
Abstract
Hepatocellular carcinoma (HCC) is a fatal disease without effective treatment. In recent decades, numbers of molecular profiling studies have improved our understanding of critical oncogenic events driving hepatocarcinogenesis and identified potential molecular targets for drug development in HCC. However, these studies have also revealed the heterogeneous nature of this disease and underscore the impact of intertumoral and/or intratumoral heterogeneity of HCC on a successful treatment. In this chapter, we will summarize common HCC-associated molecular alterations, review data related to molecular heterogeneity, understand what drives the evolution of HCC heterogeneity and how this knowledge would lead us to generate new research directions, and improve the outcome of HCC patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hepatocellular carcinoma
- Molecular alteration
- Subtype
- Heterogeneity
- Intratumoral
- Intertumoral
- Target treatment
Introduction
Hepatocellular carcinoma (HCC) is the most common form of primary hepatic tumors and is the second leading cause of global cancer-related death, responsible for more than 745,000 deaths every year [1]. Late diagnosis, high postoperative recurrence rate, and lack of effective treatment for patients with advanced disease explain the poor outcomes for most HCC patients. Identifying effective treatment for HCC has been a major research focus for decades as evidenced by the inclusion of more than 1200 clinical trials testing different interventions in the ClinitalTrials.gov database [2]. However, a majority of the above-listed clinical trials failed, and only sorafenib, regorafenib, and lenvatinib succeeded to show survival benefit in HCC, which subsequently received regulatory approval [3,4,5]. However, these treatments only provide a marginal improvement of overall survival with palliative intent. Furthermore, regardless of the final results of clinical trial, treatment response between patients varied, indicating the heterogeneous characteristic of HCC.
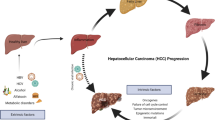
“Heterogeneity” means a state that consists of dissimilar or diverse elements [6]. Accordingly, heterogeneity of HCC means that patients given with an identical HCC diagnosis could be different. The concept describing HCC as a heterogeneous disease does not come out just in recent years; back in 1954, Edmondson introduced the well-known Edmondson-Steiner Grading system, which described that patients could be grouped by the grade of tumor differentiation, and tumors with worse differentiation were more likely to develop metastasis [7]. Using the same grading system, Kenmochi et al. demonstrated that 47.7% HCC cases had two or more areas within a tumor presented with different grade of differentiation [8]. The abovementioned studies reveal two different entities of heterogeneity; Edmondson’s work pointed out diversity among patients with the same histological type of tumor, termed as “intertumoral heterogeneity” (Fig. 14.1), while results from Kenmochi et al. demonstrated the other entity – “intratumoral heterogeneity” – referring to heterogeneity among cancer cells within a single patient.

Tumor heterogeneity of HCC. This cartoon summarized the concepts distinguishing intertumoral heterogeneity and intratumoral heterogeneity in HCC. Intertumoral heterogeneity denotes the variations among different HCC patients, and intratumoral heterogeneity refers to the differences among individual lesions and/or cells in a single patient
With the advent of molecular medicine, we know that the heterogeneity of HCC is not restricted in pathophenotypic differences but is linked to biological mechanisms driving tumor progression. In this chapter, we will start by establishing a general understanding of molecular heterogeneity linking to hepatocarcinogenesis. We will then explore the evidence of intertumoral and intratumoral molecular heterogeneity in HCC. Lastly, we will discuss the importance to integrate the knowledge of heterogeneity into tailor treatment for individual patients and potential challenges and opportunities ahead.
Molecular Alterations of HCC
Cancer is considered as a genetic disease, meaning that mutation(s) of genetic material lead to the initiation and progression of cancer [9]. According to the way by which a cancer cell obtains an alteration, genetic variances can be separated into two entities – germline mutation and somatic mutation. Germline mutation are defined as genetic variances that develop within the heritable genome and are transmitted from parent to offspring, whereas somatic mutations, which account for most genetic aberrations found in cancer, are acquired de novo by cancer cells [10]. Up to 90% of genetic variances in tumors are somatic mutations, which arise from replication errors or from DNA damage with incorrect repair [11]. Common factors triggering the development of somatic mutation in HCC could be divided into two categories: the exogenous mutagens include oxidative and hypoxia stress plus exposure of chemicals or radiation such as UV, while endogenous mutagens include defective DNA repair machinery or other factors related to genomic stability. Notably, among all the mutations identified, only a fraction of mutations, termed driver mutations, can confer a survival advantage to cancer cells, leading to preferential growth, survival, and metastasis.
In the following section, we will review data related to different types of molecular alterations and learn how they affect the biology of HCC.
Genomic Alteration
In HCC, several different kinds of genomic alterations have been reported and are summarized as follows: change of gene copy numbers, chromosomal rearrangement, mutation, and viral genome insertion [12].
Copy Number Variation
Copy number variation (CNV) refers to a DNA segment of one kilobase or larger with variable copy number (could be number gain, loss, or amplification) as compared to a reference genome [13]. The presence of CNV may change the physical arrangement of genes on chromosomes, leading to functional alterations of involved genes. Compared to healthy populations, CNVs are more frequently found in patients with cancer, and they are more likely to occur in regions containing cancer driver genes and/or tumor suppressor genes [14, 15]. In HCC, CNVs had been found to affect many important oncogenes, such as MYC, MET, MDM4, and YY1AP1, and tumor suppressor genes, like PTEN and RB1 [12, 15,16,17,18,19]. Totoki et al. used a bioinformatic algorism to assess CNVs and showed that recurrent focal amplification was more frequently observed than homozygous deletion, and a fraction of patients (28.9% in his cohort) presented with concurrent high CNVs and ploidy change, suggesting a high degree of structural changes in the whole genome [20].
Somatic Mutation
With the increasing use of next-generation sequencing, researchers are able to explore the human genome in more depth and advance our knowledge of cancer biology. There are two major platforms, whole-exome sequencing and whole-genome sequencing, being widely adapted for large-scale DNA sequencing studies today. Whole-exome sequencing captures and sequences the DNA fragments containing exonic regions, which enable the comprehensive detection of somatic alterations in the protein-coding regions and has led to the discovery of many novel genes implicated in carcinogenesis. However, whole-exome sequencing covers only about 1% of human genome and may miss important information in the noncoding regions. In contrast, whole-genome sequencing provides a full coverage of human genome, allowing identification of all genetic events, such as substitutions, structural rearrangement, and viral genome integrations, that may occur in coding and noncoding regions. Both methods have been widely adapted for HCC genomic studies, providing valuable information about the genetic mechanism of hepatocarcinogenesis.
Nucleotide Substitution Signature
There are six patterns of somatic base substitutions, namely, C > A/G > T, C > G/G > C, C > T/G > A, T > A/A > T, T > C/A > G, and T > G/A > C, which could occur and cause a point mutation of a gene. Notably, the choice of substitution is not made by random selection; several studies suggested that the patterns of substitutions could be indicative of a specific mutagenesis mechanism occurring in tumor cells. Totoki et al. reported the first whole sequencing study and showed that C > T/G > A and T > C/A > G substitutions are dominant in a HCV-related HCC patient [21]. Similar substitution patterns were shown in larger cohorts and in patients with hepatitis B virus (HBV)-related HCC [22, 23]. Interestingly, the substitution patterns change in patients with HCC related to nonviral etiologies; A > T/T > A transversions at [C|T]AG trinucleotide motifs were associated with aristolochic acid (a plant-derived carcinogen), and G > T/C > A was highly correlated with aflatoxin B1 exposure, which may lead to TP53 249S mutations [24,25,26], though Guichard et al. showed that G > T/C > A substitution was also enriched in well-differentiated tumors and tumors that developed on non-cirrhotic livers [27].
Significant Mutated Genes
Based on the original idea of tumor development, mutational activation of oncogene and loss-of-function mutation of tumor suppressor gene lead to cancer initiation and progression. Therefore, genes with higher degrees of mutation are more likely to be critical in cancer biology.
Genome-wide sequencing studies provided landscape views of genetic alterations and identified recurrent mutated genes in HCC [22,23,24, 27, 28]. Several genes, such as TERT promoter, TP53, and CTNNB, were commonly mutated in different cohort, suggesting that these genes may be functionally critical in HCC.
HBV Genome Integration
Chronic HBV infection is an important etiology associated with the development of HCC, particularly in China and other HBV endemic areas [29]. HBV is a DNA virus, and integration of a viral DNA into the host genome is one of the mechanisms by which HBV promotes hepatocarcinogenesis [30]. Around 85% of HBV-infected HCC patients exhibited evidence of HBV DNA integration in the host genome, and interestingly, the occurrence of integration is more enriched in the tumor part than in the adjacent normal liver [24, 30]. The HBV integration breakpoints can be found across the whole genome, and approximately 50% of them occur within several particular genes, such as TERT and MLL4 [22,23,24, 30]. Insertion of HBV DNA results in change of gene expression (mostly upregulation) and may alter chromosomal stability and trigger CNVs [22, 30].
Epigenomic Alteration
Epigenetic alterations refer to the molecular changes, such as DNA methylation, chromatin remodeling, and noncoding RNAs, that affect gene function independent of changing the DNA sequence of a gene [31]. Epigenetic changes are highly prevalent in many different types of cancer, including HCC. Several studies had addressed the importance of epigenetic regulation in affecting hepatocarcinogenesis. Genome-wide DNA methylation profiles showed that a tumor exerted a significant increment in both hypo- and hypermethylation in comparison to a paired non-tumor, and tumor-specific hypermethylation determined the expressions of CDKN2A, HHIP, PTGR1, TMEM106A, MT1M, MT1E, and CPS1 [24]. On the other hand, emerging evidence showed that dysregulation of microRNAs, a class of short, noncoding RNA, contributed to activation of oncogenic signaling in HCC; for instance, Meng et al. showed that overexpression of miR-21 inhibits the expression of the phosphatase and tensin homolog (PTEN) tumor suppressor [32], and Coulourn et al. described that loss of miR-122 expression in liver tumor significantly enhances metastatic properties of cancer cell through upregulation of a network involving VEGF, HIF1A, RAC1, RHOA, and vimentin [33]. Lastly, studies also showed that genes related to chromatin modification were frequently altered in HCC [20, 24].
Key Driving Genes and Pathways in HCC
In recent decades, comprehensive studies on liver cancer genome had identified many recurrently mutated genes in HCC and improve our understanding of hepatocarcinogenesis. By exploring aggregation of altered genes, important oncogenic pathways in HCC were recognized, which provided a more functional understanding regarding the development and progression of HCC. Here, we will introduce the most frequently altered pathway in HCC.
TP53 Pathway
Somatic mutations in the TP53 gene are the most frequently altered events in human cancer [34]. In HCC, mutations in TP53, mostly inactivation mutation, could be found in 18–37% of patients [20, 24, 30]. Notably, some patients exert a mutation-independent p53 inactivation mechanism [35]; 23% of patients in the TCGA cohort exhibited downregulation of p53 target genes (surrogate for p53 inactivation) without detectable TP53 mutations [24]. Furthermore, tumors with low p53 activity inferred by the p53 target signature were associated with increased copy number instability, higher pathological grade, reduced expression of mature hepatocyte signature, and increased risk of tumor recurrence [24]. On the other hand, alterations of several other genes within the TP53 network, namely, IRF2, MDM2/MDM4, ATM, RPS6KA3, CDKN2A, RB1, CDK4, CCND1, and CCNE, were also identified, resulting in a high prevalence (up to 72%) of TP53 signaling alterations in HCC [20, 24].
Wnt/β-Catenin Pathway
Aberrant activation of Wnt signaling is a critical molecular event driving hepatocarcinogenesis. There are several different genes involving this signaling pathway. Somatically acquired missense mutation in exon 3 of the CTNNB1 (β-catenin) gene, which leads to constant activation of β-catenin by preventing phosphorylation of β-catenin, is the most common molecular change related to this signaling pathway (frequency ranging from 10% to 32.8% in genome-wide sequencing studies) [12, 30]. Other alterations, such as epigenetic inactivation of SFRPs and SOX, inactivating mutation of AXIN1 or APC, and upregulation of FGF19, MYC, and CCND1, were also reported in HCC cohorts [20, 24]. Collectively, alterations of the Wnt-associated signaling pathway are observed in 44–66% of patients with HCC.
TERT Pathway
To obtain the ability of infinite replication, activation of telomerase (encoded by TERT gene) is required for cancer cells [12]. Tokoki et al. reported that 54% of HCC patients in their cohort had somatic mutation of TERT gene at its promoter region, and the percentage of TERT mutation was higher in HCV-positive cases (64%) in comparison to nonviral (59%) and HBV-positive cases (37%) [20]. Similar results were shown in the TCGA cohort; TERT mutation was found in 44% of patients and enriched in HCV-related HCC patients [24]. Additionally, the occurrence of TERT promoter mutation was frequently found with CDKN2A silencing, which further enhanced the expression of TERT through downregulation of p16INK4A [24, 36].
Chromatin Remodeling Pathway
Chromatin refers to the DNA-protein complex within the nucleus that helps to package DNA into a more compact and denser structure [37]. DNA interacts with histone protein via covalent bonding and forms a nucleosome, the basic structure of chromatin. Thus, modification of histone protein, including acetylation or methylation, would affect the DNA-histone structure (e.g., open or closed), leading to change of gene expression [12].
In HCC, alterations of ARID1A, ARID1B, and ARID2 are frequently observed in HCC patients [20, 24]. The ARID family genes encode the core proteins of a nucleosome remodeling complex, SWI/SNF (switch/sucrose non-fermentable); alterations of these genes, such as frameshift mutations, copy number loss, and homozygous deletion, lead to dysregulation of chromatin [38]. In addition to ARID family, mutations of BAP1, KMT2D CREBBP were reported in HCC patients. In sum, alterations of chromatin modifier genes could be identified in nearly 50% of HCC patients.
PI3K-mTOR Pathway
The phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling pathway is critical to cell growth and angiogenesis; MET, FGFR, VEGFA, and several other growth factors can activate PI3K/AKT and mTOR signaling, while PTEN counteracts with activation of this signaling pathway [39]. Aberrant activation of PI3K-related pathway is a common driving mechanism in many types of cancers. In HCC, oncogenic activation of PI3K- mTOR signaling affects about 45% of patients, and inactivating mutation of TSC1-TSC2 is the most common contributing factor [20].
Nrf2-KEAP1 Pathway
Nuclear factor-like 2 (encoded by NFE2L2, also known as NRF2) is a transcriptional factor that regulates many genes associated with antioxidation and metabolism [40]. NRF2 is regulated by KEAP1 via the ubiquitin-proteasome pathway; activation of the missense mutation of NFE2L2 and inactivation of the mutation of the KEAP1 gene are recurrently seen in HCC [12].
Metabolic Alteration and Metabolomic Investigation
Metabolic reprogramming, which describes serial changes involving nutrition uptake and utilization, can fuel cancer cell growth and proliferation [41]. In HCC, accelerated glucose uptake and preferential activation of pyruvate kinase muscle isozyme M2 (PKM2)-mediated glycolysis had been shown to increase the proliferation and progression of liver cancer cell [42]. Also, dysregulated glutamine metabolism and increased de novo lipogenesis were also found to promote the development of therapeutic resistance and cell survival in HCC [43, 44].
Metabolomics is the global and unbiased survey of the complement of small molecules (<1 kDa) in a biological sample (could be biofluid, tissue, organ, or organism) and measures the end products of various metabolic processes happening in cells [45]. Several studies had applied this method and successfully identified specific metabolic profiles that help early diagnosis and clinical outcome prediction in HCC [46,47,48].
Intertumoral Heterogeneity in HCC
Intertumoral heterogeneity refers to the diversity between different HCC patients. Many different factors contributing to the generation of intertumoral heterogeneity in HCC have been identified and could be summarized into three major categories: environmental exposures, individual genetic predispositions, and somatic molecular alterations [26, 29, 46, 47, 49, 50].
Environmental Exposure
The development of HCC is known to be associated with chronic liver inflammation induced by various different exposures, such as hepatitis viruses, alcohol, smoking, and aflatoxin exposure [29]. Therefore, recognizing different environmental exposures is the first step in characterizing the heterogeneity of HCC among different patients.
Nearly 80% of HCC is contributed by hepatitis B virus (HBV) or hepatitis C virus (HCV) infections [51]. Though liver tumors related to chronic HBV and HCV infection are histologically similar, each virus has very distinct mechanisms driving hepatocarcinogenesis. Several large-scale genomic studies showed that liver tumors associated with different viruses had distinct mutation patterns [23, 24, 28]. HBV is a DNA virus meaning that a viral genome would integrate into the host genome and may, therefore, affect the integrity of the host genome and alter gene expression near the integration site [52]. HBV-infected tumors were characterized by increasing frequency of TP53 mutation and hyperexpression of CCND1, CCNE1, GLI2, TERT, and MLL4, which seem to be associated with viral DNA integration into the host genome [23, 24, 30]. Distinct from HBV, HCV is an RNA virus and does not integrate into the host genome and replicates within the hepatocyte cytoplasm. HCV-related HCCs were found to enrich inactivating mutation of ARID2 gene [53], silencing of CDKN2A promoter, and TERT promoter mutation [24].
Other potential environmental factors were also found to leave a distinct “molecular fingerprint” in HCC, for example, alcohol-related tumors were significantly enriched with inactive mutation of ARID1A and enriched with alterations of CTNNB1, TERT, CDKN2A, SMARCA2, and HFG [27, 28], and aflatoxin exposure was significantly related to TP53 R249S mutation [28, 54].
It is not uncommon that patients may be exposed to more than one risk factors related to HCC, and different environmental exposures often show a synergized effect in promoting the progression of hepatocarcinogenesis. For example, alcohol use doubles the risk of HCV-related HCC [55], and smoking increases the risk of alcoholism-related HCC [56]. Interestingly, evidence of synergistic interaction between different environmental factors could be found in molecular studies; for instance, HCC tumors related to aflatoxin B1 exposure and HBV infection shared similar genetic characteristics, recurrent TP53-R249S mutation, and high AFB1 signature [24], which correlated with clinical observations [57].
Individual Genetic Predispositions
Individual genetic predisposition (also called genetic susceptibility) reflects the collective effect of germline mutation(s); it influences the individual risk or tendency to develop disease and contributes to the heterogeneous biology of HCC [50, 58]. According to a number of genetic alterations involved, genetic predisposition could be further subclassified as “monogenic” and “polygenic.”
Monogenic Germline Variance
Several monogenic predispositions related to increasing risk of HCC have been reported, and alpha-1 antitrypsin (AAT) deficiency [59], hereditary tyrosinemia type 1 [60], hemochromatosis [61], and porphyrias [62] are the most well-known.
AAT deficiency is caused by mutation of SERPINA1 gene, resulting in altered protease/antiprotease balance, and associated with an increased risk of HCC, particularly in male patients [59, 63]. Hereditary tyrosinemia type 1 is caused by mutation of FAH gene, which leads to accumulation of tyrosine catabolic intermediates in the liver due to defective tyrosine metabolism, and, subsequently, results in liver inflammation and HCC development. Hemochromatosis is an iron metabolism disorder related to the mutation of HFE gene; patients harboring C282Y mutation of HFE gene have excessive gastrointestinal iron absorption and storage in the liver and many other organs. The risk of HCC in hemochromatosis patient is approximately 20-fold higher compared to the general population [61, 64]. Hepatic porphyrias are a group of diseases associated with abnormal heme biosynthesis; reduced free heme pool may increase the reactive oxygen species stress and mutation burden in patients with porphyria and consequently increases the risk of HCC [65]. Besides porphyrias, all abovementioned syndromes are inherited in an autosomal recessive fashion.
It is worth noting that HCC that developed in patients with germline mutation which we discussed here may have a different clinical presentation than those without, and their liver tumors tend to occur earlier. For example, 40% of patients with tyrosinemia type 1 develop HCC in their childhood [60]. Patients with AAT deficiency can develop pulmonary emphysema [59]. Some porphyria patients have neuropsychiatric symptoms [50]. On the other hand, we should also keep in mind that for most of the monogenetic syndromes, harboring genetic changes is not sufficient to drive the formation of HCC, suggesting a role of other modulating factors (environmental or genetic) [50].
Polygenic Risk Factors
Several conditions or diseases inherited as polygenic traits are associated with a higher risk of HCC. For example, patients with type 2 diabetes mellitus have a 2.5-fold increase of HCC risk [66], and patients with nonalcoholic steatohepatitis (NASH)-related cirrhosis have an annual 2.4–12.8% HCC incidence rate [67]. Other conditions, such as hypothyroidism, autoimmune hepatitis, and positive family history are also regarded as polygenic risk factors for HCC [50]. Apart from monogenetic risk factor, the incremental risk of HCC in patients with polygenetic predisposition is mediated by the combination of many genetic variations. Therefore, it is not surprised to see that these polygenetic conditions connect to hepatocarcinogenesis via multiple mechanisms. For instance, the development of HCC in patients with NASH may be attributed to obesity, insulin resistance, lipotoxicity, dysregulation of intestinal microflora, and genetic polymorphism [67]. Similar to monogenetic risk factor, polygenetic factors may interact and synergize with environmental factors in promoting hepatocarcinogenesis. For example, the risk of virus-related HCC is higher in patients comorbid with diabetes [68] and hypothyroidism [69].
Somatic Molecular Alterations
Somatic alterations account for 90% of molecular alterations occurring in the tumor genome. Taking the different biological function of each molecule into account, a vast amount of molecular alterations are the major contributors to tumor heterogeneity.
Characterizing Molecular Heterogeneity in HCC: A Rapidly Evolving Journey
Multiple molecular alterations and critical oncogenic signaling pathways had been identified in HCC tumors (as summarized in section “Molecular Alterations of HCC”). Each molecular feature, i.e., high vs. low CNVs or TP53 mutant vs. wild type, explains partly the trajectory of a tumor and could be taken as a reference to define heterogeneity. For example, Katoh et al. used the pattern of CNV to identify biologically distinct clusters among 87 HCC patients and showed that patients in the cluster with more dominant CNV features had worse survival [17]. Notably, different from some of the malignant diseases with strong molecular-alteration-driven phenotype, such as adenocarcinoma of the lung and epithelial growth factor receptor mutation [70] or estrogen-receptor-positive breast cancer [71], there is no dominant oncogenic molecular feature being recognized in HCC, meaning that it is difficult to define a homogenous patient or tumor clusters by single or few molecular features.
With the advent of bioinformatics and computational power, researchers are able to process and analyze a vast amount of data at the same time. Several studies showed that combining multiple “omics” data, namely, genomics, transcriptome, epigenome, and metabolome, could provide a better molecular classification in HCC and greatly enhanced our understanding of this complex disease [15, 20, 24, 72,73,74,75,76,77]. However, different omics platforms use very distinct methods to analyze different biological aspects of a subject, resulting in a huge inherited heterogeneity among the data and difficulty in combining them for analysis. To solve this problem, many studies chose to use a platform to produce a stable signature for clustering and match data generated from the other platforms to this clustering. Transcriptome profiling is widely adapted to identify stable molecular subtypes linking to tumor biology and clinical outcome in HCC [72, 74]. Evidence applying tumor transcriptome to define HCC subtypes linked to metastasis status and patient survival was first obtained by Ye et al. [78]. Lee et al. subsequently applied an unsupervised approach to define HCC molecular subtypes [79]. Boyault et al. defined six clusters among 65 samples according to their transcriptomic data and showed that these six clusters (G1–G6) link to distinct genotype and phenotype [72]. G1 tumors were typified by low copy number of HBV, AXIN1 mutation, younger age, higher serum level of AFP, and frequent origin from Africa. G2 tumors were associated with high HBV burden and mutations of PI3KCA and TP53. Similar to G2, G3 tumors were also associated with TP53 mutation but could be differentiated by lacking HBV infection and overexpression of cell cycle regulatory genes, such as CDC6, MAD2L1, CCNA2, and CCNE2. G4 was considered as a heterogeneous group that was comprised of both tumor and non-tumor sample, and a subgroup with TCF1 mutation was identified in G4. For the rest of the two clusters, G5 and G6 were both associated with activation of Wnt/β-catenin, but tumors of the G6 cluster presented with a higher degree of β-catenin activation and more satellite nodules around the main tumors based on pathologic analysis. Chaisaingmongkol et al. analyzed transcriptome data obtained from 62 Thai HCC patients by consensus clustering method and identified three different clusters (C1–C3) linking to unique genomic and metabolomic features and patients’ clinical outcomes [74]. More interestingly, in this study, the authors showed that the C1 and C2 signatures in HCC were shared with a subgroup of patients with cholangiocarcinoma in an ancestry-dependent manner (will be discussed in more detail in the following section).
A transcriptome meta-analysis that involves 603 HCC tumors identified three molecular subtypes (S1–S3) commonly observed across geographic regions and patient races/ethnicities [73]. S1 tumors are characterized with activation of transforming growth factor (TGF)-beta and Wnt pathways and associated with a more disseminative phenotype. S2 tumors are characterized by positivity of stemness markers such as AFP and EPCAM. S1 and S2 tumors collectively represent more aggressive tumors accompanied with more frequent TP53 mutations. S3 tumors are more differentiated and less aggressive compared to S1/S2 tumors (indolent subtype), in which CTNNB1-mutated tumors are accumulated. Of note, histological variants and clinical variables are associated with molecular subtypes [80]. Steatohepatitic variant and immune cell infiltrates are more frequently observed in S1 tumors. S2 tumors are associated with a macrotrabecular/compact pattern, clear cell variant, and high serum AFP. S3 tumors are associated with microtrabecular and pseudoglandular patterns. Similar correlations were confirmed in subsequent studies [81, 82].
On the other hand, several studies demonstrated the feasibility to interrogate different omics data to identify molecular subtypes [24, 75, 76]. Woo et al. combined DNA copy number and DNA methylation pattern for molecular classification and identified three subtypes in HCC (C1–C3); C1 tumors were typified by the highest frequency of CNV and methylation; recurrent BAP1 mutation; higher expression of CA9, KRT19, EPCAM, and PROM; and worse clinical survival [75]. Another good example of multi-omics study in HCC is the TCGA cohort, which comprehensively studied hundreds of HCC patients using six different platforms, namely, exome sequencing, DNA copy number, mRNA sequencing, microRNA sequencing, methylomics, and proteomics [83]. In the TCGA cohort, three subtypes (iC1–iC3) were identified based on the results of copy number and methylation change of DNA, expressions of mRNA and miRNA, and protein array. Interestingly, this molecular classification not only differentiates the molecular features of tumors but also showed a strong link with important clinical features of patients. For example, iC1 tumors, characterized by low frequency of CTNNB1 mutation, TERT promoter mutation, and DNA-methylation-mediated DKN2A silencing, were clinically linked to younger age, Asian ethnicity, female gender, and normal body weight, and iC2 tumors tended to have low-grade differentiation and less microvascular invasion. Collectively, molecular heterogeneity in HCC has been demonstrated in several studies and in various cohorts. Comprehensive molecular profiling enhances our understanding of the oncogenic events relevant to the development and progression of HCC.
Molecular Similarity of HCC and Intrahepatic Cholangiocarcinoma
Most of our current knowledge of defining specific cancer types are primarily based on pathological findings; we ask where the tumor is found, what it looks like, and if it presents with specific markers. HCC and intrahepatic cholangiocarcinoma (iCCA), the two major types of primary liver cancer, were considered as two distinct diseases in terms of tumor origin, morphology, and clinical behavior [83,84,85]. Surprisingly, with the availability of large-scale genomic studies, common molecular features were found in a subset of patients with HCC or iCCA.
In the TCGA cohort, there were four HCC patients who presented with positive IDH1/2 mutations, a genetic alteration being more frequently seen in iCCA rather than HCC [24]. Besides confirming the histopathological presentations of these four tumors as pure HCC, the authors further showed that the IDH1/2 mutation linked to a unique transcriptome and miRNA signature, an aggressive tumor behavior, and worse clinical course, suggesting a novel subclass in HCC being identified. On the other hand, the work done by Chaisaingmongkol et al. also identified similar molecular features in these two diseases [74]. They used global transcriptome expression to define molecular classes in HCC and iCC and identified three subtypes of HCC and four subtypes of ICC. Intriguingly, they compared subtypes of HCC and iCCA and found that the C1 and C2 of HCC were biologically similar with that of iCCA. The C1 subtype was enriched with PLK1 and ECT2 mutation and associated with worse clinical outcomes, where the C2 subtype presented with link to obesity, T cell infiltration, bile acid metabolism, and better outcomes. They further validated the presence of the C1/2 signature in three different HCC cohorts and two iCCA cohorts and found that these two signatures could be identified in Asian, not Caucasian, HCC/iCCA patients.
The commonality of HCC and iCCA identified by the abovementioned studies highlights the value of large-scale genomic analysis in fully addressing cancer heterogeneity and associated distinct tumor biology in different patients.
Intratumoral Heterogeneity in HCC
Evolution of Intratumoral Heterogeneity
Intratumoral heterogeneity, referring to the variations among different tumor lesions and/or tumor cells within a single patient, is largely driven by genomic instability [49]. Unstable genome in tumor cells leads to the occurrence of a wide range of mutations and, as such, fosters genetic diversity and the generation of genetically different cancer cell clones. As a tumor expands or develops metastasis, these clones would compete for survival, leading to clonal evolution of a given lesion or host. Therefore, the architecture of tumor lesion is determined by a framework composed of clonal evolution, competition, and best-fit selection. Additionally, it is worthy of note that intratumoral heterogeneity is not only limited to the uneven distribution of tumor clones across various lesions but also includes the dynamic changes of a given lesion (also termed as temporal heterogeneity) [49, 86].
Spectrum of Intratumoral Heterogeneity
Heterogeneity of Different Tumor Lesions
As we mentioned at the beginning, intratumoral heterogeneity in HCC was first characterized by Dr. Kojiro’s group who found that 47.7% of patients harbor two or more subpopulations within one tumor [8]. Later, Dr. Weber’s group analyzed 120 tumor areas obtained from 23 patients and found that 87% of patients had evidence of intratumoral heterogeneity defined by morphological presentation, immunohistochemical staining of a collection of liver cell markers, and mutation status of TP53 and CTNNB1 [87]. Notably, the percentage of detectable genetic variations among different lesions increased in patients with a larger tumor or advanced disease stage, suggesting that intratumoral heterogeneity evolves during tumor progression. In addition, 26% of patients in this study with morphologically distinct tumors showed no differences on the protein level and the mutation status of TP53 and CTNNB1, suggesting the limitation of given methodology in fully addressing heterogeneity.
Using whole-genome sequencing to analyze 43 tumor lesions from 10 HCC patients, Xue et al. showed that all the patients in this cohort presented with evidence of intratumoral heterogeneity [88]. Importantly, the extent of intratumoral heterogeneity varied among different patients. By interrogating the features of somatic mutation, hepatitis B integrations, and copy number variations, the authors supported the branched evolution of different HCC clones. Compared to primary tumors, mutation patterns of intrahepatic metastasis or tumor thrombi were more distinct than that of satellite nodules.
Collectively, the abovementioned studies showed the presence of intratumor heterogeneity linking to distinct biology in HCC and suggested that analyzing a single lesion may be underrepresented.
Heterogeneity at Single-Cell Level
As mentioned at the beginning of this section, a single cancer cell can give rise to a distinct subpopulation (also termed clone). With tumor progression, cells within one clone may develop new mutations, leading to formation of different subclone(s). Therefore, within one tumor lesion, a cell per se may be different from each other, making single-cell study a must to fully address the intratumoral heterogeneity of a tumor.
Hou et al. were the first to report heterogeneity at the single-cell level in HCC; they used a single-cell triple omics sequencing technique, which simultaneously analyze genome, DNA methylome, and transcriptome of a single cell, to analyze 25 single cells derived from an HCC patient [89]. Importantly, even in such a few number of cells being studied, they identified two subgroups of cells with distinct molecular features. The other work presented by Zheng et al. focused on exploring the heterogeneity of a specific cell population – cancer stem cell (CSC) – by analyzing 2595 CSCs obtained from one HCC patient or enriched from HuH1 and HuH7 cells [90]. Using single-cell RNA sequencing to characterize transcriptome features or flow cytometry to determine the intensities of stem cell markers expressed on cell surfaces, they showed a huge heterogeneity among these cells. Importantly, the transcriptome signatures obtained from different subpopulations were associated with the outcome of patients in different HCC cohorts. The other thing worthy of note was that if the authors mixed more than 100 tumor cells and conducted genomic analysis of this cell mixture (simulation of bulk sample), the variations identified at the single-cell level could not be recaptured, suggesting the data we obtained from bulky tumor samples might be biased in a certain degree.
Stem Cell Feature and Heterogeneity in HCC
CSC, also termed as tumor-initiating cell, refers to a subset of cancer cells with self-renewal and differentiation capabilities. Notably, the presence of CSCs in tumor is not only a demonstration of intratumor heterogeneity (a subset of cells that are functionally distinct from the rest), but it also promotes repopulation of tumor cells, resulting in greater intratumoral heterogeneity [91].
In HCC, the presence of CSC can be phenotypically identified by specific markers, such as CD133, EpCAM, and CD44, or functionally defined by the capability of tumor initiation and asymmetric differentiation [92]. Ma et al. isolated CD133-high and CD133-low expressed cells from two human HCC cell lines and showed that CD133+ HCC cells confer higher proliferation and chemoresistance [93]. In concordance, Zheng et al. showed that a specific subgroup of cells positive for CD133 and CD44 was identified in a 2-AAF-induced rat liver cancer model, and this specific CD133+CD44+ cell clone could expand and differentiate into bi-lineage cell types [94]. The ability of bi-lineage differentiation indicates that CSC can initiate branching evolution, denoting the emergence and divergent propagation of multiple subclonal tumor cell populations from a common ancestor [49]. Compared with linear evolution (sequential genetic alterations and survival-of-the-fitness selection convey a linear model of clonal evolution), branching evolution is more likely to create a more heterogeneous tumor, which had been evidenced in HCC and many other types of cancers [49, 88, 95].
On the other hand, a growing body of literature suggest that the stem cell signature is an important feature that defines intertumoral heterogeneity in HCC. Yamashita et al. showed that HCC patients could be subclassified into four groups according to the expression levels of two hepatic stem cell-associated marker, epithelial cell adhesion molecule (EpCAM) and α-fetoprotein (AFP); patients with hepatic stem cell-like HCCs (EpCAM+, AFP+) and hepatocytic progenitor-like HCCs (EpCAM−, AFP+) had worse survival in comparison to others [96]. More importantly, tumors with the hepatic stem cell-like signature were characterized by unique molecular features shown at global transcriptome, miRNA, and metabolomic levels [48, 97].
Collectively, CSCs affect clonal architecture in a tumor, permitting the development and progression of tumor heterogeneity in HCC. Tracing the life path of CSCs provides an important scope to identify critical mechanisms driving tumor progression in HCC.
Immune Heterogeneity in HCC
“No man is an island,” as stated in the famous work by John Donne; cancer cells are within a complex community comprised by various immune and stromal cells. Tumor cells interact with and are being affected by their surrounding cells, suggesting that the tumor-associated microenvironment could be an important driver of tumor heterogeneity [98]. A major portion of the tumor-associated microenvironment is immune cells, which interact frequently with tumor cells. The cross talk between tumor and immune cells is complicated; the immune system had been implicated in preventing and promoting tumor growth, and on the other hand, tumor cells were shown to be able to shape the immune contexture and escape immune attack through tumor progression [98]. Notably, the effects of the immune system on a tumor are heterogeneous both among patients and lesions.
In the TCGA cohort, about 22% of HCC patients displayed high or moderate levels of lymphocyte infiltration, and about one-third of patients exhibited high expression of immune markers, such as CTLA-4 and other immune checkpoints [24]. Additionally, the variations of immune contexture were not only observed between different patients, but evidence of a distinct tumor immune microenvironment coexisting within a single individual had been shown in ovarian cancer [99].
Taken together, tumor-associated immune microenvironment is highly heterogeneous and tightly connects with the development and progression of a tumor. Addressing the discrepancies of the immune system is needed to fully acknowledge the complex tumor ecosystem in HCC.
Adapting Tumor Heterogeneity to Personalized Treatment in HCC
Hepatocarcinogenesis is a highly complex multistep process driven by host genome alterations and chronic liver inflammation. Therefore, the most common feature of HCC patients may be “heterogeneous.” Recognizing genetic features/classes to identify a critical target for drug development, to help in outcome prediction, and to guide personalized medicine had shown to be a successful strategy in breast cancer, lung cancer, and many other malignant diseases [70, 71, 100]. With the accumulation of large-scale genomic studies in the recent decade, our understanding of HCC genome evolution, potential druggable molecular alterations, and the dynamic interaction between HCC cells and microenvironment has increased significantly. However, the improvement of knowledge has not yet been able to change the paradigm of HCC treatment like what had been shown in other malignant diseases.
Several reasons may be responsible for this disappointing situation. First, molecular heterogeneity among HCC patients was insufficiently acknowledged in the commonly adapted clinical trial design. Till present, most of the published clinical trials in HCC are still based on the “all-comer” design to test their compound of interest, including molecular target agents. The unmeasured patient heterogeneity could be a significant confounder to the results of clinical trials. A good example is the development journey of ramucirumab, a monoclonal antibody against vascular endothelial growth factor receptor 2. In its first phase III trial (REACH trial), ramucirumab was tested as a second-line treatment for HCC in a nonselected patient cohort, and the results failed to demonstrate any survival benefit in patients treated with ramucirumab in comparison to placebo [101]. But the researchers found that the response rates of patients with high AFP levels were significantly higher in the post hoc analysis of REACH trial, leading to a follow-up trial that focused only on patients with high serum AFP level. Encouragingly, researchers successfully showed a 55% of recurrence risk reduction in this subgroup of patients, which was originally diluted in a mixed patient papulation [102]. Currently, there are some ongoing studies being designed in a biomarker enrichment base, such as LY2157299 in patients with high AFP (NCI01246989) and JNJ-42756293 in patients with fibroblast growth factor 19 amplification. Additionally, there are two innovated models of clinical trial designs, the basket study design and the umbrella study design, being proposed to help researchers address intertumoral heterogeneity among patients and to improve the efficiency in testing potential druggable molecular alterations in cancer patients. The “basket trial” is designed to test the effect of a single intervention on a specific molecular mutation/features regardless of cancer types, while the “umbrella trial” is designed to test the potencies of different drugs on different mutations under the “same disease umbrella” [103, 104]. Several large ongoing clinical trials, such as the NCI-MATCH study, adapted these novel study structures to design their protocols, and some of them had already released good results [105, 106].
Second, intratumoral heterogeneity, particularly the treatment-induced dynamic clonal changes, was shown to be a critical reason for primary resistance and relapse to target therapy in many types of cancers [49, 107], but this issue has not been addressed in current HCC clinical trials. Part of the reasons could be the difficulty of obtaining adequate tissues for molecular profiling, particularly obtaining samples of multiple lesions from a patient or sequential sampling. Alternatively, the use of less invasive tests, such as circulating tumor cells, circulating DNA, or multiparametric magnetic resonance imaging, may be more feasible to be adapted into clinical trial or even routine clinical practice and help researchers to capture the diversity across different tumor lesions and/or the dynamic changes along with molecular interventions and disease progression [108, 109].
Lastly, similar to drug sensitivity, drug tolerability is heterogeneous among patients. With a high incidence of liver cirrhosis, the individual variations of drug metabolism could magnify the differences of individual drug tolerability. So far, how to predict the occurrence and/or the severity of treatment-associated adverse effects has not yet been fully addressed in HCC. In patients with renal cell carcinoma receiving sorafenib treatment, Qin et al. showed that a polymorphism of VEGF, VEGF rs2010963 CG + GG genotype, was significantly linked to a higher risk of hand-foot syndrome, a common side effect associated with sorafenib [110]. Whether this or other genomic variations may be associated with the safety profile of sorafenib in HCC remained unclear, and more studies are warranted in the future.
Conclusion
HCC is a disease of great genetic diversity. With numerous tools available today, hundreds to thousands of molecular alterations could be detected within a liver tumor, but how to capture the story leading to tumor development and progression remains to be a big challenge. Recognizing molecular heterogeneity does not aim to find differences that separate patients but, by contrast, to identify critical features to unite patients into a relatively homogeneous subgroup. The diagnosis of HCC in this “omics era” should not be restricted to image-based criteria. Interrogating molecular profiling in different patients and, even, at various regions of a given tumor is the foundation of precision medicine. Full recognition of tumor heterogeneity is required not only to improve diagnosis and outcome prediction but also, more importantly, to allow researcher and clinician to design more effective anticancer therapies to every HCC patient.
References
Wong MCS, Jiang JY, Goggins WB, Liang M, Fang Y, Fung FDH, et al. International incidence and mortality trends of liver cancer: a global profile. Sci Rep. 2017;7:45846. https://doi.org/10.1038/srep45846. https://www.nature.com/articles/srep45846#supplementary-information.
ClinicalTrials.gov. Interventional studies | hepatocellular carcinoma. Bethesda (MD): National Library of Medicine (US); 2018. https://www.clinicaltrials.gov/ct2/results?cond=Hepatocellular+Carcinoma&age_v=&gndr=&type=Intr&rslt=&Search=Apply. Accessed June 14 2018.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–90.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. https://doi.org/10.1016/S0140-6736(16)32453-9.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73. https://doi.org/10.1016/S0140-6736(18)30207-1.
Merriam-Webster. “Heterogeneity.”. 2018. www.merriam-webster.com/dictionary/heterogeneity. Accessed 14 June 2018.
Edmondson H. Tumors of the liver and intrahepatic bile ducts, atlas of tumors pathology. Washington, DC: Armed Forces Institute of Pathology; 1958.
Kenmochi K, Sugihara S, Kojiro M. Relationship of histologic grade of hepatocellular carcinoma (HCC) to tumor size, and demonstration of tumor cells of multiple different grades in single small HCC. Liver. 1987;7(1):18–26.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–8.
Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445. https://doi.org/10.1038/sj.onc.1207714.
Martincorena I, Campbell PJ. Somatic mutation in cancer and normal cells. Science (New York, NY). 2015;349(6255):1483–9. https://doi.org/10.1126/science.aab4082.
Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11(6):340.
Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7(2):85–97. https://doi.org/10.1038/nrg1767.
Shlien A, Malkin D. Copy number variations and cancer. Genome Med. 2009;1(6):62. https://doi.org/10.1186/gm62.
Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58(2):706–17. https://doi.org/10.1002/hep.26402.
Schlaeger C, Longerich T, Schiller C, Bewerunge P, Mehrabi A, Toedt G, et al. Etiology-dependent molecular mechanisms in human hepatocarcinogenesis. Hepatology. 2008;47(2):511–20.
Katoh H, Ojima H, Kokubu A, Saito S, Kondo T, Kosuge T, et al. Genetically distinct and clinically relevant classification of hepatocellular carcinoma: putative therapeutic targets. Gastroenterology. 2007;133(5):1475–86. https://doi.org/10.1053/j.gastro.2007.08.038.
Zhao X, Parpart S, Takai A, Roessler S, Budhu A, Yu Z, et al. Integrative genomics identifies YY1AP1 as an oncogenic driver in EpCAM(+) AFP(+) hepatocellular carcinoma. Oncogene. 2015;34(39):5095–104. https://doi.org/10.1038/onc.2014.438.
Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual report to the nation on the status of cancer, Part I: national cancer statistics. Cancer. 2018; https://doi.org/10.1002/cncr.31551.
Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46(12):1267–73. https://doi.org/10.1038/ng.3126.
Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43(5):464–9.
Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet. 2016;48(5):500–9. https://doi.org/10.1038/ng.3547.
Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–4.
The Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169(7):1327–41 e23. https://doi.org/10.1016/j.cell.2017.05.046.
Poon SL, Pang ST, McPherson JR, Yu W, Huang KK, Guan P, et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med. 2013;5(197):197ra01. https://doi.org/10.1126/scitranslmed.3006086.
Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–8.
Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–8. https://doi.org/10.1038/ng.2256.
Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47(5):505–11. https://doi.org/10.1038/ng.3252. http://www.nature.com/ng/journal/v47/n5/abs/ng.3252.html#supplementary-information.
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–27. https://doi.org/10.1056/NEJMra1001683.
Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44(7):765.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–54.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007;133(2):647–58. https://doi.org/10.1053/j.gastro.2007.05.022.
Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28(40):3526–36. https://doi.org/10.1038/onc.2009.211.
Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2(1):a001008. https://doi.org/10.1101/cshperspect.a001008.
Soussi T. The TP53 gene network in a postgenomic era. Hum Mutat. 2014;35(6):641–2. https://doi.org/10.1002/humu.22562.
Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396(6706):84–8. https://doi.org/10.1038/23962.
Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8(6):1409–20. https://doi.org/10.1158/1535-7163.mct-08-0860.
Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11:726–34.
Carracedo A, Pandolfi PP. The PTEN–PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527. https://doi.org/10.1038/onc.2008.247.
Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci. 1996;93(25):14960–5. https://doi.org/10.1073/pnas.93.25.14960.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. https://doi.org/10.1016/j.cmet.2007.10.002.
Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635. https://doi.org/10.1038/nrc.2016.77.
Li L, Che L, Tharp KM, Park HM, Pilo MG, Cao D, et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology. 2016;63(6):1900–13. https://doi.org/10.1002/hep.28508.
Kim MJ, Choi YK, Park SY, Jang SY, Lee JY, Ham HJ, et al. PPARdelta Reprograms Glutamine Metabolism in Sorafenib-Resistant HCC. Mol Cancer Res. 2017;15(9):1230–42. https://doi.org/10.1158/1541-7786.mcr-17-0061.
Beyoglu D, Idle JR. Metabolomics and its potential in drug development. Biochem Pharmacol. 2013;85(1):12–20. https://doi.org/10.1016/j.bcp.2012.08.013.
Patterson AD, Maurhofer O, Beyoglu D, Lanz C, Krausz KW, Pabst T, et al. Aberrant lipid metabolism in hepatocellular carcinoma revealed by plasma metabolomics and lipid profiling. Cancer Res. 2011;71:6590–600.
Yin P, Wan D, Zhao C, Chen J, Zhao X, Wang W, et al. A metabonomic study of hepatitis B-induced liver cirrhosis and hepatocellular carcinoma by using RP-LC and HILIC coupled with mass spectrometry. Mol Biosyst. 2009;5:868–76.
Budhu A, Roessler S, Zhao X, Yu Z, Forgues M, Ji J, et al. Integrated metabolite and gene expression profiles identify lipid biomarkers associated with progression of hepatocellular carcinoma and patient outcomes. Gastroenterology. 2013;144(5):1066–75 e1. https://doi.org/10.1053/j.gastro.2013.01.054.
Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. https://doi.org/10.1038/nrclinonc.2017.166.
Dragani TA. Risk of HCC: genetic heterogeneity and complex genetics. J Hepatol. 2010;52(2):252–7. https://doi.org/10.1016/j.jhep.2009.11.015.
Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13(2):123–35. https://doi.org/10.1038/nrc3449.
Neuveut C, Wei Y, Buendia MA. Mechanisms of HBV-related hepatocarcinogenesis. J Hepatol. 2010;52(4):594–604. https://doi.org/10.1016/j.jhep.2009.10.033.
Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43(9):828–9. https://doi.org/10.1038/ng.903.
Villar S, Ortiz-Cuaran S, Abedi-Ardekani B, Gouas D, Nogueira da Costa A, Plymoth A, et al. Aflatoxin-induced TP53 R249S mutation in hepatocellular carcinoma in Thailand: association with tumors developing in the absence of liver cirrhosis. PLoS One. 2012;7(6):e37707. https://doi.org/10.1371/journal.pone.0037707.
Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S87–96.
Kuper H, Tzonou A, Kaklamani E, Hsieh CC, Lagiou P, Adami HO, et al. Tobacco smoking, alcohol consumption and their interaction in the causation of hepatocellular carcinoma. Int J Cancer. 2000;85(4):498–502.
Humans IWGotEoCRt. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr Eval Carcinog Risks Hum. 2002;82:1–556.
Frank SA. Genetic predisposition to cancer — insights from population genetics. Nat Rev Genet. 2004;5:764. https://doi.org/10.1038/nrg1450.
Fairbanks KD, Tavill AS. Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol. 2008;103(8):2136–41; quiz 42. https://doi.org/10.1111/j.1572-0241.2008.01955.x.
Weinberg AG, Mize CE, Worthen HG. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J Pediatr. 1976;88(3):434–8.
Elmberg M, Hultcrantz R, Ekbom A, Brandt L, Olsson S, Olsson R, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125(6):1733–41.
Fracanzani AL, Taioli E, Sampietro M, Fatta E, Bertelli C, Fiorelli G, et al. Liver cancer risk is increased in patients with porphyria cutanea tarda in comparison to matched control patients with chronic liver disease. J Hepatol. 2001;35(4):498–503.
Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol. 1996;8(10):989–94.
Kowdley KV. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S79–86.
Andersson C, Bjersing L, Lithner F. The epidemiology of hepatocellular carcinoma in patients with acute intermittent porphyria. J Intern Med. 1996;240(4):195–201. https://doi.org/10.1046/j.1365-2796.1996.21847000.x.
Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med. 2017;5(13):270. https://doi.org/10.21037/atm.2017.04.41.
Cholankeril G, Patel R, Khurana S, Satapathy SK. Hepatocellular carcinoma in non-alcoholic steatohepatitis: current knowledge and implications for management. World J Hepatol. 2017;9(11):533–43. https://doi.org/10.4254/wjh.v9.i11.533.
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–76. https://doi.org/10.1053/j.gastro.2007.04.061.
Hassan MM, Kaseb A, Li D, Patt YZ, Vauthey JN, Thomas MB, Curley SA, Spitz MR, Sherman SI, Abdalla EK, Davila M. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. 2009;49(5):1563–70. https://doi.org/10.1002/hep.22793.
Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–92.
van’t Veer LJ, Dai H, Van de Vijver MJ, He YD, Hart AA, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–6.
Boyault S, Rickman DS, de Reynies A, Balabaud C, Rebouissou S, Jeannot E, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45(1):42–52. https://doi.org/10.1002/hep.21467.
Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69(18):7385–92. https://doi.org/10.1158/0008-5472.CAN-09-1089.
Chaisaingmongkol J, Budhu A, Dang H, Rabibhadana S, Pupacdi B, Kwon SM, et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell. 2017;32(1):57–70 e3. https://doi.org/10.1016/j.ccell.2017.05.009.
Woo HG, Choi J-H, Yoon S, Jee BA, Cho EJ, Lee J-H, et al. Integrative analysis of genomic and epigenomic regulation of the transcriptome in liver cancer. Nat Commun. 2017;8(1):839. https://doi.org/10.1038/s41467-017-00991-w.
Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24(6):1248–59. https://doi.org/10.1158/1078-0432.ccr-17-0853.
Xue R, Li J, Bai F, Wang X, Ji J, Lu Y. A race to uncover a panoramic view of primary liver cancer. Cancer Biol Med. 2017;14(4):335–40. https://doi.org/10.20892/j.issn.2095-3941.2017.0112.
Ye QH, Qin LX, Forgues M, He P, Kim JW, Peng AC, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat Med. 2003;9(4):416–23.
Lee JS, Chu IS, Heo J, Calvisi DF, Sun Z, Roskams T, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40(3):667–76. https://doi.org/10.1002/hep.20375.
Tan PS, Nakagawa S, Goossens N, Venkatesh A, Huang T, Ward SC, et al. Clinicopathological indices to predict hepatocellular carcinoma molecular classification. Liver Int. 2016;36(1):108–18. https://doi.org/10.1111/liv.12889.
Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouze E, Blanc JF, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol. 2017;67(4):727–38. https://doi.org/10.1016/j.jhep.2017.05.014.
Kurebayashi Y, Ojima H, Tsujikawa H, Kubota N, Maehara J, Abe Y, et al. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology. 2018;68(3):1025–41. https://doi.org/10.1002/hep.29904.
Lau SK, Prakash S, Geller SA, Alsabeh R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum Pathol. 2002;33(12):1175–81. https://doi.org/10.1053/hupa.2002.130104.
Chen LD, Xu HX, Xie XY, Xie XH, Xu ZF, Liu GJ, et al. Intrahepatic cholangiocarcinoma and hepatocellular carcinoma: differential diagnosis with contrast-enhanced ultrasound. Eur Radiol. 2010;20(3):743–53. https://doi.org/10.1007/s00330-009-1599-8.
Man XB, Tang L, Zhang BH, Li SJ, Qiu XH, Wu MC, et al. Upregulation of Glypican-3 expression in hepatocellular carcinoma but downregulation in cholangiocarcinoma indicates its differential diagnosis value in primary liver cancers. Liver Int. 2005;25(5):962–6. https://doi.org/10.1111/j.1478-3231.2005.01100.x.
Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer. 2013;108:479. https://doi.org/10.1038/bjc.2012.581.
Friemel J, Rechsteiner M, Frick L, Böhm F, Struckmann K, Egger M, et al. Intratumor heterogeneity in hepatocellular carcinoma. Clin Cancer Res. 2015;21(8):1951–61.
Xue R, Li R, Guo H, Guo L, Su Z, Ni X, et al. Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gastroenterology. 2016;150(4):998–1008. https://doi.org/10.1053/j.gastro.2015.12.033.
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26(3):304–19. https://doi.org/10.1038/cr.2016.23.
Zheng H, Pomyen Y, Hernandez MO, Li C, Livak F, Tang W, et al. Single cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology. 2018; https://doi.org/10.1002/hep.29778.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. https://doi.org/10.1038/35102167.
Nio K, Yamashita T, Kaneko S. The evolving concept of liver cancer stem cells. Mol Cancer. 2017;16:4. https://doi.org/10.1186/s12943-016-0572-9.
Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58.
Zheng YW, Tsuchida T, Shimao T, Li B, Takebe T, Zhang RR, et al. The CD133+CD44+ precancerous subpopulation of oval cells is a therapeutic target for hepatocellular carcinoma. Stem Cells Dev. 2014;23(18):2237–49. https://doi.org/10.1089/scd.2013.0577.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13. https://doi.org/10.1038/nature10762.
Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68(5):1451–61.
Ji J, Wang XW. Identification of cancer stem cell-related microRNAs in hepatocellular carcinoma. Methods Mol Biol. 2012;826:163–75.
Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases — elimination, equilibrium and escape. Curr Opin Immunol. 2014;27:16–25. https://doi.org/10.1016/j.coi.2014.01.004.
Jimenez-Sanchez A, Memon D, Pourpe S, Veeraraghavan H, Li Y, Vargas HA, et al. Heterogeneous tumor-immune microenvironments among differentially growing metastases in an ovarian cancer patient. Cell. 2017;170(5):927–38.e20. https://doi.org/10.1016/j.cell.2017.07.025.
Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–26.
Zhu AX, Park JO, Ryoo BY, Yen CJ, Poon R, Pastorelli D, et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859–70. https://doi.org/10.1016/S1470-2045(15)00050-9.
Zhu AX, Galle PR, Kudo M, Finn RS, Qin S, Xu Y, et al. A study of ramucirumab (LY3009806) versus placebo in patients with hepatocellular carcinoma and elevated baseline alpha-fetoprotein (REACH-2). J Clin Oncol. 2018;36(4_suppl):TPS538-TPS. https://doi.org/10.1200/JCO.2018.36.4_suppl.TPS538.
Cunanan KM, Iasonos A, Shen R, Begg CB, Gönen M. An efficient basket trial design. Stat Med. 2017;36(10):1568–79. https://doi.org/10.1002/sim.7227.
Renfro LA, Sargent DJ. Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol. 2017;28(1):34–43. https://doi.org/10.1093/annonc/mdw413.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. https://doi.org/10.1056/NEJMoa1500596.
Do K, O’Sullivan Coyne G, Chen AP. An overview of the NCI precision medicine trials-NCI MATCH and MPACT. Chin Clin Oncol. 2015;4(3):31. https://doi.org/10.3978/j.issn.2304-3865.2015.08.01.
Somasundaram R, Villanueva J, Herlyn M. Intratumoral heterogeneity as a therapy resistance mechanism: role of melanoma subpopulations. Adv Pharmacol (San Diego, Calif). 2012;65:335–59. https://doi.org/10.1016/B978-0-12-397927-8.00011-7.
Hectors SJ, Wagner M, Bane O, Besa C, Lewis S, Remark R, et al. Quantification of hepatocellular carcinoma heterogeneity with multiparametric magnetic resonance imaging. Sci Rep. 2017;7(1):2452. https://doi.org/10.1038/s41598-017-02706-z.
Sun Y-F, Guo W, Xu Y, Shi Y-H, Gong Z-J, Ji Y, et al. Circulating tumor cells from different vascular sites exhibit spatial heterogeneity in epithelial and mesenchymal composition and distinct clinical significance in hepatocellular carcinoma. Clin Cancer Res. 2018;24(3):547–59. https://doi.org/10.1158/1078-0432.ccr-17-1063.
Qin C, Cao Q, Li P, Wang S, Wang J, Wang M, et al. The influence of genetic variants of sorafenib on clinical outcomes and toxic effects in patients with advanced renal cell carcinoma. Sci Rep. 2016;6:20089. https://doi.org/10.1038/srep20089.
Acknowledgement
This work is supported by the Intramural Research Program of the Center for Cancer Research of the U.S. National Cancer Institute (Z01 BC 01313 and Z01 BC 010877) and Taipei Veterans General Hospital-National Yang-Ming University Excellent Physician Scientist Cultivation Program.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hung, M.H., Wang, X.W. (2019). Molecular Alterations and Heterogeneity in Hepatocellular Carcinoma. In: Hoshida, Y. (eds) Hepatocellular Carcinoma. Molecular and Translational Medicine. Humana, Cham. https://doi.org/10.1007/978-3-030-21540-8_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-21540-8_14
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-21539-2
Online ISBN: 978-3-030-21540-8
eBook Packages: MedicineMedicine (R0)