
Abstract
The variation between and among the many types of cancer presents a formidable challenge both to practicing clinicians and medical researchers. There are several characteristics that are common to all cancers such as unrestrained proliferation and evasion of cell death. Another common feature is that of metastasis. Metastasis is “initiated” when primary tumor cells acquire the ability to invade surrounding tissues and eventually develop secondary tumors in distant locations. This process appears to rely not only on changes at the genetic level of tumor cells themselves but also from their interaction with surrounding stromal cells and the immune system. The genetic and molecular changes that give rise to metastatic change are of special interest due to the significant decline in a patient’s prognosis after metastasis has occured. A host of genes and pathways involved in several pathways have been implicated in this process, several of which will be reviewed in detail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Our understanding of the processes of tumorigenesis and metastasis has evolved over time. During the last decade the use of automated high-throughput screening methods has become more widespread and the costs of DNA sequencing and microarray analysis have significantly declined. Large-scale studies have allowed scientists to identify genes and signalling pathways that contribute to a tumor cell’s capacity for metastasis. Perhaps the most important contribution to our understanding of metastasis has been a move away from reductionist approaches to the study of this disease process. The development of new in vivo models has significantly aided in our understanding of metastasis, a process that is likely impossible to mimic in vitro. For example, in the Rip-Tag transgenic mouse model of pancreatic islet cell tumorigenesis, forced expression of VEGF-C in tumor islet cells encourages metastasis via lymph nodes [62]. Also, improvements in in vivo live imaging techniques have the potential to provide major breakthroughs in our understanding of cancer metastasis [9, 17].
Metastasis occurs when cells from a primary tumor acquire the capacity to travel to other parts of the body and form secondary tumors. It is a complex and spectacularly inefficient process. Cancer cells escape from the primary tumor each day but only a tiny fraction of these survive. Of those that manage to survive challenges present in the general circulation, such as hydrodynamic shear forces and immune cells, even fewer will go on to colonize other parts of the body, and yet fewer still are able to successfully form metastatic lesions [11, 40]. Cells capable of metastasis may not go on to form detectable metastatic lesions immediately upon coloniziaton of another part of the body [50]. For reasons not yet clear, not all types of cancer are equal in terms of capacity to metastasize. Cancer of epithelial tissue are far more likely to become life-threatening via metastasis than cancers originating from other tissues. Metastasis is a dreaded diagnosis as it carries a very poor patient prognosis (American Cancer Society (2011). Cancer Facts and Figures 2011. Atlanta, GA: American Cancer Society). Metastasis is the cause of death in 90% of deaths from solid tumors [62].
Although the characteristics of metastasis typically vary by cancer type, there are some general trends that have been identified from large-scale analysis of patient data. Tumor size and regional lymph node involvement are among the two most important predictors of future [14]. Although tumor size being predictive of prognosis is at first glance logical, in that a larger mass of cells is mathematically more likely to have acquired genetic changes that may contribute to metastatic ability, this is not always the case. Some patients present with metastatic disease with an unidentifiable primary tumor (cancer of unknown primary or CUP). As for the predicitive ability of nodal involvement, in the case of sarcomas, nodal involvement is seen in less than 3% of patients [18]. Tumor grade, depth of invasion and lymphovascular invasion are also important predictors of metastatic risk across cancer types [10, 14]. Patterns of metastasis also differ by cancer type and can differ among individuals, however certain trends have been clearly identified. For example, in colon cancer, the most common site of metastasis is liver (via venous blood flow from the colon to the liver) and in breast cancer they are the contralateral breast tissue and lymph nodes (via lymphatic channels).
2 Models of Cancer Metastasis
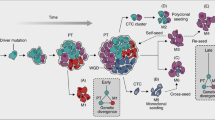
Many different models of tumorigenesis and metastasis have been put forth over the years. Both the Halsted and later Fisher models of metastasis in breast cancer were limited in their ability to explain variations observed in clinical data. Hellman suggests that a more useful view is that of breast cancer as a complex spectrum of diseases which can be explained by both predetermination and traditional progression models [28]. In the clonal dominance model, cells with metastatic ability take over and dominate the overall population of the tumor [58]. The dynamic heterogeneity model posits that metastatic variants occur at a certain frequency within the tumor cell population and are unstable. Thus their turnover limits the overall capacity of a tumor to become metastatic [26]. The ability to determine patient prognosis by DNA microarray analysis of primary tumors suggests that cells with metastatic ability may not be as rare as suggested by some models of metastasis. Such data seems to point toward a model in which genetic changes acquired relatively early on in disease progression that are necessary for tumorigenesis are also necessary for metastasis (Fig. 5.1). This would help to explain cases of cancer of unknown primary. Yet again we are confronted with clinical data at odds with this explanation, such as the success of early screening in reducing cancer mortality. Also, cases in which cancer cells remain dormant for long periods of time after removal of primary tumors only to re-appear years later in distant sites suggest that additional mutations are necessary for successful metastasis. Yet global gene expression analysis of primary and metastatic tumors reveals, time and again, very little difference between the two expression patterns. This suggests that a very small number of key genes are required to tip the scales and make metastasis possible. Another hypothesis that is gaining ground is that cancer cells, either through changes in their immunogenic properties or damage to the host immune system, acquire the ability to evade destruction by immune surveillance.

Models of breast cancer metastasis
Serving as a model of metastasis, there are several proposed pathways via which primary breast cancer tumors might metastasize. In the left-most model (1), tumor cells acquire the capacity to metastasize early in the process of tumorigenesis. Shown in the second model is the tendency for some tumors to produce different clones that each harbor different capacities for metastasis and tissue-specific metastatic proclivities. The next model (3) is a representation of the parellel evolution model. Here, metastatic tumor cells are dispersed from the primary tumor very early and develop separately from and in parallel with the primary tumor. The fourth model depicts the cancer stem cell model in which only stem cells have metastatic capacity. (Adapted from Ref. [60])
As is typically the case with considering a spectrum of diseases as complex as cancer, it is likely that no single model will suffice to explain all of metastasic cancer. What can be said with relative certainty is that metastasis follows a basic set of progressive steps. The basic steps involved in metastasis (Fig. 5.2) are as follows:
-
1.
Acquisition of the capacity to invade local tissues
-
2.
Intravasation (gaining access to the circulation)
-
3.
Extravasation (exiting from the circulation)
-
4.
Formation of micrometastasis in a new environment and colonization (growth into macrometastasis)

Stages of metastasis
Cancer is generally thought to progress in a step-wise fashion. Tumor cells that acquire the necessary characteristics to “escape” from a primary lesion and locally invade surrounding tissue may then enter into the general circulation via intravasation. From here, tumor cells that survive the harsh environment (shear forces, lack of support structure, growth signals, etc.) can take up residence in distant tissues, again making their way through the endothelial barrier via extravasation. Tumor cells here form micrometastatic colonies that may or may not go on to form macrometastases
Each of these steps require the acquisition of a host of specialized characteristics/functions. This chapter will discuss some of the genetic changes that aid cancer cells in their acquisition of these characteristics.
3 Stages of the Metastatic Process
3.1 Signalling Pathways Involved in Local Invasion
3.1.1 Epithelial to Mesenchymal Transition
More than 80% of cancers are carcinomas; that is they are of epithelial tissue origin. Carcinomas are complex masses of cells, of which as much as 90% can be non-neoplastic. This diverse collection of non-neoplastic cells compose the tumor stroma. These cells are mostly of mesenchymal origin and are either remnants of the tissue that was invaded by the neoplastic cells or are “recruited” from the surrounding tissue by the neoplastic cells to aid in their growth and survival. Hodgkin’s lymphoma is an extreme example of this phenomenon. In this disease, 99% of the cells in a tumor are non-neoplastic and surround the rare neoplastic Reed-Sternberg cells.
As is the case in normal epithelial tissue, tumors of epithelial origin rely on heterotypic signalling (signalling between different cell types) between stromal cells and the neoplastic epithelial cells for maintenance of tumor growth and architecture. As the neoplastic epithelial cells proliferate, trophic signals are released and are in turn sensed by cells of the stroma which carry receptors specific for such signals. Thus the tumor and stroma cells proliferate concurrently. These stromal cells can even be found layered within metastases originating from these primary carcinomas, highlighting the interdependence between neoplastic and non-neoplastic cells in a tumor.
The process of epithelial to mesenchymal transition (EMT) involves an alteration in both morphology and gene expression pattern of epithelial cells to that of mesenchymal cells. It is necessary during wound healing to allow re-shaping of the epithelial cell layers and also for some morphogenetic processes of embryogenesis. These are known as type II and type I EMT, respectively [33]. Growing evidence suggests that this process is “hijacked” by cancer cells and used to significantly change their morphology and motility, thereby allowing them to invade nearby tissue. This process is known as type III EMT. It has also been suggested to play a role in cancer progression through maintenance of stem cell-like properties, prevention of apoptosis and senescence, and suppression of immune responses [54]. This is triggered in part by ras oncogene activation within neoplastic tissue cells but also is contributed to by chemical signals from non-neoplastic cells outside the tumor proper.
The leading edges of carcinomas exhibit an EMT front where they are invading surrounding tissue. This can often be seen in immunostained tissue slices containing tumor and non-neoplastic tissue side-by-side. Cancer cells at the edge of the invading tumor do not express epithelial cell surface markers such as E-cadherin, a protein which is strongly expressed by cells in the center of tumors and allows epithelial cells to adhere to one another. Instead, cells express surface markers characteristic of fibroblasts such as vimentin, N-cadherin and fibronectin. Loss of E-cadherin expression through epignetic silencing or expression of mutant forms of this protein has been identified in many carcinoma types and is possibly the single most important change contributing to this type of tumor’s ability to become locally invasive. Several signaling pathways (WNT, TGF-β, FGF, EGF, STAT3 and NF-κB) suppress E-cadherin expression via the transcriptional repressors SNAIL, SLUG and TWIST [14, 56]. The expression of E-cadherin and its associated catenins can also be down-regulated via growth factor mediated-phosphorylation and subsequent proteosomal degradation. These growth factors include epidermal growth factor receptor (EGFR) [27], c-MET (hepatocyte growth factor receptor or HGFR) [31], fibroblast growth factor receptors (FGFRs) [15], Src-family kinases and insulin-like growth factor 1R (IGF-1R) [14]. The degradation of E-cadherin leads to nuclear translocation of β-catenin which affects transcription of genes including the oncogene c-myc and the cell cycle regulator cyclin D1 [56]. The expression of N-cadherin by tumor cells allows them to move into the stroma of the epithelial tissue where other N-cadherin expressing fibroblasts reside. Like E-cadherin, N-cadherin expressing cells bind to one another, however with much less strength than the bonds formed by E-cadherin.
Once these tumor cells escape from the tissue of origin and take up residence in another part of the body, they may find themselves in an environment with a different set of extracellular signals. This may result in a reversion back to the epithelial phenotype, thus becoming more like the cells in the center of the primary tumor from which they originated. This mimics the mesencymal to epithelial transition or MET, which is, like EMT, also involved in wound healing and embryogenesis and may explain why distant metastases often resemble the primary tumors from which they originated. This conversion would also allow cells to regain epithelial cell-cell adhesion and facilitate colonization at new sites [56].
Two other cell transition processes have been described and involve an ameoboid cell phenotype: the collective to ameoboid transition (CAT) and the mesencymal to ameobiod transition (MAT). CAT is caused by β1-integrin inhibition. MAT is triggered by inhibition of proteases and relies on signalling via Rac, Rho/ROCK and EphA2. Ameoboid cancer cells differ significantly from mesenchymal cancer cells. As a result of their unique transition they completely lose cell polarity, are capable of chemotaxis and have very loose attachments to extracellular matrix [56]. They also migrate significantly faster than mesenchymal cancer cells with a speed of up to 20 um/min versus 0.1–1 um/min [20]. They do so by mechanically disrupting matrix structures rather than using proteases to degrade them [21]. Ameoboid cancer cells usually are seen after a patient has been treated with integrin or protease inhibitors. Matrix metalloproteinase (MMP) inhibitors appear to have little to no effect on inhibition of cancer progression in such cases [22, 49].
Transmission of signals between the tissue stroma and tumor is achieved largely via transforming growth factor beta (TGF-β) along with tumor necrosis factor alpha (TNF-α), insulin-like growth factor 1 (IGF-1), epidermal growth factor (EGF) and hepatocyte growth factor (HGF). Interaction between TGF-β and ras oncogenes may trigger EMT. Raf, which is immediately downstream of Ras, can also trigger EMT. Phosphoinositide 3-kinase (PI3K) in turn protects cells from pro-apoptotic functions of TGF-β [62]. TNF-α, produced by inflammatory cells in the early stages of tumor progression, together with TGF-β, are important not only for the initiation but also the maintenance of EMT, via maintenance of NF-κB signalling. NF- κB is a key transcriptional regulator of the inflammatory response and is widely activated in cancer.
In the case of non-epithelial tumors, such as those of hematopoietic and connective tissue and the central nervous system (CNS), the waters are quite muddy. It is possible that an EMT-associated transcription factors are important in the case of CNS, as it is derived from an early embryonic epithelium [61].
3.1.2 Hypoxia and an Activated HIF Program
Hypoxia inducible factor-1 (HIF1) is an oxygen sensitive transcriptional activator and as such is a key regulator for induction of genes that facilitate adaptation and survival of cells from normoxia (~21% oxygen) to hypoxia (~1% oxygen). It is composed of two subunits, alpha and beta. The beta subunit is constitutively expressed and the alpha subunit is responsive to oxygen. It is key in the adaptation of cancer cells to hypoxia through its activation of a set of genes that are involved in angiogenesis, iron and glucose metabolism, and cell proliferation/survival (Fig. 5.3). Angiogenesis-associated genes such as vascular endothelial growth factor (VEGF), prostaglandin derived growth factor (PDGF) and angiopoietin-2 are upregulated by HIF-1α. Also upregulated are matrix metalloproteinases 1 and 2 (MMP-1 and MMP-2) and C-X-C chemokine receptor type 4 (CXCR4). While these genes are involved in tumorigenesis, they also serve functions specific to metastasis. MMP-1 helps dissolve the basement membrane and MMP-2 alters architecture of the extracellular matrix. Dissolution of the basement membrane is a key step in migration as it gives tumor cells access to blood and lymphatic vessels in the stroma. CXCR4 in turn causes cancer cells to migrate towards areas of angiogenesis [14]. Inactivation of the p53 signalling system, which would normally activate cell death in conditions of low oxygen, contributes to the ability of cancer cells to survive in a hypoxic environment. Evasion of cell death and the ability to revert to glycolysis for cellular respiration are essential for survival once tumor cells have entered the circulation. Thus characteristics that provide a selective advantage to some cells during tumorigenesis also come in handy once cells exit into the circulation.

Hypoxia in cancer
Due to rapid proliferation, tumors suffer from a lack of sufficient oxygenation. Cells deeper within the tumor (red and pink cells) have less access to oxygen than those found in the perimeter (green cells). As the partial pressure of oxygen (pO2) drops, HIF1 expression increases. Hypoxia leads to upregulation of many genes involved in metastasis, including CXCR4 and VEGF. CXCR4 expression causes cells to migrate toward areas of angiogenesis and may lead to chemokine-mediated organ-specific metastasis. VEGF upregulation leads to angiogenesis which increases tumor aggressivess as well as the tumor’s capacity for metastasis
HIF-1α expression and tumor hypoxia are both prognostic markers of patient outcome and metastasis in several cancer types [30, 41, 55].
3.1.3 Intravasation
The processes of intra- and extravasation are not as well understood as invasion. What is known for certain is that tumor cells encounter unique challenges upon entering the circulation. Most cells require attachment to some kind of substrate for survival and in the absence of such substrate, cells can undergo a form of apoptosis known as anoikis. These circulating cells must also be capable of surviving in the absence of the mitogenic and trophic factors that were present in the stroma from which they originated. Shear forces within vessels can simply tear cells apart. Those that manage to reach larger vessels, some of which may do so by associating with an entourage of platelets, will eventually pass through the heart, after which they will most likely become lodged within the capillaries of the lungs. However, not all metastasis occurs in lungs and thus these cells somehow manage to pass to larger passageways and travel to distant locations in the body. This is likely achieved through arterial-venous shunts. Cells may also pinch off large portions of their cytoplasm and the remaining cell size may be small enough for them to maneuver through the small capillaries. At some point, the cells will need to exit the circulation in some way or another, a process known as extravasation.
3.1.4 Extravasation
In extravasation, we encounter yet another instance of cancer cells hijacking an already existing process for their benefit. Circulating tumor cells express selectin ligands, a group of transmembrane glycoproteins that are also expressed on leukocytes. These proteins are essential for leukocyte transmigration from the circulation to sites of tissue damage or infection, an important component of the body’s adaptive and innate immune response. Selectins expressed on cells that line the vascular walls bind to selectin ligands on leukocytes and cancer cells. This binding is relatively weak and, combined with shear forces in the circulation, results in a sort of rolling movement along the vessels. At some point, a cell or group of cells may become lodged in the vessel. Cells may then proliferate, creating a small tumor that eventually bursts through the vessel wall. Expression of VEGF by cancer cells can also facilitate their extravasation via enhancing endothelial permeability and disrupting the junctions between endothelial cells. Cancer cells with an ameoboid phenotype can easily squeeze through junctions that cells normally would be prevented from traversing. Expression of CXCR4 by cancer cells may result in the selective extravasation of into organs that express CXCL12, such as liver, lung, bone and lymph nodes. Expression of CXCR4 on tumor cells leads to selective extravasation into organs that constitutively express CXCL12 such as liver, lung, bone and lymph nodes [44, 65].
In breast cancer, a gene signature associated with lung metastasis has been identified. Four of the genes in this signature (EREG, MMP1, MMP2 and COX2) have been shown to facilitate blood vessel growth and appear to be essential for extravasation into the lung. Inhibition of these genes resulted in the entrapment of cancer cells within vessels [14, 42]. Again we also see the action of Twist, in this case increasing the ability of cancer cells to migrate intravascularly and extravasate [17, 34, 53].
3.1.5 Colonization & Macrometastasis
After successful extravasation, cells must have the ability to colonize (that is, survive and proliferate) in the new tissue. Antibodies against cytokeratins are used to detect micrometastases in primary carcinoma while epithelial cell adhesion molecule (EpCAM) antibodies can be used to detect micrometastases in lymph nodes. Most extravasated cancer cells do not actually go on to form macrometastases and it can take decades for tumor cells to form clinically detectable metastases after primary tumors are removed [14]. This is referred to as dormancy [1].
The processes involved in this are not well understood. The dormancy period may reflect entry into a state of senescence or may result from active immune surveillance that is able to rid the body of most, but not all, of the cells within micrometastases.
3.2 Evading the Immune System
The body has a number of mechanisms that it uses to ward off cancer development. At the cellular level there is the pRb circuit, DNA repair mechanisms and the apoptotic machinery. At the tissue level, cells that detach from the basement membrane typically undergo anoikis. Until about a decade ago, the role of the immune system in cancer was a highly debated one but evidence of its capacity to identify and destroy cancer cells has been steadily accumulating. First, a body of work in mice provided strong indications for an important role of the immune system in defense against cancer. The development of technology to genetically engineer mice led to the creation of mouse strains deficient in genes that play specific roles in the immune system, such as IFN-γ, perforin, Rag1 and Rag2. These knock-out mice provided key advancements in our understanding of the relationship between the immune system and the development of cancer. But what about humans?
It has been observed that people with compromised immune systems are more likely to develop certain kinds of cancer. Organ transplant recipients, who receive long-term immunosuppressive therapy to prevent rejection of the transplanted tissue, have a very high increased risk of developing some kind of cancer. Cancers of viral origin occur at a much higher frequency in those who are immunocompromised. Kaposi’s sarcoma (caused by human herpes virus 8) occurs in HIV patients at a rate 3000 times higher than in the general population and tumors caused by human papilloma virus are far more frequent in organ transplant recipients and AIDS patients [61].
The immune system may also be able to recognize tumors of nonviral origin, but it is not clear whether this is indeed the case. Anti-tumor antibodies have also been detected in the blood of cancer patients but it is not known whether these antibodies function in the removal of cancer cells from the body. Another example are tumor-infiltrating lymphocytes which may be recruited to the tumor to aid in its growth or may have invaded the tumor upon recognizing it as “foreign”. The presence of these lymphocytes in several tumor types correlates with improved survival but there is no direct evidence that these are the cause of said improved survival.
The immune system can actively attack circulating tumor cells. For example, natural killer (NK) cells can engage cancer cells via TNF-related molecules such as TRAIL or CD95L, or through the perforin pathway. Both cause tumor cell death, and inhibiting TRAIL or using mice that are deficient in NK cells leads to increased metastasis [14].
3.3 The Role of Cancer Stem Cells in Metastasis
The concept of cancer stem cells (CSCs), first developed over a decade ago, was at first a controvertial hypothesis. Accumulated evidence now stongly supports the existence of such cells in a variety of cancers including several leukemias and many solid tumors [3]. The genetic characteristics of CSCs vary by cancer type and even subtype. However, they share in common a high tumorigenic and metastatic potential with unlimited self-renewal capacity. They appear to be resistant to conventional therapies and often able to enter quiescence and/or a state of slow-cycling. This characteristic may explain, at least in part, the dormancy observed in patients whose cancer re-appears decades after initial therapy [1]. It could also explain why CSCs are not as sensitive as other cancer cells to cytotoxic drugs that target actively cycling cells.
This tumor sub-population was named for their similarity to normal adult stem cells present in tissues such as the gastrointestinal mucosa and cells of the hematopoietic system. Due to genetic and epigenetic instability, the CSC population within a single primary tumor is hetergeneous. CSC are not necessarily the “cell of origin” that first gave rise to the primary tumor as cells within the tumor population may undergo changes over time that confer their “stemness”. Another characteristic of CSCs is that they tend to have high expression of EMT markers. Aktas et al. showed that, in patients with metastatic breast cancer, non-responders to treatment had significantly higher expression of EMT markers (62% vs 10% in responders) and ALDH1 (44% vs 5% in responders) [2].
The resistance that CSCs exhibit to conventional drugs may be caused by increased capacity for drug efflux, increased expression of free radical scavengers and increased DNA repair capacity [3]. A great deal of research is now focused on targeting the CSC niche as it appears to be essential for complete eradication of the disease. This has been achieved in part by gene expression profiling of CSCs to identify unique targets. An antibody therapy designed against a CSC-specific isoform of CD44 (CD44v6) resulted in severe skin toxicity in phase I trials for head and neck squamous cell carcinoma [48]. Other antibody therapies against markers such as CD123 and CD133 face challenges due to their also being expressed by normal stem cells. Such targets carry a high potential for toxic side-effects, much like traditional chemotherapeutic drugs.
Another method being developed is pre-treatment with a drug aimed at sensitizing the CSCs to conventional therapy. Francipane et al. reported sensitization of colon cancer to chemotherapy after treatment with IL-4 inhibitor [19]. Yet another means of overcoming the resistance of CSCs involves the inhibition of TGF pathway by bone morphogenetic proteins (BMPs). In a mouse xenograft model of brain cancer, this caused differentiation of the CSCs and subsequent cure [16]. Drug efflux pathways may also be targeted to sensitive CSCs to conventional chemotherapy.
3.4 New Targets in the Clinic
As our understanding of cancer has evolved so has the approach to treatment. Although classical chemotherapeutic drugs, radiotherapy and surgical resection are still the most common modes of treatment for most cancer types, there is a trend toward more targeted and individualized therapy. Here we discuss some of the recent developments in treatment specifically targeting metastasis.
Inhibitors of the CXCR4-CRCL12 chemokine axis are currently in Phase I and II clinical trials. This receptor-ligand pair is involved in cell migration during embryogenesis and wound healing. It has been implicated in cancer cell migration and its expression correlates with poor prognosis in colon, breast and gallbladder cancers [29, 46, 63, 64]. Organs and tissues that possess high levels of CRCL12, such as liver, lung, bone marrow, and lymph nodes, attract the migration of CXCR4-expressing cancer cells [13]. Upregulation of HIF1-α, which is involved in the adaptation of cancer cells to a hypoxic environment, also leads to increased gene expression of CXC4 thus contributing to the progression of cancer [47]. CXCR4 expression is currently used as a biomarker of aggressive breast cancer and represents a potentially important target for therapy.
Combination therapy with CXCR4 antagonists, such as plerixafor, disrupts the interaction between CLL and stromal cells, recirculates CLL cells into the bloodstream and exposes them to conventional drugs [8]. This same drug was effective in minimizing the invasion and metastasis of epithelial ovarian cancer cells [4]. In combination therapy with decarbazine, plerixafor significantly suppressed the metastatis of melanoma as compared with decarbazine treatment alone [36]. Study of these molecules and the pathway in which they function should lead to better and more specific inhibitors. It should be noted that successful treatment may require combined inhibition of other protein targets in this pathway.
Another interesting tack under investigation is the targeting of epigenetic mechanisms. Epigenetic changes appear to occur early in the process of tumorigenesis [25]. During TGF-β mediated EMT, there is a global reduction in the heterochromatin mark H3 Lys9 dimethylation (H3K9me2), an increase in the euchromatin mark H3 Lys4 trimethylation (H3K4me3) and an increase in the transcriptional mark H3 Lys36 trimethylation (H3K36me3) [59].
Epigenetic agents in the clinic include DNA demethylating drugs and histone deacetylase/demethylase inhibitors. The aim of treatment with DNA demethylating agents is to re-activate the expression of key regulatory genes that are silenced during cancer progression via methylation of CpG islands. The first DNA methylation inhibitor to be used in the clinic was 5-azacytidine, synthesized nearly 50 years ago and used to treat acute myelogenous leukemia [12]. It is now also approved for the treatment of myeloid dysplastic syndrome and chronic myelomonocytic leukemia. Its relative, 5-aza-2’deoxycytidine, is approved for myeloid dysplastic syndrome and acute myelogenous leukemia. The main concern with these drugs is their high level of systemic toxicity and thus there is ongoing work to identify more specific inhibitors. Gemcitabine, an analogue of pyramidine cytosine, is structurally similar to 5-aza-2’deoxycytidine and appears to reactivate several epigenetically silenced genes via destabilization and inhibition of DNA methyltransferase 1. It is used as monotherapy or in combination with cisplatin for the treatment of several solid tumors [24, 57]. RNAi techniques have shown that more specific inhibition of DNA methyltransferases may also be effective. However, these methods have not yet been tested in vivo so it remains to be seen whether these results will hold up at the organismal level [25].
Histone deacetylase inhibitors (HDACi), long used in treatment of some psychiatric disorders and as anti-epileptics, have caught the attention of researchers in other fields including those studying cancer, inflammatory and parasitic diseases [6]. HDACs affect many different physiological processes. Their inhibition in cancer cells leads to cell cycle arrest, apoptosis, autophagy and anti-angiogenesis. Their specificity toward malignant cells is of particular interest. Two drugs have been approved by the U.S. FDA for treatment of progressive, persistent or recurrent cutaneous T-cell lymphoma (Vorinostat, approved in 2006; and Romidepsin, approved in 2009) [35]. There are currently about a dozen small molecule inhibitors in on-going clinical trials for several blood cancers, as well as lung, ovarian, and breast cancers and hepatocellular carcinoma [51]. It should be noted that the autophagy triggered by HDACi may be a mechanism of resistance rather than cell death [35].
Another target of increasing interest is the TGF-β pathway, in part because it is involved in so many aspects of cancer development and progression [39, 45, 54]. However, approaches to this pathway must be considered carefully as it plays a dual role in cancer, as both tumor suppressor and tumor promoter [52]. There is a wide range of approaches being taken to inhibit TGF-β, including antisense molecules, monoclonal antibodies and TFG- β receptor kinase inhibitors (current small molecules in pre-clinical and clinical trials are reviewed in Sheen et al. [52].
Other targets of interest are cell adhesion molecules such as selectins and cadherins. Antagonists such as neutralizing monoclonal antibodies, competitive ligand inhibitors and metabolic carbohydrate mimetics have been designed to target cellular interactions with selectins [5, 37]. Selectins not only are important for the motility of cancer cells in vessels but also allow cancer cells to attach to platelets, resulting in platelet aggregation and the formation of blood clotting. Experimental models have shown a role for the coagulation pathway in metastatis and some clinical studies indicate that patients treated with anti-coagulants such as low molecular weight heparins (LMWH) tend to have better outcome, but the data is far from conclusive [32] (see Mandala et al. for anti-coagulant indications) [38]. The precise mechanism(s) involved are unclear but may be associated with platelet-covered cancer cells being able to evade immune surveillance and lysis by natural killer cells [23]. Inhibition of P-selectin and heparanase by semi-synthetic sulfated hexasaccharides were shown to inhibit metastasis in mouse xenograft models using colon carcinoma cells (MC-38GFP) and a melanoma cell line (B16-BL6). The inhibition was similar to that seen in mice deficient in P-selectin [7].
There is currently a clinical trial underway for patients with previously untreated multiple myeloma (ClinicalTrials.gov identifier: NCT01518465) that includes an anti-coagulant, dalteparin (an LMWH), which inhibits P-selectin and L-selectin binding to cancer cells [43]. Mousa PetersenPrevious studies including dalteparin suggest that it is not useful in treating metastatic disease but may be helpful in patients with better prognosis [32]. Thus, P-selectin inhibition may prove to be useful in the prevention of metastasis, while patients already suffering metastatic disease may not benefit from such treatment. However, studies with new-generation P-selectin specific inhibitors are likely necessary before a conclusion can be drawn on this matter. SelG1 is an anti-P-selectin monoclonal antibody currently in Phase II clinical trials for pain management in sickle cell disease. Inclacumab is another such antibody, also in small-scale Phase II clinical trials, that is being used to reduce myocardial damage in patients undergoing percutaneous coronary intervention (PCI). There are currently no cancer clinical trials that include these P-selectin antibodies.
4 Conclusions
While great strides forward have been made in the detection and treatment of various cancer types, cancer metastasis remains a difficult puzzle to investigate. Research on resected tumors must be focused in more closely on portions of the leading edge which likely have genetic and proteomic profiles much different from that of cells within other parts of the tumor. Epigenetic changes are likely as important as genetic changes and must be considered in concert. As global gene and protein expression microarray technology and live in vivo imaging become more widely available for basic research purposes, our understanding of metastasis will hopefully advance more rapidly.
References
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7(11):834–846. https://doi.org/10.1038/nrc2256
Aktas B, Tewes M, Fehm T, Hauch S, Kimmig R, Kasimir-Bauer S (2009) Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res 11(4):R46. https://doi.org/10.1186/bcr2333. Epub 2009 Jul 9
Baccelli I, Trumpp A (2012) The evolving concept of cancer and metastasis stem cells. J Cell Biol 198(3):281–293. https://doi.org/10.1083/jcb.201202014
Barbolina MV, Kim M, Liu Y, Shepard J, Belmadani A, Miller RJ, Shea LD, Stack MS (2010) Microenvironmental regulation of chemokine (C-X-C-motif) receptor 4 in ovarian carcinoma. Mol Cancer Res 8(5):653–664. https://doi.org/10.1158/1541-7786.mcr-09-0463
Barthel SR, Gavino JD, Descheny L, Dimitroff CJ (2007) Targeting selectins and selectin ligands in inflammation and cancer. Expert Opin Ther Targets 11(11):1473–1491. https://doi.org/10.1517/14728222.11.11.1473
Blanchard F, Chipoy C (2005) Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today 10(3):197–204. https://doi.org/10.1016/s1359-6446(04)03309-4
Borsig L, Vlodavsky I, Ishai-Michaeli R, Torri G, Vismara E (2011) Sulfated hexasaccharides attenuate metastasis by inhibition of P-selectin and heparanase. Neoplasia 13(5):445–452
Burger JA (2010) Chemokines and chemokine receptors in chronic lymphocytic leukemia (CLL): from understanding the basics towards therapeutic targeting. Semin Cancer Biol 20(6):424–430. https://doi.org/10.1016/j.semcancer.2010.09.005
Cai W, Chen X (2008) Multimodality molecular imaging of tumor angiogenesis. J Nucl Med 49(Suppl 2):113S–128S. https://doi.org/10.2967/jnumed.107.045922
Cancer Prinicples & Practice of Oncology (2008) (V. DeVita, MD, T. Lawrence, MD PhD & S. Rosenberg, MD PhD Eds. 8th ed.): Lippincott Williams & Wilkins
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2(8):563–572. https://doi.org/10.1038/nrc865
Christman JK (2002) 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene 21(35):5483–5495. https://doi.org/10.1038/sj.onc.1205699
Debnath B, Xu S, Grande F, Garofalo A, Neamati N (2013) Small molecule inhibitors of CXCR4. Theranostics 3(1):47–75. https://doi.org/10.7150/thno.5376
DeVita VT, MD (2008) Cancer principles and practice of oncology, 8th edn. Lippincott Williams & Wilkins Philadelphia
El-Hariry I, Pignatelli M, Lemoine NR (2001) FGF-1 and FGF-2 regulate the expression of E-cadherin and catenins in pancreatic adenocarcinoma. Int J Cancer 94(5):652–661
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, Nikkhah G, Dimeco F, Piccirillo S, Vescovi AL, Eberhart CG (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28(1):5–16. https://doi.org/10.1002/stem.254
Fein MR, Egeblad M (2013) Caught in the act: revealing the metastatic process by live imaging. Dis Model Mech 6(3):580–593. https://doi.org/10.1242/dmm.009282
Fong Y, Coit DG, Woodruff JM, Brennan MF (1993) Lymph node metastasis from soft tissue sarcoma in adults. Analysis of data from a prospective database of 1772 sarcoma patients. Ann Surg 217(1):72–77
Francipane MG, Alea MP, Lombardo Y, Todaro M, Medema JP, Stassi G (2008) Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res 68(11):4022–4025. https://doi.org/10.1158/0008-5472.CAN-07-6874. Review
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3(5):362–374. https://doi.org/10.1038/nrc1075
Friedl P, Wolf K (2008) Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res 68(18):7247–7249. https://doi.org/10.1158/0008-5472.can-08-0784
Giampieri S, Pinner S, Sahai E (2010) Intravital imaging illuminates transforming growth factor beta signaling switches during metastasis. Cancer Res 70(9):3435–3439. https://doi.org/10.1158/0008-5472.can-10-0466
Gil-Bernabe AM, Lucotti S, Muschel RJ (2013) Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol 162(4):433–441. https://doi.org/10.1111/bjh.12381
Gray SG, Baird AM, O’Kelly F, Nikolaidis G, Almgren M, Meunier A et al (2012) Gemcitabine reactivates epigenetically silenced genes and functions as a DNA methyltransferase inhibitor. Int J Mol Med 30(6):1505–1511. https://doi.org/10.3892/ijmm.2012.1138
Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM et al (2012) DNA methylation inhibitors in cancer: recent and future approaches. Biochimie 94(11):2280–2296. https://doi.org/10.1016/j.biochi.2012.07.025
Harris JF, Chambers AF, Hill RP, Ling V (1982) Metastatic variants are generated spontaneously at a high rate in mouse KHT tumor. Proc Natl Acad Sci USA 79(18):5547–5551
Hazan RB, Norton L (1998) The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem 273(15):9078–9084
Hellman S (2005) Premise, promise, paradigm and prophesy. Nat Clin Pract Oncol 2(7):325
Hiller DJ, Meschonat C, Kim R, Li BD, Chu QD (2011) Chemokine receptor CXCR4 level in primary tumors independently predicts outcome for patients with locally advanced breast cancer. Surgery 150(3):459–465. https://doi.org/10.1016/j.surg.2011.07.005
Hockel M, Vaupel P (2001) Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93(4):266–276
Iiizumi M, Liu W, Pai SK, Furuta E, Watabe K (2008) Drug development against metastasis-related genes and their pathways: a rationale for cancer therapy. Biochim Biophys Acta 1786(2):87–104. https://doi.org/10.1016/j.bbcan.2008.07.002
Kakkar AK, Levine MN, Kadziola Z, Lemoine NR, Low V, Patel HK et al (2004) Low molecular weight heparin, therapy with dalteparin, and survival in advanced cancer: the fragmin advanced malignancy outcome study (FAMOUS). J Clin Oncol 22(10):1944–1948. https://doi.org/10.1200/jco.2004.10.002
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119(6):1420–1428. https://doi.org/10.1172/jci39104
Khan MA, Chen HC, Zhang D, Fu J (2013) Twist: a molecular target in cancer therapeutics. Tumour Biol 34(5):2497–2506. https://doi.org/10.1007/s13277-013-1002-x
Khan O, La Thangue NB (2012) HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol 90(1):85–94. https://doi.org/10.1038/icb.2011.100
Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K et al (2010) CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res 70(24):10411–10421. https://doi.org/10.1158/0008-5472.can-10-2591
Ludwig RJ, Schon MP, Boehncke WH (2007) P-selectin: a common therapeutic target for cardiovascular disorders, inflammation and tumour metastasis. Expert Opin Ther Targets 11(8):1103–1117. https://doi.org/10.1517/14728222.11.8.1103
Mandala M, Falanga A, Roila F (2011) Management of venous thromboembolism (VTE) in cancer patients: ESMO clinical practice guidelines. Ann Oncol 22(Suppl 6):vi85–vi92. https://doi.org/10.1093/annonc/mdr392
Massague J (2008) TGFbeta in Cancer. Cell 134(2):215–230. https://doi.org/10.1016/j.cell.2008.07.001
Mehlen P, Puisieux A (2006) Metastasis: a question of life or death. Nat Rev Cancer 6(6):449–458. https://doi.org/10.1038/nrc1886
Milani M, Harris AL (2008) Targeting tumour hypoxia in breast cancer. Eur J Cancer 44(18):2766–2773. https://doi.org/10.1016/j.ejca.2008.09.025
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD et al (2005) Genes that mediate breast cancer metastasis to lung. Nature 436(7050):518–524. https://doi.org/10.1038/nature03799
Mousa SA, Petersen LJ (2009) Anti-cancer properties of low-molecular-weight heparin: preclinical evidence. Thromb Haemost 102(2):258–267. https://doi.org/10.1160/th08-12-0832
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410(6824):50–56. https://doi.org/10.1038/35065016
Pardali E, Goumans MJ, ten Dijke P (2010) Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 20(9):556–567. https://doi.org/10.1016/j.tcb.2010.06.006
Popple A, Durrant LG, Spendlove I, Rolland P, Scott IV, Deen S, Ramage JM (2012) The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br J Cancer 106(7):1306–1313. https://doi.org/10.1038/bjc.2012.49
Ramsey DM, McAlpine SR (2013) Halting metastasis through CXCR4 inhibition. Bioorg Med Chem Lett 23(1):20–25. https://doi.org/10.1016/j.bmcl.2012.10.138
Riechelmann H, Sauter A, Golze W, Hanft G, Schroen C, Hoermann K, Erhardt T, Gronau S (2008) Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol 44(9):823–829. https://doi.org/10.1016/j.oraloncology.2007.10.009. Epub 2008 Jan 18
Sabeh F, Shimizu-Hirota R, Weiss SJ (2009) Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol 185(1):11–19. https://doi.org/10.1083/jcb.200807195
Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ et al (2003) From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA 100(13):7737–7742. https://doi.org/10.1073/pnas.1331931100
Shabason JE, Tofilon PJ, Camphausen K (2010) HDAC inhibitors in cancer care. Oncology (Williston Park) 24(2):180–185
Sheen YY, Kim MJ, Park SA, Park SY, Nam JS (2013) Targeting the transforming growth factor-beta signaling in cancer therapy. Biomol Ther (Seoul) 21(5):323–331. https://doi.org/10.4062/biomolther.2013.072
Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, Klemke R (2010) Visualizing extravasation dynamics of metastatic tumor cells. J Cell Sci 123(Pt 13):2332–2341. https://doi.org/10.1242/jcs.069443
Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5):871–890. https://doi.org/10.1016/j.cell.2009.11.007
Trastour C, Benizri E, Ettore F, Ramaioli A, Chamorey E, Pouyssegur J, Berra E (2007) HIF-1alpha and CA IX staining in invasive breast carcinomas: prognosis and treatment outcome. Int J Cancer 120(7):1451–1458. https://doi.org/10.1002/ijc.22436
van Zijl F, Krupitza G, Mikulits W (2011) Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res 728(1-2):23–34. https://doi.org/10.1016/j.mrrev.2011.05.002
Voutsadakis IA (2011) Molecular predictors of gemcitabine response in pancreatic cancer. World J Gastrointest Oncol 3(11):153–164. https://doi.org/10.4251/wjgo.v3.i11.153
Waghorne C, Thomas M, Lagarde A, Kerbel RS, Breitman ML (1988) Genetic evidence for progressive selection and overgrowth of primary tumors by metastatic cell subpopulations. Cancer Res 48(21):6109–6114
Wang Y, Shang Y (2013) Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res 319(2):160–169. https://doi.org/10.1016/j.yexcr.2012.07.019
Weigelt B, Peterse JL, van’t Veer LJ (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5(8):591–602. https://doi.org/10.1038/nrc1670
Weinberg RA (2013) The biology of cancer, vol 2. Garland Science, New York
Weinberg RA (2007) The biology of cancer, 1st edn. Garland Science, New York
Yao X, Zhou L, Han S, Chen Y (2011) High expression of CXCR4 and CXCR7 predicts poor survival in gallbladder cancer. J Int Med Res 39(4):1253–1264
Zhang NH, Li J, Li Y, Zhang XT, Liao WT, Zhang JY et al (2012) Co-expression of CXCR4 and CD133 proteins is associated with poor prognosis in stage II-III colon cancer patients. Exp Ther Med 3(6):973–982. https://doi.org/10.3892/etm.2012.527
Zlotnik A (2006) Involvement of chemokine receptors in organ-specific metastasis. Contrib Microbiol 13:191–199. https://doi.org/10.1159/000092973
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Moroski-Erkul, C.A., Demir, E., Gunduz, E., Gunduz, M. (2019). Genetic Basis of Metastasis. In: De Mello, R., Mountzios, G., Tavares, Á. (eds) International Manual of Oncology Practice. Springer, Cham. https://doi.org/10.1007/978-3-030-16245-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-16245-0_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16244-3
Online ISBN: 978-3-030-16245-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)