
Abstract
Atmospheric deposition is very often the most important source of mercury (Hg) to aquatic systems. Elevated Hg deposition can lead to elevated levels of Hg in aquatic food webs, which degrades the ecosystem and elevates our exposure to Hg via fish consumption. Therefore, an understanding of the atmospheric cycling and deposition of Hg is necessary to develop effective and efficient ways to lower Hg levels in any water body. This is particularly true for the Florida Everglades where direct atmospheric inputs constitute in excess of 95% of the external inputs of Hg to the ecosystem. This chapter thus presents an overview of the atmospheric Hg cycle and discusses key aspects of the cycle that are particularly important to understanding atmospheric Hg deposition in the Florida Everglades.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Overview of the Atmospheric Hg Cycle
The atmospheric Hg cycle is complex and represents the combination of several input, output and transformation processes that govern the concentrations and dynamics of various forms of Hg in the atmosphere. The forms of Hg in the atmosphere include gaseous elemental Hg (GEM), gaseous oxidized Hg (GOM), and aerosol (particulate-bound) Hg (PBM). GOM can be further subdivided into many neutral chemical species such as gaseous oxides, hydroxides, and halogen complexes with Hg(II). PBM represents all forms of Hg that are associated with particles in the atmosphere, although the majority of the PBM is thought to be in the +2 oxidation state [Hg(II)] Subir et al. 2012).
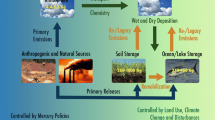
Considering the atmospheric cycle of Hg as it is implemented in the GEOS-Chem model (Fig. 3.1; from Horowitz et al. 2017), GEM accounts for about 90% of the total atmospheric Hg, GOM accounts for about 10%, and PBM accounts for less than a few percent. While GEOS-Chem treats gas-phase and particulate-bound Hg(II) as separate tracers, given the uncertainty in measurement capabilities to separate GOM and PBM (e.g., Gustin et al. 2013) the quantities are combined for model-observation comparisons. It is thought that gaseous oxidized Hg exchanges rapidly with oxidized Hg on aerosols (Amos et al. 2012), and both forms are rapidly deposited by wet and dry deposition processes.

The modern global budget for atmospheric Hg as implemented in the GEOS-Chem model (from Horowitz et al. 2017). The lower panel illustrates the chemical reactions in the atmosphere that cycle atmospheric Hg between GEM [Hg(0)] and Hg(II). Used with permission (https://creativecommons.org/licenses/by/3.0/)
The GEOS-Chem model includes primary emissions of GEM [Hg(0)] and Hg(II) (GOM plus PBM) from geogenic and anthropogenic sources, as well as GEM emissions from biomass-burning, vegetation, soil, snow, and the oceans. Primary natural emissions of Hg (from volcanic activity, crustal degassing, and weathering of geologic deposits) are on the order of 80–600 Mg/yr, primarily as GEM (Wilson et al. 2013). Estimates for current global primary anthropogenic Hg emissions include 2000 Mg/yr (Amos et al. 2013), and 2280 Mg/yr (Zhang et al. 2016). It is estimated that about 35% of the primary anthropogenic Hg emissions are in the form of Hg(II) with 65% as GEM (Zhang et al. 2016; Streets et al. 2017). Re-emission of legacy anthropogenic Hg (mostly as GEM) is estimated at around 4700 Mg/yr.
As described by Horowitz et al. (2017), in the gas phase, GEM is oxidized to Hg(II) in a two-step process involving halogen radicals (e.g., Br, Cl) or OH in the first step and a variety of oxidants in the second step (NO2, HO2, ClO, BrO, OH, Cl, Br). In the aqueous phase (in cloud water, rain, and fog), GEM is oxidized by oxidants including aqueous HOCl, OH, and ozone. Gas phase oxidation of GEM by ozone may also be important and is an area of current study. Photochemical reduction of atmospheric Hg(II) back to GEM occurs through photolysis in air and in aqueous reactions in clouds and aerosol (Horowitz et al. 2017; Saiz-Lopez et al. 2018).
The concentrations of most of these potential GEM oxidants are well known from decades of atmospheric chemistry measurements and modeling (e.g. Seinfeld and Pandis 2016). Recent advances in techniques for measuring halogens, however, have revealed higher concentrations of reactive bromine than were expected in the troposphere (Wang et al. 2015; Coburn et al. 2016). Ongoing field experiments continue to map the abundance of reactive halogens, but these high concentrations can likely be explained by emissions of biogenic halocarbons from marine algae, debromination of sea-salt aerosols, and recycling of background aerosols (Sherwen et al. 2016).
Hg deposition occurs via wet and dry deposition processes to the land (vegetation, soil, snow) and the oceans. It is estimated that about 65–80% of total Hg deposition on a global scale is due to wet and dry deposition of Hg(II), while 20–35% is due to dry deposition of GEM. Hg(II) in the lower atmosphere (the boundary layer) will adsorb (dry deposit) to vegetation and open water and be taken up by sea salt aerosols. In GEOS-Chem, on a global basis, dry deposition accounts for about 40% of total Hg(II) deposition while wet deposition of Hg(II) accounts for about 60% (Horowitz et al. 2017). Wet deposition of Hg occurs when cloud water absorbs Hg(II) and when precipitation scavenges aerosol Hg. Most of the wet deposition is concentrated in the tropics, where rain is most abundant. GEM is not a significant contributor to wet deposition due to its relatively low solubility in water.
It is essential to accurately quantify all emission sources of both GEM and Hg(II), and the potentially rapid redox cycling in the atmosphere between these forms, since deposition of Hg(II) will have significant impacts on ecosystem bioaccumulation of Hg on local and regional scales. This is critically important since ecosystem studies have shown that recently deposited Hg(II) is taken up into aquatic food webs most quickly (Branfireun et al. 2005; see Section 3.2 in Chap. 3, Volume III for a more detailed discussion).
Using the GEM and Hg(II) emission rates in Fig. 3.1, one can estimate that the atmospheric residence time of GEM is on the order of 6 months (reservoir size of 3500 Mg divided by the total input rate of 7740 Mg/yr). Because of its low solubility in water and its relatively long atmospheric residence time, GEM emissions are transported far from their sources and become more well-mixed in the atmosphere.
The residence time of Hg(II) is on the order of 25 days (400 Mg divided by net input of 5600 Mg/yr). The oxidized forms of atmospheric Hg (GOM and PBM) are very soluble in water, are easily scavenged by rainfall, and adsorb to surfaces, resulting in deposition much closer to their sources.
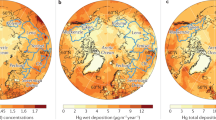
Figure 3.2a (from AMAP/UNEP 2015) shows the emissions of Hg on regional and local scales. High Hg emission rates are associated with population density and with artisanal-scale gold mining (ASGM) in South America and central and southern Africa. On a global basis, over 90% of modern Hg emission is as GEM, although GOM and PBM can account for up to 20% in certain areas (southeast Asia, Europe, and North America). Figure 3.2b shows that the modeled concentrations of GEM are highest near regions of high population and high anthropogenic emissions (China, India, Europe) and in South America and Africa due to ASGM. Atmospheric Hg concentrations are significantly lower in the southern hemisphere due to the lower population and the slow exchange of air across the tropics. Figure 3.2c shows model results for the global deposition of atmospheric Hg (which includes GEM, GOM, and PBM). The deposition patterns reveal several important factors that influence atmospheric Hg deposition. Uptake of the GEM by forested areas yields high deposition across South America and central and southern Africa, although much of that GEM is re-emitted on an annual basis. Elevated Hg deposition associated with high precipitation rates is seen along the Inter-Tropical Convergence Zones (ITCZ) in the equatorial Atlantic, Pacific, and Indian Oceans and in the Southern Ocean near Antarctica. This is interpreted as being due to more efficient oxidation of GEM to Hg(II) species which are easily scavenged during precipitation events. Combinations of regionally elevated Hg emissions and elevated rainfall yield higher Hg deposition in the southeastern US and in the western North Pacific and western North Atlantic. The highest Hg deposition is associated with high population density and high anthropogenic emissions as seen in Europe, India, and China.

As discussed above, the residence time of GEM in the atmosphere is relatively long (6–12 months), while Hg(II) has a much shorter residence time (days to weeks). As a result, GEM from all emission sources in the northern hemisphere is relatively well-mixed while Hg(II) shows strong local and regional concentration differences. Hg(II) is also quickly removed from the atmosphere by precipitation while GEM is not. Thus, local and regional sources of Hg(II) will have greater local and regional impacts on Hg deposition.
Except for a few areas where industrial effluent containing Hg contaminates the local environment, the dominant source of Hg to the global environment is from atmospheric deposition. Atmospheric deposition is the main source of Hg to Florida, especially the Florida Everglades (USEPA 1996). Modeling studies also show that the majority (>98%) of the atmospherically Hg deposited in Florida is from Hg sources outside of Florida (USEPA 2008; FDEP 2013). The model results for Hg deposition in the Everglades are reviewed in Chap. 5, this volume and Chap. 3, Volume III.
Amos et al. (2013) estimated that about 13% of the atmospheric Hg deposited globally was from natural sources. According to their calculations, the current atmospheric deposition of Hg on a global basis can be apportioned as follows: 3% primary natural; 10% legacy natural, 27% primary anthropogenic, and 60% legacy anthropogenic Hg. Variations from this global Hg deposition apportionment are to be expected on local and regional scales where anthropogenic emissions of oxidized Hg are important.
Estimates for the global budget of Hg (Table 3.1) suggest that the modern atmospheric Hg reservoir may be as much as 7.5 times larger relative to natural levels due to increasing anthropogenic Hg emissions since the start of the industrial revolution (Amos et al. 2013). This conclusion was questioned by Engstrom et al. (2014); a detailed study of Hg in lake sediment cores indicated that the magnitude of early Hg emissions from artisanal gold mining were overestimated by Amos et al. (2013), and that the modern/natural deposition ratio should be closer to 3–5. Amos et al. (2015) tried to harmonize the time periods for observed and modeled estimates of anthropogenic enrichment and reported that the mean enrichment from the precolonial era (3000 BC–1550 AD) to the twentieth century maximum (1950–1975) was 19 for varved sediment cores and 26 for peat cores. Consistent with these results, Enrico et al. (2017) estimated enrichment of 15 ± 4 from the Holocene to the present-day as reconstructed from a peat bog. Regardless of the exact value, Hg concentrations in the modern atmosphere, and atmospheric Hg deposition, are significantly elevated over pre-industrial times.
Emissions of Hg to the atmosphere from anthropogenic activities over time since the 1400s were summarized by Beal et al. (2015; Fig. 3.3). Primary anthropogenic Hg emissions were slightly higher (~500 Mg/yr) from 1450–1650, then declined to ~100–150 Mg/yr from 1650–1850. Starting around 1850, Hg emissions increased dramatically, associated with the American gold and silver rush period when amalgamation with liquid Hg was used to extract precious metals from ores. Emissions of Hg peaked again from 1940–1975 due to increased commercial and industrial use. Temporary declines in Hg deposition following those two emission peaks demonstrate the relatively rapid response time of atmospheric Hg deposition to changes in the atmospheric loading. Primary anthropogenic input has apparently declined by about 20% from 1990 to 2010 due to reductions in the use of Hg in man-made products (and the incineration of those products) and improved Hg scrubbing during coal combustion (Zhang et al. 2016).

Records of total Hg deposition in mountain glacier ice cores from Alaska (panel a) and Wyoming (panel b) with estimates for primary anthropogenic Hg emissions (panel c). (Beal et al. 2015). The red lines in panel a are smoothed estimates obtained from locally weighted polynomial regression (LOESS). (Reprinted with permission from Beal et al. (2015). Copyright 2015 American Chemical Society)
Estimates for the global distribution of atmospheric Hg deposition rates (subdivided by source region) were derived from an ensemble of three chemical transport models (GLEMOS, GEOS-Chem, GMHG) (Fig. 3.4; AMAP/UNEP 2015). The model results demonstrate strong seasonality for Hg deposition and the importance of natural and legacy Hg emissions. For example, over North America, Hg deposition peaks in the spring and summer months. Emission of Hg from North American sources only account for 5–12% of the total Hg deposition over North America (lowest during the spring and summer months). Legacy anthropogenic and natural emissions and primary natural emissions dominate the deposition of atmospheric Hg everywhere but are least important (60–70%) over East Asia and South Asia where local and regional emissions are more dominant. Further research on both modern and historical Hg deposition rates on a global scale is needed to help constrain these model estimates.

Monthly Hg deposition (2013) attributed to Hg emissions from various geographic regions. (Used with permission from AMAP/UNEP 2015)
3.2 Importance of Atmospheric Hg Speciation on Transport and Deposition
As noted above, gaseous elemental mercury (GEM) has a lifetime in the atmosphere of approximately 6–12 months. Species with such long atmospheric lifetimes are usually widely dispersed throughout the troposphere with relatively modest variations in concentration (Fig. 3.2b). The resulting dry deposition of GEM is therefore driven primarily by global sources as opposed to local emissions.
Oxidized mercury [Hg(II)], in contrast, has a lifetime of only a few weeks in the free troposphere (~2–18 km altitude), like other aerosols (e.g. sulfate) and highly soluble gases (e.g. nitric acid) (Seinfeld and Pandis 2016). However, within the planetary boundary layer (<2 km altitude), both GOM and PBM have much shorter lifetimes, around 1 day, since they are removed by dry deposition on contact with the surface. Consequently, oxidized mercury is predominantly deposited near its emission source (up to 100 km away), although it can be transported much farther if it is lifted above the boundary layer.
While very high Hg(II) concentrations have been reported around anthropogenic sources, observations from aircraft sampling campaigns and monitoring on mountain tops show that Hg(II) concentrations generally increase with altitude. This is believed to be caused by oxidation of GEM to produce Hg(II) that accumulates in the upper troposphere and is consistent with the model results shown in Fig. 3.1 where net oxidation of GEM to Hg(II) is on the order of 5000 Mg/yr. This region of the atmosphere rarely experiences rain and it can take a month or longer for air to mix down to lower altitudes. As a result, Hg(II) deposition is high where large-scale high-pressure systems transport air downwards (Shah and Jaegle 2017). This happens mainly at sub-tropical latitudes (which includes Florida). In addition, Hg(II) deposition is high in regions with frequent deep convection, like Florida, that can scavenge Hg(II) directly from high altitudes.
3.3 Modifiers to the Atmospheric Hg Cycle that Influence Hg Deposition in the Everglades
Guentzel et al. (2001) reported rainfall Hg data collected across the state of Florida from 1993 through 1996 (the Florida Atmospheric Mercury Study; FAMS) and concluded that strong convective thunderstorms in the summer months were responsible for ~84% of the annual rainfall Hg deposition vs. lower altitude rain events and winter-time frontal storms. They showed that wet deposition of Hg was nearly uniform at 20–23 μg/m2-yr from West Palm Beach to Ft. Myers and throughout the Florida Everglades and that rainfall Hg deposition reached a peak during the summer months when convective thunderstorms are very common over southern Florida. Similar results were reported by Shanley et al. (2015) who observed elevated rainfall Hg concentrations and fluxes from summertime thunderstorms at a pristine site in Puerto Rico. To explain their findings, Guentzel et al. (2001) hypothesized that there must be a rapidly-replenished reservoir of “background” Hg(II) in the free troposphere that supplies Hg(II) to these thunderstorms when the southeasterly trade winds shift north and flow across southern Florida.
When Guentzel et al. (2001) obtained their rainfall Hg data across southern Florida (1993–1996), no data had been published on Hg(II) concentrations in the free troposphere anywhere in the world, and the hypothesis could not be tested. Since then, aircraft sampling for Hg(II) in the free troposphere over the Bahamas found very high Hg(II) concentrations (50–250 pg/m3) between 1–4 km height (Landis et al. 2005; Sillman et al., 2007). Brooks et al. (2014) reported Hg(II) concentrations as high as 150 pg/m3 at 4 km during the spring and summer months over Tullahoma, TN (10–30 times higher than concurrent ground level measurements). Both aircraft Hg(II) data sets were collected using KCl-coated annular denuders that subsequently have been reported to under-collect Hg(II) by as much as a factor of 2 (Swartzendruber et al. 2009; Gustin et al. 2013); thus, the concentrations of Hg(II) in the free troposphere may be significantly higher than were reported. This reservoir of Hg(II) could easily support the summertime rainfall Hg concentrations and fluxes that Guentzel et al. (2001) reported across southern Florida. Further support for this hypothesis comes from Holmes et al. (2016) who used rainfall Hg data from the NADP-MDN program across the continental US and GridSat-B1 satellite cloud-top brightness data to conclude that thunderstorms generate 50% higher rainfall Hg concentrations relative to weak convective or stratiform events of equal precipitation depth. In a modeling study, Kaulfus et al. (2017) concluded that, after controlling for precipitation depth, the highest Hg deposition occurs in supercell thunderstorms, with decreasing deposition in disorganized thunderstorms, quasi-linear convective systems (QLCS), extratropical cyclones, light rain, and land-falling tropical cyclones. Convective morphologies (supercells, disorganized, and QLCS) enhanced wet deposition by a factor of at least 1.6 relative to non-convective morphologies.
Lacking any data on Hg(II) concentrations in the free troposphere, Guentzel et al. (2001) likely overestimated that local and regional emissions of Hg could account for 30–46% of the rainfall Hg deposition across southern Florida. More recent estimates for the importance of local and regional source of atmospheric Hg for Hg deposition in the Everglades are discussed in Chap. 5, this volume. Briefly, the Florida Mercury TDML Study (FDEP 2013) concluded that Florida sources accounted for less than 2% of the total Hg deposition at the Everglades National Park site.
3.4 Evasion of Hg
Recent modeling studies of the global Hg cycle suggest that uptake and evasion losses of GEM are roughly balanced for terrestrial ecosystems (Amos et al. 2013; Song et al. 2015; Horowitz et al. 2017; see Fig. 3.1). The very low GEM evasion estimate of Amos et al. (2014) was an exception to these other three studies, with evasion loss approximately sixfold less, although their uptake fluxes are similar. Excluding the very low evasion flux indicated by Amos et al. (2014), the global modeling studies cited above indicate that GEM evasion accounts for 50 ± 5% of the total Hg deposition to terrestrial surfaces (wet plus dry GEM, GOM, and PBM). In a more detailed model of Hg uptake by terrestrial ecosystems, Smith-Downey et al. (2010) suggest that plants take up GEM but do not release 100% of it. Plants also collect GOM and PBM, some of which is photo-reduced back to GEM. Overall, GEM evasion accounted for 67% of the total Hg deposition to terrestrial systems (Smith-Downey et al. 2010). In a field experiment, Poulain et al. (2006) measured 33–59% GEM evasion from mesocosms spiked with Hg(II). These various estimates for GEM evasion agree reasonably well and suggest that GEM evasion from wetland ecosystems is probably on the order of 50–60% of total Hg deposition.
With respect to the oceans, however, current estimates indicate that evasion losses of GEM from the ocean surface greatly exceed uptake fluxes of GEM from the atmosphere (median ratio for the four studies is 3:1). The large GEM evasion losses from the surface ocean are due to Hg(II) reduction in surface waters. The GEM flux approximately balances the deposition of GOM and PBM and influences how rapidly the Hg reservoir in the surface ocean turns over (~ 0.25 year; Amos et al. 2013; Song et al. 2015).
3.5 Conclusions
The atmospheric Hg cycle is extremely complex in part due to the variety of emission sources and emission speciation profiles (GEM vs. Hg(II)) and their geographic distribution. It is also complex because transformations of GEM to GOM are driven by a variety of atmospheric oxidants with different sources and distributions in the atmosphere and because the physics of atmospheric mixing varies spatially. Significant progress has been made in recent years on modeling the in situ processes influencing GEM oxidation and Hg(II) reduction, but there is still much to learn. The residence times for GEM and Hg(II) in the troposphere with respect to their inputs are on the order of 6–12 months and 25 days, respectively, while Hg(II) in the boundary layer is deposited much more quickly than GEM. As a result, the atmospheric deposition of Hg is strongly influenced by the magnitude and speciation of Hg emissions on local and regional scales, and by the patterns of vegetation and rainfall across the globe. Both field data and model simulations suggest that convective thunderstorms yield significantly higher rainfall Hg concentrations and fluxes due to scavenging of Hg(II) from the free troposphere. Local and regional Hg emissions can have significant impacts on regional Hg deposition; the lack of accurate emission inventories and emission profiles [GEM vs. Hg(II)] however continues to affect the reliability of model predictions for Hg deposition.
The state of knowledge on the atmospheric mercury cycle continues to advance, and more recent information on Hg emissions and deposition has been summarized in the 2018 UNEP Global Mercury Assessment (GMA).
References
AMAP/UNEP (2015) Global mercury modelling: update of modelling results in the global mercury assessment 2013. Arctic Monitoring and Assessment Programme/UNEP Chemicals Branch, Oslo/Geneva. iv + 32 pp
Amos HM, Jacob DJ, Holmes CD, Fisher JA, Wang Q, Yantosca RM, Corbitt ES, Galarneau E, Rutter AP, Gustin MS, Steffen A, Schauer JJ, Graydon JA, St Louis VL, Talbot RW, Edgerton ES, Zhang Y, Sunderland EM (2012) Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition. Atmos Chem Phys 12:591–603
Amos HM, Jacob DJ, Streets DG, Sunderland EM (2013) Legacy impacts of all-time anthropogenic emissions on the global mercury cycle. Glob Biogeochem Cycles 27:410–421
Amos HM, Jacob DJ, Kocman D, Horowitz HM, Zhang YX, Dutkiewicz S, Horvat M, Corbitt ES, Krabbenhoft DP, Sunderland EM (2014) Global biogeochemical implications of mercury discharges from rivers and sediment burial. Environ Sci Technol 48:9514–9522
Amos HM, Sonke JE, Obrist D, Robins N, Hagan N, Horowitz HM, Mason RP, Witt M, Hedgecock IM, Corbitt ES, Sunderland EM (2015) Observational and modeling constraints on global anthropogenic enrichment of mercury. Environ Sci Technol 49:4036–4047
Beal SA, Osterberg EC, Zdanowicz CM, Fisher DA (2015) Ice core perspective on mercury pollution during the past 600 years. Environ Sci Technol 49:7641–7647
Branfireun BA, Krabbenhoft DP, Hintelmann H, Hunt RJ, Hurley JP, Rudd JWM (2005) Speciation and transport of newly deposited mercury in a boreal forest wetland: a stable mercury isotope approach. Water Resour Res 41:11
Brooks S, Ren XR, Cohen M, Luke WT, Kelley P, Artz R, Hynes A, Landing W, Martos B (2014) Airborne vertical profiling of mercury speciation near Tullahoma, TN, USA. Atmosphere 5:557–574
Coburn S, Dix B, Edgerton E, Holmes CD, Kinnison D, Liang Q, ter Schure A, Wang SY, Volkamer R (2016) Mercury oxidation from bromine chemistry in the free troposphere over the southeastern US. Atmos Chem Phys 16:3743–3760
Engstrom DR, Fitzgerald WF, Cooke CA, Lamborg CH, Drevnick PE, Swain EB, Balogh SJ, Balcom PH (2014) Atmospheric Hg emissions from preindustrial gold and silver extraction in the Americas: a reevaluation from lake-sediment archives. Environ Sci Technol 48:6533–6543
Enrico M, Le Roux G, Heimburger LE, Van Beek P, Souhaut M, Chmeleff J, Sonke JE (2017) Holocene atmospheric mercury levels reconstructed from peat bog mercury stable isotopes. Environ Sci Technol 51:5899–5906
Florida Department of Environmental Protection (2013) Mercury TMDL for the state of Florida, Final report, watershed evaluation and TMDL section, October 24, 2013. https://floridadep.gov/sites/default/files/Mercury-TMDL.pdf
Guentzel JL, Landing WM, Gill GA, Pollman CD (2001) Processes influencing rainfall deposition of mercury in Florida. Environ Sci Technol 35:863–873
Gustin MS, Huang JY, Miller MB, Peterson C, Jaffe DA, Ambrose J, Finley BD, Lyman SN, Call K, Talbot R, Feddersen D, Mao HT, Lindberg SE (2013) Do we understand what the mercury speciation instruments are actually measuring? Results of RAMIX. Environ Sci Technol 47:7295–7306
Holmes CD, Krishnamurthy NP, Caffrey JM, Landing WM, Edgerton ES, Knapp KR, Nair US (2016) Thunderstorms increase mercury wet deposition. Environ Sci Technol 50:9343–9350
Horowitz HM, Jacob DJ, Zhang YX, Dibble TS, Slemr F, Amos HM, Schmidt JA, Corbitt ES, Marais EA, Sunderland EM (2017) A new mechanism for atmospheric mercury redox chemistry: implications for the global mercury budget. Atmos Chem Phys 17:6353–6371
Kaulfus AS, Nair U, Holmes CD, Landing WM (2017) Mercury wet scavenging and deposition differences by precipitation type. Environ Sci Technol 51:2628–2634
Landis MS, Lynam M, Stevens RK (2005) The monitoring and modeling of mercury species in support of local regional and global modeling. In: Pirrone N, Mahaffey KR (eds) Dynamics of mercury pollution on regional and global scales. Kluwer Academic, New York, pp 123–151
Poulain AJ, Orihel DM, Amyot M, Paterson MJ, Hintelmann H, Southworth GR (2006) Relationship to aquatic between the loading rate of inorganic mercury ecosystems and dissolved gaseous mercury production and evasion. Chemosphere 65:2199–2207
Saiz-Lopez A, Sitkiewicz SP, Roca-Sanjuán D, Oliva-Enrich JM, Dávalos JZ, Notario R et al (2018) Photoreduction of gaseous oxidized mercury changes global atmospheric mercury speciation, transport and deposition. Nat Commun 9(1):1–9. https://doi.org/10.1038/s41467-018-07075-3
Seinfeld JH, Pandis SN (2016) Atmospheric chemistry and physics: from air pollution to climate change, 3rd edn. Wiley, New York. Published 2016, ISBN-10: 1118947401
Shah V, Jaegle L (2017) Subtropical subsidence and surface deposition of oxidized mercury produced in the free troposphere. Atmos Chem Phys 17:8999–9017
Shanley JB, Engle MA, Scholl M, Krabbenhoft DP, Brunette R, Olson ML, Conroy ME (2015) High mercury wet deposition at a “clean air” site in Puerto Rico. Environ Sci Technol 49:12474–12482
Sherwen T, Schmidt JA, Evans MJ, Carpenter LJ, Grossmann K, Eastham SD, Jacob DJ, Dix B, Koenig TK, Sinreich R, Ortega I, Volkamer R, Saiz-Lopez A, Prados-Roman C, Mahajan AS, Ordonez C (2016) Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem. Atmos Chem Phys 16:12239–12271
Sillman S, Marsik FJ, Al-Wali KI, Keeler GJ, Landis MS (2007) Reactive mercury in the troposphere: model formation and results for Florida, the Northeastern United States, and the Atlantic Ocean. J Geophys Res-Atmos 112:17
Smith-Downey NV, Sunderland EM, Jacob DJ (2010) Anthropogenic impacts on global storage and emissions of mercury from terrestrial soils: insights from a new global model. J Geophys Res Biogeosci 115:11
Song S, Selin NE, Soerensen AL, Angot H, Artz R, Brooks S, Brunke EG, Conley G, Dommergue A, Ebinghaus R, Holsen TM, Jaffe DA, Kang S, Kelley P, Luke WT, Magand O, Marumoto K, Pfaffhuber KA, Ren X, Sheu GR, Slemr F, Warneke T, Weigelt A, Weiss-Penzias P, Wip DC, Zhang Q (2015) Top-down constraints on atmospheric mercury emissions and implications for global biogeochemical cycling. Atmos Chem Phys 15:7103–7125
Streets DG, Horowitz HM, Jacob D, Lu ZF, Levin L, ter Schure AFH, Sunderland EM (2017) Total mercury released to the environment by human activities. Environ Sci Technol 51:5969–5977
Subir M, Ariya PA, Dastoor AP (2012) A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry – a missing link. Atmos Environ 46:1–10
Swartzendruber PC, Jaffe DA, Finley B (2009) Development and first results of an aircraft-based, high time resolution technique for gaseous elemental and reactive (oxidized) gaseous mercury. Environ Sci Technol 43:7484–7489
UN Environment (2019) Global mercury assessment 2018. UN Environment Programme, Chemicals and Health Branch, Geneva
United States Environmental Protection Agency (USEPA) (1996) South Florida ecosystem assessment interim report. Monitoring for adaptive management: implications for ecosystem restoration. Region 4 SESD and ORD. EPA-904-96-008. December 1996
USEPA REMSAD (2008) Model-based analysis and tracking of airborne mercury emissions to assist in watershed planning. Watershed branch (4503-T), Office of Wetlands, Oceans, and Watersheds, U.S. Environmental Protection Agency, 1200 Pennsylvania Avenue, NW, Washington, DC 20460 (2009)
Wang SY, Schmidt JA, Baidar S, Coburn S, Dix B, Koenig TK, Apel E, Bowdalo D, Campos TL, Eloranta E, Evans MJ, DiGangi JP, Zondlo MA, Gao RS, Haggerty JA, Hall SR, Hornbrook RS, Jacob D, Morley B, Pierce B, Reeves M, Romashkin P, ter Schure A, Volkamer R (2015) Active and widespread halogen chemistry in the tropical and subtropical free troposphere. Proc Natl Acad Sci U S A 112:9281–9286
Wilson S, Kindbom K, Yaramenka K, Steenhuisen F, Telmer K, Munthe J (2013) Global emissions of mercury to the atmosphere, in technical background report for the global mercury assessment 2013. Arctic Monitoring and Assessment Programme, Geneva, pp 4–37
Zhang YX, Jacob DJ, Horowitz HM, Chen L, Amos HM, Krabbenhoft DP, Slemr F, St Louis VL, Sunderland EM (2016) Observed decrease in atmospheric mercury explained by global decline in anthropogenic emissions. Proc Natl Acad Sci U S A 113:526–531
Acknowledgments
We would like to thank Curt Pollman and Hannah Horowitz for their very helpful reviews of this chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Landing, W.M., Holmes, C.D. (2019). Overview of the Atmospheric Mercury Cycle. In: D. Pollman, C., Rumbold, D., Axelrad, D. (eds) Mercury and the Everglades. A Synthesis and Model for Complex Ecosystem Restoration . Springer, Cham. https://doi.org/10.1007/978-3-030-20070-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-20070-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-20069-5
Online ISBN: 978-3-030-20070-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)