
Abstract
Combinations of antimicrobial agents are often used in the management of infectious diseases. Antimicrobial agents used as part of combination therapy are often selected empirically. As regrowth and the emergence of polymyxin (either colistin or polymyxin B) resistance has been observed with polymyxin monotherapy, polymyxin combination therapy has been suggested as a possible means by which to increase antimicrobial activity and reduce the development of resistance. This chapter provides an overview of preclinical and clinical investigations of CMS/colistin and polymyxin B combination therapy. In vitro data and animal model data suggests a potential clinical benefit with many drug combinations containing clinically achievable concentrations of polymyxins, even when resistance to one or more of the drugs in combination is present and including antibiotics normally inactive against Gram-negative organisms. The growing body of data on the emergence of polymyxin resistance with monotherapy lends theoretical support to a benefit with combination therapy. Benefits include enhanced bacterial killing and a suppression of polymyxin resistant subpopulations. However, the complexity of the critically ill patient population, and high rates of treatment failure and death irrespective of infection-related outcome make demonstrating a potential benefit for polymyxin combinations extremely challenging. Polymyxin combination therapy in the clinic remains a heavily debated and controversial topic. When combinations are selected, optimizing the dosage regimens for the polymyxin and the combinatorial agent is critical to ensure that the benefits outweigh the risk of the development of toxicity. Importantly, patient characteristics, pharmacokinetics, the site of infection, pathogen and resistance mechanism must be taken into account to define optimal and rational polymyxin combination regimens in the clinic.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
16.1 Introduction
Combinations of antimicrobial agents have been used in the management of infectious diseases since the 1940s [160]. Reasons for the use of antimicrobial combinations include prevention of resistance selection during treatment, decreased dose-related toxicity as a result of reduced dosage, broadening of spectrum in polymicrobial infections, and ‘synergy’ [183]. However, it remains controversial whether combination therapy , given empirically or as definitive treatment for many infection types, is warranted. There are also potential disadvantages with combination therapy including a greater risk of drug toxicity, increased cost, and superinfection with even more resistant bacteria [119]. Clinicians often resort to antibiotic combinations as a consequence of limited therapeutic options in the hope of improving the activity of available agents. In clinical practice, antimicrobial agents used as part of combination therapy are often selected empirically by clinicians, mainly by trial and error or based on personal experience. This approach is poorly guided and may not be optimal for patient care.
Polymyxin (colistin [administered as colistin methanesulphonate ; CMS] or polymyxin B ) combination therapy is increasingly used clinically [10, 11, 30, 51, 62, 78, 120, 139, 141, 142, 162]. However, systematic investigations of such combinations are a relatively recent phenomenon. As outlined in Chap. 15, the emerging pharmacodynamic (PD) and pharmacokinetic (PK) data on CMS/colistin and polymyxin B suggest that caution is required with monotherapy. Given this situation, polymyxin combination therapy has been suggested as a possible means by which to increase antimicrobial activity and reduce the development of resistance [63, 72, 99, 151].
The growing body of data on the emergence of polymyxin resistance with monotherapy lends theoretical support to a benefit with combination therapy. As discussed in Chap. 8, a consistent finding of both in vitro and in vivo studies is regrowth with colistin or polymyxin B monotherapy, even with concentrations far exceeding those which can be safely achieved clinically [12, 13, 16, 25,26,27, 39, 67, 88, 89, 93, 103, 104, 128, 147, 165, 173, 174, 201, 208]. Amplification of colistin-resistant subpopulations in heteroresistant isolates, i.e. isolates that are susceptible to polymyxins based upon their minimum inhibitory concentrations (MICs) but which contain resistant subpopulations, has been shown to contribute to the observed regrowth following polymyxin monotherapy [13, 14, 16, 24, 45, 50, 84, 89, 103, 123, 147, 173, 174, 188]. Studies undertaken in in vitro pharmacokinetic/pharmacodynamic (PK/PD) models simulating clinically achievable unbound plasma concentration-time profiles of colistin or polymyxin B in critically ill patients demonstrated early regrowth of heteroresistant strains of Pseudomonas aeruginosa [16, 103, 173], Klebsiella pneumoniae [45, 208] and Acinetobacter baumannii [84, 89], with population analysis profiles (PAPs) revealing substantial increases in the proportion of polymyxin-resistant subpopulations; PAPs after 72 h (colistin) or 96 h (polymyxin B) were substantially different from the PAPs prior to polymyxin therapy and those for the growth controls. Similar increases in the proportion of colistin-resistant bacteria with monotherapy have been observed in other in vitro studies (both static and dynamic time-kill infection models) [1, 13, 14, 24, 123, 128, 143, 147, 174] and, for A. baumannii, murine thigh and lung infection models [50]; many of these studies include polymyxin concentrations well above the MIC of the organism. These observations suggest that the susceptible bacterial populations were selectively eradicated, resulting in unopposed growth of resistant subpopulations (such as LPS-deficient A. baumannii [114]; discussed in detail in Chap. 5) and consequently the emergence of resistance over time. Heteroresistance notwithstanding, adaptive resistance (see Chap. 5) may also contribute to regrowth as evidenced by reversion to the susceptible state following serial passaging on drug-free plates of one of three isolates in the study by Tam et al. [173]. Finally, a recent study demonstrated that amino acid alterations in two-component systems such as PmrAB, PhoPQ and ParRS involved in polymyxin resistance (due to modifications of lipopolysaccharides in the Gram-negative cell wall) occur rapidly in vitro in the presence of colistin within the period of selection of single-step mutants [32]. This suggests polymyxin treatment may provoke genetic mutations related to resistance as a mutagen within a short period in addition to the selection of pre-existing resistant subpopulations. Such observations highlight the importance of polymyxin combinations to minimize the emergence of polymyxin resistance.
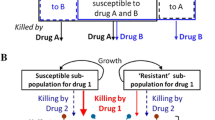
In addition to a reduction in the emergence of polymyxin resistance, combination therapy has the potential to increase bacterial killing via ‘synergy ’. Two mechanisms have been proposed whereby polymyxin combinations may provide an enhanced PD effect. As regrowth with polymyxin monotherapy is due, at least in part, to amplification of pre-existing polymyxin-resistant subpopulations in heteroresistant strains, it has been suggested that polymyxin combinations may give rise to so-called subpopulation synergy , the process whereby one drug kills the resistant subpopulation(s) of the other drug, and vice versa (Fig. 16.1) [23]. Additionally mechanistic synergy, whereby two drugs acting on different cellular pathways increase the rate or extent of killing of the other drug, has been suggested as a mechanism by which polymyxin combinations may lead to an enhanced antimicrobial effect (Fig. 16.1) [23]. The ability of colistin to increase the permeability of the outer membrane of many Gram-negative bacteria (Chap. 4) represents one possible mechanism for mechanistic synergy, potentially allowing better access of other antimicrobial agents to their target sites within the pathogen and thereby improving activity. Potential examples of each type of synergy are discussed subsequently in the PK/PD time-kill studies section. Mechanisms of subpopulation and mechanistic synergy are not mutually exclusive and both may operate simultaneously.

Schematic representations for subpopulation synergy (Panel A) and mechanistic synergy (Panel B). In subpopulation synergy, drug A kills the resistant subpopulations of drug B, and vice versa. In mechanistic synergy for drugs acting on different cellular pathways, drug A increases the rate or extent of killing by drug B, and vice versa. (Figure adapted from Bulitta et al. [23], with permission)
An important observation of some recent studies which investigated colistin susceptibility has been the substantially increased susceptibility of colistin-resistant isolates of several Gram-negative species to many antibiotics, including some normally considered inactive against Gram-negative organisms (e.g., rifampicin, macrolides, glycopeptides and daptomycin) [25, 61, 66, 86, 92, 109, 187, 190]. For example, Li et al. [92] examined the antibiograms of paired colistin-susceptible and -resistant strains of multidrug-resistant (MDR) A. baumannii against a broad range of antibiotics. In that study, the MICs of most colistin-resistant strains were substantially lower against a number of antibiotic classes typically used against Gram-negative organisms than their colistin-susceptible counterparts (e.g. >16 times lower in some cases against the penicillin class and carbapenems). Additionally, the colistin-resistant strains had substantially increased susceptibility to many antibiotics that are typically inactive against Gram-negative bacteria (e.g., rifampicin, fusidic acid, and erythromycin). The authors suggested that this may be due to substantial changes in the outer membrane of A. baumannii which occur as a result of resistance to colistin, thereby allowing antibiotics such as rifampicin and the lipopeptides, macrolides and streptogramins greater access to their target sites. This unexpected finding further emphasises the need for rational, systematic examination of polymyxin combination therapy. This chapter will provide an overview of both preclinical and clinical investigations of CMS/colistin and polymyxin B combination therapy.
16.2 Preclinical Studies of CMS/Colistin or Polymyxin B Combination Therapy
16.2.1 In Vitro Studies
In vitro studies examining combination therapy most commonly define the pharmacodynamic (PD) interaction of the agents in terms of additivity, synergy, indifference or antagonism, with the method used to determine such interactions dependent upon the experimental system employed [144]. For example, with the checkerboard microbroth dilution method the fractional inhibitory concentration (FIC) index is used. The FIC is calculated as follows [144]:
Though various definitions are used throughout the literature, synergy with this method has traditionally been defined as an FIC index of ≤0.5, additivity as an FIC index of 1.0, and antagonism as an FIC index of 2.0. However, more recent criteria suggest that an FIC index of >4 should be applied to definitions of antagonism to account for inherent imprecision of the technique when twofold dilutions are used and because an FIC index of 2.0 is probably indicative of an indifferent, rather than a true antagonistic, effect [6]. Though widely used, the checkerboard method is less discriminatory than other more sophisticated in vitro methods (e.g., static or PK/PD time-kill models; discussed below) for assessing the interactions of antimicrobial agents [28, 126, 194]. Discordance between results derived from combination testing using Etest and time-kill methods has also been reported for polymyxins [175]. Consequently, results derived from FIC and Etest methods will not be discussed here.
Time-kill methods have important advantages over the checkerboard technique. Primarily, the time-kill method measures the bactericidal activity of the combination being tested and provides a picture of antimicrobial action over time (based on serial viable counts); in contrast, the checkerboard technique provides only inhibitory data and is usually examined at a single time point (after 16–24 h of incubation) [144]. Time-kill models can be subdivided into static and PK/PD models. In static time-kill models, with the exception of a small degree of loss in drug activity due to bacterial metabolism or inactivation, bacteria are exposed to static (fixed) concentrations of an antibacterial agent over a defined period of time. PK/PD models essentially fall into one of two categories: one-compartment (1-CM) or two-compartment (2-CM) models [65, 186]. In these models, the test organism is presented with a dynamic concentration of drug designed to mimic in vivo PK. PK/PD models typically consist of a central reservoir containing the organism, a diluent reservoir and a waste reservoir. Drug is added to the central reservoir to achieve the desired peak concentration and the elimination profile is mimicked by addition of sterile, drug-free media to the central reservoir and removal of an equal volume of drug-containing media into the waste reservoir; various adaptations of this standard model are available to simultaneously mimic the in vivo PK of two or more drugs with differing half-lives [21]. Though 1-CM are most common, the 2-CM hollow-fibre infection model (HFIM) – which prevents bacterial elimination by physically separating bacteria from the central reservoir – is now considered gold standard for detailed examination of the effects of different regimens and PK on the time-course of bacterial killing and emergence of resistance [22].
For both static and PK/PD time-kill methods synergy has traditionally been defined as a 100-fold increase in killing at 24 h (as measured by colony counts; i.e. a ≥2-log10 lower CFU/mL) with the combination relative to its most active component (Fig. 16.2) [144]; antagonism is defined as a 100-fold decrease (i.e. a ≥2-log10 higher CFU/mL) in killing at 24 h with the combination compared with the most active single drug alone. While a strict application of these definitions requires that at least one of the drugs being tested produces no significant inhibition or killing alone, there are no established criteria with which to evaluate interactions when using two or more drugs, each of which has significant activity alone [144]. Consequently, these definitions are commonly applied in practice even when more than one drug displays significant bacterial killing. Variations on, and additions to, these definitions abound in the literature however, complicating comparisons of effect between studies. A typical example is that synergy is sometimes reported as described above, with the qualification that the number of surviving organisms in the presence of the combination must be ≥2-log10 CFU/mL below the starting inoculum [53, 61, 134, 149, 167]. In this way, an interaction described as synergistic by the former definition may not be synergistic by the latter. These definitions are also commonly applied at times other than 24 h.

Effects of antimicrobial combinations as measured with the time-kill method. A + B, synergism; C + D, antagonism; E + F, indifference. (Figure adapted from Pillai et al. [144], with permission)
Numerous in vitro studies have used the static or PK/PD time-kill method to examine polymyxin combination therapy, with the majority of studies utilising CMS or colistin (sulphate). However, as discussed in Chap. 3, CMS is an inactive prodrug of colistin and undergoes conversion to colistin in aqueous media [15, 91]. Administration of CMS will therefore result in a variable formation of active colistin over time, making the administering CMS in these in vitro systems inappropriate. Unfortunately, as for animal studies discussed above, it is not always possible to ascertain whether the ‘colistin’ administered was colistin (sulphate) or CMS (sodium). Antimicrobial agents combined with polymyxins in time-kill models include both agents with and without usual activity against Gram-negative pathogens. Studies have included polymyxins combined with rifampicin [8, 9, 19, 60, 82, 84, 94, 124, 177, 179, 180], carbapenems [8, 13, 16, 34, 36, 39, 45, 60, 82, 83, 87, 89, 96, 100, 102, 103, 111, 127, 134, 135, 137, 149, 161, 167, 168, 176,177,178, 180, 184], tigecycline [4, 18, 19, 27, 37, 40, 44, 60, 68, 80, 116, 122, 137, 148, 177], ampicillin/sulbactam [89, 180], ceftazidime [67], ciprofloxacin [8, 67], aminoglycosides [8, 40, 131, 152, 171], glycopeptides [18, 60, 66, 140, 187, 190], fosfomycin [5, 40, 46, 80, 87, 166, 177, 188, 201, 208] and others [1, 34, 39, 61, 97, 127, 129, 143, 153, 155, 175, 177, 187, 193, 201]; rifampicin, the carbapenems and tigecycline are the most commonly studied antibiotics in combination with colistin. The most common organisms studied are P. aeruginosa, A. baumannii and K. pneumoniae, and these will be the primary focus of the remainder of this section.
Despite a relatively large number of published studies examining polymyxin (primarily colistin) combination therapy there are a number of deficiencies with much of the existing information in addition to the lack of certainty around the form of ‘colistin’ administered; these deficiencies apply to both static and dynamic (PK/PD) models. Firstly, the vast majority of studies employ a single, generally lower inoculum (~105–106 CFU/mL). However, as the antibacterial activity of both colistin and polymyxin B is subject to an inoculum effect [24, 173], and as high bacterial densities can be found in some infections [107, 169], it is important to examine the antibacterial activity of combination therapy at multiple inocula. Second, many studies present antibiotic concentrations as multiples of the MIC with little reference to, or discussion of, the clinical relevance of the actual concentrations used. Further to this, many authors judge the ‘success’ of a particular combination only by whether synergy was attained rather than examining the overall antimicrobial activity of the combination. However, a combination that attains synergy may still achieve poor overall antimicrobial activity and may even be less active overall than another combination considered antagonistic. Third, consideration of polymyin heteroresistance and the effect of combinations on the development of polymyxin resistance have only been examined in a small number of recent studies [1, 5, 13, 16, 27, 39, 45, 68, 84, 89, 100, 102, 103, 143, 201, 208]. As discussed above heteroresistance is known to contribute to regrowth observed following colistin or polymyxin B monotherapy, although its clinical significance is unclear. Given the status of the polymyxins as agents of last resort and reports of increasing polymyxin resistance, it is crucial to systematically examine the effect of combination therapy on the emergence of polymyxin resistance, including on heteroresistant strains, in order to design optimal dosage regimens. Finally, remarkably few studies utilise PK/PD models, the introduction of which has been an important advancement in antimicrobial research, to investigate polymyxins in combination.
The next two sections of this chapter will discuss significant recent static and dynamic (PK/PD) time-kills investigations with polymyxins (colistin or polymyxin B) and will focus primarily on studies involving P. aeruginosa, A. baumannii and K. pneumoniae. Although polymyxins have been reported to be synergistic against a variety of pathogenic fungi including a variety of Candida, Aspergillus and other species in combination with echinocandins, azoles and amphotericin B [2, 105, 117, 133, 158, 205, 206], these studies will not be discussed here.
16.2.2 Static Time-Kill Studies
Pseudomonas aeruginosa
Few studies have examined polymyxin combinations against P. aeruginosa using either static or dynamic (the latter discussed below) time-kill models. Two studies by Pankuch et al. combined colistin with either meropenem [134] or doripenem [135] against clinical isolates of P. aeruginosa; the proportion of MDR strains was not stated. Sub-MIC concentrations of colistin (0.12–1 mg/L) and meropenem (0.06–8 mg/L) were synergistic against 13 (25.5%) of 51 isolates (all isolates colistin-susceptible; 6 [11.8%] isolates meropenem-resistant) at 24 h, whereas colistin (0.12–16 mg/L) and doripenem (0.03–128 mg/L) demonstrated synergy against 19 (76.0%) of 25 isolates (1 [4%] colistin-resistant isolate; 14 [56%] isolates doripenem-resistant). Urban et al. examined antibiotic combinations using polymyxin B, doripenem, and rifampicin against five MDR isolates of P. aeruginosa (one K. pneumoniae carbapenemase [KPC]-producing and four non-metallo-β-lactamase [MBL] or KPC-β-lactamase producing) [184]. All isolates were carbapenem-resistant and one polymyxin resistant, and antibiotics were used at a concentration of 0.25× MIC. As monotherapy, none of the tested antibiotics was bactericidal (defined as a ≥3-log10 CFU/mL decrease in 24 h). Triple therapy with the combination of polymyxin B, doripenem and rifampicin was most effective, with bactericidal activity achieved against all isolates at 24 h. Combinations utilising only two antibiotics were less effective, with polymyxin B plus doripenem or rifampicin bactericidal against only one isolate. Despite examining combination therapy ‘synergy’ was not directly examined in this investigation.
Bergen et al. systematically investigated bacterial killing and resistance emergence with colistin alone and in combination with imipenem against P. aeruginosa [13]. Conducted over 48 h this study included five clinical isolates and an ATCC reference strain representing a mixture of colistin and imipenem susceptible and resistant strains, colistin heteroresistant and non-heteroresistant strains, and MDR and non-MDR strains; one isolate contained IMP- and CTX-M-type β-lactamases. Importantly, of the static time-kill studies discussed in this chapter only this study examined the effect of combinations at multiple inocula (~106 and ~108 CFU/mL); it was also the first study to specifically incorporate colistin-heteroresistant strains and investigate the emergence of polymyxin resistance with polymyxin combination therapy. In combination experiments both antibiotics were studied at concentrations of 0.5×, 4× and 16× MIC for susceptible isolates and 1, 4 and 32 mg/L for colistin and 1, 8 and 32 mg/L for imipenem for resistant isolates; the majority of concentrations for colistin and all concentrations for imipenem can be considered clinically achievable. In total nine colistin/imipenem combinations were examined for each isolate at each inoculum. Regrowth of all isolates was observed with colistin monotherapy even with colistin concentrations well above those which can be safely achieved clinically. The addition of imipenem to colistin at both inocula generally resulted in substantial improvements in bacterial killing over equivalent monotherapy against MDR P. aeruginosa isolates resistant to either antibiotic. The improvements in activity against these isolates were observed across the 48-h duration and with all colistin concentrations at the low inoculum, and 4× and 16× MIC (or 4 and 32 mg/L) colistin at the high inoculum. Notably, the total reductions in log10 CFU/mL achieved with combinations containing lower colistin concentrations (0.5× and 4× MIC or 1 and 4 mg/L) were on many occasions similar in magnitude to the reductions achieved with combinations containing 16× MIC colistin, particularly at the 106 inoculum. Benefits in overall antibacterial activity for this combination were less pronounced against the three isolates susceptible to both antibiotics, although substantial improvements in initial kill (i.e., up to 6 h) were present. As for the emergence of colistin resistance, colistin monotherapy against the five colistin-susceptible isolates generally led to increases in colistin-resistant subpopulations at both the low and high inocula, with combination therapy generally resulting in a similar proportion of colistin-resistant subpopulations at 48 h as with equivalent monotherapy. While this result would appear to negate one of the major theoretical attractions of colistin combination therapy, namely a reduction in the emergence of colistin resistance, the same authors subsequently conducted a similar experiment with two of these isolates in a dynamic (PK/PD) model combining colistin with doripenem and achieved a very different result. The potential reason for this and the implications for antimicrobial combination testing are discussed in detail in the section examining PK/PD time-kill studies.
Two studies have examined colistin [46] or polymyxin B [188] combined with fosfomycin against P. aeruginosa. Di et al. examined this combination against 5 carbapenem-resistant but colistin-susceptible clinical isolates over 24 h [46]. Antibiotics were administered at concentrations of 0.5× and 1× MIC (range, 0.25–4 mg/L for colistin and 16–256 mg/L for fosfomycin). Neither antibiotic produced substantial bacterial killing as monotherapy with regrowth to ~108 CFU/mL. However, in combination at both 0.5× and 1× MIC, in all but one case no viable bacteria were detected after no later than 12 h; the only exception was against the isolate with the highest colistin MIC (4 mg/L) and only with the combination with both antibiotics at 0.5× MIC. With polymyxin B (0.5, 1 and 2 mg/L) and fosfomycin (30, 150 or 300 mg/L) combinations, Walsh et al. similarly observed enhanced bacterial killing over 24 h against 3 polymyxin B-susceptible heteroresistant isolates [189]. Though synergy was observed in only 39 (48.1%) of 81 cases (9 combinations across 3 isolates at 3 time points), this was much higher (28 [51.9%] of 54 cases) when only combinations containing polymyxin B at 1 or 2 mg/L are considered. Against 2 colistin-resistant isolates, bacterial killing was not substantially enhanced with the combination.
A. baumannii
In the two studies by Pankuch et al. discussed above, colistin was also combined with either meropenem [134] or doripenem [135] against clinical isolates of A. baumannii; the proportion of MDR strains was not stated. Colistin (0.06–8 mg/L) and meropenem (0.03–64 mg/L) showed synergy against 49 (94.2%) of 52 isolates (13 [25%] isolates colistin-resistant; 15 [28.8%] isolates meropenem-resistant) at 24 h, whereas colistin (0.12–16 mg/L) and doripenem (0.06–32 mg/L) showed synergy against 25 (100%) of 25 isolates of A. baumannii (11 [44%] isolates colistin-resistant; 9 [36%] isolates doripenem-resistant). Shields et al. examined colistin plus doripenem against five XDR isolates (defined as resistant to all agents except polymyxins and tigecycline) of A. baumannii taken from patients who had received solid organ transplants [163]; all isolates were colistin-susceptible based on MICs. Against all five isolates doripenem monotherapy at sub-MIC concentrations resulted in virtually no antimicrobial activity, whereas colistin monotherapy (0.25× to 1× MIC) was bacteriostatic (inhibiting growth of the inocula without causing significant killing) (Fig. 16.3). However, the combination of colistin (0.125× to 0.25× MIC) plus doripenem (8 mg/L) resulted in undetectable bacterial levels at 8 h without evidence of regrowth by 24 h. Interestingly, based on this, in vitro data combinations of CMS (5 mg/kg/day of CBA in 2–4 divided doses) and doripenem (500 mg 8-hourly) were recommended for use in their institution for patients who have received solid organ transplants and were infected with XDR A. baumannii. At the time of publication four patients had been treated with this combination with a fifth patient receiving meropenem plus colistin; 4 (80%) of the 5 patients had a positive clinical response and survived.

Representative time-kill curves with colistin and doripenem alone, and in combination, against an extensively drug-resistant (XDR) isolate of A. baumannii. (DOR doripenem, COL colistin. Doripenem MIC alone = 64 μg/mL, Colistin MIC alone = 2 μg/mL) (Figure adapted from Shields et al. [163], with permission)
In a follow-up study to that of Shields et al. discussed above [163], the same group compared the in vitro killing effects of colistin (2 mg/L), doripenem (8 mg/L) and sulbactam (4 mg/L) alone, and in combination, against isolates of XDR A. baumannii collected from patients with recurrent respiratory tract infections prior to (initial) and following (recurrent) treatment with intravenous CMS plus doripenem [127]; 4 (44%) of the 9 patients received additional CMS via inhalation . Patients had received the combination (doses were not stated) for a minimum of 13 days (median duration, 31 days; range, 13 to 74 days) with the median time between collection of initial and recurrent isolates being 65 days (range, 28–188 days). Nine initial and recurrent isolates (1 of each from each patient) were collected (18 isolates in total), with 8 (89%) of 9 pairs genetically indistinguishable. Time-kill studies revealed synergy at 24 h was more frequent when colistin was combined with doripenem (16 [89%] of 18 isolates) than sulbactam (9 [50%] of 18 isolates). The killing effects of the colistin/doripenem combination was attenuated against isolates previously exposed to the combination in vivo (mean log kill [CFU/mL] at 24 h of −5.08 log10 versus −2.88 log10 for initial and recurrent isolates, respectively), although there was no difference in the mean log kills against the initial and recurrent isolates exposed to colistin plus sulbactam. The triple combination of these agents achieved greater log kills than either colistin/doripenem or colistin/sulbactam combination among recurrent isolates (mean log10 kills [CFU/mL] at 24 h of −5.74 versus −2.88 and −1.51, respectively), including those that did not respond to the colistin/doripenem combination. Interestingly, although only one of nine initial isolates was colistin-resistant, five isolates were colistin-resistant following treatment. However, although colistin MICs influenced the extent of killing somewhat, colistin/doripenem combinations were equally active against colistin-susceptible and –resistant isolates. The MICs of doripenem rather than colistin were associated with the extent of killing by colistin and doripenem in combination, with each of the isolates that failed to respond to treatment having a doripenem MIC >64 mg/L. Such an association has also been demonstrated for colistin/doripenem combinations in KPC-producing K. pneumoniae [36] (discussed below).
Tan et al. examined colistin (at 1× MIC; range: 0.5–2 mg/L), minocycline (at 1× MIC for susceptible isolates [n = 9] and 4 mg/L for resistant isolates [n = 4]; range, 0.06–16 mg/L) and their combination against 13 imipenem-resistant isolates (MIC >8 mg/L) of A. baumannii across 24 h [175]. As monotherapy neither antibiotic demonstrated bactericidal activity at any time but the combination was bactericidal against 9 (69%) isolates at 24 h. Synergy was detected in 1 (8%), 2 (15%), 2 (15%) and 12 (92%) of isolates at 2, 4, 6, and 24 h, respectively. Tripodi et al. examined colistin (6 mg/L), rifampicin (5 mg/L), imipenem (20 mg/L) and ampicillin/sulbactam (50 mg/L) alone or in double (colistin plus each of the second drugs) or triple (colistin plus rifampicin plus imipenem, or colistin plus rifampicin plus ampicillin/sulbactam) combinations against nine isolates of MDR A. baumannii producing OXA-58 carbapenemase [180]. Colistin was the most active agent as monotherapy with double and triple combinations generally showing similar activity to that of colistin monotherapy. However, triple therapy with the combination of polymyxin B, doripenem and rifampicin was more effective against five non-MBL or KPC-producing isolates when compared to monotherapy or double combination therapy [184]. In another study, colistin at concentrations of 0.25×, 0.5× and 1× MIC plus daptomycin 10 mg/L was synergistic against ten MDR-colistin-susceptible isolates of A. baumanni in 16 (53.3%) of 30 cases at 24 h, although no benefit with the combination was seen against a further four MDR-colistin-resistant isolates [61]; however, it is not clear whether colistin (sulphate) or CMS was used in this investigation.
One laboratory examined colistin (1 mg/L) alone and in combination with the glycopeptide antibiotics vancomycin (20 mg/L) [66] or teicoplanin (20 mg/L) [190] against five MDR-colistin-susceptible isolates of A. baumannii. Colistin as monotherapy was rapidly bactericidal against all isolates with rapid regrowth to control values by 24 h. However, when combined with vancomycin regrowth was suppressed in four of the five isolates even at 48 h, with ~5–7-log10 CFU/mL greater killing at this time compared to colistin monotherapy. The colistin/teicoplanin combination suppressed regrowth against all isolates at 24 h, with >8-log10 CFU/mL greater killing compared with colistin monotherapy and a ≥ 4-fold log reduction compared with the starting inoculum at this time. Surprisingly, although experiments were conducted for 48 h only the 24 h results were reported. Despite the substantially improved bacterial killing with both glycopeptides the authors noted that, given the potential of both colistin and vancomycin to cause nephrotoxicity when either agent is used alone, there may be concern about the suitability of this combination in the clinic. Although teicoplanin has a similar mechanism of action to vancomycin, it has a more favourable effect profile including a lower incidence of renal toxicity which may make such a combination more acceptable to clinicians [29, 172]. A colistin/telavancin combination was synergistic at 24 h against a single MDR clinical isolate (representative of the epidemic UK lineage OXA-23 clone 1) of A. baumanni [73]. However, in contrast to teicoplanin above, the incidence of renal toxicity with telavancin is higher than that of vancomycin which may limit the utility of this combination [185]. Similarly, a polymyxin B/rifampicin combination was synergistic at 24 h against two MDR isolates of A. baumanni positive for OXA-23 and OXA-51 and an Acinetobacter sp. positive for OXA-58 and IMP-type carbapenemases [95].
Phee et al. examined colistin (1-2 mg/L) combined with fusidic acid (1 mg/L; 16 mg/L for the colistin-resistant isolate) against six isolates of A. baumannii across 24 h [143]. All but a single colistin-resistant isolate were colistin-heteroresistant, and all but the reference strain were either MDR, XDR or pandrug-resistant [PDR] according to the classification of Magiorakos et al. [106]. The majority of isolates contained OXA-23 clone 1 or 2, OXA-51 and OXA-23. Though bacterial killing with colistin monotherapy was virtually superimposable with that of the combination across the first 6 h for all heteroresistant isolates, by 24 h substantial regrowth had occurred with monotherapy but remained suppressed with combination therapy; bacterial killing and suppression of regrowth was also observed with the combination against the colistin-resistant isolate. Synergy was observed in all cases at 24 h (enhanced bacterial killing of ~3–8 log10 CFU/mL). The combination also prevented the emergence of colistin resistance, with little increase in MIC above baseline after 7 days of serial passage in the presence of both drugs compared with monotherapy. Park et al. examined the combination of colistin (2 mg/L) with doripenem (8 mg/L) or tigecycline (2 mg/L; concentration representative of achievable tissue levels) against 69 isolates of A. baumannii [137]. Of the isolates, 28 were MDR (100%, 0% and 25% susceptible to colistin, doripenem, and tigecycline, respectively) and 41 XDR (51.2%, 7.3%, and 29.3% susceptible to colistin, doripenem, and tigecycline, respectively). Of 35 isolates tested for the presence of the OXA carbapenemase gene, 34 (97.1%) contained OXA-23 whereas only 2 (5.7%) carried the ISAba-OXA-51 gene. At 24 h, the colistin/doripenem combination showed the highest rate of synergy in both the MDR (15 [53.6%] of 28 cases) and XDR (22 [53.7%] of 41 cases) groups; the equivalent values for the colistin/tigecycline combination were 10 (35.7%) of 28 cases and 18 (43.9%) of 41 cases.
Finally, Ozbek and Mataraci [129] examined the activity of antibiotic lock therapy (ALT) with colistin plus clarithromycin against biofilm-embedded A. baumannii using an in vitro antibiotic lock model involving segments of central venous catheters; ALT involves the instillation of high concentrations of an antimicrobial agent into the lumen of an infected central venous catheter for extended periods to overcome the relative antimicrobial resistance of biofilm-embedded bacteria. Using two isolates of colistin-susceptible A. baumannii they found that against both strains colistin at 400× MIC completely eradicated biofilm bacteria within 3 days, whereas the combination of colistin (400× MIC) plus clarithromycin (200 mg/mL; ~100× serum concentration) sterilized the biofilm in 2 days.
K. pneumoniae and Other Enterobacteriaceae
A small number of studies have examined polymyxin combinations specifically against KPC-producing bacteria, primarily K. pneumoniae [36, 60, 83, 148, 166, 184, 208]. Pournaras et al. examined colistin and tigecycline alone and in combination against eight KPC-producing enterobacterial clinical strains (four K. pneumoniae, two Escherichia coli, one E. cloacae and one Serratia marcescens) [148]; all produced KPC-2 carbapenemase and were colistin-susceptible. Each antibiotic was tested at 1×, 2× and 4× MIC (range, 0.5–4 mg/L for colistin and 0.25–16 mg/L for tigecycline) and experiments conducted over 24 h. The colistin/tigecycline combinations substantially improved bacterial killing across 24 h and was synergistic at 1× and 2× MIC against most organisms at 4 and 8 h; at 4× MIC, synergy was maintained at 24 h against all strains. Similar improvements in bacterial killing against four KPC-3-producing K. pneumoniae isolates were reported by Lee and Burgess with colistin or polymyxin B (both at 2× MIC; range, 0.125–0.5 mg/L for colistin and 0.25–0.5 mg/L for polymyxin B) combined with doripenem (6 mg/L) [83]; all isolates were polymyxin-susceptible and doripenem-resistant. In that study, none of the monotherapy regimens sustained bactericidal killing at 24 h. However, colistin or polymyxin B plus doripenem combinations maintained bactericidal activity across 24 h against all isolates, achieving synergy at this time; synergy was maintained at 48 h in 2 (50%) of 4 isolates with colistin and all isolates with polymyxin B. MIC measurements were additionally repeated at 24 h on all isolates following exposure to colistin or polymyxin B monotherapy. All isolates developed polymyxin resistance (MICs, 8–128 mg/L) and cross resistance between colistin and polymyxin B was observed. In another study triple therapy with polymyxin B, doripenem and rifampicin (all at 0.25× MIC) was most effective against five MDR isolates each of K. pneumoniae (two with KPC and three with ACT-1 [AMPC-type] β-lactamases) and E. coli (one KPC-3 and four KPC-2 β-lactamases) [184]; all isolates were polymyxin B-susceptible and doripenem-resistant. Bactericidal activity was achieved against 4 (80%) of 5 isolates of K. pneumoniae and 5 (100%) of 5 isolates of E. coli at 24 h. Monotherapy with any agent failed to produce bactericidal activity, whereas combinations utilising only two antibiotics were less effective with polymyxin B plus rifampicin bactericidal against only 1–2 (20–40%) of 5 isolates of each species; polymyxin B plus doripenem was bactericidal against only 1 (20%) of 5 K. pneumoniae isolates but 4 (80%) of 5 E. coli isolates. In another study, the combination of colistin (5 mg/L) plus fosfomycin (100 mg/L) was synergistic at 24 h against only 1 (6%) of 17 KPC-2-producing K. pneumoniae isolates [166].
Clancy et al. examined colistin (2 mg/L) in combination with doripenem (8 mg/L) against 23 KPC-2-producing strains of K. pneumoniae [36]; each strain contained a variant mutant opmK35 porin gene. The median colistin and doripenem MICs were 4 mg/L (range, 0.125–128 mg/L) and 32 mg/L (range, 4–256 mg/L), respectively. Colistin MICs were > 2 mg/L against 14 (63%) of 23 strains. The colistin/doripenem combination was significantly more active at 12 and 24 h than either monotherapy against the four strains with doripenem MICs of ≤8 mg/L, with synergy at 24 h against all 4 strains. In contrast, there was no overall difference in median bacterial killing for strains with doripenem MICs >8 mg/L, with synergy reported at 24 h in 6 (32%) of 19 strains. There was no difference in synergy between strains with colistin MICs of ≤2 mg/L and > 2 mg/L at either 12 or 24 h. Notably, insertions encoding glycine and aspartic acid at amino acid (aa) positions 134 and 135 (ins aa134–135 GD; n = 8) and ompK36 promoter IS5 mutations (n = 7) were associated with significantly higher doripenem MICs and diminished efficacy of colistin/doripenem combinations; in these cases, bacterial killing more closely resembled colistin monotherapy. However, other mutant/wild-type ompK36 strains demonstrated increased killing with the combination, even with elevated doripenem MICs. The authors suggested that doripenem MICs and ompK36 genotyping of KPC-K. pneumoniae may be useful for identifying strains most likely to respond to colistin/doripenem combination therapy. These results suggest that despite membrane permeabilization by a polymyxin potentially increasing access of doripenem to target sites and allowing it to overcome hydrolysis by KPC, OmpK36 porins may also be necessary for synergy .
While the majority of studies (checkerboard and time-kill) examining polymyxin combination therapy against K. pneumoniae addressed KPC-producing strains, fewer studies address MBL-producing strains. Souli et al. examined colistin (5 mg/L) in combination with imipenem (10 mg/L) against 42 unique clinical isolates of blaVIM-1-type MBL-producing K. pneumonia [167]. After 24 h exposure to the combination, synergy was reported against 12 (50%) of 24 colistin-susceptible isolates, but antagonism was observed against 10 (55.6%) of 18 colistin-resistant isolates. Interestingly, resistance to colistin (MICs 64–256 mg/L) was observed in 7 (58.3%) of 12 isolates that were initially susceptible to colistin. In contrast, none of four isolates initially susceptible to imipenem and which showed regrowth at 24 h developed resistance to imipenem. Tangden et al. conducted more than 200 time-kill experiments with 24 antibiotic regimens including colistin (4.0 mg/L) in double and triple combinations with meropenem (6.8 mg/L), aztreonam (17 mg/L), fosfomycin (83 mg/L) and rifampicin (1.7 mg/L) against two VIM-1-type and two NDM-1-type K. pneumoniae strains (all colistin-susceptible; susceptibilities to the other antibiotics varied substantially) [177]. At 24 h, colistin plus fosfomycin was bactericidal and synergistic against three of the four strains (both NDM-1-types [each fosfomycin resistant] and one VIM-1-type), while the triple combination of colistin/fosfomycin/meropenem was bactericidal against three strains and synergistic against all strains. While colistin plus rifampicin was only synergistic at this time against both NDM-1-type strains, the addition of meropenem to this regimen resulted in bactericidal and synergistic activity against all strains; this triple combination was the most effective regimen overall. Double combinations of colistin with either meropenem or aztreonam produced synergy in only one strain, although the triple combination produced synergy in three of the four strains. Albur et al. reported that colistin or CMS in combination with tigecycline did not increase bacterial killing against a range of NDM-1-producing Enterobacteraceae [4]; however, the concentrations chosen in this investigation were extremely low (e.g. the maximum concentration of colistin used was 0.29 mg/L). Abdul Rahim et al. examined polymyxin B (0.5, 1 or 2 mg/L) plus chloramphenicol (8, 16 or 32 mg/L) combinations against four NDM-producing K. pneumoniae strains (all polymyxin B-susceptible and -heteroresistant; three susceptible to chloramphenicol) [1]. Combination therapy significantly delayed regrowth, with synergy observed in 25 (89.3%) of 28 cases at both 6 and 24 h; at 24 h, no viable bacteria were detected in 15 (53.4%) of 28 cases with various combinations across all strains. The emergence of polymyxin-resistant bacteria was also completely suppressed with combination therapy. In another study, colistin/tigecycline combinations were synergistic against a single isolate of VIM-1- and SHV-12-producing K. pneumoniae, although colistin/ciprofloxacin combinations were indifferent against the same isolate [37].
Corvec et al. combined colistin with tigecycline, fosfomycin or gentamicin (each at 0.5×, 1×, and 4× MIC) against a single strain of ESBL-producing E. coli [40]. Colistin combined with tigecycline decreased bacterial counts at 24 h by ~4.5- and 7-log10 CFU/mL compared with the initial inoculum and monotherapy, respectively. The colistin/fosfomycin combination was synergistic at 6 h with no viable bacteria detected at or subsequent to this time. Colistin plus gentamicin was no better than either monotherapy alone (regrowth with both monotherapies had reached control values by 24 h). A similar study by Ku et al. that employed nine ESBL-producing K. pneumoniae isolates (five carbapenem-resistant and four – susceptible; one colistin-resistant) examined colistin combined with either tigecycline or fosfomycin (all antibiotics at 0.25× or 0.5× MIC) [80]. With concentrations of 0.5× MIC, synergy at 24 h was reported in 8 (88.9%) and 6 (66.6%) of 9 cases for the combinations with tigecycline and fosfomycin, respectively. However, synergy was absent with both combinations when concentrations of 0.25× MIC were used.
In two further studies the combination of colistin and tigecycline had no benefit over equivalent monotherapy against a single isolate of OXA-48-producing carbapenem-resistant K. pneumoniae susceptible to both drugs [44], and only marginal benefit against six carbapenem-resistant isolates of Enterobacter (E. coli [n = 2], K. pneumoniae [n = 2], E. aerogenes [n = 1] and E. cloacae [n = 1]) with varying resistance determinants [18].
Other Bacteria
Against one reference strain and three clinical isolates of S. maltophilia (all with elevated MICs to each antibiotic), colistin (2 mg/L) combined with tigecycline (1 mg/L) or rifampicin (8 mg/L) was synergistic at 24 h in all cases except against one isolate and only with the colistin/tigecycline combination (a 1.7 log10 CFU/mL reduction) [19].
16.2.3 PK/PD Time-Kill Studies
To date few studies have utilized PK/PD models to examine colistin in combination, while only one has employed polymyxin B. Gunderson et al. was the first to utilise a one-compartment PK/PD model to examine colistin in combination [67]. In that study colistin (steady-state peak concentration [C max] of 6 or 18 mg/L every 24 h; half-life, 3 h) was combined with either ceftazidime (constant concentration of 50 mg/L) or ciprofloxacin (C max 5 mg/L every 12 h; half-life, 3 h) against two colistin-susceptible MDR isolates of P. aeruginosa; experiments were conducted over 48 h with an inoculum of ~106 CFU/mL. Although the combination of colistin plus ciprofloxacin generally produced poorer bacterial killing than with either drug alone, the authors reported the combination of colistin plus ceftazidime was synergistic. However, in light of more recent understanding of colistin pharmacokinetics in both critically ill patients [63, 75, 108, 115, 146] and patients with CF [90] (Chap. 15), only one maximal concentration of colistin (6 mg/L) employed by Gunderson et al. can be considered potentially clinically achievable [67]. Additionally, although the simulated 3 h half-life of colistin is representative of that observed in patients with CF [90], colistin was administered as a single dose every 24 h. Given colistin is typically administered intermittently to patients every 8–12 h, the colistin PK profile generated across a 24-h period was not representative of that observed in CF or critically ill patients. Moreover, although synergy was defined as a ≥2-log10 decrease in colony count relative to the count obtained with the more active of the two antibiotics alone at 24 h, it appears that only changes in log10 CFU/mL between colistin monotherapy and combination therapy were considered; when data for ceftazidime monotherapy (which was performed for only one of the two isolates tested) is considered, synergy was not observed.
A small number of conference abstracts have appeared examining colistin in combination with meropenem [168], amikacin [131], and rifampicin [9] against A. baumannii utilising PK/PD models. While combinations with meropenem and rifampicin were reported to be synergistic, there are significant limitations with all these investigations, not least of which is that it is unclear whether ‘colistin’ (which was dosed every 12 h) was administered as colistin (sulphate) or CMS (sodium). Additionally, in the two studies where PK data were reported [9, 168], ‘colistin’ concentrations were determined using microbiological assays; as discussed in Chap. 6, microbiological assays are incapable of differentiating between colistin present in a sample at the time of collection and colistin formed in vitro from administered CMS during the incubation period of the microbiological assay. Finally, as for the majority of investigations examining colistin combinations using time-kill methodology, experiments were conducted for 24 h and used a single, generally lower inoculum (~5 × 105–106 CFU/mL). Given these limitations, while the synergy observed in these dynamic systems is interesting it is difficult to draw any firm conclusions from these studies.
More recent studies have systematically investigated polymyxin combination therapy, including the emergence of polymyxin resistance, using in vitro PK/PD models [5, 16, 27, 39, 45, 68, 84, 89, 100, 102, 103, 178, 201, 208]. Unfortunately, as was the case for Gunderson et al. discussed earlier [67], a number of recent studies simulated a colistin half-life more representative of that observed in patients with CF (range: 4–4.7 h), not critically ill patients (Chap. 15) [5, 27, 39, 178, 201]. Two studies were conducted over 24 h at a single, low inoculum (106 CFU/mL) [68, 100]. Consequently, these studies will not be considered below. Three studies utilized a 1-CM to examine colistin combinations against planktonic MDR isolates of P. aeruginosa [16], K. pneumoniae [45], and A. baumannii [84]. Two additional studies utilized a HFIM to examine colistin combinations against planktonic MDR isolates of P. aeruginosa [103] and a single KPC-producing isolate of K. pneumoniae [208]; one study utilized polymyxin B against a single MDR isolate of A. baumannii [89]. Of these six studies, three combined colistin (constant concentrations of 0.5, 2 or 5 mg/L across the studies) with doripenem (C max of 2.5 or 25 mg/L every 8 h; half-life, 1.5 h) against P. aeruginosa (one heteroresistant reference strain and one colistin-resistant MDR clinical isolate in the 1-CM study; two heteroresistant strains and one colistin-resistant MDR clinical isolate in the HFIM study; all strains across the two studies doripenem-susceptible) [16, 103] and K. pneumoniae (one heteroresistant reference strain and three MDR clinical isolates [one each of colistin-susceptible, -heteroresistant, and -resistant]; three strains doripenem-susceptible) [45]. Against A. baumannii, one study combined colistin (constant concentrations of 0.5, 2 or 5 mg/L) with rifampicin (C max of 5 mg/L every 24 h; half-life, 3 h) against one MDR-colistin-susceptible and one MDR-colistin-resistant isolate [84], whereas one combined polymyxin B (C max of 3.61 mg/L at 0 h, then C max of 2.41 mg/L every 12 h; half-life, 8 h) with meropenem (C max of 54.8 mg/L; half-life, 1.5 h) and/or ampicillin/sulbactam (C max of 132/70.2 mg/L; half-life, 1.5 h) [103]. Colistin (C max of 0.46 mg/L; half-life, 7 h) and fosfomycin (C max of 150 mg/L mg/L; half-life, 2 h) were combined against a single KPC-2-expressing K. pneumoniae isolate (colistin- and fosfomycin-susceptible) [208]. All 1-CM studies were conducted at both a low (~106 CFU/mL) and high (~108 CFU/mL) inocula to account for the attenuated activity of colistin at higher inocula [24], the latter mimicking the high bacterial densities found in some infections [107, 169]; all HFIM studies used only a single inoculum (~106, 108 or 109 CFU/mL). One additional study examined colistin in combination with doripenem against MDR P. aeruginosa growing in a biofilm [102]. In all but one case colistin was administered as a continuous infusion to simulate the ‘flat’ profiles of formed colistin observed in critically ill patients at steady state across a CMS dosage interval [63, 146] (see Chap. 15). The concentrations of colistin employed ranged from 0.46 mg/L to 5 mg/L. Given the bound fraction of colistin in human plasma is ~50% [115], minimal binding of colistin in the growth media [12, 102], and that total (i.e. bound and unbound) plasma colistin concentrations of ~2–3 mg/L are typically achieved at steady state (with some patients achieving concentrations of up to ~10 mg/L) [63, 108, 115, 146], these dosage regimens of colistin (and also polymyxin B) reflect clinically achievable unbound (free) plasma colistin concentration-time profiles in patients. Administration of the second, or in the case of polymyxin B, third drug (doripenem, rifampicin, meropenem, or ampicillin/sulbactam) similarly reflected unbound plasma drug concentration-time profiles achieved in patients [17, 20, 79, 101, 154]. Studies were conducted across 72–96 h (1-CM) and 10–14 days (HFIM).
Across the six above studies directed specifically against planktonic bacteria, combination therapy generally resulted in substantial improvements in bacterial killing at both inocula. In many cases improvements in bacterial killing with combination therapy were dramatic. For example, against a colistin-susceptible strain of A. baumannii at the 106 CFU/mL inoculum no viable bacteria were detected at 24 h with colistin/rifampicin combinations containing colistin 0.5 or 2 mg/L, whereas regrowth to ~8 log10 CFU/mL had occurred at this time with equivalent colistin monotherapy [84]. At the 108 CFU/mL inoculum colistin (at either 2 or 5 mg/L) plus rifampicin increased bacterial killing across 72 h by as much as ~8 log10 CFU/mL and, with the highest dose colistin combination regimen (5 mg/L), resulted in no viable bacteria being detected following commencement of treatment. Similar improvements were observed against the colistin-resistant isolate. In the HFIM (108 CFU/mL inoculum), while double polymyxin B combinations were largely ineffective against a single isolate of A. baumannii resistant to all investigated antibiotics, the triple combination (polymyxin B plus meropenem and ampicillin/sulbactam) resulted in no viable bacteria being detected from 96 h onwards [89]. Against a colistin-susceptible (MIC 1 mg/L) doripenem-resistant (MIC 8 mg/L) isolate of K. pneumoniae, the combination of colistin at 0.5 mg/L plus doripenem at C max of 2.5 mg/L at the low inoculum produced ~4- to 5-log10-greater killing than equivalent monotherapy at 48 and 72 h, whereas colistin at 0.5 or 2 mg/L plus doripenem at C max of 25 mg/L at the high inoculum produced ~5- to 7-log10-greater killing at 48 and 72 h (with no viable colonies detected across the 72-h period on at least one occasion) [45]. Similar improvements were observed against two colistin-heteroresistant (MIC 1 mg/L) doripenem-susceptible (MIC<0.125) isolates, although only colistin at 2 mg/L plus doripenem at C max of 25 mg/L resulted in enhanced bacterial killing of the colistin-resistant isolate and only at the low inoculum. In the HFIM (inoculum 106 CFU/mL), a single KPC-2-producing K. pneumoniae isolate was completely eradicated by a colistin (C max of 0.46 mg/L)/fosfomycin (C max of 150 mg/L) combination [208]. Against P. aeruginosa, combinations containing colistin 0.5 or 2 mg/L plus doripenem at C max of 25 mg/L resulted in eradication of the colistin-resistant MDR isolate at the low inoculum and substantial reductions in regrowth (including to below the limit of detection at ~50 h) at the high inoculum (Fig. 16.4) [16]. For the same combination in the HFIM (colistin 2 or 5 mg/L plus doripenem at C max of 25 mg/L), markedly enhanced bacterial killing was observed with each combination against both heteroresistant (and MDR) isolates across 10 days, with only the combination containing colistin at 2 mg/L and only against one isolate failing to eradicate the bacteria [103]. Against the colistin-resistant isolate, both combinations enhanced bacterial killing by ~5–6 log10 cfu/mL on 3 days, with regrowth then occurring; regrowth approached control values by 10 days.

Time-kill curves for colistin and doripenem monotherapy (Panels A and C) and the combination (Panels B and D) against a non-mucoid MDR-colistin-resistant clinical isolate (19147 n/m) of P. aeruginosa at an inoculum of ~106 CFU/mL (left-hand panels) and ~108 CFU/mL (right-hand panels). The y axis starts from the limit of detection and the limit of quantification (LOQ) is indicated by the horizontal broken line. (Figure adapted from Bergen et al. [16], with permission)
While subpopulation synergy may have contributed to enhance bacterial killing against some isolates in the above investigations, it cannot explain enhanced activity against all isolates. For example, greater bacterial killing of P. aeruginosa was observed with the colistin/doripenem combination against a colistin-resistant MDR isolate with near complete resistance to colistin (MIC, 128 mg/L) and which contained enzymes active against carbapenems [16, 103], and similarly with the colistin/rifampicin combination against A. baumannii despite rifampicin ordinarily being inactive against Gram-negative pathogens [84]. The triple combination of polymyxin B/meropenem/ampicillin/sulbactam eradicated also eradicated a clinical isolate of A. baumannii resistant to all antibiotics investigated [89]. In each case it may be that a form of mechanistic synergy was operative due to permeabilization of the outer membrane by colistin [207]. It is possible that increasing the permeability of the outer membrane resulted in substantially increased concentrations of β-lactam in the periplasm, facilitating access to the cytoplasmic membrane where they act on penicillin-binding proteins [125, 199]. Similarly for rifampicin, which ordinarily does not effectively penetrate the Gram-negative outer membrane [191], increased membrane permeabilization may improve access to its target site within the cytoplasm. In this latter case, the substantial changes to the outer membrane of A. baumannii associated with the development of colistin resistance [71, 114] may additionally facilitate access to intracellular target sites.
An important feature common to the above six studies was the substantial reduction or, in some cases, complete suppression of the emergence of colistin-resistant subpopulations with combination therapy. As observed previously against all three bacterial species monotherapy with colistin generally resulted in substantial increases in the proportion of colistin-resistant subpopulations in colistin-susceptible or -heteroresistant isolates at both high and low inocula, often by as early as 24 h. However, the addition of doripenem to colistin eliminated the emergence of colistin-resistant colonies of K. pneumoniae [45] except at the lowest concentration combination tested (colistin 0.5 mg/L plus doripenem 2.5 mg/L) at the high (~108 CFU/mL) inocula. Against P. aeruginosa, resistant colonies were greatly reduced in number and emerged later (following 72–96 h of treatment) with all colistin/doripenem regimens at both inocula in the 1-CM [16], with the most resistant subpopulations (i.e., those growing in the presence of colistin at 10 mg/L on the PAP plates) absent with combination therapy. In the HFIM, the same combination against P. aeruginosa completely eliminated colistin-resistant subpopulations [103]. All three colistin/rifampicin regimens (colistin 0.5, 2 or 5 mg/L plus rifampicin 5 mg/L) completely suppressed the emergence of colistin-resistant subpopulations in a MDR-colistin-susceptible clinical isolate of A. baumannii such that at 72 h no colistin-resistant colonies were detected with any colistin/rifampicin combination at either inoculum (Fig. 16.5) [84]. Two important observations arise from these investigations. First, although combination therapy with doripenem had no effect on colistin resistance of MDR-colistin-resistant isolates of P. aeruginosa [16, 103] and K. pneumoniae [45], against A. baumannii the colistin/rifampicin combinations containing 2- or 5-mg/L colistin reduced the pre-existing colistin-resistant subpopulations of a colistin-resistant isolate to below the limit of detection at the low inocula, indicating that this combination may suppress the emergence of de novo colistin resistance. Second, on the few occasions where extensive regrowth (even up to ~7-log10 CFU/mL) occurred with combination therapy (with both doripenem and rifampicin), no colistin-resistant colonies were detected. While the reason for the observed regrowth despite an apparent lack of colistin resistance is unknown, this important finding suggests that combining doripenem or rifampicin with colistin may reduce the emergence of colistin-resistant subpopulations.

(Left) Time-kill curves with various clinically relevant dosage regimens of colistin (Col) and rifampicin (Rif) alone and in combination at an inoculum of ~106 CFU/mL (Panel A) and ~108 CFU/mL (Panel B) against a colistin-susceptible MDR clinical isolate (FADDI-AB030) of A. baumannii. (Right) Population analysis profiles (PAPs) at baseline (0 h) and after 72-h exposure to colistin monotherapy, colistin-rifampicin combination therapy, or neither antibiotic (control). The y axis starts from the limit of detection and the limit of quantification (LOQ) is indicated by the horizontal broken line. (Figure adapted from Lee et al. [84], with permission)
An interesting observation to come out of the studies by Bergen et al. [16] and Ly et al. [103] and which has implication for future rational testing of antibiotic combinations generally concerns the use of dynamic antibiotic concentrations simulating human PK when assessing the efficacy of combination therapy, and the duration over which such experiments are conducted. As discussed in the static time-kill section Bergen et al. previously examined the combination of colistin and imipenem at multiple inocula (~106 and ~108 CFU/mL) against multiple strains of P. aeruginosa using a static time-kill model [13]. In two subsequent PK/PD (dynamic) studies investigating colistin/doripenem, both isolates investigated in the 1-CM study [16] and two of three isolates (the third isolate being an additional colistin-heteroresistant strain) in the HFIM study [103] were included in this earlier investigation. While the antibiotics and their concentrations between the three studies are not directly comparable, the activity of colistin combined with either imipenem or doripenem was broadly similar across 48 h (the duration of the earlier study) at each inoculum against heteroresistant strains. However, substantial differences were evident against a colistin-resistant MDR isolate. In the static model, combinations with concentrations as high as 32 mg/L colistin plus 16× MIC imipenem failed to reduce bacterial numbers of this isolate to below the limit of detection at any time (maximum bacterial killing of ~3.5 log10 CFU/mL). In stark contrast, bacterial eradication was achieved in the 1-CM (duration, 96 h) with combinations containing colistin (0.5 or 2 mg/L) and doripenem 25 mg/L no later than 24 h at the low inoculum, and bacteria reduced to below detectable levels at approximately 48 h with the same combinations at the high inoculum. With the higher initial inoculum in the HFIM (109 CFU/mL), progressive bacterial killing occurred over 72–96 h (maximum bacterial killing of ~6 log10 CFU/mL), but slow regrowth ultimately close to control values occurred over the subsequent 7 days. Likewise, changes in PAPs with colistin/imipenem combinations against heteroresistant isolates in the static time-kill model generally mirrored those observed with equivalent colistin monotherapy, whereas the emergence of colistin resistance was greatly reduced (1-CM) or completely suppressed (HFIM) with colistin/doripenem combinations in the PK/PD models. Loss of imipenem due to degradation in the static experiments may have contributed to this result (colistin is stable under these conditions) [16], whereas intermittent dosing of doripenem in the PK/PD models replenished concentrations and avoided the combination effectively becoming colistin monotherapy over time. These observations highlight the importance of simulating PK profiles when assessing the activity and emergence of resistance to antimicrobial therapy. Additionally, the regrowth that occurred in the HFIM following substantial initial killing across the first 72–96 h of therapy highlights the importance of longer durations of therapy to fully assess the effectiveness of combinations.
While the above studies examined bacterial killing against planktonic cells, bacteria growing in a biofilm are protected from environmental, immune system and antimicrobial threats, making them substantially more resistant to antibiotic treatment. Such resistance is evidenced by substantial increases in MICs and MBCs [41, 70, 107]. The need for very high concentrations of colistin when used as monotherapy to achieve any substantial killing of biofilm-embedded bacterial cells has been demonstrated both in vitro [69, 72, 132] and in vivo [70]. Using a mouse lung infection biofilm model, Hengzhuang et al. [70] reported a colistin serum concentration of 64× MIC (i.e. 128 mg/L) was required to achieve a 1 log10 decrease in CFU/lung. Such concentrations are unattainable clinically and necessitate alternative strategies such as antibiotic combinations in order to adequately treat biofilm infections.
Only one study has examined polymyxin combination therapy using dynamic antibiotic concentrations against bacteria growing in a biofilm. Using a CDC biofilm reactor Lora-Tamayo et al. examined colistin (constant concentrations of 1.25 mg/L and 3.50 mg/L) in combination with doripenem (C max 25 mg/L every 8 h; half-life, 1 h) over 72 h against P. aeruginosa [102]. One colistin-susceptible reference strain and two MDR-colistin-susceptible-carbapenem-resistant clinical isolates were employed, with bacterial killing of both biofilm-embedded and planktonic bacteria examined; each clinical isolate had been the cause of outbreaks in the Hospital Universitario de Bellvitge in Barcelona, Spain, and contained either a VIM-2 metallo-β-lactamase or a PSE-1 β-lactamase plus a MexXY-OprM efflux-pump. Against biofilm-embedded bacteria monotherapy with colistin at 1.25 mg/L was ineffective against the reference strain and produced only modest, non-bactericidal killing of the clinical isolates; colistin at 3.5 mg/L produced greater and more rapid initial killing against all three strains, but with subsequent regrowth by 72 h such that bactericidal activity was only observed at this time against one clinical strain. The combination of colistin 1.25 mg/L plus doripenem showed some additive effects against biofilm-embedded bacteria during the first 24–32 h of treatment (Fig. 16.6, top panels), but was generally no better than colistin monotherapy against the clinical isolates. The combination of colistin 3.5 mg/L plus doripenem resulted in greater and more sustained killing than either corresponding monotherapy across 72 h. Notably, against both clinical isolates greater initial killing (of ~2–3 log10 CFU/cm2 compared to equivalent monotherapy) was observed and the combination remained synergistic at 72 h (Fig. 16.6, top panels). Importantly, both colistin/doripenem combinations eliminated the emergence of colistin resistance against biofilm -embedded bacteria observed with the highest colistin monotherapy (3.50 mg/L) (Fig. 16.6, lower panels), and substantially reduced (colistin 1.25 mg/L plus doripenem) or eliminated (colistin 3.5 mg/L plus doripenem) the emergence of resistance in planktonic bacteria.

Upper panels: Bacterial killing by colistin (Col) alone at two different clinically relevant concentrations, doripenem (Dor) alone, and in combination against biofilm -embedded cells of three different P. aeruginosa strains; results expressed using the log change method (log change = log10[CFUt] − log10[CFU0]). Lower panels: Emergence of colistin resistance (i.e. colonies able to grow in the presence of ≥4 mg/L colistin) among biofilm-embedded P. aeruginosa across the treatment period with the same treatment regimens. Results expressed as the absolute number of recovered bacteria. For the lower panels, the y axis starts from the limit of quantification. Data are presented as means ± standard deviation of the mean. (Figure adapted from Lora-Tamayo et al. [102], with permission)
16.3 Animal Studies
Only a small number of animal studies have examined polymyxin combination therapy, providing mixed results. All these studies have utilized colistin (or CMS). Unfortunately, there are a number of shortcomings with the existing literature which makes the results difficult to interpret. Specifically, it is not always possible to ascertain whether the ‘colistin’ administered in these studies was colistin (sulphate) or CMS (sodium). In patients colistin is administered in the form of its inactive derivative, CMS, with the active species colistin forming in vivo following CMS administration (Chap. 7). However, in animal models the administration of colistin sulphate is preferable as it permits greater control over the PK profile of the active species, colistin. In a number of studies, it is unclear whether colistin or CMS was administered [33,34,35, 58, 64, 136, 195, 200]. Importantly, irrespective of the form of ‘colistin’ utilised, few studies provide a rationale for the doses of CMS/colistin administered with the majority of administered doses apparently chosen to reflect human doses on a mg/kg basis. However, such dosing fails to recognise the importance of animal scaling that results in PK dissimilarities across species [202], resulting in substantially lower plasma concentrations in the preclinical models. Adding to this difficulty is that PK data for CMS/colistin and second antibiotic are absent from virtually all investigations, preventing comparisons with PK profiles achieved in patients; such comparisons are crucial to adequately assess the likely value of the combination in the clinical setting. Where concentrations of antibiotics are measured, antimicrobial assays are generally used for quantification of antibiotic concentrations. As previously discussed, such assays are incapable of providing accurate information on the time-course of plasma concentrations of the prodrug (CMS) and the active entity (colistin). Given these shortcomings results from animal studies will only be considered briefly here.
Yamagishi et al. used a murine thigh infection model to examine ‘colistin’ (16 mg/kg/12 h administered intraperitoneally [IP]) combined with aztreonam (400 mg/8 h; administered subcutaneously [SC]) against five clinical isolates (two MDR) of P. aeruginosa [195]. Though the authors’ state the administered dosing regimens produce antimicrobial exposures similar to humans following IV administration of standard doses, the achieved concentrations of each agent were not reported. Compared to monotherapy, the combination at 24 h produced greater bacterial killing (maximum additional killing ~1 log10 CFU) against four of five isolates. Using mouse [34] and rat [33] sepsis models Cirioni et al. examined ‘colistin’ (CMS or colistin sulphate not specified; 1 mg/kg) in combination with either imipenem (mouse model; 20 mg/kg) or rifampicin (rat model; 10 mg/kg) against a reference strain and MDR clinical isolate of P. aeruginosa; all antibiotics were administered IV and once only. ‘Colistin’ plus either imipenem or rifampicin resulted in significant reductions in bacterial counts across 72 h when compared with monotherapy with either drug, although only the colistin/imipenem combination resulted in significantly lower mortality. Aoki et al. examined the effect of CMS (administered either intranasally (5 mg/kg/12 h) or subcutaneously (10 mg/kg/12 h) in combination with either imipenem (30 mg/kg/12 h SC) or rifampicin (25 mg/kg/24 h orally) against a reference strain and MDR clinical isolate of P. aeruginosa using a mouse pneumonia model [8]; treatment was continued for 48 h. Whereas all control mice and mice treated with CMS, imipenem or rifampicin monotherapy died within 42 h of infection with the reference strain, the CMS plus imipenem or rifampicin combinations increased survival to 62.5% and 75% at 72 h, respectively. A clear difference was observed in survival between mice treated with intranasal or SC CMS plus rifampicin (100% vs. 14%; P < 0.01); intranasal CMS was also superior to CMS administered SC when combined with imipenem. Similar trends were observed with the MDR clinical isolate.
Against MDR A. baumannii, two studies found no differences in survival or bacterial clearance from the lungs in mouse pneumonia models with rifampicin monotherapy (IP: 25 mg/kg/6 h or 25 mg/kg/24 h; rifampicin was the most active monotherapy) and rifampicin/CMS (IM; 20 mg/kg/8 h or 40 mg/kg/6 h) combination therapy [118, 130]. However, in the same model Yang et al. observed significantly fewer bacteria at 24 h in the lungs of mice treated IP with ‘colistin’ (10 mg/kg) and minocycline (50 mg/kg) compared to monotherapy, with the combination producing substantially greater survival at 7 days [200]. Against a single MDR isolate of A. baumannii, Pantopoulou et al. found little difference in survival with CMS (3 mg/kg IM) or rifampicin (5 mg/kg IV) as mono- or combination therapy in a neutropenic rat thigh infection model, although in this investigation both antibiotics were administered as single doses only at the beginning of the experiment [136]. In a much larger study in a murine thigh infection model involving 15 extensively drug-resistant (XDR) isolates of A. baumannii, reductions in bacterial counts of >2log10 CFU compared to monotherapy at 48 h were observed with the combinations of ‘colistin’ (20 mg/kg/8 h) and fusidic acid (500 mg/kg/8 h) or rifampicin (25 mg/kg/6 h) [58]; these combinations were superior to colistin combined with meropenem (200 mg/kg/8 h), tigecycline (50 mg/kg/24 h), fosfomycin (100 mg/kg/4 h), and sulbactam (120 mg/kg/12 h). In a mouse sepsis model, the addition of sulbactam (240 mg/kg/12 h IP) to CMS (5 mg/kg/12 h IP) had no significant effect on bacterial counts of a single carbapenem-resistant (OXA-51-, OXA-58- and PER-1-positive) isolate of A. baumanni [47]. However, in a mouse sepsis model involving two clinical isolates (1 MDR) of A. baumannii, Cirioni et al. recently showed a single a dose of ‘colistin’ (1 mg/kg) plus either daptomycin (7 mg/kg) or teicoplanin (7 mg/kg) administered IP substantially enhanced survival at 72 h [35]. In that study lethality rates against the susceptible isolates were 100% in the control group, 80% with daptomycin or teicoplanin alone, 50% with colistin alone, 10% with colistin/daptomycin and 15% with colistin/teicoplanin; lethality rates were similar against the MDR isolate. The combinations also significantly reduced the number of bacteria in intraabdominal fluid.
Giacometti et al. examined ‘colistin’ (CMS or colistin sulphate not specified; 1 mg/kg) in combination with piperacillin (60 mg/kg) against E. coli in a rat intraperitoneal infection model [64]. Following a single IP administration of antibiotics, mortality at 48 h was 93.3%, 33.3%, 33.3%, and 0% for controls, ‘colistin’ monotherapy, piperacillin monotherapy, and the ‘colistin’ plus piperacillin combination, respectively. More recently, Michail et al. examined several combinations of tigecycline (50 mg/kg/24 h SC) including with CMS (40 mg/kg/8 h SC) against eight clinical isolates of K. pneumoniae and two isolates (one clinical isolate and one reference strain) of E. coli in a murine thigh infection model [113]; all organisms produced KPC-2 carbapenemase and were susceptible to colistin. As monotherapy, CMS exhibited substantially less bacterial killing than tigecycline. In combination, bacterial killing at 48 h was either essentially the same as tigecycline monotherapy or, in 4 (40%) of 10 cases, antagonistic. However, as antagonism was broadly defined as simply a lower log10 CFU reduction with combination therapy compared to monotherapy, the magnitude of this antagonism is unclear. Demiraslan et al. similarly examined the combination of CMS (5 mg/kg/12 h IP) and tigecycline (20 mg/kg/12 h IP) against a single OXA-48-producing carbapenem-resistant isolate of K. pneumoniae using a sepsis mouse model [44]; this strain was also positive for bla TEM-1 and bla CTX-M-15 genes and was susceptible to both colistin and tigecycline. The combination was tested against both immunocompetent and immunosuppressed mice. In both sets of mice, bacterial counts at 24 and 48 h in liver and lung samples were decreased by both CMS and tigecycline monotherapy compared to controls, however there was no significant difference between the most active monotherapy (CMS) and combination therapy at this time. Mutlu Yilmaz et al. likewise found no differences in efficacy between CMS (1.25 mg/kg/6 h IP) and tigecycline (10 mg/kg/12 h IP) monotherapy and combination therapy across 48 h against a single MDR strain of A. baumannii using a rat pneumonia model [122].
Only one study has specifically examined polymyxin combinations against biofilms in vivo [102], most likely due to a lack of suitable models. Bacterial cells growing within a biofilm are often substantially more resistant than planktonic cells to antibiotic treatment due to the self-produced polymeric matrix that protects the cells from environmental, immune system and antimicrobial threats [41, 48, 107, 121]. With increased MICs and minimum bactericidal concentrations (MBCs) of polymyxins associated with biofilm infections [69, 70], and increasing multidrug-resistance generally, alternative strategies such as polymyxin combination therapy have been suggested for treatment of biofilm infections [102]. Corvec et al. employed a foreign-body infection model involving the implantation of Teflon cages into guinea pigs (four cages/guinea pig) to investigate the activity of antibiotic combinations including colistin (15 mg/kg) in combination with fosfomycin (150 mg/kg), gentamicin (10 mg/kg) and tigecycline (10 mg/kg) [40]; all antibiotics were administered 12-hourly IP for 4 days. A single extended-spectrum-β-lactamase (ESBL)-producing clinical strain of E. coli susceptible to all antibiotics tested was employed. Although the authors reported significantly lower (>3 log10 CFU/mL) bacterial counts (and therefore greater bacterial killing) with each combination immediately and 5 days after the treatment period against planktonic bacteria aspirated from cage fluid, it appears that comparisons of combination therapy were only made against gentamicin or tigecycline monotherapy without including colistin or fosfomycin monotherapy. When the latter are included the differences in bacterial killing appear not to be as great immediately following therapy, although in all cases combinations did result in substantially improved bacterial killing relative to monotherapy 5 days following cessation of treatment. Against biofilm-embedded bacteria 5 days following discontinuation of antibiotic therapy, only monotherapy with fosfomycin was able to eradicate some biofilms (cure rate of 17%; cure rate defined as the percentage of total cages without E. coli growth). However, the combinations of colistin with fosfomycin, tigecycline and gentamicin significantly increased the cure rate to 67%, 50% and 33%, respectively.
Two research group have employed an invertebrate model of the wax moth caterpillar Galleria mellonella which has been proposed as an inexpensive an easy alternative to mammalian models to generate reliable and reproducible data on microbial virulence similar to that obtained using higher animals [31, 76, 159]. One group examined the activities of colistin (2.5 mg/kg)/glycopeptide (vancomycin and teicoplanin, 10 mg/kg) [74] or colistin (2.5 mg/kg)/telavancin (10 mg/kg) [73] combinations against A. baumannii-infected caterpillars (1 ATCC reference strain and 1 MDR clinical isolate) over 96 h. In 5 (83%) of 6 cases (3 combinations across 2 isolates) combinations significantly enhanced the survival of larvae compared with monotherapy. Other similar experiments by the same group examined a colistin (0.25 mg/kg)/tigecycline (1 mg/kg) combination against carbapenem-resistant Enterobacteriaceae (six strains comprising E. coli (n = 2), Enterobacter aerogenes (n = 1), Enterobacter cloacae (n = 1) and K. pneumoniae (n = 2)) [18], and the same combination plus a colistin (0.25 mg/kg)/rifampicin (10 mg/kg) combination against two strains of Stenotrophomonas maltophilia [19]. The colistin/tigecycline combination significantly improved survival against all Enterobacteriaceae isolates and 1 (50%) of 2 S. maltophilia isolates, while the colistin/rifampicin combination significantly improved survival in both S. maltophilia isolates. More recently, another group has undertaken similar experiments over 96 with colistin (2.5 mg/kg) combined with vancomycin (15 mg/kg; n = 4) [198], levofloxacin (6.7 mg/kg; n = 4) [192] and daptomycin (4 mg/kg; n = 2) [196] against A. baumannii and colistin (2.5 mg/kg) combined with imipenem (15 mg/kg; n = 2) against E. cloacae [197]; all studies included at least 1 MDR isolate. With the exception of the colistin/vancomycin combination that was less effective than vancomycin monotherapy against a colistin-resistant isolate, in all cases combination therapy significantly improved survival compared to monotherapy.
Clearly, future animal studies investigating polymyxin combination therapy which administer colistin (sulphate) or polymyxin B and which provide the crucial PK data currently lacking in existing studies are urgently required. Such investigations will be crucial to build on the knowledge gained from in vitro studies (discussed above) and are essential to optimise polymyxin therapy.
16.4 Clinical Studies of CMS or Polymyxin B Combination Therapy
Very few studies have formally assessed the benefit of CMS (the sulphomethylated derivative of colistin and the form administered intravenously [IV]) or polymyxin B combinations, and those that have are commonly retrospective in nature. Although a small number of investigations have been undertaken prospectively, these tend to contain small patient numbers and are thus low powered. Additionally, the doses of antibiotics administered, including polymyxins, are often not stated and PK data is absent. The majority of the data reviewed here is taken from studies seeking to ascertain their general benefit in patients. Studies that have assessed CMS or polymyxin B for a variety of MDR Gram-negative pathogens and infection sites combined into single studies have been inconclusive in differentiating between the value of monotherapy and combination therapy [54,55,56,57, 145, 181]. This section will focus on those studies that provide the greatest insight into specific situations where polymyxin combination therapy appears to be of promise or significant value.
Klebsiella pneumoniae
A retrospective cohort analysis by Qureshi et al. examined the utility of combination therapy in treating KPC-producing K. pneumoniae bacteraemia [150]. In total, 41 patients with genetically confirmed infections were included with a majority (32 [78%] of 41) being hospital acquired and the remainder (9 [22%] of 41) health care associated. The primary outcome was 28-day mortality which, among all patients that received definitive antibiotic therapy for >48 h, was 38.2% (12/34; 7 patients did not receive definitive antibiotic therapy). Treatments varied extensively. Nineteen patients received monotherapy with most receiving CMS or polymyxin B (n = 7), tigecycline (n = 5), or a carbapenem (imipenem or meropenem; n = 4); 15 patients received combination antibiotics. For combination therapy, CMS or polymyxin B were combined with unspecified carbapenems (n = 5), tigecycline (n = 1) or a fluoroquinolone (n = 1) while the most common polymyxin-free combination was tigecycline with either a carbapenem (n = 3) or aminoglycoside (n = 2). The doses of each antibiotic administered were not reported. Combination treatment was the only significant predictor of survival (p = 0.02) with a 28-day mortality of 13.3% (2/15) compared to 57.8% (11/19) for monotherapy. Of specific interest, 1 patient receiving CMS or polymyxin B (which polymyxin was not stated) in combination died compared with 4 (57.1%) of 7 patients that received polymyxin monotherapy. The incidence of mortality in patients receiving polymyxin monotherapy was higher than that reported by Dubrovskaya et al. with polymyxin B monotherapy against KPC producing K. pneumoniae (57.1% [4/7] vs. 18% [7/40]) [49]. This difference is likely due to the greater severity of infection in the patients in the former study who were mostly critically ill. All of the deaths in this studied occurred despite K. pneumoniae having MICs within the susceptible range for each of the respective antibiotics administered, highlighting the suboptimal use of CMS and polymyxin B especially as monotherapy.
A case control study conducted in Greece produced similar results for KPC producing K. pneumoniae bloodstream infections. Zarkotou et al. identified 35 patients that received appropriate antimicrobial therapy (considered susceptible to the respective antibiotic using EUCAST Clinical Breakpoints ), a subset of which received CMS [203]. None of 20 patients administered multiple antibiotics died compared to 7 (46.7%) of 15 patients receiving monotherapy. Of the patients that received combination treatment, 14 were administered CMS whereas 7 received CMS as monotherapy; in this latter group mortality was 66.7% (4/7). The most common combination was CMS plus tigecycline (n = 9), while unspecified carbapenems were combined with CMS in an additional 2 patients. Unfortunately, the doses of each antibiotic administered were not specified. Nevertheless, this data provides qualified support for the use of combination regimens including colistin (administered as CMS) against KPC-producing K. pneumoniae bacteraemia. Another study conducted in Italy similarly compared monotherapy to combination treatment in a larger population of 125 patients with KPC-producing K. pneumoniae bacteraemia [182]. CMS was administered as monotherapy in 22 patients and in combination in 51 patients (combined with [No. of patients]: tigecycline [23], gentamicin [7], meropenem [4], tigecycline plus meropenem [16], gentamicin plus meropenem [1]). The dose of CMS administered in both groups was six to nine million international units (IU; equivalent to 180–270 mg of colistin base activity [CBA]) IV every 8–12 h following an unspecified loading dose. Thirty-day mortality was significantly reduced with combination therapy (34.1%; 27/79) compared to monotherapy (54.3%; 25/46). Of the 22 patients that received CMS monotherapy, 11 (50%) died; unfortunately, individual mortality rates for each combination regimen were not stated. The triple combination of colistin, tigecycline, and meropenem was the only drug regimen reported as significantly more common in the survivor group. However, it must not be overlooked that this finding may be the result of the triple combination also being the most common carbapenem-containing combination regimen. This study again confirms the importance of combination therapy in KPC-producing K. pneumoniae bacteraemia and emphasizes the benefit of including a carbapenem with CMS. Further studies are warranted to optimize specific combination regimens.
Overall, the available clinical data supports the use of combination antibiotic regimens over monotherapy for KPC-producing K. pneumoniae bacteraemia, especially those containing either CMS or polymyxin B in combination with a carbapenem or tigecycline [150, 182, 203]. Since KPC strains hydrolyze carbapenems, the evidence that mortality is reduced by the combination of a carbapenem and a polymyxin is of interest. Results from an investigation by Daikos et al. further support the use of a carbapenem in addition to another agent to treat KPC-producing K. pneumonia e, suggesting that if the infecting pathogen has a carbapenem MIC of ≤4 mg/L, combination therapy may reduce mortality compared to other non-carbapenem combinations [42]. The type and severity of infection caused by KPC-producing K. pneumoniae may be an important factor in dictating the utility of polymyxin combination therapy. More severe infections (i.e. bacteraemia) have benefited from combinations with these drugs [150, 182, 203], whereas the cumulative assessment of all types of infection including patients who were considerably less ill, suggests monotherapy with a polymyxin may be sufficient [49]. Further, KPC-producing K. pneumoniae pneumonia and bacteraemia with pneumonia as its source of infection have both been associated with higher mortality and underline clinical scenarios where monotherapy appears insufficient for most patients. This lack of success in treating pneumonia based infections with monotherapy may be the result of low polymyxin concentrations at the site of infection in the lungs where supplemental antibiotics would in theory be useful [75, 209]. Further studies in this regard are warranted.
Pseudomonas aeruginosa
Conway et al. prospectively treated patients with cystic fibrosis (CF) chronically colonized with P. aeruginosa and experiencing an acute respiratory tract exacerbation with CMS monotherapy or combination therapy in an effort to define the benefit of multiple P. aeruginosa coverage in these patients [38]. Patients treated with monotherapy (n = 36) received 160 mg CMS (two million IU [equivalent to 60 mg CBA]) IV every 8 h while those receiving combination therapy (n = 35) received the same CMS dose with additional aztreonam, azlocillin, piperacillin, ceftazidime, imipenem or ciprofloxacin. By Day 12 all patients showed clinical improvement based on clinical measurement, patient weight, Shwachman-Kulczycki score, Chrispin-Norman and Northern chest radiograph scores. However, combination treatment resulted in significantly more patients returning to a normal C-reactive protein level at this time suggesting less inflammatory activity in the lungs. The authors concluded that IV CMS was effective in treating acute respiratory exacerbations of P. aeruginosa as monotherapy or combination therapy.
Linden et al. conducted a prospective study that compared treatment efficacy of CMS monotherapy (n = 10) and combination therapy (n = 13) in 23 patients infected with MDR P. aeruginosa [98]; 21 patients were critically ill, defined as having at least 2 major organ system failures during the study. The types of infection varied with the most common being pneumonia (n = 18), bacteraemia (n = 8) and intra-abdominal infections (n = 6). For patients in both monotherapy and combination treatment groups, CMS was administered IV based on ideal body weight and estimated creatinine clearance (range: ~2.7–13.3 mg/kg/day; equivalent to ~33,000–167,000 IU/kg/day or 1–5 mg CBA/kg/day). Amikacin or an antipseudomonal β-lactam was added to CMS for patients in the combination group. An unfavourable response, defined as persistence or worsening of presenting signs and symptoms or death, was reported for 4 (40%) of 10 patients receiving only CMS and 5 (38.5%) of 13 patients on combination therapy. However, 11 patients had other co-infecting pathogens which may have confounded the results. Based on this data it is evident that colistin provides an important ‘salvage’ option for patients who have failed or are resistant to other antipseudomonal therapies, but it cannot support the use of combination treatment. In a similar study by Furtado et al., polymyxin B combinations (most commonly combined with imipenem) did not provide additional benefit over polymyxin B monotherapy for pneumonia caused by MDR P. aeruginosa [59]. Polymyxin B was dosed based on creatinine clearance (1.5–2.5 mg/kg/day when CrCl ≥80 mL/min; 2.5 mg/kg on Day 1, then 1.0–1.5 mg/kg/day thereafter when CrCl 30–80 mL/min; 2.5 mg/kg on Day 1, then 1.0–1.5 mg/kg every 2–3 days thereafter when CrCl <30 mL/min; 2.5 mg/kg on Day 1, then 1.0 mg/kg every 5–7 days thereafter) and, unusually, was administered by continuous infusion over 24 h rather than in divided intervals (usually every 12 h). There was no difference in favourable outcomes (defined as partial resolution of signs and symptoms by the end of treatment; unfavourable was the persisting or worsening of signs and symptoms or death during treatment) between the groups (14 [50.0%] of 28 vs. 21 [45.7%] of 46 in patients receiving combination therapy and monotherapy, respectively). Based on these data, the authors suggested polymyxin B monotherapy would be an appropriate ‘salvage’ option for MDR P. aeruginosa pneumonia, although the overall low favourable outcome rate (47.3%) relative to other studies may suggest against administering it as a continuous infusion.
To our knowledge, no clinical studies to date support the use of CMS or polymyxin B based combinations in favour of polymyxin monotherapy for treatment of infections caused by MDR P. aeruginosa. Existing data regarding CMS or polymyxin B combinations in humans is limited with studies frequently pooling patients with many types and sites of infection and varying degrees of severity, limiting the usefulness of the results obtained [38, 59, 98]. Further, more focussed studies are warranted which may assist to identify subsets of patients that benefit from combination therapy.
Acinetobacter baumannii
In a recent prospective study, Aydemir et al. compared CMS monotherapy (n = 22) to a combination of CMS and rifampicin (n = 21) for ventilator-associated pneumonia (VAP) caused by carbapenem resistant A. baumannii [10]. CMS was administered at 300 mg CBA/day IV in three divided doses adjusted for renal impairment based on the manufacturer’s recommendations; rifampicin was administered nasogastrically at a dose of 600 mg/day. No difference in the primary endpoint of clinical response was observed between the two groups (40.9% for monotherapy, 52.4% for combination; p = 0.654), however microbiological clearance (a secondary endpoint) was obtained significantly more quickly with combination therapy (4.5 ± 1.7 days for monotherapy, 3.1 ± 0.5 days for combination; P = 0.029).
It is important to note that in the studies discussed above, CMS was dosed according to the product information which likely cannot achieve high enough plasma concentrations to optimally treat severe infections for all patients. In order to more rapidly attain higher plasma concentrations recent studies have suggested the use of a loading dose of nine million IU of CMS (equivalent to ~270 mg of CBA) followed by nine million IU per day in divided doses instead of the six million IU (equivalent to ~180 mg of CBA) received by many of the patients reviewed above [43, 55, 108]; administration of higher doses of polymyxin B have also been suggested [52]. Non-traditional ‘front loaded’ or ‘burst’ polymyxin regimens (e.g. high dose, short duration polymyxin at the start of therapy, with lower overall exposure), especially in combination, require further analysis in patients in order to fully define their therapeutic role in the management of MDR Gram-negative infections. Since the mortality rate remains high for infections with KPC-producing K. pneumoniae, P. aeruginosa and A. baumannii, it is critical to continue to investigate optimal dosing strategies for polymyxins, including the role of combination therapy. Given the limitations associated with existing clinical data future randomized controlled trials with robust study designs are urgently required to more fully understand the utility of CMS or polymyxin B based combinations [138].
16.5 Randomized Controlled Trials Evaluating Polymyxin Combinations
Although there are no adequately powered published randomized controlled trials (RCTs) to examine whether therapy with polymyxins (polymyxin B or colistin) administered in combination with another active agent is superior to polymyxin B or colistin monotherapy against carbapenem-resistant Enterobacteriaceae or carbapenem-resistant P. aeruginosa infections, there are recent RCTs in evaluating polymyxin combinations against MDR A. baumannii. The first open label RCT comparing synergistic combinations with monotherapy was a prospective study by Durante-Mangoni et al. who conducted a larger (n = 209) multi-centre prospective study examining CMS/rifampicin combinations against extensively drug resistant A. baumannii [51]; extensively drug resistant was defined as an MIC ≥16 mg/L for carbapenems and resistant to all other antibiotics except colistin. Patients were allocated to receive either CMS (160 mg or two million units; equivalent to ~60 mg CBA; n = 105) every 8 h IV as monotherapy or CMS (same dose) plus rifampicin 600 mg every 12 h IV (n = 105). Most patients had VAP (69.8%) while the remainder had bloodstream infections (20.1%), hospital acquired pneumonia (8.6%), or intra-abdominal infections (2.4%). Although there was no difference between monotherapy and combination therapy for the primary endpoint of 30-day mortality, eradication of A. baumannii was significantly higher with the addition of rifampicin (60.6% vs 44.8%, P = 0.034). Additionally, the risk of death within 30 days was similar between combination therapy and monotherapy (OR = 0.88, 95% CI 0.46–1.69; P = 0.71) despite a significantly improved microbiological cure rate in patients receiving colistin + rifampin (P = 0.034) with no resistance developing in either arm. No colistin loading dose was administered and the maximum daily maintenance dose was low by current standards.
In another, open label, prospective, randomized trial of 94 patients with carbapenem-resistant A. baumannii (CRAB) infections, subjects were randomised to receive colistin alone or colistin + fosfomycin for 7–14 days [164]. Some patients in both groups received other antibiotics; for example, 17.0% and 8.5% of patients in the monotherapy and combination groups, respectively, received a carbapenem. There was no difference between monotherapy and combination therapy arms in infection-related (23.1% vs. 16.3%; P = 0.507) or all-cause mortality (57.4% vs. 46.8%; P = 0.41). However, the patients who received combination therapy had a significantly more favourable microbiological response than those who received colistin alone. Interestingly, microbiological cure in the first 72 h (65.7% vs. 78.8%; P = 0.028) and at the end of treatment (84.5% vs. 100%; P = 0.023) was greater in the combination arm.
Recently, Paul et al. conducted a randomized controlled superiority trial in 406 patients comparing colistin monotherapy with colistin (nine MIU or 300 mg CBA/day) + high dose extended infusion meropenem combination therapy for the treatment of carbapenem-resistant Gram-negative bacilli [139]. Patients with bacteraemia, ventilator-associated pneumonia, hospital-acquired pneumonia , or urosepsis caused by carbapenem-non-susceptible Gram-negative bacteria were included. Patients received either intravenous colistin (9-million unit loading dose, followed by 4.5 million units twice per day) or colistin with meropenem (2-g prolonged infusion three times per day). The primary outcome was clinical failure, defined as not meeting all success criteria by intention-to-treat analysis, at 14 days after randomisation. Most infections were caused by A. baumannii (312/406, 77%), although some infections were due to CRE and carbapenem-resistant P. aeruginosa. No significant difference between colistin monotherapy (156/198, 79%) and combination therapy (152/208, 73%) was observed for clinical failure at 14 days (risk difference − 5.7%, 95% CI -13.9 to 2.4; risk ratio [RR] 0.93, 95% CI 0.83–1.03). Results were similar among patients with A. baumannii infections (RR 0.97, 95% CI 0.87–1.09). No differences were noted in clinical failure (76% vs. 71%; P = 0.22) or 28-day mortality (41% vs. 41%; P = 0.84) in comparing monotherapy and combination therapy arms. All-cause 28-day mortality was 86 (43%) of 198 patients treated with colistin monotherapy and 94 (45%) of 208 patients treated with combination therapy. Combination therapy increased the incidence of diarrhea (56 [27%] vs 32 [16%] patients) and decreased the incidence of mild renal failure (37 [30%] of 124 vs 25 [20%] of 125 patients at risk of or with kidney injury). There were no significant differences (6% for monotherapy versus 5% for combination therapy; P = 0.77) noted as it relates to colistin-resistance during or after therapy or isolation of new carbapenem-resistant bacteria. As it relates to infection type, most patients had hospital-acquired or ventilator associated pneumonia or bacteraemia (355/406, 87%).
Finally, there is an ongoing RCT comparing colistin monotherapy to colistin plus meropenem combination therapy for the management of invasive infections due to carbapenem-resistant Gram-negative organisms (https://clinicaltrials.gov/ct2/show/NCT01597973). Data from this study, should further elucidate the role of polymyxin combinations. Furthermore, given the potential advantages of polymyxin B over colistin, clinical data assessing the impact of polymyxin B-based combination regimens are needed. Future studies should also address the impact of infection site and resistance mechanisms on the effectiveness of combination therapy.
16.6 Conclusions and Future Directions
In general, the in vitro data for polymyxin combination therapy suggests a potential benefit with many drug combinations, particularly so when only the more sophisticated PK/PD models are considered. A common finding is that low, sub-MIC (yet clinically achievable) concentrations of polymyxins (e.g. 0.5 mg/L) in combination with another agent may significantly enhance bacterial killing even when resistance to one or more of the drugs in combination is present. This may be true not only when the second drug would normally be active against the particular bacterial species but also with agents such as the glycopeptides that should ordinarily have no effect on Gram-negative organisms due to the relative impermeability of the outer membrane. Such an observation is important as total (i.e. bound and unbound) plasma concentrations of colistin (following IV administration of CMS) and polymyxin B are typically in the range of ~2–3 mg at steady state, although a proportion of patients will achieve lower plasma concentrations (Chap. 15) [63, 81, 85, 90, 115, 146, 156, 157, 204]. Given this situation it may nevertheless be possible to enhance bacterial killing with polymyxin combination therapy even in patients who achieve low plasma concentrations with standard dosage regimens. Alternatively, it may be possible to take advantage of increased bacterial killing at low plasma concentrations by using lower-than-normal doses of polymyxins, especially given the toxicity concerns associated with their use (discussed in Chap. 17).
A close look at the existing in vitro data on combination therapy also reveals that even when improvements in bacterial killing were not observed at later time points (e.g. 24 or 48 h), in many cases there were improvement in initial killing (e.g. up to 6 h). While regrowth obviously occurred in these situations it must be remembered that the in vitro models used lack the immune components present in vivo. Thus, in an immunocompetent host combination therapy at the commencement of treatment may help to quickly reduce bacterial levels to facilitate clearance by the immune system. Importantly, the few studies undertaken in PK/PD models have shown a substantial reduction in the emergence of polymyxin-resistant subpopulations. Given the increasing emergence of polymyxin resistance since their reintroduction into clinical practice [3, 7, 77, 86, 110, 112, 170] and their role as a last-line therapeutic option, combination therapy could potentially play an important role in minimising further resistance development.
Finally, the data would suggest that a ‘one-size-fits-all’ approach to identifying optimal combination regimens is not appropriate. This can be illustrated by the study conducted by Clancy et al. where isolates of K. pneumoniae responded differently to a colistin/doripenem combination depending on the presence or absence of particular resistance mechanisms [36]. Thus, the specific resistance mechanisms manifested by different isolates of a bacterial species may dictate the efficacy of particular combination regimens. Ultimately the true value of combination therapy must be evaluated in well-designed, well powered, randomized clinical trials in critically ill patients which are urgently required in order to define the clinical benefit of polymyxin combination therapy. Future advances in rapid diagnostics and next-generation omics technologies will guide optimal use of polymyxin combinations which are precise to each patient, infection site, pathogen and resistance mechanism.
References
Abdul Rahim N, Cheah SE, Johnson MD, Yu H, Sidjabat HE, Boyce J, Butler MS, Cooper MA, Fu J, Paterson DL, Nation RL, Bergen PJ, Velkov T, Li J (2015) Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two ‘old’ antibiotics-polymyxin B and chloramphenicol. J Antimicrob Chemother 70:2589–2597
Adams EK, Ashcraft DS, Pankey GA (2016) In vitro synergistic activity of caspofungin plus polymyxin B against fluconazole-resistant Candida glabrata. Am J Med Sci 351:265–270
Al-Sweih NA, Al-Hubail MA, Rotimi VO (2011) Emergence of tigecycline and colistin resistance in acinetobacter species isolated from patients in Kuwait hospitals. J Chemother 23:13–16
Albur M, Noel A, Bowker K, Macgowan A (2012) Bactericidal activity of multiple combinations of tigecycline and colistin against NDM-1-producing Enterobacteriaceae. Antimicrob Agents Chemother 56:3441–3443
Albur MS, Noel A, Bowker K, Macgowan A (2015) The combination of colistin and fosfomycin is synergistic against NDM-1-producing Enterobacteriaceae in in vitro pharmacokinetic/pharmacodynamic model experiments. Int J Antimicrob Agents 46:560–567
Anonymous (2011) Instructions to authors. Antimicrob Agents Chemother 55:1–23
Antoniadou A, Kontopidou F, Poulakou G, Koratzanis E, Galani I, Papadomichelakis E, Kopterides P, Souli M, Armaganidis A, Giamarellou H (2007) Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: first report of a multiclonal cluster. J Antimicrob Chemother 59:786–790
Aoki N, Tateda K, Kikuchi Y, Kimura S, Miyazaki C, Ishii Y, Tanabe Y, Gejyo F, Yamaguchi K (2009) Efficacy of colistin combination therapy in a mouse model of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. J Antimicrob Chemother 63:534–542
Attridge RT, Carden MF, Padilla S, Nathisuwan S, Burgess DS (2008) Colistin and rifampicin alone and in combination against multidrug-resistant Acinetobacter baumannii using an in vitro PK-PD model (abstract C1-1053, p69). In: Abstracts of the 48th annual interscience conference on antimicrobial agents and chemotherapy (ICAAC), Washington, DC, October 25–28. American Society for Microbiology
Aydemir H, Akduman D, Piskin N, Comert F, Horuz E, Terzi A, Kokturk F, Ornek T, Celebi G (2013) Colistin vs. the combination of colistin and rifampicin for the treatment of carbapenem-resistant Acinetobacter baumannii ventilator-associated pneumonia. Epidemiol Infect 141:1214–1222
Batirel A, Balkan I, Karabay O, Agalar C, Akalin S, Alici O, Alp E, Altay FA, Altin N, Arslan F, Aslan T, Bekiroglu N, Cesur S, Celik AD, Dogan M, Durdu B, Duygu F, Engin A, Engin DO, Gonen I, Guclu E, Guven T, Hatipoglu CA, Hosoglu S, Karahocagil MK, Kilic AU, Ormen B, Ozdemir D, Ozer S, Oztoprak N, Sezak N, Turhan V, Turker N, Yilmaz H (2014) Comparison of colistin-carbapenem, colistin-sulbactam, and colistin plus other antibacterial agents for the treatment of extremely drug-resistant Acinetobacter baumannii bloodstream infections. Eur J Clin Microbiol Infect Dis 33(8):1311–1322
Bergen PJ, Bulitta JB, Forrest A, Tsuji BT, Li J, Nation RL (2010) Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob Agents Chemother 54:3783–3789
Bergen PJ, Forrest A, Bulitta JB, Tsuji BT, Sidjabat HE, Paterson DL, Li J, Nation RL (2011) Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother 55:5134–5142
Bergen PJ, Li J, Nation RL, Turnidge JD, Coulthard K, Milne RW (2008) Comparison of once-, twice- and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J Antimicrob Chemother 61:636–642
Bergen PJ, Li J, Rayner CR, Nation RL (2006) Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:1953–1958
Bergen PJ, Tsuji BT, Bulitta JB, Forrest A, Jacob J, Sidjabat HE, Paterson DL, Nation RL, Li J (2011) Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 55:5685–5695
Betrosian AP, Frantzeskaki F, Xanthaki A, Georgiadis G (2007) High-dose ampicillin-sulbactam as an alternative treatment of late-onset VAP from multidrug-resistant Acinetobacter baumannii. Scand J Infect Dis 39:38–43
Betts JW, Phee LM, Hornsey M, Woodford N, Wareham DW (2014) In vitro and in vivo activity of tigecycline/colistin combination therapies against carbapenem resistant Enterobacteriaceae. Antimicrob Agents Chemother 58:3541–3546
Betts JW, Phee LM, Woodford N, Wareham DW (2014) Activity of colistin in combination with tigecycline or rifampicin against multidrug-resistant Stenotrophomonas maltophilia. Eur J Clin Microbiol Infect Dis 33(9):1565–1572
Bhavnani SM, Hammel JP, Cirincione BB, Wikler MA, Ambrose PG (2005) Use of pharmacokinetic-pharmacodynamic target attainment analyses to support phase 2 and 3 dosing strategies for doripenem. Antimicrob Agents Chemother 49:3944–3947
Blaser J (1985) In-vitro model for simultaneous simulation of the serum kinetics of two drugs with different half-lives. J Antimicrob Chemother 15(Suppl A):125–130
Bulitta JB, Landersdorfer CB, Forrest A, Brown SV, Neely MN, Tsuji BT, Louie A (2011) Relevance of pharmacokinetic and pharmacodynamic modeling to clinical care of critically ill patients. Curr Pharm Biotechnol 12:2044–2061
Bulitta JB, Li J, Poudyal A, Yu HH, Owen RJ, Tsuji BT, Nation RL, Forrest A (2009) Quantifying synergy of colistin combinations against MDR Gram-negatives by mechanism-based models (abstract A1-573, p41). In: Abstracts of the 49th annual interscience conference of antimicrobial agents and chemotherapy (ICAAC), San Francisco, CA, September 12–15. American Society for Microbiology
Bulitta JB, Yang JC, Yohonn L, Ly NS, Brown SV, D’Hondt RE, Jusko WJ, Forrest A, Tsuji BT (2010) Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother 54:2051–2062
Bulman ZP, Ly NS, Lenhard JR, Holden PN, Bulitta JB, Tsuji BT (2017) Influence of rhlR and lasR on polymyxin pharmacodynamics in Pseudomonas aeruginosa and implications for quorum sensing inhibition with azithromycin. Antimicrob Agents Chemother 61(4):pii: e00096-16
Bulman ZP, Sutton MD, Ly NS, Bulitta JB, Holden PN, Nation RL, Li J, Tsuji BT (2015) Emergence of polymyxin B resistance influences pathogenicity in Pseudomonas aeruginosa mutators. Antimicrob Agents Chemother 59:4343–4346
Cai X, Yang Z, Dai J, Chen K, Zhang L, Ni W, Wei C, Cui J (2017) Pharmacodynamics of tigecycline alone and in combination with colistin against clinical isolates of multidrug-resistant Acinetobacter baumannii in an in vitro pharmacodynamic model. Int J Antimicrob Agents 49:609–616
Cappelletty DM, Rybak MJ (1996) Comparison of methodologies for synergism testing of drug combinations against resistant strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother 40:677–683
Cavalcanti AB, Goncalves AR, Almeida CS, Bugano DD, Silva E (2010) Teicoplanin versus vancomycin for proven or suspected infection. Cochrane Database Syst Rev. https://doi.org/10.1002/14651858.CD007022.pub2
Ceccarelli G, Falcone M, Giordano A, Mezzatesta ML, Caio C, Stefani S, Venditti M (2013) Successful ertapenem-doripenem combination treatment of bacteremic ventilator-associated pneumonia due to colistin-resistant KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother 57:2900–2901
Champion OL, Cooper IA, James SL, Ford D, Karlyshev A, Wren BW, Duffield M, Oyston PC, Titball RW (2009) Galleria mellonella as an alternative infection model for Yersinia pseudotuberculosis. Microbiology 155:1516–1522
Choi MJ, Ko KS (2013) Mutant prevention concentrations of colistin for Acinetobacter baumannii, Pseudomonas aeruginosa and Klebsiella pneumoniae clinical isolates. J Antimicrob Chemother 69(1):275–277
Cirioni O, Ghiselli R, Orlando F, Silvestri C, Mocchegiani F, Rocchi M, Chiodi L, Abbruzzetti A, Saba V, Scalise G, Giacometti A (2007) Efficacy of colistin/rifampin combination in experimental rat models of sepsis due to a multiresistant Pseudomonas aeruginosa strain. Crit Care Med 35:1717–1723
Cirioni O, Ghiselli R, Silvestri C, Kamysz W, Orlando F, Mocchegiani F, Di Matteo F, Riva A, Lukasiak J, Scalise G, Saba V, Giacometti A (2007) Efficacy of tachyplesin III, colistin, and imipenem against a multiresistant Pseudomonas aeruginosa strain. Antimicrob Agents Chemother 51:2005–2010
Cirioni O, Simonetti O, Pierpaoli E, Barucca A, Ghiselli R, Orlando F, Pelloni M, Trombettoni MM, Guerrieri M, Offidani A, Giacometti A, Provinciali M (2016) Colistin enhances therapeutic efficacy of daptomycin or teicoplanin in a murine model of multiresistant Acinetobacter baumannii sepsis. Diagn Microbiol Infect Dis 86:392–398
Clancy CJ, Chen L, Hong JH, Cheng S, Hao B, Shields RK, Farrell AN, Doi Y, Zhao Y, Perlin DS, Kreiswirth BN, Nguyen MH (2013) Mutations of the ompK36 porin gene and promoter impact responses of sequence type 258, KPC-2-producing Klebsiella pneumoniae strains to doripenem and doripenem-colistin. Antimicrob Agents Chemother 57:5258–5265
Cobo J, Morosini MI, Pintado V, Tato M, Samaranch N, Baquero F, Canton R (2008) Use of tigecycline for the treatment of prolonged bacteremia due to a multiresistant VIM-1 and SHV-12 beta – lactamase-producing Klebsiella pneumoniae epidemic clone. Diagn Microbiol Infect Dis 60:319–322
Conway SP, Pond MN, Watson A, Etherington C, Robey HL, Goldman MH (1997) Intravenous colistin sulphomethate in acute respiratory exacerbations in adult patients with cystic fibrosis. Thorax 52:987–993
Cordoba J, Coronado-Alvarez NM, Parra D, Parra-Ruiz J (2015) In vitro activities of novel antimicrobial combinations against extensively drug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 59:7316–7319
Corvec S, Furustrand Tafin U, Betrisey B, Borens O, Trampuz A (2013) Activities of fosfomycin, tigecycline, colistin, and gentamicin against extended-spectrum-beta-lactamase-producing Escherichia coli in a foreign-body infection model. Antimicrob Agents Chemother 57:1421–1427
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
Daikos G, Markogiannakis A (2011) Carbapenemase-producing Klebsiella pneumoniae: (when) might we still consider treating with carbapenems? Clin Microbiol Infect 17:1135–1141
Dalfino L, Puntillo F, Mosca A, Monno R, Spada ML, Coppolecchia S, Miragliotta G, Bruno F, Brienza N (2012) High-dose, extended-interval colistin administration in critically ill patients: is this the right dosing strategy? A preliminary study. Clin Infect Dis 54:1720–1726
Demiraslan H, Dinc G, Ahmed SS, Elmali F, Metan G, Alp E, Doganay M (2013) Carbapenem-resistant Klebsiella pneumoniae sepsis in corticosteroid receipt mice: tigecycline or colistin monotherapy versus tigecycline/colistin combination. J Chemother 26(5):276–281. https://doi.org/10.1179/1973947813Y.0000000143
Deris ZZ, Yu HH, Davis K, Soon RL, Jacob J, Ku CK, Poudyal A, Bergen PJ, Tsuji BT, Bulitta JB, Forrest A, Paterson DL, Velkov T, Li J, Nation RL (2012) The combination of colistin and doripenem is synergistic against Klebsiella pneumoniae at multiple inocula and suppresses colistin resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 56:5103–5112
Di X, Wang R, Liu B, Zhang X, Ni W, Wang J, Liang B, Cai Y, Liu Y (2015) In vitro activity of fosfomycin in combination with colistin against clinical isolates of carbapenem-resistant Pseudomas aeruginosa. J Antibiot (Tokyo) 68:551–555
Dinc G, Demiraslan H, Elmali F, Ahmed SS, Metan G, Alp E, Doganay M (2014) Efficacy of sulbactam and its combination with imipenem, colistin and tigecycline in an experimental model of carbapenem-resistant Acinetobacter baumannii sepsis. Chemotherapy 59:325–329
Donlan RM (2002) Biofilms: microbial life on surfaces. Emerg Infect Dis 8:881–890
Dubrovskaya Y, Chen TY, Scipione MR, Esaian D, Phillips MS, Papadopoulos J, Mehta SA (2013) Risk factors for treatment failure of polymyxin B monotherapy for carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob Agents Chemother 57:5394–5397
Dudhani RV, Turnidge JD, Nation RL, Li J (2010) fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J Antimicrob Chemother 65:1984–1990
Durante-Mangoni E, Signoriello G, Andini R, Mattei A, De Cristoforo M, Murino P, Bassetti M, Malacarne P, Petrosillo N, Galdieri N, Mocavero P, Corcione A, Viscoli C, Zarrilli R, Gallo C, Utili R (2013) Colistin and rifampicin compared with colistin alone for the treatment of serious infections due to extensively drug-resistant Acinetobacter baumannii: a multicenter, randomized clinical trial. Clin Infect Dis 57:349–358
Elias LS, Konzen D, Krebs JM, Zavascki AP (2010) The impact of polymyxin B dosage on in-hospital mortality of patients treated with this antibiotic. J Antimicrob Chemother 65:2231–2237
Entenza JM, Moreillon P (2009) Tigecycline in combination with other antimicrobials: a review of in vitro, animal and case report studies. Int J Antimicrob Agents 34(8):e1–e9
Falagas ME, Bliziotis IA, Kasiakou SK, Samonis G, Athanassopoulou P, Michalopoulos A (2005) Outcome of infections due to pandrug-resistant (PDR) Gram-negative bacteria. BMC Infect Dis 5:24
Falagas ME, Rafailidis PI, Ioannidou E, Alexiou VG, Matthaiou DK, Karageorgopoulos DE, Kapaskelis A, Nikita D, Michalopoulos A (2010) Colistin therapy for microbiologically documented multidrug-resistant Gram-negative bacterial infections: a retrospective cohort study of 258 patients. Int J Antimicrob Agents 35:194–199
Falagas ME, Rafailidis PI, Kasiakou SK, Hatzopoulou P, Michalopoulos A (2006) Effectiveness and nephrotoxicity of colistin monotherapy vs. colistin-meropenem combination therapy for multidrug-resistant Gram-negative bacterial infections. Clin Microbiol Infect 12:1227–1230
Falagas ME, Rafailidis PI, Matthaiou DK, Virtzili S, Nikita D, Michalopoulos A (2008) Pandrug-resistant Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii infections: characteristics and outcome in a series of 28 patients. Int J Antimicrob Agents 32:450–454
Fan B, Guan J, Wang X, Cong Y (2016) Activity of colistin in combination with meropenem, tigecycline, fosfomycin, fusidic acid, rifampin or sulbactam against extensively drug-resistant Acinetobacter baumannii in a murine thigh-infection model. PLoS One 11:e0157757
Furtado GHC, D’Azevedo PA, Santos AF, Gales AC, Pignatari ACC, Medeiros EAS (2007) Intravenous polymyxin B for the treatment of nosocomial pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. Int J Antimicrob Agents 30:315–319
Gaibani P, Lombardo D, Lewis RE, Mercuri M, Bonora S, Landini MP, Ambretti S (2014) In vitro activity and post-antibiotic effects of colistin in combination with other antimicrobials against colistin-resistant KPC-producing Klebsiella pneumoniae bloodstream isolates. J Antimicrob Chemother 69(7):1856–1865
Galani I, Orlandou K, Moraitou H, Petrikkos G, Souli M (2014) Colistin/daptomycin: an unconventional antimicrobial combination synergistic in vitro against multidrug-resistant Acinetobacter baumannii. Int J Antimicrob Agents 43:370–374
Garnacho-Montero J, Amaya-Villar R, Gutierrez-Pizarraya A, Espejo-Gutierrez De Tena E, Artero-Gonzalez ML, Corcia-Palomo Y, Bautista-Paloma J (2013) Clinical efficacy and safety of the combination of colistin plus vancomycin for the treatment of severe infections caused by carbapenem-resistant Acinetobacter baumannii. Chemotherapy 59:225–231
Garonzik SM, Li J, Thamlikitkul V, Paterson DL, Shoham S, Jacob J, Silveira FP, Forrest A, Nation RL (2011) Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 55:3284–3294
Giacometti A, Cirioni O, Ghiselli R, Orlando F, Mocchegiani F, D’Amato G, Silvestri C, Riva A, Del Prete MS, Saba V, Scalise G (2003) Antiendotoxin activity of antimicrobial peptides and glycopeptides. J Chemother 15:129–133
Gloede J, Scheerans C, Derendorf H, Kloft C (2010) In vitro pharmacodynamic models to determine the effect of antibacterial drugs. J Antimicrob Chemother 65:186–201
Gordon NC, Png K, Wareham DW (2010) Potent synergy and sustained bactericidal activity of a vancomycin-colistin combination versus multidrug-resistant strains of Acinetobacter baumannii. Antimicrob Agents Chemother 54:5316–5322
Gunderson BW, Ibrahim KH, Hovde LB, Fromm TL, Reed MD, Rotschafer JC (2003) Synergistic activity of colistin and ceftazidime against multiantibiotic-resistant Pseudomonas aeruginosa in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 47:905–909
Hagihara M, Housman ST, Nicolau DP, Kuti JL (2014) In vitro pharmacodynamics of polymyxin B and tigecycline alone and in combination against carbapenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 58:874–879
Hengzhuang W, Wu H, Ciofu O, Song Z, Hoiby N (2011) Pharmacokinetics/pharmacodynamics of colistin and imipenem on mucoid and nonmucoid Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother 55:4469–4474
Hengzhuang W, Wu H, Ciofu O, Song Z, Hoiby N (2012) In vivo pharmacokinetics/pharmacodynamics of colistin and imipenem in Pseudomonas aeruginosa biofilm infection. Antimicrob Agents Chemother 56:2683–2690
Henry R, Vithanage N, Harrison P, Seemann T, Coutts S, Moffatt JH, Nation RL, Li J, Harper M, Adler B, Boyce JD (2012) Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids, and poly-beta-1,6-N-acetylglucosamine. Antimicrob Agents Chemother 56:59–69
Herrmann G, Yang L, Wu H, Song Z, Wang H, Hoiby N, Ulrich M, Molin S, Riethmuller J, Doring G (2010) Colistin-tobramycin combinations are superior to monotherapy concerning the killing of biofilm Pseudomonas aeruginosa. J Infect Dis 202:1585–1592
Hornsey M, Phee L, Longshaw C, Wareham DW (2013) In vivo efficacy of telavancin/colistin combination therapy in a Galleria mellonella model of Acinetobacter baumannii infection. Int J Antimicrob Agents 41:285–287
Hornsey M, Wareham DW (2011) In vivo efficacy of glycopeptide-colistin combination therapies in a Galleria mellonella model of Acinetobacter baumannii infection. Antimicrob Agents Chemother 55:3534–3537
Imberti R, Cusato M, Villani P, Carnevale L, Iotti GA, Langer M, Regazzi M (2010) Steady-state pharmacokinetics and BAL concentration of colistin in critically Ill patients after IV colistin methanesulfonate administration. Chest 138:1333–1339
Jander G, Rahme LG, Ausubel FM (2000) Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol 182:3843–3845
Johansen HK, Moskowitz SM, Ciofu O, Pressler T, Hoiby N (2008) Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J Cyst Fibros 7:391–397
Kasiakou SK, Michalopoulos A, Soteriades ES, Samonis G, Sermaides GJ, Falagas ME (2005) Combination therapy with intravenous colistin for management of infections due to multidrug-resistant Gram-negative bacteria in patients without cystic fibrosis. Antimicrob Agents Chemother 49:3136–3146
Kaye KS, Pogue JM, Tran TB, Nation RL, Li J (2016) Agents of last resort: polymyxin resistance. Infect Dis Clin N Am 30:391–414
Ku YH, Chen CC, Lee MF, Chuang YC, Tang HJ, Yu WL (2017) Comparison of synergism between colistin, fosfomycin and tigecycline against extended-spectrum beta-lactamase-producing Klebsiella pneumoniae isolates or with carbapenem resistance. J Microbiol Immunol Infect 50(6):931–939
Kwa AL, Lim TP, Low JG, Hou J, Kurup A, Prince RA, Tam VH (2008) Pharmacokinetics of polymyxin B1 in patients with multidrug-resistant Gram-negative bacterial infections. Diagn Microbiol Infect Dis 60:163–167
Lagerback P, Khine WW, Giske CG, Tangden T (2016) Evaluation of antibacterial activities of colistin, rifampicin and meropenem combinations against NDM-1-producing Klebsiella pneumoniae in 24 h in vitro time-kill experiments. J Antimicrob Chemother 71:2321–2325
Lee GC, Burgess DS (2013) Polymyxins and doripenem combination against KPC-producing Klebsiella pneumoniae. J Clin Med Res 5:97–100
Lee HJ, Bergen PJ, Bulitta JB, Tsuji B, Forrest A, Nation RL, Li J (2013) Synergistic activity of colistin and rifampin combination against multidrug-resistant Acinetobacter baumannii in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 57:3738–3745
Lee J, Han S, Jeon S, Hong T, Song W, Woo H, Yim DS (2013) Population pharmacokinetic analysis of colistin in burn patients. Antimicrob Agents Chemother 57:2141–2146
Lee JY, Song JH, Ko KS (2011) Identification of nonclonal Pseudomonas aeruginosa isolates with reduced colistin susceptibility in Korea. Microb Drug Resist 17:299–304
Leite GC, Oliveira MS, Perdigao-Neto LV, Rocha CK, Guimaraes T, Rizek C, Levin AS, Costa SF (2016) Antimicrobial combinations against pan-resistant Acinetobacter baumannii isolates with different resistance mechanisms. PLoS One 11:e0151270
Lenhard JR, Smith NM, Bulman ZP, Tao X, Thamlikitkul V, Shin BS, Nation RL, Li J, Bulitta JB, Tsuji BT (2017) High dose ampicillin/sulbactam combinations combat polymyxin-resistant Acinetobacter baumannii in a hollow-fiber infection model. Antimicrob Agents Chemother 61(3):pii: e01268-16
Lenhard JR, Thamlikitkul V, Silveira FP, Garonzik SM, Tao X, Forrest A, Soo Shin B, Kaye KS, Bulitta JB, Nation RL, Li J, Tsuji BT (2017) Polymyxin-resistant, carbapenem-resistant Acinetobacter baumannii is eradicated by a triple combination of agents that lack individual activity. J Antimicrob Chemother 72:1415–1420
Li J, Coulthard K, Milne R, Nation RL, Conway S, Peckham D, Etherington C, Turnidge J (2003) Steady-state pharmacokinetics of intravenous colistin methanesulphonate in patients with cystic fibrosis. J Antimicrob Chemother 52:987–992
Li J, Milne RW, Nation RL, Turnidge JD, Coulthard K (2003) Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob Agents Chemother 47:1364–1370
Li J, Nation RL, Owen RJ, Wong S, Spelman D, Franklin C (2007) Antibiograms of multidrug-resistant clinical Acinetobacter baumannii: promising therapeutic options for treatment of infection with colistin-resistant strains. Clin Infect Dis 45:594–598
Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, Liolios L (2006) Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50:2946–2950
Liang W, Liu XF, Huang J, Zhu DM, Li J, Zhang J (2011) Activities of colistin- and minocycline-based combinations against extensive drug resistant Acinetobacter baumannii isolates from intensive care unit patients. BMC Infect Dis 11:109
Lim TP, Tan TY, Lee W, Sasikala S, Tan TT, Hsu LY, Kwa AL (2009) In vitro activity of various combinations of antimicrobials against carbapenem-resistant Acinetobacter species in Singapore. J Antibiot (Tokyo) 62:675–679
Lin KH, Chuang YC, Lee SH, Yu WL (2010) In vitro synergistic antimicrobial effect of imipenem and colistin against an isolate of multidrug-resistant Enterobacter cloacae. J Microbiol Immunol Infect 43:317–322
Lin L, Nonejuie P, Munguia J, Hollands A, Olson J, Dam Q, Kumaraswamy M, Rivera H Jr, Corriden R, Rohde M, Hensler ME, Burkart MD, Pogliano J, Sakoulas G, Nizet V (2015) Azithromycin synergizes with cationic antimicrobial peptides to exert bactericidal and therapeutic activity against highly multidrug-resistant Gram-negative bacterial pathogens. EBioMedicine 2:690–698
Linden PK, Kusne S, Coley K, Fontes P, Kramer DJ, Paterson D (2003) Use of parenteral colistin for the treatment of serious infection due to antimicrobial-resistant Pseudomonas aeruginosa. Clin Infect Dis 37:e154–e160
Lister PD, Wolter DJ, Hanson ND (2009) Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610
Liu X, Zhao M, Chen Y, Bian X, Li Y, Shi J, Zhang J (2016) Synergistic killing by meropenem and colistin combination of carbapenem-resistant Acinetobacter baumannii isolates from Chinese patients in an in vitro pharmacokinetic/pharmacodynamic model. Int J Antimicrob Agents 48:559–563
Loos U, Musch E, Jensen JC, Mikus G, Schwabe HK, Eichelbaum M (1985) Pharmacokinetics of oral and intravenous rifampicin during chronic administration. Klin Wochenschr 63:1205–1211
Lora-Tamayo J, Murillo O, Bergen PJ, Nation RL, Poudyal A, Luo X, Yu HY, Ariza J, Li J (2014) Activity of colistin combined with doripenem at clinically relevant concentrations against multidrug-resistant Pseudomonas aeruginosa in an in vitro dynamic biofilm model. J Antimicrob Chemother 69(9):2434–2442
Ly NS, Bulitta JB, Rao GG, Landersdorfer CB, Holden PN, Forrest A, Bergen PJ, Nation RL, Li J, Tsuji BT (2015) Colistin and doripenem combinations against Pseudomonas aeruginosa: profiling the time course of synergistic killing and prevention of resistance. J Antimicrob Chemother 70:1434–1442
Ly NS, Bulman ZP, Bulitta JB, Baron C, Rao GG, Holden PN, Li J, Sutton MD, Tsuji BT (2016) Optimization of polymyxin B in combination with doripenem to combat mutator Pseudomonas aeruginosa. Antimicrob Agents Chemother 60:2870–2880
Maccallum DM, Desbois AP, Coote PJ (2013) Enhanced efficacy of synergistic combinations of antimicrobial peptides with caspofungin versus Candida albicans in insect and murine models of systemic infection. Eur J Clin Microbiol Infect Dis 32:1055–1062
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL (2012) Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281
Mah TF, O’Toole GA (2001) Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39
Markou N, Markantonis SL, Dimitrakis E, Panidis D, Boutzouka E, Karatzas S, Rafailidis P, Apostolakos H, Baltopoulos G (2008) Colistin serum concentrations after intravenous administration in critically ill patients with serious multidrug-resistant, Gram-negative bacilli infections: a prospective, open-label, uncontrolled study. Clin Ther 30:143–151
Mendes RE, Fritsche TR, Sader HS, Jones RN (2008) Increased antimicrobial susceptibility profiles among polymyxin-resistant Acinetobacter baumannii clinical isolates. Clin Infect Dis 46:1324–1326
Menuet M, Bittar F, Stremler N, Dubus JC, Sarles J, Raoult D, Rolain JM (2008) First isolation of two colistin-resistant emerging pathogens, Brevundimonas diminuta and Ochrobactrum anthropi, in a woman with cystic fibrosis: a case report. J Med Case Rep 2:373
Mezzatesta ML, Caio C, Gona F, Zingali T, Salerno I, Stefani S (2016) Colistin Increases the cidal activity of antibiotic combinations against multidrug-resistant Klebsiella pneumoniae: an in vitro model comparing multiple combination bactericidal testing at one peak serum concentration and time-kill method. Microb Drug Resist 22:360–363
Mezzatesta ML, Gona F, Caio C, Petrolito V, Sciortino D, Sciacca A, Santangelo C, Stefani S (2011) Outbreak of KPC-3-producing, and colistin-resistant, Klebsiella pneumoniae infections in two Sicilian hospitals. Clin Microbiol Infect 17:1444–1447
Michail G, Labrou M, Pitiriga V, Manousaka S, Sakellaridis N, Tsakris A, Pournaras S (2013) Activity of tigecycline in combination with colistin, meropenem, rifampin, or gentamicin against KPC-producing Enterobacteriaceae in a murine thigh infection model. Antimicrob Agents Chemother 57:6028–6033
Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD (2010) Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977
Mohamed AF, Karaiskos I, Plachouras D, Karvanen M, Pontikis K, Jansson B, Papadomichelakis E, Antoniadou A, Giamarellou H, Armaganidis A, Cars O, Friberg LE (2012) Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother 56:4241–4249
Moland ES, Craft DW, Hong SG, Kim SY, Hachmeister L, Sayed SD, Thomson KS (2008) In vitro activity of tigecycline against multidrug-resistant Acinetobacter baumannii and selection of tigecycline-amikacin synergy. Antimicrob Agents Chemother 52:2940–2942
Moneib NA (1995) In-vitro activity of commonly used antifungal agents in the presence of rifampin, polymyxin B and norfloxacin against Candida albicans. J Chemother 7:525–529
Montero A, Ariza J, Corbella X, Domenech A, Cabellos C, Ayats J, Tubau F, Borraz C, Gudiol F (2004) Antibiotic combinations for serious infections caused by carbapenem-resistant Acinetobacter baumannii in a mouse pneumonia model. J Antimicrob Chemother 54:1085–1091
Moore NM, Flaws ML (2011) Treatment strategies and recommendations for Pseudomonas aeruginosa infections. Clin Lab Sci 24:52–56
Morelli P, Ferrario A, Tordato F, Piazza A, Casari E (2014) Successful treatment of post-neurosurgical multidrug-resistant Pseudomonas aeruginosa meningo-encephalitis with combination therapy of colistin, rifampicin and doripenem. J Antimicrob Chemother 69:857–859
Moskowitz SM, Foster JM, Emerson J, Burns JL (2004) Clinically feasible biofilm susceptibility assay for isolates of Pseudomonas aeruginosa from patients with cystic fibrosis. J Clin Microbiol 42:1915–1922
Mutlu Yilmaz E, Sunbul M, Aksoy A, Yilmaz H, Guney AK, Guvenc T (2012) Efficacy of tigecycline/colistin combination in a pneumonia model caused by extensively drug-resistant Acinetobacter baumannii. Int J Antimicrob Agents 40:332–336
Napier BA, Band V, Burd EM, Weiss DS (2014) Colistin heteroresistance in Enterobacter cloacae is associated with cross-resistance to the host antimicrobial lysozyme. Antimicrob Agents Chemother 58:5594–5597
Nastro M, Rodriguez CH, Monge R, Zintgraff J, Neira L, Rebollo M, Vay C, Famiglietti A (2013) Activity of the colistin-rifampicin combination against colistin-resistant, carbapenemaseproducing Gram-negative bacteria. J Chemother 26(4):211–216
Nicolau DP (2008) Carbapenems: a potent class of antibiotics. Expert Opin Pharmacother 9:23–37
Norden CW, Wentzel H, Keleti E (1979) Comparison of techniques for measurement of in vitro antibiotic synergism. J Infect Dis 140:629–633
Oleksiuk LM, Nguyen MH, Press EG, Updike CL, O’Hara JA, Doi Y, Clancy CJ, Shields RK (2014) In vitro responses of Acinetobacter baumannii to two- and three-drug combinations following exposure to colistin and doripenem. Antimicrob Agents Chemother 58:1195–1199
Owen RJ, Li J, Nation RL, Spelman D (2007) In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J Antimicrob Chemother 59:473–477
Ozbek B, Mataraci E (2013) In vitro effectiveness of colistin, tigecycline and levofloxacin alone and combined with clarithromycin and/or heparin as lock solutions against embedded Acinetobacter baumannii strains. J Antimicrob Chemother 68:827–830
Pachon-Ibanez ME, Docobo-Perez F, Lopez-Rojas R, Dominguez-Herrera J, Jimenez-Mejias ME, Garcia-Curiel A, Pichardo C, Jimenez L, Pachon J (2010) Efficacy of rifampin and its combinations with imipenem, sulbactam, and colistin in experimental models of infection caused by imipenem-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 54:1165–1172
Padilla S, Carden MF, Attridge RT, Burgess DS (2008) In-vitro activity of colistin, amikacin and in combination against multi-drug resistant Acinetobacter baumannii in a pharmacodynamic-pharmacokinetic model. 4th annual Louis C. Littlefield celebrating pharmacy research excellence day in professional, graduate and postgraduate programs, University of Texas, Austin, TX, April 17
Pamp SJ, Gjermansen M, Johansen HK, Tolker-Nielsen T (2008) Tolerance to the antimicrobial peptide colistin in Pseudomonas aeruginosa biofilms is linked to metabolically active cells, and depends on the pmr and mexAB-oprM genes. Mol Microbiol 68:223–240
Pankey G, Ashcraft D, Kahn H, Ismail A (2014) Time-kill assay and Etest evaluation for synergy with polymyxin B and fluconazole against Candida glabrata. Antimicrob Agents Chemother 58:5795–5800
Pankuch GA, Lin G, Seifert H, Appelbaum PC (2008) Activity of meropenem with and without ciprofloxacin and colistin against Pseudomonas aeruginosa and Acinetobacter baumannii. Antimicrob Agents Chemother 52:333–336
Pankuch GA, Seifert H, Appelbaum PC (2010) Activity of doripenem with and without levofloxacin, amikacin, and colistin against Pseudomonas aeruginosa and Acinetobacter baumannii. Diagn Microbiol Infect Dis 67:191–197
Pantopoulou A, Giamarellos-Bourboulis EJ, Raftogannis M, Tsaganos T, Dontas I, Koutoukas P, Baziaka F, Giamarellou H, Perrea D (2007) Colistin offers prolonged survival in experimental infection by multidrug-resistant Acinetobacter baumannii: the significance of co-administration of rifampicin. Int J Antimicrob Agents 29:51–55
Park GC, Choi JA, Jang SJ, Jeong SH, Kim CM, Choi IS, Kang SH, Park G, Moon DS (2016) In vitro interactions of antibiotic combinations of colistin, tigecycline, and doripenem against extensively drug-resistant and multidrug-resistant Acinetobacter baumannii. Ann Lab Med 36:124–130
Paul M, Carmeli Y, Durante-Mangoni E, Mouton JW, Tacconelli E, Theuretzbacher U, Mussini C, Leibovici L (2014) Combination therapy for carbapenem-resistant Gram-negative bacteria. J Antimicrob Chemother 69(9):2305–2309. https://doi.org/10.1093/jac/dku168
Paul M, Daikos GL, Durante-Mangoni E, Yahav D, Carmeli Y, Benattar YD, Skiada A, Andini R, Eliakim-Raz N, Nutman A, Zusman O, Antoniadou A, Pafundi PC, Adler A, Dickstein Y, Pavleas I, Zampino R, Daitch V, Bitterman R, Zayyad H, Koppel F, Levi I, Babich T, Friberg LE, Mouton JW, Theuretzbacher U, Leibovici L (2018) Colistin alone versus colistin plus meropenem for treatment of severe infections caused by carbapenem-resistant Gram-negative bacteria: an open-label, randomised controlled trial. Lancet Infect Dis 18:391–400
Percin D, Akyol S, Kalin G (2014) In vitro synergism of combinations of colistin with selected antibiotics against colistin-resistant Acinetobacter baumannii. GMS Hyg Infect Control 9(2):Doc14
Petrosillo N, Giannella M, Antonelli M, Antonini M, Barsic B, Belancic L, Inkaya AC, De Pascale G, Grilli E, Tumbarello M, Akova M (2014) Clinical experience of colistin-glycopeptide combination in critically ill patients infected with Gram-negative bacteria. Antimicrob Agents Chemother 58:851–858
Petrosillo N, Ioannidou E, Falagas ME (2008) Colistin monotherapy vs. combination therapy: evidence from microbiological, animal and clinical studies. Clin Microbiol Infect 14:816–827
Phee LM, Betts JW, Bharathan B, Wareham DW (2015) Colistin and fusidic acid: a novel potent synergistic combination for the treatment of multi-drug resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother 59(8):4544–4550
Pillai SK, Moellering RC, Eliopoulos GM (2005) Antimicrobial combinations. In: Lorian V (ed) Antibiotics in laboratory medicine, 5th edn. Lippincott Williams & Wilkins, Philadelphia
Pintado V, San Miguel LG, Grill F, Mejia B, Cobo J, Fortun J, Martin-Davila P, Moreno S (2008) Intravenous colistin sulphomethate sodium for therapy of infections due to multidrug-resistant gram-negative bacteria. J Infect 56:185–190
Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris I, Karaiskos I, Poulakou G, Kontopidou F, Armaganidis A, Cars O, Giamarellou H (2009) Population pharmacokinetic analysis of colistin methanesulphonate and colistin after intravenous administration in critically ill patients with Gram-negative bacterial infections. Antimicrob Agents Chemother 53:3430–3436
Poudyal A, Howden BP, Bell JM, Gao W, Owen RJ, Turnidge JD, Nation RL, Li J (2008) In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother 62:1311–1318
Pournaras S, Vrioni G, Neou E, Dendrinos J, Dimitroulia E, Poulou A, Tsakris A (2011) Activity of tigecycline alone and in combination with colistin and meropenem against Klebsiella pneumoniae carbapenemase (KPC)-producing Enterobacteriaceae strains by time-kill assay. Int J Antimicrob Agents 37:244–247
Principe L, Capone A, Mazzarelli A, D’Arezzo S, Bordi E, Di Caro A, Petrosillo N (2013) In vitro activity of doripenem in combination with various antimicrobials against multidrug-resistant Acinetobacter baumannii: possible options for the treatment of complicated infection. Microb Drug Resist 19:407–414
Qureshi ZA, Paterson DL, Potoski BA, Kilayko MC, Sandovsky G, Sordillo E, Polsky B, Adams-Haduch JM, Doi Y (2012) Treatment outcome of bacteremia due to KPC-producing Klebsiella pneumoniae: superiority of combination antimicrobial regimens. Antimicrob Agents Chemother 56:2108–2113
Rahal JJ (2006) Novel antibiotic combinations against infections with almost completely resistant Pseudomonas aeruginosa and Acinetobacter species. Clin Infect Dis 43(Suppl 2):S95–S99
Rodriguez-Avial I, Pena I, Picazo JJ, Rodriguez-Avial C, Culebras E (2015) In vitro activity of the next-generation aminoglycoside plazomicin alone and in combination with colistin, meropenem, fosfomycin or tigecycline against carbapenemase-producing Enterobacteriaceae strains. Int J Antimicrob Agents 46:616–621
Rodriguez CH, Nastro M, Vay C, Famiglietti A (2015) In vitro activity of minocycline alone or in combination in multidrug-resistant Acinetobacter baumannii isolates. J Med Microbiol 64:1196–1200
Ruslami R, Nijland HM, Alisjahbana B, Parwati I, Van Crevel R, Aarnoutse RE (2007) Pharmacokinetics and tolerability of a higher rifampin dose versus the standard dose in pulmonary tuberculosis patients. Antimicrob Agents Chemother 51:2546–2551
Safarika A, Galani I, Pistiki A, Giamarellos-Bourboulis EJ (2015) Time-kill effect of levofloxacin on multidrug-resistant Pseudomonas aeruginosa and Acinetobacter baumannii: synergism with imipenem and colistin. Eur J Clin Microbiol Infect Dis 34:317–323
Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Bordinhao RC, Wang J, Forrest A, Nation RL, Li J, Zavascki AP (2013) Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis 57:524–531
Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, Behle TF, Saitovitch D, Wang J, Forrest A, Nation RL, Zavascki AP, Li J (2013) Pharmacokinetics of polymyxin B in patients on continuous venovenous haemodialysis. J Antimicrob Chemother 68:674–677
Schemuth H, Dittmer S, Lackner M, Sedlacek L, Hamprecht A, Steinmann E, Buer J, Rath PM, Steinmann J (2013) In vitro activity of colistin as single agent and in combination with antifungals against filamentous fungi occurring in patients with cystic fibrosis. Mycoses 56:297–303
Seed KD, Dennis JJ (2008) Development of Galleria mellonella as an alternative infection model for the Burkholderia cepacia complex. Infect Immun 76:1267–1275
Selwyn S (1983) The history of antimicrobial agents. In: Clinical chemotherapy. fundamentals, vol 1. Thieme-Stratton, New York
Sheng WH, Wang JT, Li SY, Lin YC, Cheng A, Chen YC, Chang SC (2011) Comparative in vitro antimicrobial susceptibilities and synergistic activities of antimicrobial combinations against carbapenem-resistant Acinetobacter species: Acinetobacter baumannii versus Acinetobacter genospecies 3 and 13TU. Diagn Microbiol Infect Dis 70:380–386
Shields RK, Clancy CJ, Gillis LM, Kwak EJ, Silveira FP, Massih RC, Eschenauer GA, Potoski BA, Nguyen MH (2012) Epidemiology, clinical characteristics and outcomes of extensively drug-resistant Acinetobacter baumannii infections among solid organ transplant recipients. PLoS One 7:e52349
Shields RK, Kwak EJ, Potoski BA, Doi Y, Adams-Haduch JM, Silviera FP, Toyoda Y, Pilewski JM, Crespo M, Pasculle AW, Clancy CJ, Nguyen MH (2011) High mortality rates among solid organ transplant recipients infected with extensively drug-resistant Acinetobacter baumannii: using in vitro antibiotic combination testing to identify the combination of a carbapenem and colistin as an effective treatment regimen. Diagn Microbiol Infect Dis 70:246–252
Sirijatuphat R, Thamlikitkul V (2014) Preliminary study of colistin versus colistin plus fosfomycin for treatment of carbapenem-resistant Acinetobacter baumannii infections. Antimicrob Agents Chemother 58:5598–5601
Smith NM, Bulman ZP, Sieron AO, Bulitta JB, Holden PN, Nation RL, Li J, Wright GD, Tsuji BT (2017) Pharmacodynamics of dose-escalated ‘front-loading’ polymyxin B regimens against polymyxin-resistant mcr-1-harbouring Escherichia coli. J Antimicrob Chemother 72:2297–2303
Souli M, Galani I, Boukovalas S, Gourgoulis MG, Chryssouli Z, Kanellakopoulou K, Panagea T, Giamarellou H (2011) In vitro interactions of antimicrobial combinations with fosfomycin against KPC-2-producing Klebsiella pneumoniae and protection of resistance development. Antimicrob Agents Chemother 55:2395–2397
Souli M, Rekatsina PD, Chryssouli Z, Galani I, Giamarellou H, Kanellakopoulou K (2009) Does the activity of the combination of imipenem and colistin in vitro exceed the problem of resistance in metallo-beta-lactamase-producing Klebsiella pneumoniae isolates? Antimicrob Agents Chemother 53:2133–2135
Srisupha-Olarn W, Burgess DS (2010) Activity of meropenem and colistin alone and in combination against multidrug-resistant Acinetobacter baumannii in a pharmacokinetic-pharmacodynamic model (abstract E-1591, p125). In: Abstracts of the 50th annual interscience conference on antimicrobial agents and chemotherapy (ICAAC), Boston, MA, September 12–15. American Society for Microbiology
Stressmann FA, Rogers GB, Marsh P, Lilley AK, Daniels TW, Carroll MP, Hoffman LR, Jones G, Allen CE, Patel N, Forbes B, Tuck A, Bruce KD (2011) Does bacterial density in cystic fibrosis sputum increase prior to pulmonary exacerbation? J Cyst Fibros 10:357–365
Suh JY, Son JS, Chung DR, Peck KR, Ko KS, Song JH (2010) Nonclonal emergence of colistin-resistant Klebsiella pneumoniae isolates from blood samples in South Korea. Antimicrob Agents Chemother 54:560–562
Sun HY, Shields RK, Cacciarelli TV, Muder RR, Singh N (2010) A novel combination regimen for the treatment of refractory bacteremia due to multidrug-resistant Pseudomonas aeruginosa in a liver transplant recipient. Transpl Infect Dis 12:555–560
Svetitsky S, Leibovici L, Paul M (2009) Comparative efficacy and safety of vancomycin versus teicoplanin: systematic review and meta-analysis. Antimicrob Agents Chemother 53:4069–4079
Tam VH, Schilling AN, Vo G, Kabbara S, Kwa AL, Wiederhold NP, Lewis RE (2005) Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:3624–3630
Tan CH, Li J, Nation RL (2007) Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 51:3413–3415
Tan TY, Ng LS, Tan E, Huang G (2007) In vitro effect of minocycline and colistin combinations on imipenem-resistant Acinetobacter baumannii clinical isolates. J Antimicrob Chemother 60:421–423
Tang HJ, Ku YH, Lee MF, Chuang YC, Yu WL (2015) In vitro activity of imipenem and colistin against a carbapenem-resistant Klebsiella pneumoniae isolate coproducing SHV-31, CMY-2, and DHA-1. Biomed Res Int 2015:568079
Tangden T, Hickman RA, Forsberg P, Lagerback P, Giske CG, Cars O (2014) Evaluation of double- and triple-antibiotic combinations for VIM- and NDM-producing Klebsiella pneumoniae by in vitro time-kill experiments. Antimicrob Agents Chemother 58:1757–1762
Tangden T, Karvanen M, Friberg LE, Odenholt I, Cars O (2017) Assessment of early combination effects of colistin and meropenem against Pseudomonas aeruginosa and Acinetobacter baumannii in dynamic time-kill experiments. Infect Dis (Lond) 49:521–527
Tascini C, Gemignani G, Ferranti S, Tagliaferri E, Leonildi A, Lucarini A, Menichetti F (2004) Microbiological activity and clinical efficacy of a colistin and rifampin combination in multidrug-resistant Pseudomonas aeruginosa infections. J Chemother 16:282–287
Tripodi MF, Durante-Mangoni E, Fortunato R, Utili R, Zarrilli R (2007) Comparative activities of colistin, rifampicin, imipenem and sulbactam/ampicillin alone or in combination against epidemic multidrug-resistant Acinetobacter baumannii isolates producing OXA-58 carbapenemases. Int J Antimicrob Agents 30:537–540
Tsioutis C, Kritsotakis EI, Maraki S, Gikas A (2010) Infections by pandrug-resistant Gram-negative bacteria: clinical profile, therapeutic management, and outcome in a series of 21 patients. Eur J Clin Microbiol Infect Dis 29:301–305
Tumbarello M, Viale P, Viscoli C, Trecarichi EM, Tumietto F, Marchese A, Spanu T, Ambretti S, Ginocchio F, Cristini F (2012) Predictors of mortality in bloodstream infections caused by Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: importance of combination therapy. Clin Infect Dis 55:943–950
Turnidge J (2014) Drug-drug combinations. In: Vinks AA, Derendorf H, Mouton J (eds) Fundamentals of antimicrobial pharmacokinetics and pharmacodynamics. Springer, New York
Urban C, Mariano N, Rahal JJ (2010) In vitro double and triple bactericidal activities of doripenem, polymyxin B, and rifampin against Multidrug-resistant Acinetobacter baumannii, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Escherichia coli. Antimicrob Agents Chemother 54:2732–2734
Vardakas KZ, Mavros MN, Roussos N, Falagas ME (2012) Meta-analysis of randomized controlled trials of vancomycin for the treatment of patients with gram-positive infections: focus on the study design. Mayo Clin Proc 87:349–363
Velkov T, Bergen PJ, Lora-Tamayo J, Landersdorfer CB, Li J (2013) PK/PD models in antibacterial development. Curr Opin Microbiol 16(5):573–579
Vidaillac C, Benichou L, Duval RE (2012) In vitro synergy of colistin combinations against colistin-resistant Acinetobacter baumannii, Pseudomonas aeruginosa, and Klebsiella pneumoniae isolates. Antimicrob Agents Chemother 56:4856–4861
Walsh CC, Landersdorfer CB, McIntosh MP, Peleg AY, Hirsch EB, Kirkpatrick CM, Bergen PJ (2016) Clinically relevant concentrations of fosfomycin combined with polymyxin B, tobramycin or ciprofloxacin enhance bacterial killing of Pseudomonas aeruginosa, but do not suppress the emergence of fosfomycin resistance. J Antimicrob Chemother 71:2218–2229
Walsh CC, McIntosh MP, Peleg AY, Kirkpatrick CM, Bergen PJ (2015) In vitro pharmacodynamics of fosfomycin against clinical isolates of Pseudomonas aeruginosa. J Antimicrob Chemother 70(11):3042–3050
Wareham DW, Gordon NC, Hornsey M (2011) In vitro activity of teicoplanin combined with colistin versus multidrug-resistant strains of Acinetobacter baumannii. J Antimicrob Chemother 66:1047–1051
Wehrli W (1983) Rifampin: mechanisms of action and resistance. Rev Infect Dis 5(Suppl 3):S407–S411
Wei W, Yang H, Hu L, Ye Y, Li J (2015) Activity of levofloxacin in combination with colistin against Acinetobacter baumannii: in vitro and in a Galleria mellonella model. J Microbiol Immunol Infect 50(6):821–830
Wei WJ, Yang HF (2017) Synergy against extensively drug-resistant Acinetobacter baumannii in vitro by two old antibiotics: colistin and chloramphenicol. Int J Antimicrob Agents 49:321–326
Wootton M, Hedges AJ, Bowker KE, Holt HA, Reeves DS, Macgowan AP (1995) A critical assessment of the agar dilution chequerboard technique for studying in-vitro antimicrobial interactions using a representative beta-lactam, aminoglycoside and fluoroquinolone. J Antimicrob Chemother 35:569–576
Yamagishi Y, Hagihara M, Kato H, Hirai J, Nishiyama N, Koizumi Y, Sakanashi D, Suematsu H, Nakai H, Mikamo H (2017) In vitro and in vivo pharmacodynamics of colistin and aztreonam alone and in combination against multidrug-resistant Pseudomonas aeruginosa. Chemotherapy 62:105–110
Yang H, Chen G, Hu L, Liu Y, Cheng J, Li H, Ye Y, Li J (2015) In vivo activity of daptomycin/colistin combination therapy in a Galleria mellonella model of Acinetobacter baumannii infection. Int J Antimicrob Agents 45:188–191
Yang H, Chen G, Hu L, Liu Y, Cheng J, Ye Y, Li J (2016) Enhanced efficacy of imipenem-colistin combination therapy against multiple-drug-resistant Enterobacter cloacae: in vitro activity and a Galleria mellonella model. J Microbiol Immunol Infect 51(1):70–75
Yang H, Lv N, Hu L, Liu Y, Cheng J, Ye Y, Li J (2016) In vivo activity of vancomycin combined with colistin against multidrug-resistant strains of Acinetobacter baumannii in a Galleria mellonella model. Infect Dis (Lond) 48:189–194
Yang Y, Bhachech N, Bush K (1995) Biochemical comparison of imipenem, meropenem and biapenem: permeability, binding to penicillin-binding proteins, and stability to hydrolysis by beta-lactamases. J Antimicrob Chemother 35:75–84
Yang YS, Lee Y, Tseng KC, Huang WC, Chuang MF, Kuo SC, Lauderdale TL, Chen TL (2016) In vivo and in vitro efficacy of minocycline-based combination therapy for minocycline-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 60:4047–4054
Yu W, Zhou K, Guo L, Ji J, Niu T, Xiao T, Shen P, Xiao Y (2017) In vitro pharmacokinetics/pharmacodynamics evaluation of fosfomycin combined with amikacin or colistin against KPC2-producing Klebsiella pneumoniae. Front Cell Infect Microbiol 7:246
Zak O, O’Reilly T (1991) Animal models in the evaluation of antimicrobial agents. Antimicrob Agents Chemother 35:1527–1531
Zarkotou O, Pournaras S, Tselioti P, Dragoumanos V, Pitiriga V, Ranellou K, Prekates A, Themeli-Digalaki K, Tsakris A (2011) Predictors of mortality in patients with bloodstream infections caused by KPC-producing Klebsiella pneumoniae and impact of appropriate antimicrobial treatment. Clin Microbiol Infect 17:1798–1803
Zavascki AP, Goldani LZ, Cao G, Superti SV, Lutz L, Barth AL, Ramos F, Boniatti MM, Nation RL, Li J (2008) Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 47:1298–1304
Zeidler U, Bougnoux ME, Lupan A, Helynck O, Doyen A, Garcia Z, Sertour N, Clavaud C, Munier-Lehmann H, Saveanu C, D’Enfert C (2013) Synergy of the antibiotic colistin with echinocandin antifungals in Candida species. J Antimicrob Chemother 68:1285–1296
Zhai B, Zhou H, Yang L, Zhang J, Jung K, Giam CZ, Xiang X, Lin X (2010) Polymyxin B, in combination with fluconazole, exerts a potent fungicidal effect. J Antimicrob Chemother 65:931–938
Zhang L, Dhillon P, Yan H, Farmer S, Hancock RE (2000) Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob Agents Chemother 44:3317–3321
Zhao M, Bulman ZP, Lenhard JR, Satlin MJ, Kreiswirth BN, Walsh TJ, Marrocco A, Bergen PJ, Nation RL, Li J, Zhang J, Tsuji BT (2017) Pharmacodynamics of colistin and fosfomycin: a ‘treasure trove’ combination combats KPC-producing Klebsiella pneumoniae. J Antimicrob Chemother 72(7):1985–1990
Ziv G, Nouws JF, Van Ginneken CA (1982) The pharmacokinetics and tissue levels of polymyxin B, colistin and gentamicin in calves. J Vet Pharmacol Ther 5:45–58
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bergen, P.J., Smith, N.M., Bedard, T.B., Bulman, Z.P., Cha, R., Tsuji, B.T. (2019). Rational Combinations of Polymyxins with Other Antibiotics. In: Li, J., Nation, R., Kaye, K. (eds) Polymyxin Antibiotics: From Laboratory Bench to Bedside. Advances in Experimental Medicine and Biology, vol 1145. Springer, Cham. https://doi.org/10.1007/978-3-030-16373-0_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-16373-0_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16371-6
Online ISBN: 978-3-030-16373-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)