
Abstract
Pancreatic ductal adenocarcinoma is an overwhelming fatal disease that often presents with overt metastases and ultimately causes the majority of cancer-associated deaths. The mechanisms underlying the metastatic cascade are complex, and research in recent years has begun to provide insights into the underlying drivers of this phenomenon. It has become clear that cancer cells, in particular pancreatic cancer cells, possess properties of plasticity involving bidirectional transition between epithelial and mesenchymal identities. Furthermore, recent work has begun to establish that there are distinct hybrid states between purely epithelial and purely mesenchymal states that cancer cells may reside, in order to thrive at different stages of carcinogenesis. We discuss how this plasticity is important for different phases of the metastatic cascade, from delamination to colonization, and how different epithelial–mesenchymal states may affect metastatic organotropism. In this review, we summarize the current understanding of pancreatic cancer cell plasticity and metastasis, and highlight current model systems that can be used to study these phenomena.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pancreatic cancer
- Epithelial-mesenchymal transition (EMT)
- Cellular plasticity
- Metastasis
- Metastatic organotropism
PDAC Metastasis and Outcomes
Pancreatic ductal adenocarcinoma (PDAC) is a major health issue in the USA, accounting for over 55,000 new cases and 44,000 deaths annually [1]. With fewer than 7% of patients surviving beyond 5 years and a projected increase in PDAC-associated deaths, PDAC will become the second leading cause of cancer deaths by 2020 [2]. Despite a significant emphasis being placed on improving early detection methods in PDAC patients, the majority of patients present with metastatic disease. The nearly universal mortality rate observed in PDAC is likely due to this unchecked metastatic potential. This dismal prognosis has not improved significantly for decades, likely due to the recently observed phenomenon that pancreatic cancer tumor cells are able to metastasize very early on during neoplastic transformation, before frank carcinoma is observed [3]. This observation, gleaned from genetically engineered mouse models (GEMMs) of PDAC, has rejuvenated the field of metastasis biology and provides an ideal model system for biologists to study the molecular underpinnings driving the metastatic cascade in this disease. The prevailing view in the field is that metastasis is facilitated through a process called epithelial-to-mesenchymal transition (EMT), where tumor cells lose their epithelial cell identity and begin to gain mesenchymal characteristics. In this chapter, we will discuss the role of epithelial cell plasticity in pancreatic cancer metastasis and introduce new paradigms thought to drive the metastatic cascade in this disease.
Models of Metastasis
Genetically Engineered Mouse Models of Metastatic PDAC
A landmark genetic mouse model of pancreatic cancer has become a mainstay in the field and has been a vital tool to shape our understanding of the molecular pathogenesis of PDAC [4]. Hingorani and colleagues looked to mimic the genetic mutations in human PDAC patients, which are dominated by activating mutations in KRAS codon 12, 13, or 61 (up to 95% of patients) and TRP53 gain-of-function mutations (approximately 70% of patients) [5]. The resulting KPC mice, with inducible endogenous expression of mutant KrasG12D and mutant Trp53R172H driven by the pancreatic epithelial cell-specific Pdx1-Cre transgene, developed invasive and metastatic PDAC that mirrors the human disease. These mice succumb to disease with a median survival of approximately 5 months, at which point metastasis to the liver and lung is evident. KPC mice have re-shaped the field and revolutionized our understanding of PDAC biology, including seminal studies that have utilized the KPC mouse to establish roles for the TGF-ß pathway [6,7,8], Hedgehog signaling [9, 10], Ink4a/Arf [11, 12], Brca1/2 [13,14,15], and other genes in PDAC tumorigenesis and progression. This now nearly ubiquitous model has provided an ideal model system to study the effect that genetic knockout of countless genes has on primary tumor formation and disease progression .
This model was expanded upon by Andrew Rhim in the laboratory of Ben Stanger through the introduction a Cre-inducible YFP lineage label driven by the same Pdx1-Cre transgene used to activate mutant KrasG12D and mutant Trp53R172H [3]. The subsequently named KCPY mice were used to show that dissemination occurs prior to frank malignancy and is driven by an underlying activation of an EMT program within the tumor cells. This model allows one to trace elegantly the metastatic process in pancreatic cancer, and is an invaluable resource in the field of metastasis biology. In particular, these mice grant us the ability to study of all stages of the metastatic cascade in vivo, from early invasion and growth into the surrounding tissue, to intravasation into the vasculature, travel through circulation, extravasation at the metastatic site, and colonization of the secondary tumor .
Orthotopic Models of Pancreatic Cancer Metastasis
The field has also adopted an orthotopic model of pancreatic cancer to quickly address the roles that a given gene may have during tumorigenesis and metastasis. Orthotopic injection of pancreatic tumor cells directly into the pancreas is an elegant approach that has been utilized for many years to efficiently test the effect of genetic alterations in PDAC tumor cells. Direct injection of KPC tumor cell cultures into the pancreas of syngeneic mice (or nude mice if cells are derived from a mixed background) yields primary tumors within days and metastatic lesions within weeks (protocol for orthotopic injection reviewed in [16]). This relatively easy and reliable in vivo method of pancreatic tumor formation is easier to genetically manipulate than autochthonous GEMMs , and yields reproducible primary tumor growth and metastasis kinetics. Injection of KCPY cultures gives the added advantage of being able to study metastasis in vivo, as previously discussed. Many studies in the pancreatic cancer field utilize both GEMMs and orthotopic models of cancer to address different aspects of tumor formation and metastatic disseminations, and both tools are great resources for studying the effect of genetic perturbation or pharmacological inhibition.
Mechanisms of Metastasis
EMT-MET Axis and Cellular Plasticity
The KPC and KPCY autochthonous pancreatic cancer models have been used by many in the field to determine the role of epithelial plasticity in PDAC metastasis. The original KCPY study demonstrated that YFP-tagged tumor cells undergo EMT and invade at an early stage to form micrometastases at the secondary site [3]. Subsequent studies have shown that a reversion to epithelial morphology, a phenomenon termed mesenchymal-to-epithelial transition (MET), is required to thrive and form macrometastases at the secondary site [17]. Conventionally, PDAC tumor cells are thought to gain more mesenchymal characteristics by undergoing EMT within the primary tumor, giving them the ability to invade into the tumor parenchyma, as they look for vasculature to begin their metastatic journey. After intravasating into blood vessels, tumor cells maintain their mesenchymal status and travel through the circulatory system, until they reach their eventual metastatic site, at which point they must extravasate into the secondary organ and undergo mesenchymal-to-epithelial transition (MET) to re-establish their epithelial identity. Once returned to an epithelial state, the tumor cells will begin to colonize and proliferate in the metastatic site. The prevailing thought is that the EMT–MET axis is very plastic, with tumor cells existing at various states throughout the spectrum in order to survive and thrive in their new environments (reviewed in [18]). Epithelial status is thought to be a pro-proliferative state, while mesenchymal status is a migratory state. Furthermore, cells undergoing EMT are thought to be more drug-resistant with a certain degree of stemness that allows them to thrive in adverse conditions, such as in circulation and when first arriving in foreign organ.
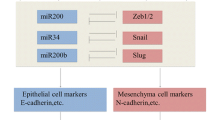
The EMT process is largely regulated at the transcriptional level through various transcription factors such as zinc finger E-box-binding homeobox 1 (ZEB1), twist-related protein 1 (TWIST), zinc finger protein SNAI1 (SNAIL), zinc finger protein SNAI2 (SLUG), and paired related homeobox protein 1 (PRRX1) (reviewed in [18]), although post-translational mechanisms have been invoked recently [19]. Conventionally, these transcription factors are thought to activate an EMT program as well as a stem cell-like program. However, the two processes are not completely linked, as is the case for PRRX1, which uncouples EMT and stemness [20]. Furthermore, PRRX1 has been shown to regulate other forms of epithelial plasticity within the pancreas, such as acinar-to-ductal metaplasia (ADM) [21], which indicate that these transcription factors have various context-specific roles for regulating plasticity at multiple levels. Perhaps more interesting, PRRX1 has two main isoforms, PRRX1A and PRRX1B, which promote MET and EMT, respectively [22]. This ability for these two isoforms to regulate both epithelial and mesenchymal states highlights the complex nature of the EMT–MET axis and showcases the plasticity inherent to the system. This has become especially evident in recent years, as various groups have begun to describe partial EMT intermediate or hybrid states. The epithelial state has been historically defined by E-CADHERIN (here E-CAD) and cytokeratins, and the mesenchymal state primarily through N-CADHERIN (herein N-CAD) and vimentin [23]. Therefore, EMT has classically been defined as loss of E-CAD and gain of N-CAD. However, the intermediate EMT states that have been described (i.e., partial loss of E-CAD or co-expression of both epithelial and mesenchymal markers within the same cell), the so-called hybrid epithelial and mesenchymal phenotypes, have broadened the definition of an EMT cell. Importantly, these hybrid cells can undergo further EMT to become more mesenchymal, or the reverse MET to re-establish their epithelial identity. Indeed, one recent publication posited that EMT is dispensable for metastasis [24].
Metastatic Organotropism: The Role of Exosomes and EMT Modulators
Metastatic organotropism may have first been described over a century ago by the English surgeon, Stephen Paget, when he elegantly described a non-random pattern of metastasis in over 700 women who died from breast cancer [25]. Paget went on to postulate that tumor cells are a seed that will only propagate when they fall on congenial soil. This provocative idea continued to perplex biologists for much of the last century, as some have argued that tumor cells have intrinsic properties that help it choose the secondary organ, while others have shown that the metastatic site itself provides an environment that is favorable for growth. This debate is particularly well suited for PDAC, as it is one of the most metastatic cancers, with upwards of 80% of pancreatic cancer patients presenting with metastases [26]. In PDAC patients, the two most common sites of metastases are the liver and the lung, and recent evidence has begun to elucidate mechanisms that may determine liver-versus lung-tropic programs. This may be of particular clinical importance for PDAC patients, as those with isolated pulmonary recurrence after pancreaticoduodenectomy have significantly increased overall survival compared with patients who have metastases to other sites (40.3 months versus 20.9 months, respectively) [27]. A separate study corroborated this finding in an independent cohort by demonstrating that PDAC patients with lung-only metastases had a median survival of 31.8 months, while those with liver-only metastases survived 9.1 months [28]. Therefore, we will delve into the underlying processes that have been proposed to control metastatic organotropism in PDAC.
Tumor exosomes, small membrane vesicles (30–100 nm) secreted by tumor cells, are packaged with a plethora of biological molecules including, but not limited to, DNA, RNA, miRNA, and protein (reviewed in [29]). Exosomes produced by the primary tumor are able to enter the circulation and have been shown to set up a pre-metastatic niche in secondary organs prior to tumor cell arrival and seeding. This pre-metastatic niche alters the local microenvironment to make it conducive for growth of the parental exosome-producing cell. Thus, to extend Paget’s analogy, exosomes are secreted by the seed and fundamentally change the soil, thus priming it for implantation of the seed. In PDAC, this has been shown elegantly by David Lyden’s group, where they established that tumor-derived exosomes are taken up by resident cells at the metastatic site to prepare the pre-metastatic niche [30]. Specifically, they demonstrated that the exosomal integrins α6β4 and α6β1 were associated with lung metastasis, while exosomal integrin αvβ5 with liver metastasis. Thus, their work has established that pre-metastatic niche formation by exosomal education is a potential mechanism of metastatic organotropism in PDAC. Furthermore, this work has led to the premise that identification of sub-populations of tumor-derived exosomes in circulation may help to predict eventual sites of metastases, and that increased monitoring to those sites may be beneficial. A related study by Achim Krüger’s group identified Tissue Inhibitor of Metalloproteinases-1 (TIMP-1) as a pre-metastatic niche modifier in the hepatic microenvironment and showed that patients with high plasma TIMP-1 have increased incidence of liver metastases [31]. It is not yet undetermined if TIMP-1 is a global pro-metastatic molecule (i.e., promotes all metastases, and not just liver-tropic metastases), as the authors did not discuss potential roles in other metastatic sites, outside of the liver. However, the proposed mode of action for promoting liver metastasis by TIMP-1 is that it activates hepatic stellate cells to prime the liver for metastases, and we speculate that this is a liver-tropic mechanism of metastasis and would not apply to the resident lung environment. Future studies might identify that other secreted factors are able to act on resident lung cells as potential lung-tropic mechanisms.
Another proposed mechanism of liver versus lung metastatic tropism is that internal mechanisms within the tumor cell give it unique characteristics that will determine the liver- or lung-tropic phenotype. Specifically, we have shown that genetic deletion of either P120CTN (encoded by Ctndd1) or E-CAD (encoded by Cdh1) in KPCY mice forces tumor cells to undergo EMT and dramatically increases metastatic load specifically to the lung [32]. This is in stark contrast to control KPCY mice, which primarily metastasize to the liver, at a much lower rate. We propose that tumor cells that metastasize to the liver require P120CTN-mediated re-stabilization of membranous E-CAD, in order to undergo MET and colonize the liver. Cells that lack P120CTN or E-CAD are unable to undergo MET and exclusively metastasize to the lung, which allows for metastatic colonization in cells that maintain a mesenchymal state (i.e., cells that have undergone EMT, but cannot revert to an epithelial state through MET). Taken in the context of the potential hybrid EMT cells discussed earlier, it is likely that, in an evolving tumor, sub-populations of cells exist that are at various stages of the EMT–MET axis, and that these cells may seed the lung if they are more mesenchymal and the liver if they are more epithelial.
We do not propose that these are the only mechanisms of metastatic organotropism, and likely that many systems are likely at play de novo in PDAC patients. It is tantalizing to speculate that the cell-intrinsic mechanisms that drive tropism (i.e., P120CTN or E-CAD protein loss in the primary tumor) could give rise to two different cell populations that interact with their pre-metastatic niche in unique ways. In this scenario, cells which have undergone EMT, and are therefore possessing mesenchymal characteristics, might have increased expression of lung-tropic exosomes with integrins α6β4 and α6β1. On the opposite side of the spectrum, cells that can undergo MET and thus possess epithelial characteristics might express exosomes with αvβ5 and metastasize to the liver. This may, in part, explain why some patients present with both liver and lung metastases, as their heterogenous primary tumor may be comprised of cells with both epithelial and mesenchymal characteristics, which simultaneously send both liver-tropic and lung-tropic exosomes to their secondary sites, preparing both for colonization. It is likely that these and many other mechanisms of metastatic organotropism are working simultaneous and that their interplay is what ultimately determines the site of metastasis.
Epigenetic and Post-Transcriptional Regulators of EMT and Metastasis
As previously mentioned, EMT has classically been described as a transcriptionally regulated process through EMT-transcription factors (EMT-TFs herein). However, recent data in PDAC have started to shift this paradigm by establishing that EMT can also be regulated through epigenetic and post-transcriptional mechanisms.
The primary means of regulating PDAC EMT at the epigenetic level have been through histone modifications, DNA methylation, and miRNA-mediated control of canonical EMT-TFs (reviewed in [33, 34]). Histone Deacetylase 1 and 2 (HDAC1 and HDAC2 herein) have been the most well-studied deacetylases that facilitate the epithelial plasticity observed PDAC, mostly in regard to their silencing of E-cadherin (CDH1) expression. This silencing is observed in highly metastatic PDAC cells and is mediated through a transcriptional repressor complex between SNAIL and HDAC1/2 that hones to the CDH1 regulatory elements [35]. A similar study showed that recruitment of the HDAC1/2 to the CDH1 promoter can also be accomplished by ZEB1/HDAC repressor complexes [36], indicating that this may be a more generalized mechanism of regulating epithelial identity in PDAC. In the absence of SNAIL or ZEB1, CDH1 remains acetylated and silenced, as HDACs are unable to be recruited to the promoter [35, 36]. Clinically, the class I HDAC inhibitor, mocetinostat, has the ability to reverse EMT by interfering with ZEB1 function [37]. This is not unique to HDAC inhibitors, as various small molecular inhibitors of epigenetic readers, writers, and erasers have shown promise in PDAC GEMMs previously discussed (reviewed in [38]). High expression of the histone methyltransferase enhancer of zeste homologue 2 (herein EZH2), in particular, has been shown to promote PDAC cell plasticity and to be a poor prognosis indicator in PDAC patients [39]. Genetic deletion or pharmacologic inhibition of EZH2: enhanced the anti-proliferative effect of gemcitabine, reversed EMT, and inhibited cellular migration in PDAC cells [40, 41]. Collectively, these studies have demonstrated that many different histone modifiers have the ability to modulate cellular plasticity in PDAC and targeting these molecules may be an Achilles heel in the EMT cascade. One of the most well-studied routes of miRNA-mediated epigenetic regulation of EMT in PDAC is through the miR-200 family. The p53-miR-200c axis has been studied in PDAC, where loss of p53 downregulates miR-200c, which alleviates normally represses the EMT program by degrading EMT-TF mRNAs [42, 43]. The broader miR-200 family appears to have similar roles in negatively regulating EMT [44,45,46], and overexpression of miR-200a or miR-200b in PDAC cells downregulated EMT-TFs [47], suggesting a conserved mechanism. All of these processes being described fundamentally alter the expression levels of critical EMT regulators, and either suppress or enhance EMT.
Ben Stanger’s group has recently provided some provocative work demonstrating that regulation of E-CAD protein level and localization is important for the degree of plasticity achieved during EMT. To that end, they established that two distinct EMT programs exist in PDAC: complete EMT (C-EMT) and partial EMT (P-EMT) [19, 54]. C-EMT was shown to be primarily driven through transcriptional repression of the epithelial program (i.e., downregulated expression of classical epithelial genes like Cdh1), while P-EMT maintained epithelial cell identity transcripts, but functionally altered the protein products of these genes. Specifically, P-EMT is mediated by re-localization of E-CAD from the membrane to the cytoplasm, causing cells to lose their epithelial cell qualities. Ultimately, both sub-types turn on mesenchymal gene programs during their respective EMTs, but repressed their epithelial cell identity through different mechanisms. Interestingly, both types of EMT cells were able to undergo MET, but differed in their invasive and metastatic qualities. P-EMT cells maintained their cell-to-cell contacts and invaded as clusters of cells, while C-EMT cells completely lost the ability to form cell junctions and invaded as single cells. This is important in vivo, as it has been known for nearly 40 years that circulating tumor cells (CTCs) that form clusters are more metastatic [48,49,50,51,52], and suggests that P-EMT cell clusters will have enhanced metastatic potential. Furthermore, this model of partial EMT fits well with the previously GEMM data in that P120CTN loss causes re-localization of E-CAD protein from the membrane to the cytoplasm, akin to the P-EMT [32]. Importantly, others have shown that PDAC patients who have cytoplasmic staining of P120CTN have significantly decreased survival relative to those with membranous P120CTN [53], which may mark a patient population undergoing partial EMT.
Concluding Remarks
We have discussed various means of regulating epithelial cell identity in PDAC and have introduced emerging paradigms that are re-shaping the broader EMT field. The publications reviewed herein have truly re-invigorated the field of EMT research in pancreatic cancer, and new insights will hopefully translate into new approaches for early detection, risk stratification, and therapy.
References
Siegel, R. L., Miller, K. D., & Jemal, A. (2018). Cancer statistics, 2018. CA: a Cancer Journal for Clinicians, 68(1), 7–30.
Ma, J., & Jemal, A. (2013). The rise and fall of cancer mortality in the USA: Why does pancreatic cancer not follow the trend? Future Oncology, 9(7), 917–919.
Rhim, A. D., et al. (2012). EMT and dissemination precede pancreatic tumor formation. Cell, 148(1-2), 349–361.
Hingorani, S. R., et al. (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell, 7(5), 469–483.
Biankin, A. V., et al. (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature, 491(7424), 399–405.
Bardeesy, N., et al. (2006). Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes & Development, 20(22), 3130–3146.
Ijichi, H., et al. (2006). Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes & Development, 20(22), 3147–3160.
Izeradjene, K., et al. (2007). Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell, 11(3), 229–243.
Olive, K. P., et al. (2009). Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science, 324(5933), 1457–1461.
Rhim, A. D., et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell, 25(6), 735–747.
Aguirre, A. J., et al. (2003). Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes & Development, 17(24), 3112–3126.
Bardeesy, N., et al. (2006). Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proceedings of the National Academy of Sciences of the United States of America, 103(15), 5947–5952.
Skoulidis, F., et al. (2010). Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell, 18(5), 499–509.
Shakya, R., et al. (2011). BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science, 334(6055), 525–528.
Rowley, M., et al. (2011). Inactivation of Brca2 promotes Trp53-associated but inhibits KrasG12D-dependent pancreatic cancer development in mice. Gastroenterology, 140(4), 1303–1313.e1-3.
Aiello, N. M., Rhim, A. D., & Stanger, B. Z. (2016). Orthotopic injection of pancreatic cancer cells. Cold Spring Harbor Protocols, 2016(1), pdb.prot078360.
Aiello, N. M., et al. (2016). Metastatic progression is associated with dynamic changes in the local microenvironment. Nature Communications, 7, 12819.
Nieto, M. A., et al. (2016). Emt: 2016. Cell, 166(1), 21–45.
Aiello, N. M., et al. (2018). EMT subtype influences epithelial plasticity and mode of cell migration. Developmental Cell, 45(6), 681–695.e4.
Ocana, O. H., et al. (2012). Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell, 22(6), 709–724.
Reichert, M., et al. (2013). The Prrx1 homeodomain transcription factor plays a central role in pancreatic regeneration and carcinogenesis. Genes & Development, 27(3), 288–300.
Takano, S., et al. (2016). Prrx1 isoform switching regulates pancreatic cancer invasion and metastatic colonization. Genes & Development, 30(2), 233–247.
Thiery, J. P., et al. (2009). Epithelial-mesenchymal transitions in development and disease. Cell, 139(5), 871–890.
Zheng, X., et al. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 527(7579), 525–530.
Paget, S. (1989). The distribution of secondary growths in cancer of the breast. 1889. Cancer and Metastasis Reviews, 8(2), 98–101.
Data, S. R. (2018). Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) Research Data (1973–2015). National Cancer Institute, DCCPS, Surveillance Research Program, released April 2018, based on the November 2017 submission.
Yamashita, K., et al. (2015). Survival impact of pulmonary metastasis as recurrence of pancreatic ductal adenocarcinoma. Digestive Surgery, 32(6), 464–471.
Decoster, C., et al. (2016). Heterogeneity of metastatic pancreatic adenocarcinoma: Lung metastasis show better prognosis than liver metastasis-a case control study. Oncotarget, 7(29), 45649–45655.
Azmi, A. S., Bao, B., & Sarkar, F. H. (2013). Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Reviews, 32(3-4), 623–642.
Hoshino, A., et al. (2015). Tumour exosome integrins determine organotropic metastasis. Nature, 527(7578), 329–335.
Grunwald, B., et al. (2016). Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via CD63 signaling to create a premetastatic niche in the liver. Gastroenterology, 151(5), 1011–1024 e7.
Reichert, M., et al. (2018). Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Developmental Cell, 45(6), 696–711.e8.
Skrypek, N., et al. (2017). Epithelial-to-mesenchymal transition: Epigenetic reprogramming driving cellular plasticity. Trends in Genetics, 33(12), 943–959.
Bedi, U., et al. (2014). Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget, 5(8), 2016–2029.
von Burstin, J., et al. (2009). E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology, 137(1), 361–71, 371 e1-5.
Aghdassi, A., et al. (2012). Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut, 61(3), 439–448.
Meidhof, S., et al. (2015). ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Molecular Medicine, 7(6), 831–847.
Hessmann, E., et al. (2017). Epigenetic treatment of pancreatic cancer: Is there a therapeutic perspective on the horizon? Gut, 66(1), 168–179.
Toll, A. D., et al. (2010). Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Human Pathology, 41(9), 1205–1209.
Ougolkov, A. V., Bilim, V. N., & Billadeau, D. D. (2008). Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clinical Cancer Research, 14(21), 6790–6796.
Avan, A., et al. (2012). Molecular mechanisms involved in the synergistic interaction of the EZH2 inhibitor 3-deazaneplanocin A with gemcitabine in pancreatic cancer cells. Molecular Cancer Therapeutics, 11(8), 1735–1746.
Singh, S. K., et al. (2015). Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. The EMBO Journal, 34(4), 517–530.
Ma, C., et al. (2015). MicroRNA-200c overexpression plays an inhibitory role in human pancreatic cancer stem cells by regulating epithelial-mesenchymal transition. Minerva Medica, 106(4), 193–202.
Bao, B., et al. (2011). Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Letters, 307(1), 26–36.
Wu, X., et al. (2016). MiR-200a suppresses the proliferation and metastasis in pancreatic ductal adenocarcinoma through downregulation of DEK gene. Translational Oncology, 9(1), 25–31.
Zhong, X., et al. (2016). Suppression of MicroRNA 200 family expression by oncogenic KRAS activation promotes cell survival and epithelial-mesenchymal transition in KRAS-driven cancer. Molecular and Cellular Biology, 36(21), 2742–2754.
Li, Y., et al. (2009). Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Research, 69(16), 6704–6712.
Fidler, I. J. (1973). The relationship of embolic homogeneity, number, size and viability to the incidence of experimental metastasis. European Journal of Cancer, 9(3), 223–227.
Liotta, L. A., Kleinerman, J., & Saidel, G. M. (1974). Quantitative relationships of intravascular tumor cells, tumor vessels, and pulmonary metastases following tumor implantation. Cancer Research, 34(5), 997–1004.
Thompson, S. C. (1974). The colony forming efficiency of single cells and cell aggregates from a spontaneous mouse mammary tumour using the lung colony assay. British Journal of Cancer, 30(4), 332–336.
Liotta, L. A., Saidel, M. G., & Kleinerman, J. (1976). The significance of hematogenous tumor cell clumps in the metastatic process. Cancer Research, 36(3), 889–894.
Lione, A., & Bosmann, H. B. (1978). Quantitative relationship between volume of tumour cell units and their intravascular survival. British Journal of Cancer, 37(2), 248–253.
Melo, S. A., et al. (2015). Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature, 523(7559), 177–182.
Aiello, N. M., et al. (2017). Upholding a role for EMT in pancreatic cancer metastasis. Nature, 547(7661), E7–E8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Pitarresi, J.R., Rustgi, A.K. (2019). Mechanisms Underlying Metastatic Pancreatic Cancer. In: Rhim, J., Dritschilo, A., Kremer, R. (eds) Human Cell Transformation. Advances in Experimental Medicine and Biology, vol 1164. Springer, Cham. https://doi.org/10.1007/978-3-030-22254-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-22254-3_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-22253-6
Online ISBN: 978-3-030-22254-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)