379:134–142
Whitesell L, Linquist S (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Yang C, Rahimpour S, Lu J et al (2013) Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc Natl Acad Sci U S A 110:966–971
Yu X, Guo ZS, Marcu MG et al (2002) J Natl Cancer Inst 94:504–513
Zhang H, Burrows F (2004) Targeting multiple signal transduction pathways through inhibition of Hsp90. J Mol Med 82:488–499
de Zoeten E, Wang L, Butler K et al (2011) Histone deacetylase 6 and heat shock protein 90 control the functions of foxp3+ T-regulatory cellsMol. Cell Biol 31:2066–2078
Zou JY, Guo YL, Guettouche T et al (1998) Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94(4):471–480
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Westlake, T., Sun, M., Rosenblum, B.C., Zhuang, Z., Rosenblum, J.S. (2019). Targeting Hsp-90 Related Disease Entities for Therapeutic Development. In: Asea, A., Kaur, P. (eds) Heat Shock Protein 90 in Human Diseases and Disorders. Heat Shock Proteins, vol 19. Springer, Cham. https://doi.org/10.1007/978-3-030-23158-3_10
Download citation
DOI: https://doi.org/10.1007/978-3-030-23158-3_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-23157-6
Online ISBN: 978-3-030-23158-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)