
Abstract
ROS1 is a receptor tyrosine kinase with ROS1 gene fusions identified in 0.9–2.1% of non-small cell lung cancers (NSCLC), as well as a number of other malignancies. These fusions are constitutively activated, leading to significant changes in cell differentiation, proliferation, growth, and survival. The fusions can be identified by a number of methods including fluorescent in situ hybridization, immunohistochemistry, real-time PCR, and next-generation sequencing. The tyrosine kinase inhibitor crizotinib is a potent inhibitor of ROS1 and was approved by the FDA for treatment of metastatic non-small cell lung cancer (NSCLC) with ROS1 rearrangement in March 2016. However, as with other oncogenes, patients treated with crizotinib eventually develop resistance and progressive disease. A number of different resistance mutations have been discovered, the mechanisms of which can be broken down into two major categories: mutations within the ROS1 kinase domain and bypass signaling pathways. Several additional tyrosine kinase inhibitors are under development with varying degrees of CNS penetration and efficacy against resistance mutations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- ROS1 rearrangement
- ROS1 inhibitor
- Lung cancer
- Non-small cell lung cancer
- Oncogene
- Crizotinib
- Ceritinib
- Tyrosine kinase inhibitor (TKI) resistance
- Targeted therapy
- Tyrosine kinase inhibitors
Introduction
Though gene fusions in the receptor tyrosine kinase ROS1 are present in only a small percentage of non-small cell lung cancer (NSCLC), it represents a viable therapeutic target with impressive clinical benefit to ROS1 inhibitors. The first ROS1 inhibitor, crizotinib, was approved for advanced NSCLC harboring ROS1 fusions in 2016. Subsequently a number of additional ROS1 inhibitors have been in development and seek to overcome resistance mutations that develop in response to crizotinib. In this chapter we provide an overview of the ROS1 gene including its history, biology, methods of detection, and role of targeted treatment against ROS1-positive NSCLC, including the development and management of resistance mutations with newer agents targeting ROS1.
Structure and Function
Human c-ros oncogene 1 (ROS1) was originally discovered as a homolog of the transforming sequence of the UR2 avian sarcoma virus [1, 2] (Fig. 1). ROS1 is located on chromosome 6q22 [4, 5]. It is a receptor tyrosine kinase that shares structural similarities to the insulin receptor tyrosine kinase, leukocyte tyrosine kinase (LTK), and anaplastic lymphoma kinase (ALK) families [6]. ROS-1 encodes a receptor tyrosine kinase that consists of a large N-terminal extracellular domain, a hydrophobic single pass transmembrane region, and a C-terminal intracellular tyrosine kinase domain. It is relatively unique, in that its extracellular domain is composed of six repeat motifs that have high homology to the extracellular matrix and plasma protein fibronectin type III repeats, almost resembling a cell adhesion molecule. Unlike most adhesion molecules, however, the intracellular kinase domain enables ROS to directly translate adhesion engagement along intracellular signaling pathways [3].

ROS fusion kinases. Schematic representation of selected ROS fusions and their corresponding protein fusion product. With the exception of FIG-ROS, all of the fusion kinases are predicted to be plasma membrane bound. SLC34A2-ROS and CD740ROS fusion proteins are here illustrated to be bimembrane spanning receptors, though this has not been confirmed experimentally. (Figure from Acquaviva et al. [3])
The ROS receptor tyrosine kinase gene is evolutionarily conserved across multiple organisms. In Drosophila melanogaster, SEVENLESS (a ROS orthologue) is associated with a seven-transmembrane G-protein-coupled cell called BOSS (bride of sevenless). It is of particular importance in the developing Drosophila eye, where BOSS is required for differentiation of cells into photoreceptors [7, 8]. ROS expression has been examined in mouse, chicken, and rat tissue throughout various stages of development, with expression of c-Ros found in kidneys, small intestines, heart, lung, and male reproductive cells with restriction seen to epithelial cells [3]. In mice, testicular expression of c-Ros was only detected in adults with in situ hybridization of the adult testes showing expression in mature stages of development (spermatids, spermatozoa) only [9, 10]. In addition, c-Ros mutated male mice were noted to be infertile, though otherwise healthy. The defect was noted to be in development of the epithelia in the epididymis, especially in regionalization and terminal differentiation. No such impairment in fertility was noted in female mice. This suggests that expression may be linked to male fertility [11]. In humans, however, research has been hindered by the fact that it remains an orphan tyrosine kinase receptor, without a known ligand. Another barrier is the inability to express the full-length wild-type receptor in cellular models. There is some speculation that c-ROS1 expression may be involved in epithelial-mesenchymal interactions as well as in the cellular differentiation cascade of epithelial tissues [3].
Gene Fusion and Cancer
ROS1 gene fusions were first identified in the human glioblastoma cell line U-118 MG [12, 13]. An intra-chromosomal homozygous microdeletion of 240 kilobases on chromosome 6q21 was found to lead to fusion of the 5′ region of the FIG gene (fused in glioblastoma; a Golgi apparatus-associated protein) to the 3′ kinase domain of ROS [14,15,16] (Fig. 1). This fusion led to constitutive activation that was dependent on its localization to the Golgi apparatus [16]. This fusion has also been identified in a number of other malignancies, including ovarian cancer [17], cholangiocarcinoma [18], and NSCLC [19]. Additional ROS1 gene fusions have been found in a number of other malignancies, including inflammatory myofibroblastic tumors [20, 21], gastric adenocarcinoma [22], colorectal cancer [23], angiosarcoma [24], thyroid cancer [25], atypical meningioma [26], and spitzoid melanomas [27].
Following the identification of ROS1 fusions in glioblastomas, NSCLC was the second solid tumor in which these rearrangements were identified. This landmark study characterized tyrosine kinase signaling across 41 NSCLC cell lines and over 150 NSCLC tumors using a phosphoproteomic approach. High-level ROS kinase activity was noted in one cell line and one tumor sample. When these samples were sequenced, two novel ROS1 fusions were identified. In the HCC78 cell line, ROS was found to be fused to the transmembrane solute carrier protein SLC34A2. This protein was found to localize to membrane fractions and display a constitutive kinase activity. siRNA against SLC34A2-ROS was found to induce apoptosis, suggesting that ROS signaling is critical for survival of these NSCLC cells. In the solid tumor, c-ROS was found to be fused to the N-terminal half a type II transmembrane protein, CD74 [28] (Fig. 1).
Since this initial publication, multiple other fusion partners have been reported in NSCLC. The CD74-ROS1 fusion remains the most common, occurring with an estimated frequency of 32% in NSCLC. Other common fusion partners include SLC34A2-ROS1 (17%), TMP3-ROS1 (tropomyosin 3; 15%), SDC4-ROS1 (syndecan 4; 11%), EZR-ROS1 (ezrin; 6%), and FIG-ROS1 (3%). Less common fusions, occurring at frequencies of 1% or less, include CCDC6-ROS1 (coiled-coil domain containing 6), LRIG3-ROS1 (leucine-rich repeats and immunoglobulin-like domains 3), KDELR2-ROS1 (KDEL endoplasmic reticulum protein retention receptor 2 gene), MSN-ROS1 (moesin gene), CLTC-ROS1 (clathrin heavy chain gene), TPD5L1-ROS1 (tumor protein D52-like gene), TMEM106B-ROS1 (transmembrane protein 106B gene), and LIMA1-ROS (LIM domain and actin-binding 1 gene) [29].
Signaling Pathway
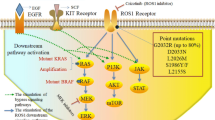
Once ROS1 becomes activated, either by its (unknown) ligand or via constitutive action from a fusion, a number of signaling pathways are triggered. The key rate-limiting step for this process is thought to be autophosphorylation of ROS1 and phosphorylation of the SH-2 domain-containing phosphatase-2 (SHP-2) [30]. Constitutive activation from a fusion then leads downstream signaling via several oncogenic pathways, including MAPK/ERK, PI3K/AKT, JAK/STAT3, and VAV3; this leads to significant changes in cell differentiation, proliferation, growth, and survival [3] (Fig. 2). Preclinical work has suggested that the activation of downstream signaling pathways depends on the fusion partner of ROS1. CD74-ROS1 but not FIG-ROS1 has been found to lead to phosphorylation of E-Syt1, which in turn led to an invasive phenotype in the CD74-ROS1 cells [31].

ROS signaling pathways. (Figure from Acquaviva et al. [3])
Epidemiology
Patients with ROS1 fusions have been found to be more likely to be younger, female, and never-smokers than ROS1-negative patients—a similar profile to patients with EGFR activating mutations and ALK rearrangements [32,33,34]. Interestingly, however, the pattern of spread for ROS1 rearranged disease appears to be different than that for ALK; ROS fusions are associated with lower rates of extrathoracic disease, including brain metastases, at initial metastatic diagnosis [35]. The vast majority of cases are adenocarcinoma, although there are rare reports of other histologies such as squamous or large cell [19, 36, 37]. The most common histologic patterns associated with ROS1 fusion adenocarcinoma are solid growth with hepatoid cytology and acinar growth with cribriform structure. Other pathologic characteristics include mucinous features, signet ring cells, and psammomatous calcifications [38]. The ROS1 rearrangement usually occurs without other known oncogenic drivers, although there have been rare reported cases of concurrent mutations such as EGFR, KRAS, BRAF, MET, and PIK3CA [19, 27, 39,40,41,42].
Prevalence of NSCLC ROS1 fusion tumor ranges in the literature from 0.9% to 2.1%, though studies are always limited by their screening technique and therefore may miss cases with rare or new fusions. Worldwide prevalence is 1.9% [29].
Detection
FISH
Detection of ROS1 rearrangements by fluorescence in situ hybridization (FISH) has been considered the gold standard. It was used in the landmark phase I study that resulted in approval of crizotinib for ROS-1 rearranged NSCLC [42]. The centromeric (3′) part of the fusion breakpoint is labeled with a one fluorochrome and the telomeric (5′) part with another of a different color. The criteria for ROS1 FISH identification in NSCLC is the same as that for ALK rearrangement. The first positive pattern is a classic break-apart pattern, in which there is a single fusion signal and two separated 3′ and 5′ signals. The second is an atypical pattern, with an isolated 3′ signal—usually one fusion signal and one isolated 3′ signal without the corresponding 5′ signal [43]. FISH testing may be performed either on biopsy or cytologic specimens. To be considered FISH positive, at least 15% of evaluated tumor cells must contain split or isolated 3′ signals [44, 45]. Interestingly, a limitation of FISH has been found to be an inability to detect small intrachromasomal deletions, which can lead to false-negative or false-positive results [39, 46].
IHC
Immunohistochemistry (IHC) can be used as a screening technique. It is less expensive and faster than performing FISH. The D4D6 rabbit monoclonal assay is commercially available (Cell Signaling Technology, Danvers, MA, USA). It is applied at different dilutions ranging from 1:50 to 1:1000. In a number of different studies, IHC has been found to have a sensitivity of near 100% and a specificity of between 85% and 100%; specificity varies depending on interpretive cutoffs and method used [19, 43, 45, 47,48,49].
Unfortunately, ROS1 IHC can be somewhat challenging to interpret. Expression can be seen in osteoclast-like giant cells adjacent to ROS-1 un-mutated tumor cells, as well as in reactive pneumocytes and alveolar macrophages [43, 49]. Staining patterns can vary depending on different intracellular localization of the ROS1 fusions [19, 43]. Results can vary depending on the performing laboratory [50]. Because of this, while it makes for an excellent screening tool, it is important to perform confirmatory testing with FISH or another testing modality.
RT-PCR
Real-time PCR (RT-PCR) utilizes specific primer sets to detect and identify known fusion variants. RT-PCR-based detection of some of the most common ROS1 fusion genes (SLC34A2, SDC, CD74, EZR, TPM3, LRIG3, GOPC) at exons 32, 34, 35, and 36 has been successfully performed with a sensitivity of 100% and specificity of 85–100% with respect to FISH [41, 51]. While this is a relatively easy, rapid, and inexpensive test, it does have some drawbacks. As the list of ROS1 fusion proteins is large and growing, RT-PCR is likely to miss rare or previously unknown variants. It can also be challenging to obtain sufficient good quality RNA from the formalin-fixed paraffin-embedded tissue samples (FFPE) [52].
In recent years, the nCounter platform (NanoString Technologies; Seattle, WA) has emerged as a clinical option. It is a multiplexed assay that can identify known fusion gene variants via the interrogation of imbalanced 5′/3′ expression levels as well as the direct detection of fusion transcript variants. It has shown a good concordance with both IHC and FISH results for ROS1 fusion detection [53, 54].
NGS
Next-generation sequencing, or NGS, enables sensitive and specific assessments of multiple genomic regions at once, allowing for detection of both known and novel fusions [45]. Several ROS1 fusions have been identified using NGS [55,56,57,58,59]. A recent study performed next-generation sequencing on 319 FFPE samples and found 100% sensitivity and specificity when compared to reference FISH assays [60]. This method allows for multiplexed detection of molecular aberrations in NSCLC in a single test instead of multiple assays; however, hybrid-based capture NGS methods may have decreased sensitivity in detecting gene fusions [45].
Targeted Therapies
Crizotinib
Crizotinib (previously PF-0234106; brand name Xalkori, Pfizer, New York, USA) is a small molecule multikinase inhibitor (Table 1). It was initially developed as an inhibitor of c-MET but was further explored against a panel of over 120 diverse kinases and was found to be almost 20 times more selective for ALK and MET as compared to other evaluated kinases [61]. Following a phase I trial and initial efficacy results from a phase II trial (PROFILE 1001) that showed 50% response rates, it was approved by the FDA for use in metastatic NSCLC with ALK rearrangements in 2011 [62, 63]. Preclinical investigation of NSCLC cell lines, including HCC78 (SLC34A2-ROS1), revealed dose-dependent inhibition with crizotinib; inhibition of ROS1 led to subsequent inhibition of its downstream targets and apoptosis of the cell line [64]. This combined with ALK and ROS1’s known homology [6] with shared high-binding affinity to crizotinib [65] and case reports of response to crizotinib in patients with ROS1-mutated NSCLC [33, 37] led to the incorporation of patients with ROS-1 rearranged NSCLC into the expansion cohort of the phase I PROFILE 1001 study. This landmark study included 50 patients with ROS1 rearranged NSCLC. Overall response rate (ORR) to crizotinib was 72% with a median duration of response (DOR) of 17.6 months, median progression-free survival (PFS) of 19.2 months, and a disease control rate (DCR) of 90% [42]. Interestingly DOR to crizotinib in ALK-rearranged patients is only 49.1 weeks, with a median PFS of 9.7 months, suggesting that crizotinib may be a more potent inhibitor of ROS1 than ALK [42, 66]. Toxicities in this study were similar to those previously described; the most common grade 3 events were hypophosphatemia (10%), neutropenia (10%), and an elevated aminotransferase activity (10%). No grade 4 or 5 events were seen [42]. Based on this study, crizotinib was approved by the FDA for treatment of metastatic NSCLC with ROS1 rearrangement in March 2016.
There have been several other studies that yielded overall similarly promising results. The retrospective EUROS1 study identified 32 patients with advanced NSCLC who had positive ROS1 rearrangement by FISH and who had received crizotinib, 30 of whom were evaluable. ORR was 80% and DCR 86.7%, and median PFS was 9.1 months; PFS at 1 year was 44% [67]. Preliminary results from the French phase II ACSé trial prospectively looked at 34 patients with ROS1-rearranged NSCLC who were given crizotinib, 24 of whom were evaluable at the time of preliminary analysis. ORR was 63%, and DCR 88% [68]. Preliminary results from the prospective European phase II EUCROSS study similarly looked at 34 patients with ROS1-rearranged NSCLC (by FISH) who were given crizotinib, 29 of whom were eligible for efficacy assessment and 20 of whom had tumor tissue available for further sequencing. Of the patients who underwent additional sequencing, 19 tested positive for the ROS1 fusion. ORR was 69% in the overall trial population and 83% in those ROS-1 positive by next-generation sequencing [69]. A phase II study in East Asian patients included 127 patients with ROS1-rearranged NSCLC who received crizotinib. ORR was 71.7%, with a median PFS of 15.9 months and a median duration of response of 19.7 months [70]. All studies included patients who had received varying numbers of prior therapies and who were overall fairly heavily pretreated, though crizotinib was their first tytyrosine kinase inhibitor (TKI).
Resistance to ROS1 TKI
While ROS-1 mutated tumors initially respond well to targeted therapy with crizotinib, most patients inevitably develop resistance. The mechanism of resistance can be broken down into two major categories: mutations within the ROS1 kinase domain or bypass signaling pathways [35].
There are two major mechanisms by which kinase domain mutations appear to confer crizotinib resistance. The first is by a gatekeeping mechanism that directly interferes with the combination of ROS1 tyrosine kinase and crizotinib, leading to resistance. The second is a solvent front mutation in the kinase domain adjacent to the crizotinib-binding site; these confer resistance via steric interference [71] (Table 2).
The first kinase domain mutation described is also the one that has been most frequently observed. A 48-year-old woman with CD74-ROS1-rearranged NSCLC was started on crizotinib with excellent response. However after 3 months of therapy she was found to have progressive disease. Biopsy upon progression revealed the persistence of the ROS1 rearrangement by FISH but RT-PCR sequencing revealed a c6094G→A, p.Gly2032Arg (G2032R) mutation that was not noted on her pretreatment biopsy. This mutation is analogous to the ALK G1202R mutation. Biopsy was repeated at autopsy, and all sites examined harbored this mutation, suggesting that it was an early event in the clonal evolution of resistance [72]. Crizotinib was designed to bind ROS1 at the ATP-binding site that sits within the cleft between the N and C terminal domains of the kinase [73]. Crystal structure analysis revealed that the G2032 residue sits at the solvent front of the kinase hinge (solvent-exposed region of the kinase). G2032R causes steric hindrance with the piperidine ring of crizotinib while still allowing for ATP binding and therefore oncogenic kinase activity [72]. One recent evaluation of 16 patients with ROS1-positive advanced NSCLC with a total of 17 repeat biopsies after progression identified G2032R mutations in 41% of the biopsy specimens [35].
Following the identification of this initial resistance mutation, a multitude of others have been identified in clinical samples. A patient with CD74-ROS1-rearranged NSCLC who progressed on crizotinib was found to acquire the solvent-front mutation D2033N. Upon crystal modeling, this mutation was noted to interfere with the favorable interaction of the ATP-binding site with the protonated piperidine region of crizotinib. It is analogous to the ALK D1203N mutation. This patient proceeded to respond to cabozantinib; upon crystal modeling, cabozantinib was not found to interact with this altered 2033 residue [74]. Multiple other mutations have been discovered in vitro but not yet been replicated in the clinical setting.
Additional kinase domain mutations have been reported. These include S1986Y/F, which leads to alterations in the alpha C helix of the kinase domain and therefore steric interference with drug binding [35, 75]. It is analogous to the ALK C1156Y substitution [76]. L1951R is a solvent front mutation without an analogous ALK mutation. L2026M is a gatekeeper mutation in the ATP pocket that impedes drug binding and is analogous to ALK L1196M [71, 77]. Interestingly when the L2026M, L1951R, and G2032R mutations were evaluated in vivo, the mutations associated with highest crizotinib resistance were those located close to the crizotinib-binding domain—G2032R and L1951 [71].
Less information is available about off-target mechanisms of crizotinib resistance. The best described is a mechanism by which cancer cells achieve resistance via activation of an alternative signaling pathway (bypass pathway). One case report describes the appearance of a BRAF V600E mutation in a woman with SDC-ROS1 fusion NSCLC who had developed resistance to crizotinib; this mutation was not present on her initial biopsy. She was started on dabrafinib and trametinib but died 11 days later [78]. Similarly a new KIT p.D816G mutation was found after progression on crizotinib in a patient with ROS1 fusion NSCLC [79]. Two case reports describe patients with alterations in KRAS; one described a new point mutation in KRAS pG12D accompanied by KRAS gene amplification found on progression biopsy. Patient was treated with the MEK inhibitor selumetinib as well as pemetrexed and was alive at the time of article submission [80]. The second report only noted focal KRAS amplification seen in a ROS1 fusion tumor biopsy of a patient who had progressed on crizotinib, though this analysis was hampered by the lack of pretreatment sample to see if this was truly a bypass mutation [81]. Another patient with CD74-ROS1 fusion NSCLC who had progressed on crizotinib was found on next-generation sequencing to have a novel point mutation of the PIK3CA gene (pL531P) that led to activation of the mTOR signaling pathway; patient was placed on an mTOR signaling pathway inhibitor but passed shortly thereafter [82]. One study that performed next-generation sequencing on 12 ROS1 fusion patients who had progressed on crizotinib identified the same KIT D816G mutation previously characterized. It also noted a HER2 (ERBB2) mutation, though no pre-crizotinib samples were available for comparison. It also noted a β-catenin CTNNB1 S45F mutation that had previously been hypothesized as a potential oncogenic driver, though they were not able to evaluate pretreatment tissue in this patient to prove its presence as a bypass mutation [77, 83]. Another study created a cell line from a patient who had developed acquired resistance to crizotinib; the cell line revealed a switch in the control of growth and survival signaling pathways from ROS1 to EGFR in the resistant cell line, though this has not yet been verified in the clinic [84, 85].
Less is known about the potential for phenotypic changes leading to resistance. In EGFR and ALK fusion cancers, histologic transformation from adenocarcinoma to small cell cancer has been observed as a mechanism for TKI resistance, but this has yet to be demonstrated in ROS1 fusion NSCLC [86]. One preclinical study that took tumor tissue from NSCLC patients who had progressed on crizotinib noted evidence of epithelial to mesenchymal transition by way of upregulated vimentin and downregulated E-cadherin. A similar finding was noted in HCC78CR1-2 cell clones, though they also harbored a L2155S mutation that had previously been found to confer crizotinib resistance in cell lines [85].
Ceritinib
Ceritinib (previously LDK378l, brand name Zykadia; Novartis Pharmaceuticals) is an oral small molecule tyrosine kinase inhibitor of ALK [87] (Table 1). Preclinical studies suggested that it would inhibit ROS1 as well [88, 89]. A Korean phase II study evaluated 32 patients with advanced NSCLC who tested positive for ROS1 rearrangement by FISH. All but two of them were crizotinib-naïve. They received ceritinib 750 mg daily. ORR was 62%, with 1 complete response (CR) and 19 partial responses (PR). DOR was 21 months, with a DCR of 81%. Median PFS was 9.3 months overall and 19.3 months in crizotinib-naïve patients. Median overall survival (OS) was 24 months. Eight patients entered the trial with metastases to the brain; intracranial disease control was obtained in five (63%) of them, with an intracranial ORR of 25%. Of note, at the beginning of the trial two patients who had previously received treatment with crizotinib were enrolled. Neither were available for objective response—one passed due to suspected leptomeningeal disease, and one withdrew from the trial 2 weeks after their first dose due to grade 3 weakness and anorexia. However, neither of them showed signs of clinical improvement after initiation of ceritinib, and the protocol was subsequently amended to only enroll crizotinib-naïve patients who had previously been treated with at least one chemotherapeutic agent [89].
Adverse events in this study were primarily grade 1-2, the most common of which were diarrhea (78%), nausea (59%), and anorexia (56%)—all of which occurred at higher frequencies than with crizotinib [42, 89]. A recent randomized phase I study of 137 patients with metastatic ALK-mutated NSCLC found that ceritinib 450 mg taken with a low-fat meal resulted in fewer GI toxicities as compared with the standard 750 mg taken fasting and was associated with comparable plasma levels when assessed pharmacokinetically [90]. Ceritinib is not FDA approved for management of ROS1-rearranged advanced NSCLC, but it is noted as an option for front-line therapy per NCCN guidelines.
Cabozantinib
Cabozantinib (PF-06463922; brand name Cabometyx; Exelixis, Alameda, CA) is an oral multikinase inhibitor with CNS penetration (Table 1). It is FDA approved for use in medullary thyroid cancer and as a second-line agent in advanced renal cell carcinoma. In vitro studies found it to exhibit excellent activity against both the wild-type ROS1 fusion and the G2032R and G2026M mutations at concentrations less than 30 nmol/L—a dose much lower than what is clinically achievable [71, 91]. It has been found to inhibit CD74-ROS1-transformed Ba/F3 cells with more potency than entrectinib, brigatinib, lorlatinib [92], or foretinib [71].
One case report described a 50-year-old woman with metastatic NSCLC who progressed after platinum-based therapy and was found to have a ROS1 fusion. She was treated with crizotinib and progressed, at which point she was found to have the ROS1 D2033N mutation within her ROS1 kinase domain. She was started on crizotinib 60 mg orally daily and achieved PR by 4 weeks and near CR by 12 weeks (92% reduction in disease burden). At the time of paper publication, she remained on therapy (near 8 months duration). In vitro analysis of CD74-ROS1 cells with D2033N mutation found significantly more suppression with cabozantinib than ceritinib, brigatinib, and lorlatinib, though they remained highly sensitive to foretinib [74]. A recent abstract evaluated HCC78R cell lines with SLC34A2-ROS1 and ABC-20 cell lines harboring CD74-ROS1 (resistant to crizotinib). NGS evaluation found both an upregulation of HB-EGF and activation of the EGFR signaling pathway as well as an upregulation of AXL. The combination of cabozantinib and gefitinib was found to inhibit the growth of HCC28R tumors in an in vivo NOG mice model [93].
Unfortunately, cabozantinib is associated with a number of toxicities. The landmark METEOR trial which evaluated its use in renal cell carcinoma noted that 71% of patients experienced grade 3 or 4 events, the most common of which were hypertension (15%), diarrhea (13%), fatigue (11%), and palmar-plantar erythrodysesthesia syndrome (8%). Sixty-two percent of patients required dose reductions [94].
Entrectinib
Entrectinib (RXDX-101, Ignyta Pharmaceuticals, San Diego, CA, USA) is a small molecule that inhibits the tyrosine kinases TRKA/B/C, ROS1, and ALK (Table 1). It has a preclinical median inhibitory concentration (IC50) of 7 nm against ROS1, higher than crizotinib [95, 96]. Entrectinib was specifically designed to cross the blood-brain barrier [95]. Two recent phase I studies (ALKA-372-001 and STARTRK-1) evaluated entrectenib in patients with advanced solid tumors. Fourteen patients with ROS1-rearranged solid tumors (all NSCLC except one melanoma) were evaluated. These patients were all crizotinib-naïve. ORR was 86%, with an intracranial ORR of 100% (in the two ROS1 fusion patients evaluated). Median PFS was 19 months. Interestingly, six patients with ROS-1-rearranged disease who had previously received crizotinib were not observed to have any response to entrectinib [97]. Preliminary phase II data was recently reported, in which 32 patients with ROS1 fusion proven NSCLC (by NGS) who were naïve to prior TKI therapy were given 600 mg by mouth of entrectinib daily in 4 weeks cycles. ORR was 75% with three complete responses, intracranial ORR 71%, median PFS 19.1 months. The most common treatment-related adverse events were fatigue/asthenia (34%), dysguisia (34%), and dizziness (24%) [98]. There has been no preclinical activity demonstrated against ROS1 resistance mutations G2032 or L2026; this combined with the lack of response in crizotinib-pretreated patients as noted above suggests that entrectinib’s role in treating crizotinib-resistant disease may be limited unless progression is only in the CNS [92].
Lorlatinib
Lorlatinib (PF-06463922, Pfizer Oncology, Groton, CT, USA) is an oral TKI that targets both ALK and ROS1 with high affinity and good CNS penetration [91] (Table 1). Phase I data has been published looking at lorlatinib in NSCLC with ALK or ROS1 rearrangement; patients were allowed to have both CNS disease and prior TKI therapy. In this study, 12 patients had ROS1 rearrangements, 7 of whom were pretreated with crizotinib. ORR was 50% [99]. Preliminary data has been presented from the phase II component of this study; the ROS1 cohort contained 47 patients, each of whom was treated with lorlatinib 100 mg daily. Regardless of prior treatment, ORR was 36%, with intracranial ORR 56%. The most common treatment-related adverse events and grade 3/4 adverse events were hypercholesterolemia (81%/16%) and hypertriglyceridemia (60%/16%) [100].
Lorlatinib is intriguing because of its activity against several crizotinib-resistant mutations. Dong et al. published a case report of a 57-year-old gentleman with a history of stage IIIB lung adenocarcinoma who initially went into remission following platinum-based chemotherapy but then relapsed and was found to have an EZR-ROS1 mutation. He initially responded well to crizotinib, with PFS of 6 months. After disease progression, he was started on lorlatinib 100 mg daily with favorable response after 3 months; he remained on drug at time of article publication [101]. Mutation type was not assessed in that publication, but another case report described an excellent response to lorlatinib in a patient who had the crizotinib- and ceritinib-resistant mutations S1986Y/F [75]. Additional cell-based assays have described sensitivity in the setting of D2033N [74] and L2026M [91] mutations. It is less clear what the role of lorlatinib is in the setting of the G2032 mutation; in preclinical studies this mutation has been found to significantly reduce lorlatinib’s potency though activity still remained overall robust. ROS1-rearranged BA/F3 cells with the G2032 mutation have been found to have an IC50 of 508 nM as compared to 0.5 nM in wild-type ROS1 [91].
Ropotrectinib
Ropotrectinib (TPX-0005; TP Therapeutics, San Diego, CA, USA) is a next-generation ROS1 inhibitor, a novel three-dimensional macrocyle with a much smaller size (MW <370) than current ROS1 inhibitors (Table 1). It was specifically designed to overcome resistance mutations. Preclinical studies have shown activity against gatekeeper and solvent mutations, including G2032R, D2033N, L2026M, S1986F/Y, L1951R, and kinases involved in bypass signaling such as focal adhesion kinase, SRC proto-oncogene, and non-receptor tyrosine kinase [102, 103]. Preliminary results have been reported from the phase I TRIDENT study. It included patients with ALK, ROS1, or NTRK1-3 fusion-positive advanced solid tumors. Patients could be either TKI pretreated or naïve, and brain metastases were allowed. At the time of report of preliminary results, 29 ROS1 patients were enrolled. Confirmed PR have been observed in both TKI-naïve and pre-treated ROS1/NTRK+ patients at all dose levels, including one crizotinib refractory ROS1 G2032R+ patient with untreated CNS metastases. Median duration of clinical PR was 6.7 months with 88% (7 out of 8) responses ongoing. Toxicities have been tolerable, with the majority of adverse events remaining at grade 1–2; most common include dysgusia (38%), dizziness (35%), paresthesia (24%), and nausea (12%) [104].
DS-6051b
DS-6051b (Daiichi Sankyo, Japan) is an oral small molecule tyrosine kinase inhibitor that has demonstrated preclinical activity against ROS1 and NTRK1-3 rearrangements [105] (Table 1). A phase I trial evaluated 15 Japanese patients with NSCLC harboring ROS1 fusions. ORR was 58.3% in the 12 patients with target lesions and 66.7% in the 9 patients who were crizotinib-naïve; DCR was 100%. Common toxicities included transaminitis (80%), diarrhea (53.3%), and nausea (46.7%). Maximal tolerated dose and recommended phase II dose was 600 mg by mouth daily [106]. Preliminary data was recently presented for a phase I trial of DS-6051b in advanced solid tumors conducted in the United States. 35 patients were enrolled, with 31 tumors evaluable. Nine patients had ROS1 fusions, including seven patients who had NSCLC and who had previously received crizotinib. Of the six evaluable NSCLC ROS1-rearranged patients who had previously received crizotinib, two patients had PR, and two had stable disease (SD). DS-6051b was noted to be tolerable up to 800 mg by mouth daily, with the primary adverse events being gastrointestinal (89%) [107].
Brigatinib
Brigatinib (AP26113, brand name Alunbrig; ARIAD Pharmaceuticals, Cambridge, MA, USA) is an inhibitor of both ALK and ROS1 fusion NSCLC (Table 1). In preclinical studies, it was found to inhibit viability of CD4-ROS1-expressing Ba/F3 cells with an IC50 of 7.5 nM, as compared to a IC50 of 9.8 nM in EMLA4-ALK cells [108, 109]. It was FDA approved for use in metastatic crizotinib-resistant ALK fusion NSCLC in April 2017. A single armed phase I/II trial evaluated patients with advanced malignancies including ALK-rearranged NSCLC refractory to currently available therapies. Three patients in this study had ROS1-rearranged NSCLC. Two of these patients had previously received crizotinib; one had progressive disease (PD), and one had SD. The single crizotinib-naïve ROS1-rearranged NSCLC patient experienced a partial response and was continuing to receive brigatinib at the time of data cutoff (21.6 mo of therapy) [110]. In a phase II trial of 222 patients with advanced ALK fusion NSCLC that had progressed on crizotinib, common treatment-related adverse events were noted to be nausea (33/44%), diarrhea (19/38%), headache (28/27%), and cough (13/34%) (brigatinib 90 mg daily/180 mg daily). A subset of patients were noted to have early onset pulmonary events (all grades 6%; grade ≥ 3 3%) [111]. Preclinical work examining CD74-ROS1 transformed BA/F3 cells in vivo has revealed that brigatinib exhibits activity against L2026M [92], but does not fare as well against D2033N [74], G2032R [71, 92, 109], or L1951R [71].
Foretenib
Foretinib (GSK1363089; GlaxoSmithKline) is an oral multikinase inhibitor that targets MET, VEGFR-2, RON, KIT, and AXL kinases. Preclinical data suggested that it was a potent inhibitor of ROS1 fusions. It also demonstrated effective inhibition against the G2032 mutation at clinically feasible concentrations [112]. However it has been found to be less potent and effective than cabozantinib, and further development of the drug was discontinued [113].
Chemotherapy and Immunotherapy
Pemetrexed (formerly LY231514, brand name Alimta, Eli Lilly, Indianapolis, Indiana, USA) is a folate-based antimetabolite that exerts its activity via inhibition of enzymes critical in purine and pyrimidine synthase. These include thymidine synthase (TS), dihydrofolate reductase, and glycinamine ribonucleotide formyltransferase [114]. Multiple studies have found that patients with ALK fusions have improved outcomes as compared to their wild-type colleagues [115]. The same appears to be true for patients with ROS1 gene rearrangements. One retrospective study of 25 patients who had received pemetrexed (with or without bevacizumab) for 12 months or longer as therapy for their advanced stage non-squamous NSCLC included 5 patients with a ROS1 gene rearrangement. Median OS was 42.2 months with median PFS of 22.1 months; patients with an oncogenic driver mutation (including but not limited to ROS1) had a statistically significant improvement in their PFS (p = 0.006) and OS (p = 0.001) compared to wild type [115]. Another retrospective study looked at four patients with metastatic NSCLC and FISH-detected ROS1 rearrangement who received pemetrexed. PFS ranged from 18 to more than 47 months [116]. A different retrospective study evaluated 253 patients with advanced NSCLC who were screened for driver mutations using RT-PCR. 19 patients (7.5%) had ROS1 fusions. These patients were noted to have a better ORR (57.9%, p = 0.026), DCR (89.5%, p = 0.033), and PFS (7.5 mo; p = 0.003) as compared to patients with other driver mutations. Interestingly, while low levels of TS have historically been considered a favorable marker for pemetrexed efficacy in NSCLC, in this population this effect was not seen [117].
Although, PD(L)1 blockade has revolutionized the treatment of advanced NSCLC both as single agent and in combination with chemotherapy, no clear data currently exists suggesting the efficacy of immunotherapy specifically in patients with ROS1 gene rearrangements. A phase I/II study evaluating the safety and tolerability of nivolumab plus crizotinib in the treatment of patients with metastatic NSCLC and ALK fusions was stopped early due to the degree of toxicity observed [118]. A recent phase II trial evaluated the use of pembrolizumab in NSCLC patients with both EGFR mutated and PD-L1-positive disease; study was similarly terminated early due to lack of efficacy even in patients with PD-L1 expression ≥50% [119]. Extrapolating from EGFR-mutant NSCLC which, like ROS1, is also associated with patients who have not smoked, a recent meta-analysis evaluating three trials found that the use of single agent PD(L)1 inhibitors failed to improve overall survival in the EGFR mutant NSCLC, though survival was improved in wild-type lung cancers [120]. Another meta-analysis similarly revealed that in EGFR-mutated patients with metastatic NSCLC, PD-1/PD-L1 therapy is inferior to EGFR TKI in terms of progression-free survival [121]. Malignancies associated with tobacco smoking are frequently associated with a higher tumor mutational load and smoking-associated signatures that may underlie their improved response to immune checkpoint blockade [122]. As patients with driver oncogene mutations such as ROS1 are much less likely to have a history of tobacco smoking and low tumor mutational burden compared to patients with smoking-associated lung cancers, it is possible that this may explain inferior outcomes to single agent immunotherapy in these oncogene-driven tumors.
Interestingly, the recent IMpower150 trial that evaluated the addition of atezolizumab to the combination of carboplatin, paclitaxel, and bevacizumab (BCP) in patients with metastatic non-squamous NSCLC found a significantly improved PFS and OS as compared to the non-atezolizumab arm. This included patients who had received TKIs, irrespective of ALK or EGFR mutational status [123]. In the subgroup analysis, addition of atezolizumab in patients with EGFR exon19 deletion or L858R mutation led to improved PFS (HR 0.41; 95% CI 0.22–0.78) vs BCP alone [124]. Hopefully, one would expect comparable results in ROS1 NSCLC.
Conclusions
With the data currently available, first-line therapy for patients with advanced ROS1-rearranged NSCLC should be crizotinib. While resistance mutations such as G2032R can pose a treatment challenge, there are a number of next-generation TKIs that may assist in the management of these patients. Although immunotherapy likely does not appear to provide benefit as a monotherapy, immune therapy combinations warrant further study. Additional studies will need to be performed to fully define the role of next-generation TKIs, combination-targeted therapies against ROS1, and bypass tract mechanisms or resistance in ROS1-rearranged NSCLC that become resistant to crizotinib.
References
Matsushime H, Wang LH, Shibuya M. Human c-ros-1 gene homologous to the v-ros sequence of UR2 sarcoma virus encodes for a transmembrane receptorlike molecule. Mol Cell Biol. 1986;6(8):3000–4.
Birchmeier C, et al. Characterization of an activated human ros gene. Mol Cell Biol. 1986;6(9):3109–16.
Acquaviva J, Wong R, Charest A. The multifaceted roles of the receptor tyrosine kinase ROS in development and cancer. Biochim Biophys Acta. 2009;1795(1):37–52.
Satoh H, et al. Regional localization of the human c-ros-1 on 6q22 and flt on 13q12. Jpn J Cancer Res. 1987;78(8):772–5.
Nagarajan L, et al. The human c-ros gene (ROS) is located at chromosome region 6q16—6q22. Proc Natl Acad Sci U S A. 1986;83(17):6568–72.
Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19(49):5548–57.
Raabe T. The sevenless signaling pathway: variations of a common theme. Biochim Biophys Acta. 2000;1496(2–3):151–63.
Hart AC, et al. Induction of cell fate in the Drosophila retina: the bride of sevenless protein is predicted to contain a large extracellular domain and seven transmembrane segments. Genes Dev. 1990;4(11):1835–47.
Sonnenberg E, et al. Transient and locally restricted expression of the ros1 protooncogene during mouse development. EMBO J. 1991;10(12):3693–702.
Tessarollo L, Nagarajan L, Parada LF. c-ros: the vertebrate homolog of the sevenless tyrosine kinase receptor is tightly regulated during organogenesis in mouse embryonic development. Development. 1992;115(1):11–20.
Sonnenberg-Riethmacher E, et al. The c-ros tyrosine kinase receptor controls regionalization and differentiation of epithelial cells in the epididymis. Genes Dev. 1996;10(10):1184–93.
Birchmeier C, et al. Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc Natl Acad Sci U S A. 1990;87(12):4799–803.
Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci U S A. 1987;84(24):9270–4.
Charest A, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer. 2003;37(1):58–71.
Charest A, et al. Association of a novel PDZ domain-containing peripheral Golgi protein with the Q-SNARE (Q-soluble N-ethylmaleimide-sensitive fusion protein (NSF) attachment protein receptor) protein syntaxin 6. J Biol Chem. 2001;276(31):29456–65.
Charest A, et al. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc Natl Acad Sci. 2003;100(3):916–21.
Birch AH, et al. Chromosome 3 anomalies investigated by genome wide SNP analysis of benign, low malignant potential and low grade ovarian serous tumours. PLoS One. 2011;6(12):e28250.
Gu TL, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6(1):e15640.
Rimkunas VM, et al. Analysis of receptor tyrosine kinase ROS1-positive tumors in non-small cell lung cancer: identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18(16):4449–57.
Lovly CM, et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014;4(8):889–95.
Yamamoto H, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016;69(1):72–83.
Lee J, et al. Identification of ROS1 rearrangement in gastric adenocarcinoma. Cancer. 2013;119(9):1627–35.
Aisner DL, et al. ROS1 and ALK Fusions in colorectal cancer, with evidence of intratumoral heterogeneity for molecular drivers. Mol Cancer Res. 2014;12(1):111–8.
Giacomini CP, et al. Breakpoint analysis of transcriptional and genomic profiles uncovers novel gene fusions spanning multiple human cancer types. PLoS Genet. 2013;9(4):e1003464.
Ritterhouse LL, et al. ROS1 rearrangement in thyroid cancer. Thyroid. 2016;26(6):794–7.
Rossing M, et al. Genomic diagnostics leading to the identification of a TFG-ROS1 fusion in a child with possible atypical meningioma. Cancer Genet. 2017;212–213:32–7.
Wiesner T, et al. Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat Commun. 2014;5:3116.
Rikova K, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–203.
Pal P, Khan Z. Ros1-1. J Clin Pathol. 2017;70(12):1001–9.
Charest A, et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006;66(15):7473–81.
Jun HJ, et al. The oncogenic lung cancer fusion kinase CD74-ROS activates a novel invasiveness pathway through E-Syt1 phosphorylation. Cancer Res. 2012;72(15):3764–74.
Wu S, et al. Clinicopathological characteristics and outcomes of ROS1-rearranged patients with lung adenocarcinoma without EGFR, KRAS mutations and ALK rearrangements. Thorac Cancer. 2015;6(4):413–20.
Bergethon K, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30(8):863–70.
Marchetti A, et al. ROS1 gene fusion in advanced lung cancer in women: a systematic analysis, review of the literature, and diagnostic algorithm. JCO Precis Oncol. 2017;1:1–9.
Gainor JF, et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis Oncol. 2017:1–13.
Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res. 2013;19(15):4040–5.
Davies KD, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18(17):4570–9.
Zhao J, et al. Advanced lung adenocarcinomas with ROS1-rearrangement frequently show hepatoid cell. Oncotarget. 2016;7(45):74162–70.
Lin JJ, et al. ROS1 fusions rarely overlap with other oncogenic drivers in non-small cell lung cancer. J Thorac Oncol. 2017;12(5):872–7.
Scheffler M, et al. ROS1 rearrangements in lung adenocarcinoma: prognostic impact, therapeutic options and genetic variability. Oncotarget. 2015;6(12):10577–85.
Cao B, et al. Detection of lung adenocarcinoma with ROS1 rearrangement by IHC, FISH, and RT-PCR and analysis of its clinicopathologic features. Onco Targets Ther. 2016;9:131–8.
Shaw AT, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71.
Bubendorf L, et al. Testing for ROS1 in non-small cell lung cancer: a review with recommendations. Virchows Arch. 2016;469(5):489–503.
Yoshida A, et al. ROS1-rearranged lung cancer: a clinicopathologic and molecular study of 15 surgical cases. Am J Surg Pathol. 2013;37(4):554–62.
Lindeman NI, et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018;142(3):321–46.
Savic S, Bubendorf L. Role of fluorescence in situ hybridization in lung cancer cytology. Acta Cytol. 2012;56(6):611–21.
Su Y, et al. Immunohistochemical detection of ROS1 fusion. Am J Clin Pathol. 2017;147(1):77–82.
Yoshida A, et al. Immunohistochemical detection of ROS1 is useful for identifying ROS1 rearrangements in lung cancers. Mod Pathol. 2014;27(5):711–20.
Sholl LM, et al. ROS1 immunohistochemistry for detection of ROS1-rearranged lung adenocarcinomas. Am J Surg Pathol. 2013;37(9):1441–9.
Fischer AH, et al. Immunohistochemistry practices of cytopathology laboratories: a survey of participants in the College of American Pathologists Nongynecologic Cytopathology Education Program. Arch Pathol Lab Med. 2014;138(9):1167–72.
Shan L, et al. Detection of ROS1 gene rearrangement in lung adenocarcinoma: comparison of IHC, FISH and real-time RT-PCR. PLoS One. 2015;10(3):e0120422.
Ribeiro-Silva A, Zhang H, Jeffrey SS. RNA extraction from ten year old formalin-fixed paraffin-embedded breast cancer samples: a comparison of column purification and magnetic bead-based technologies. BMC Mol Biol. 2007;8:118.
Lira ME, et al. A single-tube multiplexed assay for detecting ALK, ROS1, and RET fusions in lung cancer. J Mol Diagn. 2014;16(2):229–43.
Reguart N, et al. Identification of ALK, ROS1, and RET fusions by a multiplexed mRNA-based assay in formalin-fixed, paraffin-embedded samples from advanced non–small-cell lung cancer patients. Clin Chem. 2017;63(3):751–60.
Zhu VW, et al. TPD52L1-ROS1, a new ROS1 fusion variant in lung adenosquamous cell carcinoma identified by comprehensive genomic profiling. Lung Cancer. 2016;97:48–50.
Zhu YC, et al. CEP72-ROS1: a novel ROS1 oncogenic fusion variant in lung adenocarcinoma identified by next-generation sequencing. Thorac Cancer. 2018;9(5):652–5.
Hartmaier RJ, et al. High-throughput genomic profiling of adult solid tumors reveals novel insights into cancer pathogenesis. Cancer Res. 2017;77(9):2464–75.
Ou SH, et al. Identification of a novel TMEM106B-ROS1 fusion variant in lung adenocarcinoma by comprehensive genomic profiling. Lung Cancer. 2015;88(3):352–4.
Govindan R, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150(6):1121–34.
Zheng Z, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20(12):1479–84.
Christensen JG, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–22.
Kwak EL, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol. 2009;27(15S):3509.
Crinò L, et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol. 2011;29(15_suppl):7514.
Yasuda H, et al. Preclinical rationale for use of the clinically available multitargeted tyrosine kinase inhibitor crizotinib in ROS1-translocated lung cancer. J Thorac Oncol. 2012;7(7):1086–90.
Huber KVM, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature. 2014;508:222.
Camidge DR, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–9.
Mazières J, et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33(9):992–9.
Moro-Sibilot D, et al. Crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer (NSCLC). Preliminary results of the ACSé phase II trial. J Clin Oncol. 2015;33(15_suppl):8065.
Michels S, et al. MA07.05 EUCROSS: a european phase II trial of crizotinib in advanced adenocarcinoma of the lung harboring ROS1 rearrangements – preliminary results. J Thorac Oncol. 2017;12(1):S379–80.
Wu YL, et al. Phase II study of crizotinib in East Asian patients with ROS1-positive advanced non-small-cell lung cancer. J Clin Oncol. 2018;36(14):1405–11.
Katayama R, et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin Cancer Res. 2015;21(1):166–74.
Awad MM, et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N Engl J Med. 2013;368(25):2395–401.
Cui JJ, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–63.
Drilon A, et al. A novel crizotinib-resistant solvent-front mutation responsive to cabozantinib therapy in a patient with ROS1-rearranged lung cancer. Clin Cancer Res. 2016;22(10):2351–8.
Facchinetti F, et al. Crizotinib-resistant ROS1 mutations reveal a predictive kinase inhibitor sensitivity model for ROS1- and ALK-rearranged lung cancers. Clin Cancer Res. 2016;22(24):5983–91.
Friboulet L, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–73.
McCoach CE, et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin Cancer Res. 2018;24(14):3334–47.
Watanabe J, Furuya N, Fujiwara Y. Appearance of a BRAF mutation conferring resistance to crizotinib in non-small cell lung cancer harboring oncogenic ROS1 fusion. J Thorac Oncol. 2018;13(4):e66–9.
Dziadziuszko R, et al. An activating KIT mutation induces crizotinib resistance in ROS1-positive lung cancer. J Thorac Oncol. 2016;11(8):1273–81.
Zhu YC, et al. Concurrent ROS1 gene rearrangement and KRAS mutation in lung adenocarcinoma: a case report and literature review. Thorac Cancer. 2018;9(1):159–63.
Cargnelutti M, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6(7):5182–94.
Xu CW, et al. Patient harboring a novel PIK3CA point mutation after acquired resistance to crizotinib in an adenocarcinoma with ROS1 rearrangement: a case report and literature review. Thorac Cancer. 2017;8(6):714–9.
Shigemitsu K, et al. Genetic alteration of the beta-catenin gene (CTNNB1) in human lung cancer and malignant mesothelioma and identification of a new 3p21.3 homozygous deletion. Oncogene. 2001;20(31):4249–57.
Davies KD, et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One. 2013;8(12):e82236.
Song A, et al. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res. 2015;21(10):2379–87.
Lin JJ, Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer. 2016;2(7):350–64.
Shaw AT, et al. Ceritinib in ALK-rearranged non–small-cell lung cancer. N Engl J Med. 2014;370(13):1189–97.
Kim HR, et al. The frequency and impact of ROS1 rearrangement on clinical outcomes in never smokers with lung adenocarcinoma. Ann Oncol. 2013;24(9):2364–70.
Lim SM, et al. Open-label, multicenter, phase II study of ceritinib in patients with non-small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol. 2017;35(23):2613–8.
Cho BC, et al. ASCEND-8: a randomized phase 1 study of ceritinib, 450 mg or 600 mg, taken with a low-fat meal versus 750 mg in fasted state in patients with anaplastic lymphoma kinase (ALK)-rearranged metastatic non-small cell lung cancer (NSCLC). J Thorac Oncol. 2017;12(9):1357–67.
Zou HY, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A. 2015;112(11):3493–8.
Chong CR, et al. Identification of existing drugs that effectively target NTRK1 and ROS1 rearrangements in lung cancer. Clin Cancer Res. 2017;23(1):204–13.
Kato Y, et al. Combined effect of cabozantinib and gefitinib in crizotinib-resistant lung tumors harboring ROS1 fusions. Cancer Sci. 2018;109:3149.
Choueiri TK, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(7):917–27.
Ardini E, et al. Entrectinib, a Pan–TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol Cancer Ther. 2016;15(4):628–39.
Menichincheri M, et al. Discovery of entrectinib: a new 3-aminoindazole as a potent anaplastic lymphoma kinase (ALK), c-ros Oncogene 1 Kinase (ROS1), and pan-tropomyosin receptor kinases (Pan-TRKs) inhibitor. J Med Chem. 2016;59(7):3392–408.
Drilon A, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017;7(4):400–9.
Ahn M, et al. OA 14.06 entrectinib in patients with locally advanced or metastatic ROS1 fusion-positive non-small cell lung cancer (NSCLC). J Thorac Oncol. 2017;12(11):S1783.
Shaw AT, et al. Lorlatinib in non-small-cell lung cancer with ALK or ROS1 rearrangement: an international, multicentre, open-label, single-arm first-in-man phase 1 trial. Lancet Oncol. 2017;18(12):1590–9.
Solomon B, et al. OA 05.06 phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non-small-cell lung cancer. J Thorac Oncol. 2017;12(11):S1756.
Dong L, et al. Long-term progression-free survival in an advanced lung adenocarcinoma patient harboring EZR-ROS1 rearrangement: a case report. BMC Pulm Med. 2018;18(1):13.
Cui JJ, et al. TPX-0005, a novel ALK/ROS1/TRK inhibitor, effectively inhibited a broad spectrum of mutations including solvent front ALK G1202R, ROS1 G2032R and TRKA G595R mutants. Eur J Cancer. 2016;69:S32.
Cui JJ, et al. Abstract B185: TPX-0005, a supreme ROS1 inhibitor, overcomes crizotinib-resistant ROS1 mutations including solvent front mutation G2032R and gatekeeper mutation L2026M. Mol Cancer Ther. 2018;17(1 Supplement):B185.
Drilon AE, et al. A phase 1 study of the next-generation ALK/ROS1/TRK inhibitor ropotrectinib (TPX-0005) in patients with advanced ALK/ROS1/NTRK+ cancers (TRIDENT-1). J Clin Oncol. 2018;36(15_suppl):2513.
Kiga M, et al. Preclinical characterization and antitumor efficacy of DS-6051b, a novel, orally available small molecule tyrosine kinase inhibitor of ROS1 and NTRKs. Eur J Cancer. 2016;69:S35–6.
Fujiwara Y, et al. Safety and pharmacokinetics of DS-6051b in Japanese patients with non-small cell lung cancer harboring ROS1 fusions: a phase I study. Oncotarget. 2018;9(34):23729–37.
Kyriakos P, Papadopoulos LG, Janne PA, Ou S-HI, Shaw A, Goldberg TR, Greenberg J, Gu X, Tachibana M, Senaldi G, Shiga R, Zahir H, Nakamaru K, Borazanci E. First-in-human study of DS-6051b in patients (pts) with advanced solid tumors (AST) conducted in the US. J Clin Oncol. 2018;36(15_suppl):abstr 2514.
Squillace RM, et al. Abstract 5655: AP26113 possesses pan-inhibitory activity versus crizotinib-resistant ALK mutants and oncogenic ROS1 fusions. Cancer Res. 2013;73(8 Supplement):5655.
Davare MA, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci U S A. 2015;112(39):E5381–90.
Gettinger SN, et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: a single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(12):1683–96.
Kim D-W, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase–positive non–small-cell lung cancer: a randomized, multicenter phase II trial. J Clin Oncol. 2017;35(22):2490–8.
Davare MA, et al. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proc Natl Acad Sci U S A. 2013;110(48):19519–24.
Sgambato A, et al. Targeted therapies in non-small cell lung cancer: a focus on ALK/ROS1 tyrosine kinase inhibitors. Expert Rev Anticancer Ther. 2018;18(1):71–80.
Shih C, et al. LY231514, a pyrrolo[2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997;57(6):1116–23.
Liang Y, Wakelee HA, Neal JW. Relationship of driver oncogenes to long-term pemetrexed response in non–small-cell lung cancer. Clin Lung Cancer. 2015;16(5):366–73.
Riess JW, et al. A case series of lengthy progression-free survival with pemetrexed-containing therapy in metastatic non–small-cell lung cancer patients harboring ROS1 gene rearrangements. Clin Lung Cancer. 2013;14(5):592–5.
Chen YF, et al. Efficacy of pemetrexed-based chemotherapy in patients with ROS1 fusion-positive lung adenocarcinoma compared with in patients harboring other driver mutations in East Asian populations. J Thorac Oncol. 2016;11(7):1140–52.
Spigel DR, et al. Phase 1/2 study of the safety and tolerability of nivolumab plus crizotinib for the first-line treatment of anaplastic lymphoma kinase translocation positive advanced nonsmall cell lung cancer (CheckMate 370). J Thorac Oncol. 2018;13(5):682–8.
Lisberg A, et al. A phase II study of pembrolizumab in EGFR-mutant, PD-L1+, Tyrosine kinase inhibitor Naïve patients with advanced NSCLC. J Thorac Oncol. 2018;13(8):1138–45.
Lee CK, et al. Checkpoint inhibitors in metastatic EGFR-mutated non–small cell lung cancer—a meta-analysis. J Thorac Oncol. 2017;12(2):403–7.
Sheng Z, et al. The efficacy of anti-PD-1/PD-L1 therapy and its comparison with EGFR-TKIs for advanced non-small-cell lung cancer. Oncotarget. 2017;8(34):57826–35.
Kim JH, Kim HS, Kim BJ. Prognostic value of smoking status in non-small-cell lung cancer patients treated with immune checkpoint inhibitors: a meta-analysis. Oncotarget. 2017;8(54):93149–55.
Socinski MA, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Kowanetz M, et al. Abstract CT076: IMpower150: efficacy of atezolizumab (atezo) plus bevacizumab (bev) and chemotherapy (chemo) in 1L metastatic nonsquamous NSCLC (mNSCLC) across key subgroups. Cancer Res. 2018;78(13 Supplement):CT076.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Oesterich, L.G., Riess, J.W. (2019). ROS1. In: Salgia, R. (eds) Targeted Therapies for Lung Cancer. Current Cancer Research. Springer, Cham. https://doi.org/10.1007/978-3-030-17832-1_3
Download citation
DOI: https://doi.org/10.1007/978-3-030-17832-1_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-17831-4
Online ISBN: 978-3-030-17832-1
eBook Packages: MedicineMedicine (R0)