
Abstract
Myositis is a heterogeneous group of autoimmune diseases encompassing a large spectrum of clinical manifestations including some patients presenting with no muscle involvement. Thus, the clinician must be aware that arthritis, certain cutaneous features, interstitial lung disease (ILD), or dysphagia may dominate the clinical picture. Further, autoantibody detection, muscle immunohistochemistry, and advances in muscle imaging such as MRI have modified the approach to diagnosis and treatment in myositis. In the last decades, several diagnostic and classification criteria were developed for myositis, with varying strengths and limitations. In this chapter, we will review criteria developed for polymyositis (PM), dermatomyositis (DM), immune-mediated necrotizing myopathy (IMNM), overlap myositis (OM), cancer-associated myositis, and sporadic inclusion body myositis (sIBM). We will also discuss the 2017 EULAR/ACR classification criteria for juvenile and adult myositis led by international myositis experts using a robust data-driven process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Classification criteria
- Diagnostic criteria
- Idiopathic inflammatory myopathy
- Dermatomyositis
- Polymyositis
- Inclusion body myositis
- Overlap myositis
- Immune-mediated necrotizing myopathy
- Clinically amyopathic dermatomyositis
-
Myositis is a group of multisystemic diseases that can initially present with isolated arthritis, interstitial lung disease, or dermatomyositis (DM) rashes.
-
Myositis-specific autoantibodies are associated with specific disease phenotypes.
-
In myositis, early diagnosis is important to prevent organ damage such as muscle atrophy and lung fibrosis.
-
In adults, major myositis subsets include dermatomyositis, clinically amyopathic dermatomyositis, polymyositis, overlap myositis, immune-mediated necrotizing myopathy, anti-synthetase syndrome, and sporadic inclusion body myositis.
-
New classification criteria for myositis have been endorsed in 2017 by the EULAR/ACR.
Introduction
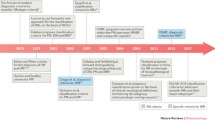
For decades, the diagnosis and classification of myositis were dependent on the presence of muscle weakness along with electromyographic changes, muscle enzyme elevation, and skeletal muscle inflammation on muscle biopsy. Further, typical skin rashes supported the diagnosis of dermatomyositis (DM). However, the past 10 years has seen a shift in the perception of myositis toward a disease characterized by multiple organ involvement with some patients manifesting no clinically evident muscle weakness. The identification of several new myositis-specific autoantibodies (MSA), often associated with distinct clinical phenotypes, has further shaped our understanding and classification of myositis as a spectrum of related diseases (Fig. 11.1).

Examples of different clinical presentations of myositis. CADM clinically amyopathic dermatomyositis, ILD interstitial lung disease, sIBM sporadic inclusion body myositis
When to Suspect Myositis
Myositis is characterized by symmetrical muscle weakness predominating in proximal limb muscles including the neck flexors. The muscle weakness is typically of low endurance rather than of low resistance type, at least at disease onset. Symptoms develop progressively over weeks (subacute) to months (chronic), with a very slow and insidious onset (i.e., years) with concomitant muscle atrophy of the knee extensors or weak finger flexors in the subset of sporadic inclusion body myositis (sIBM). Another classical presentation is the presence of a skin rash typical of DM such as Gottron papules/sign or the heliotrope rash. Myositis patients may be initially referred to an early arthritis clinic for an inflammatory arthritis mimicking rheumatoid arthritis, in a pulmonology clinic with symptoms of interstitial lung disease (ILD), or in a dermatology clinic with atypical skin rashes (Box 11.1). Lack of recognition of these atypical presentations may delay diagnosis and treatment leading to irreversible organ damage. The frequency of such patients presenting with predominantly extramuscular involvement is highly variable. For example, in a cohort of anti-Jo-1-positive subjects from Spain, isolated arthritis was noted in 18%, isolated ILD in 32%, and isolated myositis in 27% [1].
When to Suspect Myositis Spectrum of Diseases
-
Muscle weakness, low muscle endurance, or muscle enzyme elevation
-
Seronegative polyarthritis
-
Typical DM rashes or palmar papules even in the absence of muscle weakness
-
ILD associated with Raynaud phenomenon, mechanic’s hands, arthritis, or fever
-
Dysphagia
There are significant limitations when using health-care registries based on World Health Organization (WHO) classification systems to identify myositis patients as there are no International Classification of Diseases (ICD) codes for newly described myositis phenotypes such as the anti-synthetase syndrome, amyopathic DM, or autoimmune necrotizing myopathy. Therefore, myositis experts must be unified in an initiative to revise the WHO classification system and promote the inclusion of specific ICD codes for these new myositis subsets. Collaborations to create large international longitudinal registry-based studies (e.g., EuroMyositis—www.euromyositis.eu) including patients fulfilling standardized classification criteria as well as early cases failing to meet such diagnostic or classification criteria are required. More importantly, clinicians must work in multidisciplinary teams including rheumatologists, neurologists, pulmonologists, immunologists, and dermatologists to diagnose and treat early cases to prevent disease-related morbidity in the muscle, skin, joints, and lung. That is, pulmonary fibrosis patients being seen by a pulmonologist should be systematically screened for systemic autoimmune rheumatic disease symptoms such as muscle weakness, muscle enzymes elevation, mechanic’s hands or DM-associated rashes, Raynaud phenomenon, and polyarthritis, with a low threshold for rheumatology consultation and MSA screening. Likewise, patients presenting with a DM rash without objective muscle weakness may have clinically amyopathic DM, and such patients may benefit from a high-resolution computerized tomography of the lungs and MSA screening given the association of anti-MDA5 antibody with rapidly progressive ILD [2]. Similarly, patients presenting with “seronegative” (i.e., rheumatoid factor and anti-CCP negative) rheumatoid arthritis may have anti-synthetase syndrome with polyarthritis as the initial presentation.
Classification Criteria
Classification criteria are developed for research purposes in order to enroll a uniform cohort of subjects such that different published studies can be adequately compared. They require very high specificity even with a potential loss of sensitivity. Diagnostic criteria, on the other hand, aim at identifying a wider spectrum of disease cases including early cases with incomplete presentation to those with severe, advanced features and thus need to be both sensitive and specific. It is essential not to confuse classification with diagnosis, as by using classification criteria for diagnostic purposes, one may delay diagnosis and proper treatment of an individual with myositis that does not yet fulfill classification criteria. Accurate and early diagnosis is paramount to adequate myositis management.
In 1975, Bohan and Peter published the most widely used criteria for myositis, intended for both diagnosis and classification, which continued to be used to date (Table 11.1) [3]. Many large subsequent studies utilized these criteria. These were mainly based on expert opinion and included a proposal of five subgroups of myositis shown in Box 11.2. Although these criteria could differentiate PM or DM from systemic lupus erythematosus and systemic sclerosis with a sensitivity of 93% and specificity of 93% [4], the criteria lacked specificity for PM, leading to misclassification of metabolic myopathies, muscle dystrophies, and sIBM as PM. Moreover, the exclusion of other myopathies, a prerequisite to Bohan and Peter classification, was not well defined. Criteria were highly specific and worked better for DM given the requirement of characteristic rashes (heliotrope rash or Gottron papules). However, patients with less characteristic DM rashes, but all other features consistent with DM, could not be classified as having DM. Importantly, sIBM had not been recognized at the time these criteria were published. Therefore, earlier studies clearly classified sIBM as PM based on Bohan and Peter classification. In addition, the MSA were not yet discovered, and newer technologies, such as muscle MRI and sophisticated muscle immunohistochemical staining, were not available for classification purposes (Box 11.3).
Bohan and Peter Five Subgroups of Myositis
-
Primary idiopathic PM
-
Primary idiopathic DM
-
DM or PM associated with neoplasia
-
Childhood DM or PM associated with vasculitis
-
DM or PM associated with collagen-vascular disease
Box 11.3 Shortcomings of Bohan and Peter Classification Criteria
-
Lack of specificity of PM
-
Lack of newer entities such as CADM, anti-synthetase syndrome, and immune-mediated necrotizing myopathy
-
Lack of DM rashes other than heliotrope and/or Gottron papules
-
Absence of myositis-specific autoantibodies (MSA)
-
Absence of well-defined exclusion criteria
In the last decades, several groups have attempted to refine the approach of myositis classification and define various myositis subsets, proposing mostly classification criteria based on expert opinion and rarely based on data. In Table 11.2, selected criteria sets for PM/DM/IMNM are compared [3, 5,6,7,8,9,10]. Most of those include the presence of proximal muscle weakness, elevation of muscle enzymes, myopathic changes on EMG, inflammation on muscle biopsy, MSAs, and the characteristic rashes of DM. The European Neuromuscular Center (ENMC) criteria, developed by a group of myologists in 2004, provide detailed clinical, histopathologic, and laboratory criteria including MSA and muscle MRI [9]. The eight phenotypes described were definite PM, probable PM, definite DM, probable DM, amyopathic DM, possible DM sine myositis, nonspecific myositis, and, for the first time, immune-mediated necrotizing myopathy (IMNM). They defined IMNM with the same clinical and laboratory criteria as PM or DM but with the presence on muscle biopsy of predominantly necrotic muscle fibers with sparse inflammatory cells. However, these histopathological features are not specific for IMNM and can also be found in patients with, e.g., cancer-associated myopathies or muscle dystrophies. This subgroup of patients with IMNM, also termed necrotizing autoimmune myopathy (NAM), has been associated with the presence of one of two specific autoantibodies [anti-signal recognition particle (anti-SRP) and anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR)], suggesting involvement of the immune system in this subset of myositis. It is clinically important to identify IMNM cases as they are often difficult to treat and may need more aggressive or alternative immunosuppressive treatment as discussed in Chap. 24.
Cancer-associated myositis is another subgroup of myositis. It is well recognized that, in adults, DM more than PM is associated with the presence of a malignancy, but this is not well established for other myositis subsets. There is no general agreement on the definition of cancer-associated DM, but one frequently used is the occurrence of malignancy within 3 years from DM diagnosis. Recently, two MSAs have been linked with cancer-associated DM, anti-transcriptional intermediary factor 1-gamma (anti-TIF1-gamma), and antinuclear matrix protein 2 (anti-NXP2). Please see Chap. 21 for a more comprehensive discussion of these autoantibodies. There is still controversy as to whether these autoantibodies represent an epiphenomenon associated with neoplasia or if these antibodies are truly pathogenic. Their presence in adult DM patients should however prompt clinicians to be thorough in their malignancy screening.
Myositis may appear as a disease on its own but may also present in patients diagnosed with another rheumatic disorder either at the same time or one following the other. Myositis associated with another systemic autoimmune rheumatic disorder (SARD), or overlap myositis, refers to these myositis patients that also fulfill criteria for another SARD. The most common rheumatic disorders overlapping with myositis are systemic sclerosis and Sjögren syndrome, followed by systemic lupus erythematosus. Mixed connective tissue disease (MCTD) is a rare autoimmune condition associated with anti-U1RNP antibody, where myositis is regarded as one of the characteristic clinical manifestations such that this condition represents an overlap syndrome. More rarely, patients with rheumatoid arthritis also develop myositis. Whether myositis in patients with overlap syndrome is different from myositis as a single entity is still unknown.
The EULAR/ACR Myositis Classification Criteria
To overcome the limitations of Bohan and Peter’s classification criteria as well as other proposed criteria (empirically derived, based on small/single-center cohorts, lack of appropriate controls/validation), a group of international myositis experts including adult and pediatric rheumatologists, neurologists, dermatologists, epidemiologists, and biostatisticians developed data and consensus-driven new myositis classification criteria following the recommendations endorsed and published by EULAR/ACR [5, 11, 12]. These criteria were developed and validated using a collaborative data-driven methodology. Data on 93 variables were collected from 976 myositis patients and 624 patients with conditions mimicking myositis (74% adults, 26% children). Two models, with or without muscle biopsy results, were developed to better reflect some clinical settings such as pediatrics, where muscle biopsy may not be regarded as standard of care. Based on statistical performance and best specificity and sensitivity, a set of 16 variables weighted depending on their importance was identified (Table 11.3). The final score, which is the sum of scores achieved for various individual clinical features, corresponds to a certain probability of having myositis, which gives flexibility to the investigators to decide on threshold, depending on the types of study they are conducting (e.g., clinical trial vs. epidemiological). The criteria are based on two steps: (1) to identify a myositis patient compared to a non-myositis patient and (2) to identify subgroups of myositis (Fig. 11.2). A web-based calculator has been developed and can be used off-line in electronic devices. These proposed criteria have been endorsed by the European League Against Rheumatism (EULAR) and by the American College of Rheumatology (ACR) and demonstrate strong sensitivity and specificity for a probable myositis diagnosis of 93% and 88%, respectively, when biopsy results are included. Nevertheless, there are limitations as the heterogeneity of myositis limited the number of the rare subgroups (e.g., IMNM, hypomyopathic DM, and juvenile PM) that could be recruited, and therefore the criteria cannot be used to define these subsets. Moreover, only one MSA, anti-Jo-1, was documented in enough subjects to be included as a final variable, so with the wider study of other MSAs and their clinical phenotypes, these autoantibodies could be incorporated in future EULAR/ACR classification criteria. Muscle MRI, only available in 38% of cases, was also excluded from the analysis. Thus, these criteria will soon require revision using a cohort with further data on MSAs, MRI, as well as validation on an external cohort with myositis cases and comparators.

Subgroups of myositis according to the 2017 EULAR/ACR classification criteria [5]. *The polymyositis (PM) subset includes immune-mediated necrotizing myopathies (IMNM). IBM inclusion body myositis, ADM amyopathic dermatomyositis, DM dermatomyositis, JDM juvenile dermatomyositis
IBM Diagnostic/Classification Criteria
In the last decades, sIBM diagnostic criteria have shifted from Griggs et al. criteria [13], with a strong emphasis on histopathological variables to an approach based more on clinical phenotypes (Table 11.4). The MRC Centre for Neuromuscular Disease [14] and the ENMC [15] have both developed new sets of diagnostic criteria, with the goal of capturing sIBM patients at an earlier stage of their disease to allow them to access specialized care rapidly and to be included in clinical trials. This can be particularly challenging as clinical manifestations in IBM are often subtle at onset with suggestive pathological findings appearing later in the disease evolution. The ENMC criteria also reflected the advances in immunostaining of abnormal protein aggregates and the recognition of MHC-1 expression as well as mitochondrial abnormalities as markers of sIBM. Some of these newer stains as well as electron microscopy (EM) are not routinely used, making those criteria difficult to apply outside of specialized centers. By investigating the sensitivity and specificity of different “categories” of sIBM criteria defined as Boolean algebraic combinations of features (e.g., definite, probable) in patients diagnosed with sIBM by neuromuscular specialists, it was demonstrated that the available criteria for sIBM have a high specificity (>97%) but that some pathologic and clinical items, such as muscle strength comparison between different muscle groups, had a low sensitivity [16]. Those less sensitive items would exclude many patients with otherwise clinically typical sIBM from trials. The authors instead proposed a triad of data-derived criteria with 90% sensitivity and 96% specificity as shown in Box 11.4.
Box 11.4 Triad of Features Highly Specific for sIBM
-
Finger flexor or quadriceps weakness
-
Endomysial inflammation
-
Invasion of non-necrotic muscle fibers or rimmed vacuoles on histopathology
Conclusion
In summary, myositis is a heterogeneous group of diseases where muscle weakness may predominate. However, it is now clear that multiple organs are also commonly affected and that extramuscular involvement such as pharyngeal muscle weakness, skin rash, ILD, arthritis, and cardiac involvement should be systematically screened for. The detection of MSA is a new useful tool that both supports diagnosis and orients the clinicians to different myositis subgroups characterized by specific organ manifestations. New classification criteria for adult and juvenile myositis as well as myositis subgroups have been developed and recently endorsed by the EULAR and the ACR.
References
Trallero-Araguas E, Grau-Junyent JM, Labirua-Iturburu A, Garcia-Hernandez FJ, Monteagudo-Jimenez M, Fraile-Rodriguez G, et al. Clinical manifestations and long-term outcome of anti-Jo1 antisynthetase patients in a large cohort of Spanish patients from the GEAS-IIM group. Semin Arthritis Rheum. 2016;46(2):225–31.
Moghadam-Kia S, Oddis CV, Sato S, Kuwana M, Aggarwal R. Antimelanoma differentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44(3):319–25.
Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–7.
Oddis CV, Medsger TA Jr. Inflammatory myopathies. Baillieres Clin Rheumatol. 1995;9(3):497–514.
Lundberg IE, Tjarnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–64.
Tanimoto K, Nakano K, Kano S, Mori S, Ueki H, Nishitani H, et al. Classification criteria for polymyositis and dermatomyositis. J Rheumatol. 1995;22(4):668–74.
Targoff IN, Miller FW, Medsger TA Jr, Oddis CV. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 1997;9(6):527–35.
Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet. 2003;362(9388):971–82.
Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14(5):337–45.
Mahler M, Miller FW, Fritzler MJ. Idiopathic inflammatory myopathies and the anti-synthetase syndrome: a comprehensive review. Autoimmun Rev. 2014;13(4–5):367–71.
Johnson SR, Goek ON, Singh-Grewal D, Vlad SC, Feldman BM, Felson DT, et al. Classification criteria in rheumatic diseases: a review of methodologic properties. Arthritis Rheum. 2007;57(7):1119–33.
Singh JA, Solomon DH, Dougados M, Felson D, Hawker G, Katz P, et al. Development of classification and response criteria for rheumatic diseases. Arthritis Rheum. 2006;55(3):348–52.
Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, et al. Inclusion body myositis and myopathies. Ann Neurol. 1995;38(5):705–13.
Hilton-Jones D, Miller A, Parton M, Holton J, Sewry C, Hanna MG. Inclusion body myositis: MRC Centre for Neuromuscular Diseases, IBM workshop, London, 13 June 2008. Neuromuscul Disord. 2010;20(2):142–7.
Rose MR, Group EIW. 188th ENMC international workshop: inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–55.
Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. 2014;83(5):426–33.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Leclair, V., Lundberg, I.E. (2020). Making the Diagnosis of Myositis: Definition and Classification of Myositis. In: Aggarwal, R., Oddis, C. (eds) Managing Myositis. Springer, Cham. https://doi.org/10.1007/978-3-030-15820-0_11
Download citation
DOI: https://doi.org/10.1007/978-3-030-15820-0_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-15819-4
Online ISBN: 978-3-030-15820-0
eBook Packages: MedicineMedicine (R0)