
Abstract
DNA methylation is an epigenetic modification that spatially and temporally regulates gene expression and has essential roles in controlling neuronal development and function. DNA methylation is generally associated with heterochromatin and repression of gene transcription. Methylated cytosine residues can also undergo demethylation by ten-eleven translocation (TET) enzymes, resulting in 5-hydroxymethylation and its downstream derivatives. Once thought of as an intermediary in the demethylation process, 5-hmC has been found to be a unique and stable epigenetic mark. 5-hmC is more highly enriched in mammalian brains than in other somatic cells, indicating its critical roles in the central nervous system. Unlike methylation, hydroxymethylation is usually associated with euchromatin and gene activation. Much progress has been made in the past few decades in defining the roles of methylation and hydroxymethylation, and their molecular machinery in the brain and nervous system. In this chapter, we provide a comprehensive review of the roles of methylation and hydroxymethylation in brain development, functions and their dysregulation in brain disorders. First, we discuss the current understanding of these epigenetic marks in normal neuronal development as well as brain function. DNA methylation and hydroxymethylation have also been implicated in the development and progression of neurodegenerative diseases, such as Alzheimer’s and Parkinson’s diseases. We discuss consistencies and inconsistencies of the available data in human, mouse and in vitro studies that link methylation and hydroxymethylation to neurodegeneration. Finally, we explore the potential of these neuronal epigenetic marks and their molecular machinery to provide novel therapeutic targets in neurodegenerative diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
5-methylcytosine (5-mC), the addition of a methyl group to the cytosine nucleotide, is an epigenetic modification that plays an intrinsic and essential role in biological development and processes. 5-mC was first reported as identified in cow thymus in 1948 and was referred to as “epi-cytosine” (Hotchkiss 1948). In early studies of DNA methylation, restriction enzymes were used to investigate DNA methylation patterns, since certain enzymes were found to be selective in cutting nucleotide sites only if they were unmodified (Singer et al. 1979; Waalwijk and Flavell 1978). More advanced techniques have since been developed and have rapidly expanded our understanding of epigenetic functions and mechanisms. In mammals, 5-mC appears most often at CpG dinucleotides [~70–80% are methylated (Ehrlich et al. 1982; Doskocil and Sorm 1962)], but they are also found at non-CpG sites (e.g., CpA) (Xie et al. 2012). 5-mC is enriched in repetitive regions, gene bodies and intergenic regions and its functions depend on the location (Wen et al. 2014; Szulwach et al. 2011). Some of the major functions include regulation of gene expression by inhibiting transcription, genomic stability and imprinting (Zhou and Robertson 2016; Brandeis et al. 1993). The frequent inverse correlations between methylation of CpG islands and gene expression led to the understanding that 5-mC plays mostly a repressive role in gene regulation (Keshet et al. 1985), especially when at the promoter region of a gene (Langner et al. 1984). DNA methylation is associated with heterochromatin (tightly packed genetic material that is not readily accessible to transcription factors) and thus inhibits gene activation (Choy et al. 2010). DNA methylation maintains genomic stability by silencing repeat elements (Zhou and Robertson 2016; Nichol and Pearson 2002) and transposable elements through methylation, which would otherwise disrupt genomic integrity if they were expressed (Yoder et al. 1997). DNA methylation controls imprinting, in which gene expression from one parental allele is repressed, allowing for expression of the allele from the other parent (Stewart et al. 2016). In addition, it is responsible for inactivation of the X chromosome, a critical genetic event that ensures that females have only one active copy of the sex chromosome instead of two (Brandeis et al. 1993). 5-mC is particularly enriched in brain tissue and its roles in the brain will be discussed in later sections (Ehrlich et al. 1982).
In contrast, DNA hydroxymethylation is primarily associated with active gene transcription and euchromatin (Ficz et al. 2011). Unlike 5-mC, 5-hmC is enriched at poised enhancers, exon-intron boundaries and gene bodies of actively transcribed genes (Wen et al. 2014; Stroud et al. 2011; Mellen et al. 2012). Elevations in 5-hmC do not always correlate with reductions in 5-mC, as would be expected since 5-hmC is a modification of methylated cytosine (Hahn et al. 2013). Thus, 5-hmC is considered to be a stable epigenetic mark in its own right and not just an intermediary step towards demethylation (Hahn et al. 2013). While 5-mC is critical to proliferating cells, 5-hmC appears to be essential to differentiating cells, although the role seems dependent on the cell type. For example, 5-hmC undergoes significant loss during differentiation of embryonic stem cells (ESCs) (Tahiliani et al. 2009; Ruzov et al. 2011; Szwagierczak et al. 2010; Kinney et al. 2011), but it increases during differentiation in adult neural stem cells (aNSCs) (Li et al. 2017b). 5-hmC is associated with cells that are in a pluripotent state and is highest in the embryo within the first 2 weeks following fertilization (Messerschmidt et al. 2014; Ruzov et al. 2011). Importantly, the brain has the highest levels of 5-hmC compared to any other tissue examined (Globisch et al. 2010; Li and Liu 2011; Kriaucionis and Heintz 2009) and it is dynamically regulated during both embryonic neurodevelopment and neurogenesis (Szulwach et al. 2011; Santiago et al. 2014).
Both 5-mC and 5-hmC regulate cellular proliferation and differentiation by temporal and spatial control of gene expression and thus are critical to organ development (Brandeis et al. 1993; Roost et al. 2017). The brain in particular has one of the highest methylation and hydroxymethylation levels of any tissue (Ehrlich et al. 1982; Li and Liu 2011), and both modifications are essential to proper neuronal and synaptic functions (Miller and Sweatt 2007; Sweatt 2016; Fasolino and Zhou 2017; Rudenko et al. 2013). It is key that we expand our knowledge of neuronal methylation in order to further understand neuronal development and disease.
2 Methylation Machinery
2.1 Methylation Writers
DNA methylation is catalyzed by DNA methyltransferases (DNMTs), also known as DNA methylation writers, which transfer a methyl group from S-adenosylmethionine to the 5′ position of the cytosine residue (Kumar et al. 1994) (Fig. 1). Although five enzymes have been described, the major players are DNMT1, DNMT3A and DNMTB (Turek-Plewa and Jagodzinski 2005). These three writers regulate DNA methylation during critical embryonic and fetal developmental periods and their expression is dynamic throughout early development (Uysal et al. 2017). By controlling DNA methylation, they regulate the inhibition or activation of genes. DNA methyltransferase activity is high in preimplantation embryos during which time global loss and subsequent re-establishment of DNA methylation occurs (Carlson et al. 1992). DNMTs are required for imprinting and X chromosome inactivation in the embryo, as well (Howell et al. 2001; Biniszkiewicz et al. 2002). DNMT1, DNMT3A and DNMT3B are expressed ubiquitously in most somatic tissues, although the expression level varies (Robertson et al. 1999).

A schematic of the DNA methylation process and the machinery involved. DNA methyltransferases (DNMTs) catalyze the addition of a methyl group to the 5′ position of the cytosine, using S-adenosylmethionine (SAM) as the methyl donor. Ten-eleven translocation enzymes catalyze the oxidation of 5-methylcytosine into 5-hydroxymethylcytosine, and catalyze further reactions into 5-formylcytosine and 5-carboxylcytosine, in the demethylation process. Methylation readers methyl-CpG-binding protein 2 (MeCP2) and methyl-CpG binding domain proteins 1–4 (MBD1–4) facilitate the functional effects of methylation on gene expression and chromatin structure
The writers differ in their cellular functions. Broadly speaking, DNMT1 is a maintenance enzyme and DNMT3A and DNMT3B are de novo enzymes. DNMT1 maintains methylation patterns in dividing cells, is involved in DNA repair, and has specific activity on hemi-methylated DNA (Bashtrykov et al. 2012). While the canonical function of DNMT1 is to maintain methylation, there is some evidence that it may have a secondary role in de novo methylation (e.g. DNMT1 shows in vitro activity on unmethylated DNA) (Jeltsch and Jurkowska 2014). Global deficiency of DNMT1 causes embryonic lethality in mice, underscoring its essential role for maintaining methylation patterns during embryonic development (Li et al. 1992). DNMT1 is also highly expressed in adult postmitotic neurons (Inano et al. 2000). On the other hand, DNMT3A and DNMT3B add on new methyl groups to DNA in response to environment or experience (Okano et al. 1999). Both DNMT3 writers are highly expressed in undifferentiated embryonic stem cells (ESCs), but their expression drops upon differentiation (Okano et al. 1998). In adulthood, both enzymes show reduced expression, with DNMT3A being ubiquitously low and DNMT3B barely detectable, in most tissues (Okano et al. 1998). There are some key differences between the de novo enzymes, however. During embryogenesis, DNMT3B is most heavily expressed in the brain, while DNMT3A is expressed throughout the entire embryo (Okano et al. 1999). Global DNMT3B knockout impairs neural tube development, resulting in embryonic death (Okano et al. 1999). Global DNMT3A knockout does not seem to impair gross development, but these mice have impaired postnatal neurogenesis and die within a month (Okano et al. 1999; Wu et al. 2010). Importantly, DNMT3A is required for methylation of non-CpG sites, in particular CpA (Guo et al. 2014). Thus, overall, DNMT3A plays a larger role in developed brain function, whereas DNMT3B expression has more critical roles in early development.
2.2 Methylation Readers
Methylation readers are required to translate the methylation code into a functional action for certain genes. The methyl-CpG binding domain (MBD) family of proteins includes MBD1-4 and methyl-CpG-binding proteins 1 and 2 (MeCP1 and MeCP2) (Ballestar and Wolffe 2001). MeCP2 was the first and most-thoroughly studied protein in the MBD family and was found to repress gene transcription by interacting with histone deacetylases and subsequently modifying the chromatin structure to a heterochromatic state (Nan et al. 1998; Jones et al. 1998). MeCP2 and MBD1 both facilitate the methylation of histone 3 lysine 9 (H3K9), which promotes heterochromatin and transcriptional silencing (Sarraf and Stancheva 2004; Fuks et al. 2003). Interestingly, MeCP2 has been demonstrated to also associate with 5-hmC and is the primary 5-hmC binding protein in brain (Mellen et al. 2012). MeCP2 is crucial to neural development and function. Mutations in MeCP2 result in a severe neurological disorder called Rett Syndrome (which occurs only in females because the MeCP2 gene is X-linked, and the mutation is lethal in males) (Pohodich and Zoghbi 2015). Rett syndrome presents as significant intellectual, language and motor impairments that appear after the individual is at least 1 year old, and then continually progress (Ip et al. 2018). In fact, a mutation of one particular residue of MeCP2 that occurs in Rett Syndrome impairs this protein’s binding specifically with 5-hmC (Mellen et al. 2012). Deletion of MBD1 in mice impairs neuronal differentiation, neurogenesis, synaptic plasticity and cognitive function (Zhao et al. 2003).
Other methyl-binding proteins have functions beyond gene repression; for example, MBD4 is a thymine glycosylase and mediates DNA repair (Bogdanovic and Veenstra 2009). While MBD3 does not directly bind to methylated DNA, it mediates methylation function by interacting with other factors, such as histone deacetylases or MBD2 (Ballestar and Wolffe 2001). MBD3 is an essential element of the methylation machinery, as deficiency of this protein is embryonically lethal (Hendrich et al. 2001). In particular, MBD3 is expressed in embryonic neuroepithelial cells (unlike its similar counterpart, MBD2) and continues to be expressed in specific forebrain structures postnatally (Jung et al. 2003). In summary, whether mediating gene repression or facilitating other mechanisms, methylation readers are essential biological components and apparently highly influential to neurological function.
2.3 Methylation Erasers
DNA methylation is both stable and reversible. Demethylation occurs through a series of steps driven by the ten-eleven translocation (TET) family of dioxygenases (TET1, TET2 and TET3; DNA methylation erasers). In the first step of this process, 5-methylcytosine is oxidized to produce 5-hydroxymethylcytosine (5-hmC) (Tahiliani et al. 2009). The demethylation process is completed through several more steps, in which TET proteins convert 5-hmC into 5-formylcytosine (5-fC) and 5-carboxylcytosine (5-caC). Both 5-fC and 5-caC can be excised by thymine DNA glycosylase (TDG) to result in an unmodified cytosine (Ito et al. 2011; Maiti and Drohat 2011). The TET enzymes vary in their substrate preference. TET1 and TET2 demonstrate greater enzymatic activity on 5-mC as compared to 5-hmC or 5-fC (Hu et al. 2015), whereas TET3 shows a stronger affinity for 5-caC (Jin et al. 2016). The three TET enzymes also show some key distinctions in the contexts in which they form 5-hmC. For example, in mouse ESCs, TET1 is responsible for 5-hmC marks near transcription start sites, while TET2 primarily maintains 5-hmC that is enriched in gene bodies (Huang et al. 2014). During the early wave of global demethylation within the zygote, TET3 is responsible for rapidly demethylating the paternal genome (Messerschmidt et al. 2014). Finally, imbalances in the TET enzymes disrupt neural function and cognitive processes (discussed in a later section), making them essential to normal brain function (Kaas et al. 2013; Rudenko et al. 2013; Zhang et al. 2013).
3 Methylation in the Brain
3.1 5-Methylcytosine
The brain is a phenomenally complex organ, with numerous regions and nuclei, each of which express a unique gene profile to carry out specific functions. In the cerebral cortex alone, there are six different layers, characterized by differences in cell morphology and in neuronal inputs and projections. Careful orchestration of gene expression at the right time, and in the right place, is paramount to the proper development of the brain.
In human embryonic stem cells, 5-mC is decreased during early differentiation into neural precursor cells (Kim et al. 2014). Although the overall abundance of 5-mC does not significantly change from neural precursor cells (NPCs) to neurons (Hahn et al. 2013), the methylation landscape is highly dynamic, controlling production and differentiation of various brain cells in a loci-specific manner. Following neurogenesis, NPCs switch to producing astrocytes (Martynoga et al. 2012) and the methylation profile of these NPCs is distinct from the profile at the time of neurogenesis (Sanosaka et al. 2017). Glial fibrillary acidic protein (GFAP; an astrocyte marker) has reduced promoter methylation at specific transcription factor binding sites following neurogenesis, allowing for increased expression of GFAP (Takizawa et al. 2001; Teter et al. 1996; Condorelli et al. 1997). 5-mC becomes enriched in the promoters of genes related to pluripotency, which are downregulated during differentiation (Kim et al. 2014). DNMT1 has been shown to be critical to the migration of interneurons to their specific destination in the cerebral cortex, in part by regulating expression of paired box 6 (PAX6) (Pensold and Zimmer 2018; Pensold et al. 2017). Knockdown of DNMT1 in embryonic neural progenitor cells disrupts the timing of astrogliogenesis, causing earlier production of these cells (Fan et al. 2005). DNMT3B seems to be critical to early neuronal development, as global knockout mouse embryos have neural tube defects and die before birth (Okano et al. 1999).
Methylation plays a critical role in the adult brain by regulating synaptic plasticity, and different neuron types show distinct methylation patterns (Mo et al. 2015). Neuronal activation induces acute changes in the methylation of genes involved in processes such as synaptic function, calcium signaling and protein phosphorylation (Guo et al. 2011). Inhibiting DNA methyltransferase activity alters the electrophysiological properties of cultured neurons (Meadows et al. 2016) and DNMT activity in the amygdala is necessary for the neural plasticity that occurs during fear learning and memory (Maddox et al. 2014). DNMT1 expression remains at a surprisingly detectable level for a maintenance enzyme, even though most neuronal cells are postmitotic and are no longer proliferating (Goto et al. 1994; Inano et al. 2000). Deficiency of DNMT1 in forebrain neurons has been shown to cause impairments in long-term potentiation of cortical neurons (Golshani et al. 2005) and progressive degeneration of neurons in the cortex and hippocampus, in one particular knockout mouse model (Hutnick et al. 2009). Deficiency of both DNMT1 and DNMT3A in forebrain neurons (using a different knockout strategy) impairs synaptic plasticity in hippocampal neurons and produces learning and memory deficits in mice (Feng et al. 2010). DNMT3A is required for maintaining methylation of CpA sites in the adult hippocampus (Guo et al. 2014). Mice lacking the methylation reader MBD1 show reduced neurogenesis, reduced synaptic plasticity and spatial learning deficits in adulthood (Zhao et al. 2003). MeCP2 is essential to adult brain function, and as mentioned previously, mutations in MeCP2 lead to severe neurological deficits (Rett Syndrome) in humans (Pohodich and Zoghbi 2015; McGraw et al. 2011). In addition, overexpression of MeCP2 enhances learning and synaptic plasticity in early adulthood, but causes seizures and premature death later on (Collins et al. 2004), underscoring the delicate balance required in DNA methylation dynamics for brain function. Thus, 5-methylcytosine and its machinery have complex roles in both neuronal development and adult brain function.
3.2 5-Hydroxymethylcytosine
5-hmC exhibits a very different pattern from 5-mC in both embryonic and adult neurons. In contrast to 5-mC, 5-hmC increases as NPCs differentiate into neurons (Kim et al. 2014; Hahn et al. 2013). 5-hmC is specifically enriched in the bodies of neurogenesis-related genes and is associated with their upregulation during differentiation (Kim et al. 2014). In embryonic mouse forebrain, 5-hmC is increased at promoters and gene bodies during neuronal differentiation (Hahn et al. 2013). TET2 and TET3 are expressed more highly than TET1 in the cortex and not surprisingly, are upregulated during neuronal differentiation. Overexpression or deficiency of these TET proteins can alter the timing of neurogenesis or cause inappropriate cellular clustering within the layers of the cortex, respectively. Although not as critical, TET1 also mediates embryonic neurogenesis and loss of TET1 in mice also alters the timing of neuronal production to a degree (Kim et al. 2016).
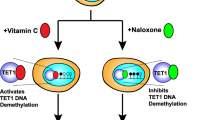
Hydroxymethylation plays a significant role in adult neuronal function. 5-hmC increases in the brain postnatally and continues to increase with aging (Wen et al. 2014; Szulwach et al. 2011; Kraus et al. 2015). It is particularly enriched in Purkinje cells of the cerebellum (Kriaucionis and Heintz 2009) and in the cortex (Kraus et al. 2012). Compared to 5-mC, it is still only 25% as abundant overall in the brain (Wagner et al. 2015). 5-hmC shows specific enrichment in genes related to synaptic function (Khare et al. 2012). In vitro studies have shown that TET1 is necessary for neurogenesis and progenitor cell proliferation in the adult brain (Zhang et al. 2013). TET1-deficient mice show reduced proliferation of neural progenitor cells (NPCs) in the hippocampus, and in vitro NPCs lacking TET1 have downregulated neurogenesis-related genes (Zhang et al. 2013). Although TET1 global knockout mice display normal brain morphology, they exhibit memory impairments as adults (Zhang et al. 2013; Rudenko et al. 2013). TET2 deficiency impairs the balance of proliferation and differentiation in mouse adult NSCs (Li et al. 2017b). TET2 is required for the increase in 5-hmC that occurs during differentiation of adult NSCs, and it controls critical neurogenic gene transcription (Li et al. 2017b). Mice that have a hippocampal deficiency of TET2 exhibit reductions in 5-hmC and cognitive impairments, both of which are rescued by replacement of TET2 (Gontier et al. 2018).
On the other hand, 5-hmC enrichment and TET enzyme expression can be regulated by neuronal activity (Kaas et al. 2013; Kremer et al. 2018). Fear conditioning in mice causes TET1 to be downregulated for several hours, and both overexpression or knockout of TET1 impairs the formation of long-term memories (Kaas et al. 2013; Rudenko et al. 2013). In primary cortical neurons, TET3 is upregulated in response to neuronal activity (Li et al. 2014). TET3 is upregulated in the cortex following fear extinction in mice (a behavioral paradigm in which fear is “un-learned”) (Li et al. 2014). In summary, 5-hydroxymethylcytosine and its machinery have critical roles in the adult brain, including neuronal differentiation and regulating cognitive processes.
3.3 Methylation and the Aging Brain
Aging cells enter into a senescent state, whereby the cell cycle is halted (for dividing cells) and progressive phenotypic changes take place, beginning with alterations in genomic methylation, the formation of heterochromatic foci and increased DNA damage (van Deursen 2014; Baker and Petersen 2018). These changes have significant effects on gene expression, such as upregulating immune factors like pro-inflammatory molecules. The overall immune response becomes gradually elevated in the aging brain (Cribbs et al. 2012), forming the “senescence-associated secretory phenotype” (Rodier et al. 2009; Baker and Petersen 2018). Microglial cells, the resident macrophages of the brain, also become activated and produce local inflammation (Samorajski 1976; Norden and Godbout 2013). With age, DNA base-excision repair is reduced, impairing genomic integrity (Gan et al. 2012). Due to an increase in double-stranded DNA breaks, the DNA damage response pathway becomes activated and sustained (Sedelnikova et al. 2004; Rodier et al. 2009). Impairments in the antioxidant pathway lead to increased oxidative stress (Droge and Schipper 2007). Consequently, signaling pathways, metabolism, synapses and neuronal circuitry all become impaired. In the hippocampus especially, neurons display mitochondrial dysfunction and dysregulated calcium homeostasis (Pandya et al. 2016; Gant et al. 2006). Regions of the cerebral cortex begin to show a reduction of synapses (Adams 1987). After the age of 60 the brain gradually begins to reduce in weight and volume due to cell loss (Samorajski 1976). Cell loss is particularly prominent in the superior temporal gyrus and precentral gyrus regions of the cerebral cortex (Samorajski 1976). Collectively, these progressive changes cause a gradual decline in cognitive function that is associated with normal aging (Poddar et al. 2018).
The widely-held opinion has been that 5-mC globally decreases with aging and that this hypomethylation may be involved in age-related disease (Unnikrishnan et al. 2018). However, this is a grossly oversimplified statement, as the dynamics of 5-mC throughout aging depend on the tissue, the gene and (within the brain) the specific brain region. In many tissues, global 5-mC does decrease with age (Unnikrishnan et al. 2018; Wilson et al. 1987; Hoal-van Helden and van Helden 1989) and this often occurs in repetitive elements (Jintaridth and Mutirangura 2010). Yet, these findings seem dependent on the method used to quantify 5-mC (Unnikrishnan et al. 2018). In the brain, 5-mC dynamics throughout aging are region-specific. One study found no difference in the level of 5-mC in the hippocampi between 2-month old (young) and 22-month old (old) mice (Chen et al. 2012), which was corroborated by another study that examined 3, 12 and 24-month old mice of both sexes (Hadad et al. 2016). This same study also demonstrated no change in hippocampal DNMT or TET enzymes with increasing age. Certain genes involved in neuronal function show age-specific changes in methylation that may lead to the cognitive decline observed in aging mammals. Methylation of Egr1, an immediate early gene that plays an important role in synaptic plasticity in the hippocampal dentate gyrus, shows increased promoter methylation and reduced transcription in aged rats as compared to young adult rats (Penner et al. 2016). In the prefrontal cortex however, a number of genes involved in synaptic plasticity exhibit increased methylation and reduced expression in aged versus younger rats (Ianov et al. 2017). Thus, although 5-mC does not appear to change globally with aging, brain region-specific or loci-specific 5-mC changes are certainly involved in the aging process of the brain and age-associated diseases.
Aging is associated with gradual reductions in the length and integrity of telomeres (Bekaert et al. 2005; Rizvi et al. 2014). Methylation levels are high in the subtelomeric regions directly adjacent to the telomeres, and this methylation supports a heterochromatic state that confers genomic stability (Ng et al. 2009). DNMTs appear to play a protective role by both controlling telomere length and preventing excessive telomeric recombination (Gonzalo et al. 2006).
Opposite to 5-mC, 5-hmC has been shown to increase with age in the brain and peripheral tissues (Wagner et al. 2015). In specific brain regions of mice, such as the cerebellum and hippocampus, 5-hmC increases from postnatal day 7 (P7) to 1 year of age (Szulwach et al. 2011). In the tree shrew, another mammal, 5-hmC is also elevated from P10 to 2 years of age in the prefrontal, parietal and occipital cortices, the hippocampus and cerebellum (Wei et al. 2017). In mice, 5-hmC increases in the hippocampus from 12 months to 22 months of age (Chen et al. 2012), although this result was not confirmed by another research group using the same quantification method (Hadad et al. 2016).
4 Methylation and Neurodegenerative Diseases
4.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is the leading cause of dementia and is characterized by progressive and profound memory loss and impairments in cognitive functioning, for which there is no cure (Scheltens et al. 2016). The neurological hallmarks of this disease include β-amyloid deposits and plaques, and neurofibrillary tangles that build up within the brain tissue, driven by the proteins β-amyloid and Tau, respectively (Goedert et al. 1991; Gorevic et al. 1986). These pathological changes lead to neuronal loss in multiple brain areas (Braak and Braak 1991). The most common form is late-onset AD, which occurs after the age of 65, while early onset, familial AD occurs before age 65 and is more strongly associated with genetic mutations (Tanzi 1999). In general, late-onset AD is marked by 6 stages of neuronal pathology (Braak and Braak 1991). β-amyloid deposits are usually the first presentation, followed by neurofibrillary changes. While the majority of β-amyloid deposits tend to appear in the cortex, neurofibrillary tangles begin accumulating in the transentorhinal cortex (in the temporal lobe), and then move into the hippocampus and the cerebral cortex (Braak and Braak 1991; Calderon-Garciduenas and Charles 2017). Not all AD cases present the exact same pattern of β-amyloid deposits and/or neurofibrillary changes. It is unknown what exactly causes AD to develop, and which markers are causal versus symptomatic of the disease.
It is estimated that at least 80% of AD cases involve a genetic component, as evidenced by family and twin studies (Heston 1989). Only 5% of AD cases are early-onset, and most of these rare cases are strongly associated with mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) (Tanzi et al. 1996). For the more common late-onset, sporadic AD, the genetic component is less clear. Polymorphisms in allele 4 of the apolipoprotein E gene (APOEε4) may contribute to over half of the late-onset cases and it is considered to be the main known genetic risk factor (Tanzi 2012). Genome-wide association studies (GWAS) have uncovered at least 11 other genes that show polymorphisms linked to AD (Tanzi 2012). Genetic variants alone however, do not fully account for the development of AD. An extensive amount of research has focused on the association of environmental factors (such as diet) in the development of the disease (Dosunmu et al. 2007). Because epigenetic modifications alter gene regulation as a result of environment or experience, methylation and hydroxymethylation have been targeted as possible risk factors in late-onset, sporadic AD (Bihaqi et al. 2012).
Postmortem studies of AD human brain tissue have revealed significant alterations in DNA methylation patterns, depending on the brain region examined. For example, both 5-mC and 5-hmC are overall significantly reduced in brains of people with AD as compared with age-matched controls with no dementia (Chouliaras et al. 2013). Furthermore, these methylation and hydroxymethylation levels strongly correlate with the build-up of β-amyloid plaques within the same brain region. Depletion of S-adenosylmethionine in vitro and in mouse models increases the proteins APP, PSEN1 and β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) (all major players in AD) while decreasing β-amyloid accumulation (Do Carmo et al. 2016). When examining specific brain regions however, the evidence is contradictory. In one study, 5-mC was decreased in the entorhinal cortex, a region which shows the earliest signs of neurofibrillary changes in people with AD (Mastroeni et al. 2010). However, two other studies reported no change of 5-mC in the entorhinal cortex (Condliffe et al. 2014; Lashley et al. 2015). There is similar contradictory evidence for changes in the level of 5-hmC, with one study showing no change (Lashley et al. 2015) and another showing decreases (Condliffe et al. 2014) in the entorhinal cortex. 5-mC and 5-hmC were increased in the medial frontal and temporal gyri of AD patients in a study that controlled for age, gender, postmortem delay and tissue storage time (Coppieters et al. 2014). Interestingly, 5-mC and 5-hmC, as well as TET1 enzyme levels, are increased in the hippocampal/parahippocampal gyrus in both preclinical and late-stage AD patients (van der Flier et al. 2011), giving support to the possibility that aberrant DNA methylation facilitates the progression of, or perhaps even plays a causal role in AD. Discordant disease phenotypes among monozygotic twins also provide evidence for an epigenetic role in AD development (Ketelaar et al. 2012). One report describes an individual with AD who had reduced 5-mC in the anterior temporal neocortex and superior frontal gyrus (regions highly vulnerable to the disease) compared to his healthy, monozygotic twin (Mastroeni et al. 2009). Although there are discrepancies in the scientific literature (possibly due to differences in age, cause of death, postmortem delay, environmental, or unknown comorbidities that were unaccounted for), it is clear that the DNA modification landscapes, including 5-mC and 5-hmC, are altered in AD and could play causal roles in disease progression even before the onset of AD pathology.
The first indication that specific AD-related genes might be affected by methylation or hydroxymethylation was reported in the 1990s using a methylation-sensitive restriction enzyme to digest the APP gene from the temporal lobe brain tissue (West et al. 1995). These results showed an altered methylation pattern of APP in the AD patient. Another gene, triggering receptor expressed on myeloid cells 2 (TREM2) has been identified as a rare variant for AD with a large effect size and has anti-inflammatory functions (Guerreiro et al. 2013; Hamerman et al. 2006). Hypermethylation of TREM2, upstream of the transcription start site, has been reported on three separate occasions by a research group using two different methods of quantification (Smith et al. 2016). Hypermethylated TREM2 was seen most strongly in the superior temporal gyrus of AD human brains as compared to control brains. Modified cytosines in neurons specifically (excluding other brain cells, such as glia or astrocytes) have been analyzed from human AD or control age-matched brain tissues (Mano et al. 2017). In this study, differentially methylated regions (DMRs) were identified in the breast cancer Type 1 susceptibility protein (BRCA1) gene promoter that were specifically hypomethylated in AD neurons. These results were confirmed with pyrosequencing, further finding that BRCA1 methylation levels correlated with the level of APOEε4 alleles, indicating a possible mechanism by which methylation may contribute to β-amyloid pathology. This coincided with increased mRNA and protein expression of BRCA1 that was present in the hippocampal and entorhinal cortex of AD brains, but not of the control brains. Interestingly, although BRCA1 is a DNA repair gene, this protein was mostly present in the cytoplasm of AD brains, suggesting its dysfunction.
Greater methylation on the β-site amyloid precursor protein cleaving enzyme 1 (BACE-1) gene promoter is associated with β-amyloid load in AD brains (Do Carmo et al. 2016). In fact, even at the level of a single CpG site, hypermethylation was associated with reduced β-amyloid in those AD patients. Data from two AD mouse models (APP/Psen1 and 3xTg-AD) are consistent with human data that show hypermethylation of the genes thromboxane A2 receptor (Tbxa2r), sorbin and SH3 domain containing 3 (Sorbs3) and spectrin beta 4 (Sptbn4) are correlated with reduced gene and protein expression in frontal cortex brain tissue (Sanchez-Mut et al. 2013). Blocking the thromboxane receptor has been shown reduce AD pathologies in mice (Lauretti et al. 2015). Sorbs3 hypermethylation in AD brains has also been previously reported (Siegmund et al. 2007) and may be a normal, age-dependent change that is accelerated with AD. Sorbs3 encodes the protein vinexin, which has roles in cell signaling, gene expression and synaptic functions (Ito et al. 2007).
Mouse models with mutations in various AD-related genes have identified clues to possible mechanisms by which methylation could regulate AD pathogenesis. However, it is important to bear in mind that transgenic mice only recapitulate key aspects of the disease and not the complete pathology. For example, J20 mice, which have a mutation in APP, exhibit plaques but no neurofibrillary tangles (Lardenoije et al. 2018). In these mice, 5-mC and the ratio of 5-mC:5-hmC decreases with age in the hippocampal CA3 region, but there is no change in 5-hmC amount. Interestingly, both 5-mC and 5-hmC levels were negatively correlated with the plaque amount in the dentate gyrus of the hippocampus (Lardenoije et al. 2018). The 3xTg-AD mouse model has mutations in Psen1, App and Tau genes and exhibits both plaques and neurofibrillary tangles. In these mice, 5-mC increases with age (from 3 to 17 months) in the dentate gyrus and in the CA1 and CA2 hippocampal regions, but it does not correlate with plaque accumulation (Lardenoije et al. 2018). Neither the J20 nor the 3xTg-Ad mouse models show any age-related change in 5-hmC. In contrast to this, a different study did find that 3xTg-AD mice show an age-related increase in 5-hmC in multiple brain regions, but found no correlation with the plaque accumulation (Cadena-del-Castillo et al. 2014). McGill-Thy1-APPTg mice (which express human APP with several mutations) have reduced global 5-mC in the cortex and hippocampus which correlate with the β-amyloid load (Do Carmo et al. 2016). The Tg5xFAD mouse (which highly expresses mutant forms of APP and PSEN1) exhibits an increase in global neuronal 5-mC, as well as increases in Dnmt3a expression (Grinan-Ferre et al. 2016). An important consideration is that 5-mC and 5-hmC were measured by multiple methods across these studies, such as immunoreactivity, LUMA, ELISA and Dot-Blot, in addition to using different mouse lines that express differing genetic risk factors for AD.
We note several interesting points beyond the mere association of methylation and neuropathologies in AD. Importantly, evidence suggests that epigenetic changes may initiate AD, given that they sometimes occur in presymptomatic patients and in AD mouse models prior to the development of cognitive dysfunction or neuronal pathology. The methylation level of BIN1 and other genes have been shown to be significantly associated with the plaque amount in AD patients—curiously, these same effect sizes between methylation levels and β-amyloid plaques were observed in elderly people with no cognitive impairment, suggesting that altered methylation pattern might be a cause, rather than an effect, of the disease (De Jager et al. 2014). In McGill-Thy1-APPTg mice, early increases in the β-amyloid peptide within neurons (which occurs prior to the β-amyloid pathology and cognitive impairment) is associated with both global hypomethylation in the hippocampus and cortex, and Bace-1 promoter hypomethylation in the cortex. Further, S-adenosylmethionine (SAM) administration could rescue the global and Bace-1 hypomethylation, reverse the cognitive impairments and reduce extracellular and intraneuronal β-amyloid pathology in the transgenic mice (Do Carmo et al. 2016). In vitro studies have demonstrated a role of β-amyloid in driving hypomethylation. For example, murine cerebral endothelial cells treated with β-amyloid have reduced global methylation (Chen et al. 2009). On the other hand, restoring methylation has been shown to reduce β-amyloid. Specifically, SAM administration to human neuroblastoma cells inhibits promoter demethylation of PSEN1 and significantly reduces β-amyloid load (Scarpa et al. 2003).
AD is a disease with multiple genetic components and complex risk factors that make untangling its developmental mechanisms difficult. Methylation studies aimed at AD genes should be carefully designed to focus on the “low-hanging fruit” of potential therapeutic targets, and on loci that are likely to be involved in the majority of AD cases. Given the evidence of certain methylation changes occurring prior to disease onset, more efforts should be made to understand the potential causal role of 5-mC and 5-hmC in AD.
4.2 Parkinson’s Disease
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease after Alzheimer’s (de Lau and Breteler 2006; Bertram and Tanzi 2005). PD is characterized by cytoplasmic aggregation of the α-synuclein protein (which creates Lewy bodies) and selective loss of dopaminergic neurons, both of which occur in the substantia nigra pars compacta of the midbrain (Wakabayashi et al. 2000; Braak et al. 2004; Gibb and Lees 1991). These pathologies lead to movement disorders including resting tremors, slow movement, rigidity, postural instability and akinesia (Erro and Stamelou 2017), and numerous other non-motor comorbidities such as constipation, sleep dysfunction, depression and cognitive impairments (Titova et al. 2017; Albers et al. 2017). The substantia nigra is a part of the basal ganglia, a group of neuronal networks that provide input to other brain regions to facilitate movement, especially by inhibiting other movements at the same time (Mink 1996). α-synuclein is a protein involved in synaptic vesicles and is typically found at the presynaptic terminal of neurons (not in the cytoplasm), which suggests its dysfunction that it accumulates in the cytoplasm (Burre et al. 2010; Wakabayashi et al. 1992). A disturbing fact is that by the time Parkinsonian symptoms appear, the neurodegeneration and loss of dopaminergic neurons is significant and irreversible (Cheng et al. 2010). Because the neuronal pathology occurs years before symptom onset, understanding early biomarkers has become key in early diagnosis (DeKosky and Marek 2003).
Like AD, the majority of PD cases are sporadic (Lill 2016). GWAS studies have confirmed loci on the genes α-synuclein (SNCA) and microtubule-associated protein tau (MAPT) as primary susceptibility factors for sporadic PD (Billingsley et al. 2018; Edwards et al. 2010; Pihlstrom et al. 2018). A large CpG island is present in the human SNCA gene that covers the transcription start sites, the promoter region and intron 1. Multiple studies support that hypomethylation of this CpG region may lead to PD pathology. For example, postmortem substantia nigra tissue from humans with sporadic PD, as well as in vitro neuronal culture experiments, show an association between SNCA intron 1 hypomethylation and increased SNCA expression (Jowaed et al. 2010; Matsumoto et al. 2010). However, a later study found no difference in the SNCA methylation in this brain region between PD and control (although these findings may be due in part to differences in the DNA isolation process prior to sequencing) (Guhathakurta et al. 2017b). A different study reported that PD individuals had lower SNCA promoter methylation, which was associated with a specific variant of SNCA in both blood and cerebral cortex tissue (Pihlstrom et al. 2015). Similar findings of SNCA methylation in blood and frontal cortex brain tissue from PD individuals have been reported, as well (Masliah et al. 2013). Other studies have failed to find differential SNCA methylation in PD blood cells (Song et al. 2014; Richter et al. 2012). Of note, only the human SNCA gene shows this many CpG sites, as compared to mice or rats (Jowaed et al. 2010; Guhathakurta et al. 2017a), underscoring the limitations of rodent data in this specific area of PD epigenetic research.
A key finding is that levodopa (l-DOPA; the primary drug used to treat Parkinsonian symptoms) increases SNCA methylation both in vitro and in vivo (Schmitt et al. 2015). The link between l-DOPA and methylation is unique in that l-DOPA is metabolized in part through O-methylation using SAM as a methyl donor (Sandler 1972). l-DOPA administration results in decreased SAM concentrations in the brain (Liu et al. 2000). Interestingly, administration of SAM into rat brains causes neuronal degeneration (Charlton and Mack 1994) and motor impairments reminiscent of PD clinical symptoms (Charlton and Crowell 1995). It is clear that methylation is involved, likely at many different aspects, of PD pathology, and it is an additional mechanism through which l-DOPA may exert its therapeutic effects. Further, the link between l-DOPA and methylation may yield insights regarding ways to improve the efficacy of this drug.
MAPT and LRRK2 are two other major genetic risk factors that show altered methylation in PD patients (Simon-Sanchez et al. 2009). PD patients have shown greater MAPT promoter methylation in the frontal cortex as compared to control individuals (Masliah et al. 2013). Another report however, demonstrated greater MAPT methylation in the cerebellum, but reduced methylation in the putamen, a basal ganglia structure near to the substantia nigra (Coupland et al. 2014). MAPT methylation in leukocytes and brain tissue also positively correlates with the age of disease onset in PD (Coupland et al. 2014). Although LRRK2 is increased in sporadic PD brains (Cho et al. 2013) and LRRK2 overexpression elevates α-synuclein pathology (Lin et al. 2009), no distinct LRRK2 promoter methylation patterns were seen in leukocytes from individuals with PD as compared to controls (Tan et al. 2014).
DNA methyltransferases play a specific role in PD neuropathology. Nuclear DNMT1 is reduced in the cerebral cortex of PD patients and shows greater cytoplasmic localization as compared to non-PD human brains (Desplats et al. 2011). Consistent with this, global 5-mC is also reduced, as quantified using immunohistochemistry and ELISA (Desplats et al. 2011). Using rat neuronal cell culture, this same group demonstrated that overexpression of α-synuclein causes Dnmt1 to shift its location to the cytoplasm. α-synuclein and Dnmt1 proteins co-immunoprecipitate (when pulling down either protein) both in cell culture and in human brain tissue, suggesting a direct association between these two proteins. Supporting this, lentiviral expression of DNMT1 partially reverses α-synuclein-induced global hypomethylation. Similar phenomena are observed in transgenic mice that overexpress human α-synuclein under the control of a Thy-1 promoter. These mice show cytoplasmic localization of Dnmt1 and global hypomethylation; but lentiviral delivery of DNMT1 increases Dnmt1 nuclear localization (Desplats et al. 2011). Additional studies further implicate DNMT activity in PD pathologies. For example, inhibiting DNA methyltransferases by 5-aza-2′-deoxycytidine in dopaminergic neuronal cell lines (human, mouse and rat) increases apoptosis (Wang et al. 2013b). 5-aza-2′-deoxycytidine also increases the degenerative effects of MPP+, 6-OHDA and rotenone (neurotoxins commonly used in PD research) and reduces SNCA promoter methylation (Wang et al. 2013b). Finally, several polymorphic variants of the DNMT3B gene correlate with PD risk in Brazilian and Chinese populations (Pezzi et al. 2017; Chen et al. 2017).
In summary, a fair amount of research has already been established regarding the role of methylation in PD. Because the neurodegeneration in PD is largely silent until after significant neuronal loss has occurred, biomarkers of early neurodegeneration would be essential to timely diagnosis and treatment. Further research may be able to identify a novel circulating factor in the blood whose methylation status might signal the onset of neurodegeneration, in advance of symptoms onset. Additional research should also be aimed at understanding the dysregulation of DNMT1 and its sequestration in the cytoplasm by α-synuclein.
4.3 Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS; also known as Lou Gehrig’s disease) is a devastating disease involving degeneration of the neurons involved in muscle control (Goetz 2000). It is considered a rare disease, as the incidence is ~5 cases per 100,000 individuals in the U.S. (Mehta et al. 2018), with a mean age of onset of around 60 years (ranging from 40 to 70 years) (Govoni et al. 2017). The neurological hallmark of this disease is the aggregation of ubiquitinated proteinaceous inclusions within the cytoplasm of both upper motor neurons of the cerebral cortex and lower motor neurons of the spinal cord (Hardiman et al. 2017; Goetz 2000). This pathology leads to reduced synapses and eventually denervation of muscle (Sasaki and Maruyama 1994). The clinical presentations of ALS are muscle atrophy, weakness, spasticity, cramps, fasciculation (muscle twitching) and dysphagia (difficulty swallowing) (Rowland and Shneider 2001; Wijesekera and Leigh 2009). Most individuals with ALS die within 3–5 years following the diagnosis due to degeneration of the neurons that lead to the muscles required to breathe (Balendra et al. 2014). Neurons in the motor cortex are lost early on in the disease, and at death more than half of the spinal motor neurons are gone (Chio et al. 2014). Astrogliosis is prominent in neurons of the ventral and dorsal horns of the spinal cord (Schiffer et al. 1996). Like Alzheimer’s and Parkinson’s diseases, only 5–10% of ALS cases are familial while the rest are sporadic (Ajroud-Driss and Siddique 2015). The cause of ALS is unknown, although an expanded hexanucleotide repeat in the chromosome 9 open reading frame 72 (C9ORF72) is the most common genetic cause of both familial and sporadic forms (DeJesus-Hernandez et al. 2011).
Spinal cord tissue from ALS patients has elevated global 5-mC levels and even greater elevations in 5-hmC levels (threefold above non-ALS individuals) (Figueroa-Romero et al. 2012). In the motor cortex of the brain, 5-mC immunoreactivity can be observed in neurons from ALS patients, while it is hard to detect in non-ALS individuals (Chestnut et al. 2011). Global methylation levels from blood samples provide conflicting evidence. 5-mC is increased in whole blood from ALS patients when measured by a restriction enzymes assay (Tremolizzo et al. 2014), and similarly ALS individuals from five families carrying a superoxide dismutase 1 (SOD1) mutation show increased 5-mC in blood using an ELISA assay (Coppede et al. 2018). However, another study reported no such increase using an ELISA assay (Figueroa-Romero et al. 2012). In general, very few studies on 5-hmC in ALS were found in a search of the literature. Regardless of the dynamics of global 5-mC and 5-hmC in ALS, methylation of specific genes may guide us to new potential therapeutic targets that could delay the progression of ALS.
The most studied loci associated with sporadic ALS is C9ORF72, which bears a hexanucleotide repeat expansion (Shatunov et al. 2010; Ahmeti et al. 2013). The expanded repeat of C9ORF72 can cause a buildup of nuclear RNA foci in individuals that carry the expanded allele (DeJesus-Hernandez et al. 2011)—a pathology that may interfere with mRNA splicing, RNA binding proteins and other mechanisms leading to cellular dysfunction. Approximately 30% of ALS patients with a C9ORF72 repeat expansion have hypermethylation of this gene’s promoter (van Blitterswijk et al. 2012). C9ORF72 promoter hypermethylation has been associated with less accumulation of protein aggregates in human ALS brains (Liu et al. 2014), suggesting that it may be a compensatory mechanism. Furthermore, the same study reported that inhibiting methylation in human cell lines using 5-aza-2′-deoxycytidine increases the susceptibility of the cells to oxidative and autophagic stress, leading the authors to hypothesize that C9ORF72 hypermethylation could be protective against neuronal toxicity. Consistent with this, another group found a negative correlation between C9ORF72 hypermethylation and neuronal loss in the hippocampus and frontal cortex of human brains (McMillan et al. 2015). Unexpectedly, ALS patients with C9ORF72 hypermethylation also have enrichment of 5-hmC at the C9ORF72 promoter (Esanov et al. 2016). There are some contrary reports regarding C9ORF72 hypermethylation, however. One set of monozygotic twins discordant for ALS showed a repeat expansion of C9ORF72 but no differences in its methylation pattern (Xi et al. 2014). Additionally, carriers of a shorter expansion of C9ORF72 show less methylation of this gene as compared to carriers with the long expansion (Gijselinck et al. 2016). Thus, it seems that C9ORF72 promoter methylation may mediate pathology in some ALS individuals with the C9ORF72 expansion but not in others.
Other candidate risk genes that have been identified for ALS, such as superoxide dismutase 1 (SOD1), vascular endothelial growth factor (VEGF), TAR DNA-binding protein 43 (TARDBP) and angiogenin (ANG) have not been reported to have differential methylation in individuals with ALS. Analysis of postmortem lateral frontal cortex tissue from sporadic ALS individuals using ChIP on Chip techniques (combining chromatin immunoprecipitation [ChIP] with whole genome scanning using a gene chip) revealed no methylation differences in any of the four candidate genes describe above (Morahan et al. 2009). Further, analysis of peripheral blood mononuclear cells (PBMCs) and brain tissue from ALS individuals using bisulfite sequencing revealed no differential methylation in the SOD1 or VEGF promoters (Oates and Pamphlett 2007). However, differential methylation has been observed in over 30 CpG sites in various genes involved in calcium dynamics, excitotoxicity and oxidative stress among ALS and control cortex samples (Morahan et al. 2009). Spinal cord tissue from ALS individuals displays over 1000 differentially expressed genes related to inflammatory and immune responses, with many of these overlapping with differentially methylated regions as well (Figueroa-Romero et al. 2012).
Studies of disease-discordant twins can be of great insight to unraveling disease etiology because it controls for genetic variation, leaving epigenetic (e.g., methylation) factors as possible mechanisms. The methylation profile of peripheral blood cells in five sets of monozygotic twins discordant for ALS revealed common differentially methylated CpG sites, including regions involved in glutamate metabolism and GABA signaling (important neurotransmitters required for synaptic transmission in many neural circuits) in the ALS siblings as compared to their unaffected twins (Young et al. 2017). Interestingly, the ALS siblings also demonstrated a more aged methylation profile compared to their non-ALS siblings [assessed by applying a Horvath algorithm to the methylation data; also known as DNAm age (Horvath 2013)] This aged DNAm profile has also been reported in another set of monozygotic twins discordant for ALS, pointing to an interesting link between epigenetic aging mechanisms and ALS (Zhang et al. 2016).
DNA methyltransferases may play a unique role in neuronal cell death in ALS. In humans with ALS, both DNMT1 and DNMT3A are upregulated in both the nucleus and mitochondria of motor cortex neurons (Chestnut et al. 2011). Transfection of DNMT3A into motor neuron-like cell culture (NSC34) causes degeneration of those cells (Chestnut et al. 2011). Interestingly, this forced expression of DNMT3A is localized to the cytoplasm not the nucleus, and frequently localizes to mitochondria. In NSC34 cells that are undergoing apoptosis, both DNMT1 and DNMT3A are significantly elevated. When the cells are induced to undergo apoptosis, inhibiting DNMTs pharmacologically reduces cell death, supporting the hypothesis that DNMTs can drive apoptosis in neuronal cell culture. Spinal cord lesions in mice also upregulate DNMT1 and DNMT3A, with DNMT3A expression localizing in mitochondria and spinal neuron synapses, and 5-mC is also upregulated upon motor neuron apoptosis. Collectively, these reports highlight a possible role for DNMTs in neuronal death occurring in ALS.
In summary, understanding C9ORF72 methylation mechanisms, contexts and regulation is an area of ALS research that should continue to be deeply explored. Judging from previously published results, it seems that epigenetic analyses of spinal tissue may yield more insight than that of blood samples. Finally, it is clear that the dynamics of DNMTs in motor neuron apoptosis is an area that should be further exploited for potential therapeutic targets against motor neuron loss.
4.4 Multiple Sclerosis
Multiple sclerosis (MS) is a chronic inflammatory disease that attacks the central nervous system (CNS) and is the most common cause of neurological disability in younger adults (Smith and McDonald 1999; Compston and Coles 2002; Grigoriadis et al. 2015). MS pathology begins as inflammatory lesions that cause demyelination of neuronal axons, and eventual transection and degeneration of axons (Trapp et al. 1998; Trapp and Nave 2008; Kraft and Wessman 1974). Because MS affects axonal conduction signals throughout the CNS, the symptoms are broad and include numbness, tingling, spasticity, fatigue/weakness, difficulty walking, vision problems, sexual dysfunction and pain (Samkoff and Goodman 2011; Schwendimann 2006). MS is diagnosed by several lesions in the central nervous system (CNS) that are separated by time and space, which are typically observed using MRI (De Angelis et al. 2019). This disease has “silent symptoms,” as evidenced by the development of many new lesions that are seen using MRI, with no simultaneous change in clinical symptoms (Isaac et al. 1988). The general course of MS has two stages: (1) relapsing and remitting MS (RRMS) and secondary progressive MS (SPSS). RRMS is characterized by bouts of symptoms (relapses; that can last several months and occur several times a year) followed by a period of recovery, in which remyelination, inflammation reduction, or compensatory mechanisms to axonal function may occur (Smith and McDonald 1999). Fewer than 10–20% of individuals with MS do not follow this typical course, but instead have primary progressive MS (PPMS) without the prolonged bouts of RRMS (Lublin and Reingold 1996; D’Amico et al. 2018). The age of onset for most MS cases is in the 20s and 30s (later for individuals with PPMS) and the symptom course and disease progression can be quite variable. The RRMS phase can last 8–20 years, but typically 50% of individuals with MS are unable to walk 25 years after disease onset (Trapp and Nave 2008).
Unlike the previously discussed neurodegenerative diseases, the heritability of MS is estimated to be 10–20 times greater for siblings of an individual with MS as compared to the general population (Didonna and Jorge 2017; Sadovnick and Baird 1988). The primary genetic risk factor has been localized to the major histocompatibility complex (MHC) region on chromosome 6p21.3 (Sawcer et al. 2005). This region encompasses 160 genes, most of which are involved in the immune response (Kalman and Lublin 1999). The human leukocyte antigen (HLA) loci within this region is the largest known MS susceptibility factor (Kalman and Lublin 1999; Isobe et al. 2016). Specific HLA haplotypes associate with age of disease onset and gender in MS patients (Hensiek et al. 2002; Moutsianas et al. 2015).
A majority of methylation research in MS has been conducted on blood samples, which is important given that MS may be initiated by circulating inflammatory factors that infiltrate the blood brain barrier (Frischer et al. 2009; Dendrou et al. 2015). Methylation patterns vary depending on the blood fraction analyzed [e.g., whole blood, peripheral mononuclear blood cells (PBMCs; containing a mixture of lymphocytes and leukocytes) or specific sub-groups of lymphocytes (Li et al. 2017a)]. Therefore, the scope and specificity of methylation analyses in blood cells is key to forming accurate conclusions. A good illustration of this is the interleukin 2 receptor subunit a (IL2RA) gene, which is elevated in PBMCs of MS patients, but shows no change in promoter methylation (Field et al. 2017). However, both IL2RA expression and promoter methylation changes are apparent when analyzing isolated T-cells. In PBMCs of MS patients, 5-hmC is decreased globally (Calabrese et al. 2014). Consistent with this, TET2 is downregulated in PBMCs (as is DNMT1), and both of these genes show aberrant promoter methylation in MS patients (Calabrese et al. 2014). PBMCs from individuals with the less common PPMS form of the disease show more differentially methylated sites than do individuals with RRMS (Kulakova et al. 2016). Alu and LINE-1 repetitive elements are hypermethylated in whole blood from MS patients, and this methylation level correlates with a more severe disability score (Neven et al. 2016). In the monocyte fraction of the blood, 19 hypomethylated CpG sites have been found in the exon 2 of the HLA DRB1*15:01 loci of MS individuals (Kular et al. 2018). Interestingly, individuals with MS that carried two copies of this allele showed less methylation of HLA DRB1*15:01 than those with one or no copies of the allele (Kular et al. 2018). When analyzing just CD8+ T cells from patients in the RRMS disease phase, significantly more hypermethylated CpG sites were observed as compared to controls (Bos et al. 2015).
The methylation profile of CD8+ T cells is starkly different from that of CD4+ T cells, with very little overlap (which is not surprising given the different functions of these two cell types in immunity (Maltby et al. 2015; Bos et al. 2015). CD4+ T cells show differentially methylated regions in the HLA-DRB loci of individuals with RRMS (Graves et al. 2014), a finding which has been replicated several times by the same group (Maltby et al. 2017). In MS patients taking the anti-inflammatory drug dimethyl fumarate, CD4+ T cells show over 900 differentially methylated positions, most of which are hypermethylated, as compared to MS patients not taking the drug (Maltby et al. 2018). The question has been posed whether methylation status of certain genes could be used as a distinguishing factor between MS individuals who were relapsing versus MS individuals who were in remittance. Eight genes were analyzed that were involved in CD4+/CD8+ T cell differentiation, oligodendrocyte differentiation or neuroinflammation (Sokratous et al. 2018). However, methylation status differed only between MS versus healthy individuals on the genes runt-related transcription factor 3 (RUNX3; which regulates CD4/CD8 T cell differentiation) and cyclin-dependent kinase inhibitor 2A (CDKN2A; which is indirectly involved in the regulation of oligodendrocyte apoptosis).
Oligodendrocytes, which make up the myelin sheath around neuronal axons, become repeatedly destroyed and regenerated during RRMS (Lee et al. 2015). DNA methylation is known to be a critical component of oligodendrocyte proliferation in brain development (Moyon and Casaccia 2017), but it is also important to the differentiation of oligodendrocyte progenitors after demyelination has occurred in the adult brain (Moyon et al. 2017). 5-mC levels are elevated in oligodendrocytes during remyelination of mice that have been given spinal cord lesions (Moyon et al. 2017). Using an inducible knockout strategy in mice, this same study identified Dnmt1 as being critical to the early phase of remyelination in lesioned mice, and Dnmt3a critical to the later phase. An analysis of lesion-free regions of MS brains showed hypermethylation and reduced expression of genes Bcl2-like protein 2 (BCL2L2) and n-myc downstream-regulated gene 1 (NDRG1), which play a role in the survival of oligodendrocytes (Huynh et al. 2014).
Environmental factors such as vitamin D deficiency and smoking also confer major risk in MS development (Michel 2018). T cells from RRMS patients show hypermethylation of an alternative promoter at exon 1C of the vitamin D receptor (VDR) gene, which surprisingly is associated with upregulation of the receptor, rather than the expected downregulation typically associated with promoter methylation (Ayuso et al. 2017). Another known environmental MS risk factor, smoking, is known to alter methylation patterns and does so most strongly in those individuals who are HLA haplotype carriers (Marabita et al. 2017). It is possible that the effect of smoking load on methylation may be increased in MS individuals, although the evidence for this is not robust (Marabita et al. 2017).
In conclusion, MS is a disease characterized by complex changes in the immune system, both in the periphery and the brain. DNA methylation should be a major focus of MS research, given the strong environmental risk factors that are already known to cause aberrant gene methylation. The focus on methylation in blood cells is important to MS research, given the myriad of circulating immune factors, and that many of these factors can infiltrate the brain in MS. The number of potential questions that can be asked regarding the role of methylation in MS pathology are limitless, and the continuous published research in this area is reflective of that. Understanding the methylation dynamics in inflammatory factors of AD patients will also greatly affect our understanding of other diseases that have major inflammatory components.
4.5 Huntington’s Disease
Huntington’s disease (HD) is the most common monogenic neurological disease and is caused by an autosomal dominant expanded repeat of the trinucleotide CAG in the huntingtin (HTT) gene (Sun et al. 2017; Gusella and MacDonald 1995). This expansion results in an excessively long polyglutamine sequence on the N-terminus of the HTT protein, which becomes fragmented and forms nuclear aggregates, conferring neuronal toxicity (DiFiglia et al. 1997). The primary clinical symptoms of HD are motor dysfunction (specifically involuntary movements and impaired voluntary movements), cognitive impairments, and frequent psychiatric comorbidities (Ghosh and Tabrizi 2018; Roos 2010). Individuals with HD have between 35 and 55 copies of the repeat (Bates et al. 2015). Greater than 55 CAG copies results in juvenile HD, in which the disease develops before the age of 20, and is almost always inherited through the father (Farrer et al. 1992; Gordon 2003). Typical disease diagnosis occurs between 30 and 50 years of age (Roos 2010). Medium spiny GABAergic neurons in the striatum (a part of the basal ganglia) are particularly vulnerable to the disease (Sieradzan and Mann 2001), especially those expressing adenosine A2A receptors (A2AR), which show alterations before symptom onset (Glass et al. 2000; Orru et al. 2011). HD is a progressive and fatal disease (Bates et al. 2015).
While the cause of HD is known to be completely genetic, understanding methylation patterns that correspond to neuronal changes in the HD brain could still be advantageous to therapeutic development. Global hydroxymethylation dynamics have been studied at various ages of an HD mouse model. In the YAC128 transgenic mouse (which contains 128 CAG repeats under control of human endogenous regulatory elements), deep sequencing of 5-hmC-enriched DNA showed no differences in the overall 5-hmC landscape in the striatum and cortex (Wang et al. 2013a). However, there were 747 differentially hydroxymethylated regions (DhMRs; mostly downregulated) that were related to gene pathways such as GABA/glutamate receptor signaling, synaptic long-term potentiation and axonal guidance signaling. Importantly, global 5-hmC was reduced in YAC128 mice (compared to wildtype) by 6 weeks of age, which is well before the onset of disease pathology. Although still lower than wildtype mice, hydroxymethylation in YAC128 mice shows an age-dependent increase until 3 months, in the striatum and hippocampus (Wang et al. 2013a). Thus, decreases in 5-hmC may be potentially useful as an HD biomarker. Surprisingly, despite lower 5-hmC observed in 3-month old HD mice, age-associated reductions in heterochromatin are enhanced in more aged YAC128 mice (Park et al. 2017).
The reports on gene-specific methylation changes in HD are underwhelming. An analysis of whole blood methylation between symptomatic HD, presymptomatic HD and non-HD individuals did not yield robust findings, perhaps due to lack of power (Zadel et al. 2018). Other studies have been consistent with this, in that no differential methylation has been observed in the blood (Hamzeiy et al. 2018) nor the cortex of HD individuals as compared to control (De Souza et al. 2016). An in vitro study of striatal cells expressing polyglutamine-expanded HTT identified a number of differentially methylated regions (most of which were low in CpG sites), using reduced representation bisulfite sequencing (Ng et al. 2013). Only SRY-box 2 (SOX2), PAX6 and nestin (NES) (all involved in neurogenesis and differentiation) showed increased methylation of regulatory regions as well as reduced expression. The D4S95 loci is closely linked to the HTT gene, and methylation of this loci shows a high degree of variability (Wasmuth et al. 1988; Pritchard et al. 1989). It has been proposed to use the methylation status of D4S95 as a predictor of HD (Theilmann et al. 1989), although no further literature on this has been found. Another study has examined whether D4S95 methylation correlates with age of onset, but discovered that it only correlates with age, in general (Reik et al. 1993).
Humans with HD, even at the earliest stage of the disease, show a decrease in adenosine A2A receptor (A2AR) protein in the putamen, a part of the striatum. Consistently, an increase of methylation at exon 1e of the A 2A R gene was observed and validated using MeDIP and bisulfite sequencing in these tissues (Villar-Menendez et al. 2013). These findings were recapitulated in two mouse models of HD, the R/61 mouse (containing 145 CAG repeats in the HTT gene) and the R6/2 mouse (115 repeats). Although no change in A 2A R promoter methylation was observed in either mouse model, exon m2 showed reduced methylation in 30-week old R6/1 mice, but not in the younger diseased mice. In the R6/2 mice, exon 2 showed reduced hydroxymethylation.
Methylation may also regulate DNA damage and repair in HD. In patient-derived HD cell lines, the APEX1 gene (involved in DNA repair) is downregulated and shows promoter hypermethylation as compared to control cells (Mollica et al. 2016). Inhibiting DNA methyltransferases rescues APEX1 gene expression and further, induces stability of the trinucleotide repeats in dividing cells. Similar to individuals with ALS, individuals with HD also show an accelerated epigenetic aging in the frontal and parietal lobes and cingulate cortex regions of the brain (Horvath et al. 2016).
Overall, there does not seem to be a huge consensus in the literature regarding a specific role for DNA methylation or hydroxymethylation in HD. Because HD has a known monogenic cause, the emphasis on methylation research in this area is not as strong as for other neurodegenerative diseases that are strongly affected by environmental factors. Nonetheless, 5-hmC may be a likely biomarker to target in order to achieve an early diagnosis (Table 1).
5 Conclusions and Future Research
Neurodegenerative diseases vary greatly in their pathologies, course of progression and impact on quality of life, although each is devastating in its own way. While the cause of Huntington’s disease may be clearly known, the instigating factors for other diseases have not been identified. Further, even though we understand that β-amyloid is responsible for creating much of the pathology in AD, it is not clear what causes β-amyloid dysfunction, or how to delay or prevent it. Unraveling the DNA methylation and hydroxymethylation dynamics in neurodegenerative diseases adds another dimension of possible answers regarding causal, mediating, and symptomatic factors in Alzheimer’s and other diseases.
A major focus in neurodegenerative disease research is the use of new models that can produce results that are more translatable to humans. The majority of non-human neurodegenerative research to date has relied on mouse models. Despite the strong genetic similarity between mice and humans, therapies that are effective in mouse models often fail to show the same effect in human patients, especially for Alzheimer’s disease (Li et al. 2013; Anand et al. 2017; Godyn et al. 2016). Even though there are conserved epigenetic loci between mice and humans that associate with Alzheimer’s disease (Sanchez-Mut et al. 2013; Gjoneska et al. 2015), there are apparently other unknown significant differences that prevent drug efficacy from translating to humans. One contributing factor is that mouse models of Alzheimer’s disease are unable to entirely recapitulate the disease pathology, typically only exhibiting several but not all major presentations (Van Dam and De Deyn 2011; Li et al. 2013; Esquerda-Canals et al. 2017). Performing studies on tissue that is genetically human (either human tissue sample or human-derived iPSCs) has become a key focus. Brain organoids (small spheroids of neurons that harbor properties of various brain regions) grown from iPSCs will be of great significance to the field of neurodegenerative disease (Wang 2018). These 3-dimensional organoids can be cultured from human fibroblasts (either healthy or diseased humans), and can provide more complex neuronal material than standard cell culture. In another example, the pig has recently been highlighted as a more relevant model of Huntington’s disease as compared to smaller mammals, such as mice (Yan et al. 2018). Transgenic HD pigs display a more severe Huntington’s disease phenotype than do mice, and exhibit other HD symptoms such as breathing difficulties. These new models may yield greater translatability into effective therapies for humans.
This review has highlighted several key findings. First is the novel finding that amyotrophic lateral sclerosis and Huntington’s disease show accelerated epigenetic age (DNAm age). Because many of the age-related brain changes are similar to neurodegenerative alterations (albeit at a milder level), it is critical that we understand the factors that can switch the normal progression of brain aging into neurodegenerative disease. DNAm age would be an important analysis to carry out for all of the neurodegenerative diseases, and not just ALS and HD. Secondly, there is a unique interaction between methylation and the primary Parkinson’s disease drug, l-DOPA, in that the drug’s metabolism requires a methyl donor from SAM and thus impacts endogenous DNA methylation dynamics. This interaction could be further explored to develop secondary drugs that may enhance the efficacy of l-DOPA for PD symptoms. Third, given the strong risk that environmental factors like vitamin D deficiency and smoking confer upon the development of multiple sclerosis, aberrant de novo methylation and the stability of that methylation, should be thoroughly investigated with regards to these factors.
In summary, exploring the roles of DNA methylation and hydroxymethylation in neurodegenerative diseases provides another dimension through which more complex questions regarding mechanism can be addressed, and especially it can supply us with insight into how environmental factors can predispose certain individuals to such diseases.
References
Adams I (1987) Comparison of synaptic changes in the precentral and postcentral cerebral cortex of aging humans: a quantitative ultrastructural study. Neurobiol Aging 8:203–212
Ahmeti KB, Ajroud-Driss S, Al-Chalabi A et al (2013) Age of onset of amyotrophic lateral sclerosis is modulated by a locus on 1p34.1. Neurobiol Aging 34:357 e357–357 e319
Ajroud-Driss S, Siddique T (2015) Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta 1852:679–684
Albers JA, Chand P, Anch AM (2017) Multifactorial sleep disturbance in Parkinson’s disease. Sleep Med 35:41–48
Anand A, Patience AA, Sharma N et al (2017) The present and future of pharmacotherapy of Alzheimer’s disease: a comprehensive review. Eur J Pharmacol 815:364–375
Ayuso T, Aznar P, Soriano L et al (2017) Vitamin D receptor gene is epigenetically altered and transcriptionally up-regulated in multiple sclerosis. PLoS One 12:e0174726
Baker DJ, Petersen RC (2018) Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest 128:1208–1216
Balendra R, Jones A, Jivraj N et al (2014) Estimating clinical stage of amyotrophic lateral sclerosis from the ALS Functional Rating Scale. Amyotroph Lateral Scler Frontotemporal Degener 15:279–284
Ballestar E, Wolffe AP (2001) Methyl-CpG-binding proteins. Targeting specific gene repression. Eur J Biochem 268:1–6
Bashtrykov P, Jankevicius G, Smarandache A et al (2012) Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem Biol 19:572–578
Bates GP, Dorsey R, Gusella JF et al (2015) Huntington disease. Nat Rev Dis Primers 1:15005
Bekaert S, De Meyer T, Van Oostveldt P (2005) Telomere attrition as ageing biomarker. Anticancer Res 25:3011–3021
Bertram L, Tanzi RE (2005) The genetic epidemiology of neurodegenerative disease. J Clin Invest 115:1449–1457
Bihaqi SW, Schumacher A, Maloney B et al (2012) Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): towards a new paradigm. Curr Alzheimer Res 9:574–588
Billingsley KJ, Bandres-Ciga S, Saez-Atienzar S et al (2018) Genetic risk factors in Parkinson’s disease. Cell Tissue Res 373:9–20
Biniszkiewicz D, Gribnau J, Ramsahoye B et al (2002) Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol 22:2124–2135
Bogdanovic O, Veenstra GJ (2009) DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma 118:549–565
Bos SD, Page CM, Andreassen BK et al (2015) Genome-wide DNA methylation profiles indicate CD8+ T cell hypermethylation in multiple sclerosis. PLoS One 10:e0117403
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Ghebremedhin E, Rub U et al (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318:121–134
Brandeis M, Ariel M, Cedar H (1993) Dynamics of DNA methylation during development. Bioessays 15:709–713
Burre J, Sharma M, Tsetsenis T et al (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667
Cadena-del-Castillo C, Valdes-Quezada C, Carmona-Aldana F et al (2014) Age-dependent increment of hydroxymethylation in the brain cortex in the triple-transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 41:845–854
Calabrese R, Valentini E, Ciccarone F et al (2014) TET2 gene expression and 5-hydroxymethylcytosine level in multiple sclerosis peripheral blood cells. Biochim Biophys Acta 1842:1130–1136
Calderon-Garciduenas ALD, Charles (2017) Alzheimer disease. In: Kovacs GA, Alafuzoff I (ed) Neuropathology, vol 145. Handbook of clinical neurology, vol 3rd series. Elsevier, Virginia Commonwealth University
Carlson LL, Page AW, Bestor TH (1992) Properties and localization of DNA methyltransferase in preimplantation mouse embryos: implications for genomic imprinting. Genes Dev 6:2536–2541
Charlton CG, Crowell B Jr (1995) Striatal dopamine depletion, tremors, and hypokinesia following the intracranial injection of S-adenosylmethionine: a possible role of hypermethylation in parkinsonism. Mol Chem Neuropathol 26:269–284
Charlton CG, Mack J (1994) Substantia nigra degeneration and tyrosine hydroxylase depletion caused by excess S-adenosylmethionine in the rat brain. Support for an excess methylation hypothesis for parkinsonism. Mol Neurobiol 9:149–161
Chen KL, Wang SS, Yang YY et al (2009) The epigenetic effects of amyloid-beta(1-40) on global DNA and neprilysin genes in murine cerebral endothelial cells. Biochem Biophys Res Commun 378:57–61
Chen H, Dzitoyeva S, Manev H (2012) Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor Neurol Neurosci 30:237–245
Chen X, Xiao Y, Wei L et al (2017) Association of DNMT3b gene variants with sporadic Parkinson’s disease in a Chinese Han population. J Gene Med 19:360–365
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67:715–725
Chestnut BA, Chang Q, Price A et al (2011) Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci 31:16619–16636
Chio A, Pagani M, Agosta F et al (2014) Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol 13:1228–1240
Cho HJ, Liu G, Jin SM et al (2013) MicroRNA-205 regulates the expression of Parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum Mol Genet 22:608–620
Chouliaras L, Mastroeni D, Delvaux E et al (2013) Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging 34:2091–2099
Choy JS, Wei S, Lee JY et al (2010) DNA methylation increases nucleosome compaction and rigidity. J Am Chem Soc 132:1782–1783
Collins AL, Levenson JM, Vilaythong AP et al (2004) Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet 13:2679–2689
Compston A, Coles A (2002) Multiple sclerosis. Lancet 359:1221–1231
Condliffe D, Wong A, Troakes C et al (2014) Cross-region reduction in 5-hydroxymethylcytosine in Alzheimer’s disease brain. Neurobiol Aging 35:1850–1854
Condorelli DF, Dell’Albani P, Conticello SG et al (1997) A neural-specific hypomethylated domain in the 5' flanking region of the glial fibrillary acidic protein gene. Dev Neurosci 19:446–456
Coppede F, Stoccoro A, Mosca L et al (2018) Increase in DNA methylation in patients with amyotrophic lateral sclerosis carriers of not fully penetrant SOD1 mutations. Amyotroph Lateral Scler Frontotemporal Degener 19:93–101
Coppieters N, Dieriks BV, Lill C et al (2014) Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain. Neurobiol Aging 35:1334–1344
Coupland KG, Mellick GD, Silburn PA et al (2014) DNA methylation of the MAPT gene in Parkinson’s disease cohorts and modulation by vitamin E in vitro. Mov Disord 29:1606–1614
Cribbs DH, Berchtold NC, Perreau V et al (2012) Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation 9:179
D’Amico E, Patti F, Zanghi A et al (2018) Late-onset and young-onset relapsing-remitting multiple sclerosis: evidence from a retrospective long-term follow-up study. Eur J Neurol 25(12):1425–1431
De Angelis F, Brownlee WJ, Chard DT et al (2019) New MS diagnostic criteria in practice. Pract Neurol 19(1):64–67
De Jager PL, Srivastava G, Lunnon K et al (2014) Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 17:1156–1163
de Lau LM, Breteler MM (2006) Epidemiology of Parkinson’s disease. Lancet Neurol 5:525–535
De Souza RA, Islam SA, McEwen LM et al (2016) DNA methylation profiling in human Huntington’s disease brain. Hum Mol Genet 25:2013–2030
DeJesus-Hernandez M, Mackenzie IR, Boeve BF et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256
DeKosky ST, Marek K (2003) Looking backward to move forward: early detection of neurodegenerative disorders. Science 302:830–834
Dendrou CA, Fugger L, Friese MA (2015) Immunopathology of multiple sclerosis. Nat Rev Immunol 15:545–558
Desplats P, Spencer B, Coffee E et al (2011) Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem 286:9031–9037
Didonna AO, Jorge R (2017) The genetics of multiple sclerosis. In: Zagon IS, McLaughlin PJ (eds) Multiple sclerosis: perspectives in treatment and pathogenesis. Codon Publications, Brisbane (AU)
DiFiglia M, Sapp E, Chase KO et al (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277:1990–1993
Do Carmo S, Hanzel CE, Jacobs ML et al (2016) Rescue of early bace-1 and global DNA demethylation by S-adenosylmethionine reduces amyloid pathology and improves cognition in an Alzheimer’s model. Sci Rep 6:34051
Doskocil J, Sorm F (1962) Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim Biophys Acta 55:953–959
Dosunmu R, Wu J, Basha MR et al (2007) Environmental and dietary risk factors in Alzheimer’s disease. Expert Rev Neurother 7:887–900
Droge W, Schipper HM (2007) Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell 6:361–370
Edwards TL, Scott WK, Almonte C et al (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74:97–109
Ehrlich M, Gama-Sosa MA, Huang LH et al (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10:2709–2721
Erro R, Stamelou M (2017) The motor syndrome of Parkinson’s disease. Int Rev Neurobiol 132:25–32
Esanov R, Belle KC, van Blitterswijk M et al (2016) C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp Neurol 277:171–177
Esquerda-Canals G, Montoliu-Gaya L, Guell-Bosch J et al (2017) Mouse models of Alzheimer’s disease. J Alzheimers Dis 57:1171–1183
Fan G, Martinowich K, Chin MH et al (2005) DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 132:3345–3356
Farrer LA, Cupples LA, Kiely DK et al (1992) Inverse relationship between age at onset of Huntington disease and paternal age suggests involvement of genetic imprinting. Am J Hum Genet 50:528–535
Fasolino M, Zhou Z (2017) The crucial role of DNA methylation and MeCP2 in neuronal function. Genes (Basel) 8
Feng J, Zhou Y, Campbell SL et al (2010) Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci 13:423–430
Ficz G, Branco MR, Seisenberger S et al (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473:398–402
Field J, Fox A, Jordan MA et al (2017) Interleukin-2 receptor-alpha proximal promoter hypomethylation is associated with multiple sclerosis. Genes Immun 18:59–66
Figueroa-Romero C, Hur J, Bender DE et al (2012) Identification of epigenetically altered genes in sporadic amyotrophic lateral sclerosis. PLoS One 7:e52672
Frischer JM, Bramow S, Dal-Bianco A et al (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132:1175–1189
Fuks F, Hurd PJ, Wolf D et al (2003) The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem 278:4035–4040
Gan W, Nie B, Shi F et al (2012) Age-dependent increases in the oxidative damage of DNA, RNA, and their metabolites in normal and senescence-accelerated mice analyzed by LC-MS/MS: urinary 8-oxoguanosine as a novel biomarker of aging. Free Radic Biol Med 52:1700–1707
Gant JC, Sama MM, Landfield PW et al (2006) Early and simultaneous emergence of multiple hippocampal biomarkers of aging is mediated by Ca2+-induced Ca2+ release. J Neurosci 26:3482–3490
Ghosh R, Tabrizi SJ (2018) Clinical features of Huntington’s disease. Adv Exp Med Biol 1049:1–28
Gibb WR, Lees AJ (1991) Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson’s disease. J Neurol Neurosurg Psychiatry 54:388–396
Gijselinck I, Van Mossevelde S, van der Zee J et al (2016) The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry 21:1112–1124
Gjoneska E, Pfenning AR, Mathys H et al (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518:365–369
Glass M, Dragunow M, Faull RL (2000) The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABA(A) receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience 97:505–519
Globisch D, Munzel M, Muller M et al (2010) Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5:e15367
Godyn J, Jonczyk J, Panek D et al (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68:127–138
Goedert M, Sisodia SS, Price DL (1991) Neurofibrillary tangles and beta-amyloid deposits in Alzheimer’s disease. Curr Opin Neurobiol 1:441–447
Goetz CG (2000) Amyotrophic lateral sclerosis: early contributions of Jean-Martin Charcot. Muscle Nerve 23:336–343
Golshani P, Hutnick L, Schweizer F et al (2005) Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat Syst 3:227–233
Gontier G, Iyer M, Shea JM et al (2018) Tet2 rescues age-related regenerative decline and enhances cognitive function in the adult mouse brain. Cell Rep 22:1974–1981
Gonzalo S, Jaco I, Fraga MF et al (2006) DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8:416–424
Gordon N (2003) Huntington’s disease of early onset or juvenile Huntington’s disease. Hosp Med 64:576–580
Gorevic PD, Goni F, Pons-Estel B et al (1986) Isolation and partial characterization of neurofibrillary tangles and amyloid plaque core in Alzheimer’s disease: immunohistological studies. J Neuropathol Exp Neurol 45:647–664
Goto K, Numata M, Komura JI et al (1994) Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation 56:39–44
Govoni V, Della Coletta E, Cesnik E et al (2017) Can the age at onset give a clue to the pathogenesis of ALS? Acta Neurol Belg 117:221–227
Graves MC, Benton M, Lea RA et al (2014) Methylation differences at the HLA-DRB1 locus in CD4+ T-Cells are associated with multiple sclerosis. Mult Scler 20:1033–1041
Grigoriadis N, van Pesch V, Paradig MSG (2015) A basic overview of multiple sclerosis immunopathology. Eur J Neurol 22(Suppl 2):3–13
Grinan-Ferre C, Sarroca S, Ivanova A et al (2016) Epigenetic mechanisms underlying cognitive impairment and Alzheimer disease hallmarks in 5XFAD mice. Aging (Albany NY) 8:664–684
Guerreiro R, Wojtas A, Bras J et al (2013) TREM2 variants in Alzheimer’s disease. N Engl J Med 368:117–127
Guhathakurta S, Bok E, Evangelista BA et al (2017a) Deregulation of alpha-synuclein in Parkinson’s disease: Insight from epigenetic structure and transcriptional regulation of SNCA. Prog Neurobiol 154:21–36
Guhathakurta S, Evangelista BA, Ghosh S et al (2017b) Hypomethylation of intron1 of alpha-synuclein gene does not correlate with Parkinson’s disease. Mol Brain 10:6
Guo JU, Ma DK, Mo H et al (2011) Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 14:1345–1351
Guo JU, Su Y, Shin JH et al (2014) Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17:215–222
Gusella JF, MacDonald ME (1995) Huntington’s disease. Semin Cell Biol 6:21–28
Hadad N, Masser DR, Logan S et al (2016) Absence of genomic hypomethylation or regulation of cytosine-modifying enzymes with aging in male and female mice. Epigenetics Chromatin 9:30
Hahn MA, Qiu R, Wu X et al (2013) Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep 3:291–300
Hamerman JA, Jarjoura JR, Humphrey MB et al (2006) Cutting edge: inhibition of TLR and FcR responses in macrophages by triggering receptor expressed on myeloid cells (TREM)-2 and DAP12. J Immunol 177:2051–2055
Hamzeiy H, Savas D, Tunca C et al (2018) Elevated global DNA methylation is not exclusive to amyotrophic lateral sclerosis and is also observed in spinocerebellar ataxia types 1 and 2. Neurodegener Dis 18:38–48
Hardiman O, Al-Chalabi A, Chio A et al (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17085
Hendrich B, Guy J, Ramsahoye B et al (2001) Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev 15:710–723
Hensiek AE, Sawcer SJ, Feakes R et al (2002) HLA-DR 15 is associated with female sex and younger age at diagnosis in multiple sclerosis. J Neurol Neurosurg Psychiatry 72:184–187
Heston LL (1989) Family studies in Alzheimer’s disease. Prog Clin Biol Res 317:195–200
Hoal-van Helden EG, van Helden PD (1989) Age-related methylation changes in DNA may reflect the proliferative potential of organs. Mutat Res 219:263–266
Horvath S (2013) DNA methylation age of human tissues and cell types. Genome Biol 14:R115
Horvath S, Langfelder P, Kwak S et al (2016) Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY) 8:1485–1512
Hotchkiss RD (1948) The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem 175:315–332
Howell CY, Bestor TH, Ding F et al (2001) Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104:829–838
Hu L, Lu J, Cheng J et al (2015) Structural insight into substrate preference for TET-mediated oxidation. Nature 527:118–122
Huang Y, Chavez L, Chang X et al (2014) Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad Sci U S A 111:1361–1366
Hutnick LK, Golshani P, Namihira M et al (2009) DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet 18:2875–2888
Huynh JL, Garg P, Thin TH et al (2014) Epigenome-wide differences in pathology-free regions of multiple sclerosis-affected brains. Nat Neurosci 17:121–130
Ianov L, Riva A, Kumar A et al (2017) DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age-related cognitive impairment. Front Aging Neurosci 9:249
Inano K, Suetake I, Ueda T et al (2000) Maintenance-type DNA methyltransferase is highly expressed in post-mitotic neurons and localized in the cytoplasmic compartment. J Biochem 128:315–321
Ip JPK, Mellios N, Sur M (2018) Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci 19:368–382
Isaac C, Li DK, Genton M et al (1988) Multiple sclerosis: a serial study using MRI in relapsing patients. Neurology 38:1511–1515
Isobe N, Keshavan A, Gourraud PA et al (2016) Association of HLA genetic risk burden with disease phenotypes in multiple sclerosis. JAMA Neurol 73:795–802
Ito H, Usuda N, Atsuzawa K et al (2007) Phosphorylation by extracellular signal-regulated kinase of a multidomain adaptor protein, vinexin, at synapses. J Neurochem 100:545–554
Ito S, Shen L, Dai Q et al (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303
Jeltsch A, Jurkowska RZ (2014) New concepts in DNA methylation. Trends Biochem Sci 39:310–318
Jin SG, Zhang ZM, Dunwell TL et al (2016) Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep 14:493–505
Jintaridth P, Mutirangura A (2010) Distinctive patterns of age-dependent hypomethylation in interspersed repetitive sequences. Physiol Genomics 41:194–200
Jones PL, Veenstra GJ, Wade PA et al (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187–191
Jowaed A, Schmitt I, Kaut O et al (2010) Methylation regulates alpha-synuclein expression and is decreased in Parkinson's disease patients’ brains. J Neurosci 30:6355–6359
Jung BP, Zhang G, Nitsch R et al (2003) Differential expression of methyl CpG-binding domain containing factor MBD3 in the developing and adult rat brain. J Neurobiol 55:220–232
Kaas GA, Zhong C, Eason DE et al (2013) TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 79:1086–1093
Kalman B, Lublin FD (1999) The genetics of multiple sclerosis. A review. Biomed Pharmacother 53:358–370
Keshet I, Yisraeli J, Cedar H (1985) Effect of regional DNA methylation on gene expression. Proc Natl Acad Sci U S A 82:2560–2564
Ketelaar ME, Hofstra EM, Hayden MR (2012) What monozygotic twins discordant for phenotype illustrate about mechanisms influencing genetic forms of neurodegeneration. Clin Genet 81:325–333
Khare T, Pai S, Koncevicius K et al (2012) 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol 19:1037–1043
Kim M, Park YK, Kang TW et al (2014) Dynamic changes in DNA methylation and hydroxymethylation when hES cells undergo differentiation toward a neuronal lineage. Hum Mol Genet 23:657–667
Kim H, Jang WY, Kang MC et al (2016) TET1 contributes to neurogenesis onset time during fetal brain development in mice. Biochem Biophys Res Commun 471:437–443
Kinney SM, Chin HG, Vaisvila R et al (2011) Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J Biol Chem 286:24685–24693
Kraft AM, Wessman HC (1974) Pathology and etiology in multiple sclerosis: a review. Phys Ther 54:716–720
Kraus TF, Globisch D, Wagner M et al (2012) Low values of 5-hydroxymethylcytosine (5hmC), the “sixth base,” are associated with anaplasia in human brain tumors. Int J Cancer 131:1577–1590
Kraus TF, Guibourt V, Kretzschmar HA (2015) 5-Hydroxymethylcytosine, the “Sixth Base”, during brain development and ageing. J Neural Transm (Vienna) 122:1035–1043
Kremer EA, Gaur N, Lee MA et al (2018) Interplay between TETs and microRNAs in the adult brain for memory formation. Sci Rep 8:1678
Kriaucionis S, Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930
Kulakova OG, Kabilov MR, Danilova LV et al (2016) Whole-genome DNA methylation analysis of peripheral blood mononuclear cells in multiple sclerosis patients with different disease courses. Acta Nat 8:103–110
Kular L, Liu Y, Ruhrmann S et al (2018) DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis. Nat Commun 9:2397
Kumar S, Cheng X, Klimasauskas S et al (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res 22:1–10
Langner KD, Vardimon L, Renz D et al (1984) DNA methylation of three 5' C-C-G-G 3' sites in the promoter and 5' region inactivate the E2a gene of adenovirus type 2. Proc Natl Acad Sci U S A 81:2950–2954
Lardenoije R, van den Hove DLA, Havermans M et al (2018) Age-related epigenetic changes in hippocampal subregions of four animal models of Alzheimer’s disease. Mol Cell Neurosci 86:1–15
Lashley T, Gami P, Valizadeh N et al (2015) Alterations in global DNA methylation and hydroxymethylation are not detected in Alzheimer’s disease. Neuropathol Appl Neurobiol 41:497–506
Lauretti E, Di Meco A, Chu J et al (2015) Modulation of AD neuropathology and memory impairments by the isoprostane F2alpha is mediated by the thromboxane receptor. Neurobiol Aging 36:812–820
Lee JY, Biemond M, Petratos S (2015) Axonal degeneration in multiple sclerosis: defining therapeutic targets by identifying the causes of pathology. Neurodegener Dis Manag 5:527–548
Li W, Liu M (2011) Distribution of 5-hydroxymethylcytosine in different human tissues. J Nucleic Acids 2011:870726
Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926
Li C, Ebrahimi A, Schluesener H (2013) Drug pipeline in neurodegeneration based on transgenic mice models of Alzheimer’s disease. Ageing Res Rev 12:116–140
Li X, Wei W, Zhao QY et al (2014) Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A 111:7120–7125
Li X, Xiao B, Chen XS (2017a) DNA methylation: a new player in multiple sclerosis. Mol Neurobiol 54:4049–4059
Li X, Yao B, Chen L et al (2017b) Ten-eleven translocation 2 interacts with forkhead box O3 and regulates adult neurogenesis. Nat Commun 8:15903
Lill CM (2016) Genetics of Parkinson’s disease. Mol Cell Probes 30:386–396
Lin X, Parisiadou L, Gu XL et al (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64:807–827
Liu XX, Wilson K, Charlton CG (2000) Effects of L-dopa treatment on methylation in mouse brain: implications for the side effects of L-dopa. Life Sci 66:2277–2288
Liu EY, Russ J, Wu K et al (2014) C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol 128:525–541
Lublin FD, Reingold SC (1996) Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46:907–911
Maddox SA, Watts CS, Schafe GE (2014) DNA methyltransferase activity is required for memory-related neural plasticity in the lateral amygdala. Neurobiol Learn Mem 107:93–100
Maiti A, Drohat AC (2011) Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem 286:35334–35338
Maltby VE, Graves MC, Lea RA et al (2015) Genome-wide DNA methylation profiling of CD8+ T cells shows a distinct epigenetic signature to CD4+ T cells in multiple sclerosis patients. Clin Epigenetics 7:118
Maltby VE, Lea RA, Sanders KA et al (2017) Differential methylation at MHC in CD4(+) T cells is associated with multiple sclerosis independently of HLA-DRB1. Clin Epigenetics 9:71
Maltby VE, Lea RA, Ribbons KA et al (2018) DNA methylation changes in CD4(+) T cells isolated from multiple sclerosis patients on dimethyl fumarate. Mult Scler J Exp Transl Clin 4:2055217318787826
Mano T, Nagata K, Nonaka T et al (2017) Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc Natl Acad Sci U S A 114:E9645–E9654
Marabita F, Almgren M, Sjoholm LK et al (2017) Smoking induces DNA methylation changes in Multiple Sclerosis patients with exposure-response relationship. Sci Rep 7:14589
Martynoga B, Drechsel D, Guillemot F (2012) Molecular control of neurogenesis: a view from the mammalian cerebral cortex. Cold Spring Harb Perspect Biol 4
Masliah E, Dumaop W, Galasko D et al (2013) Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8:1030–1038
Mastroeni D, McKee A, Grover A et al (2009) Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One 4:e6617
Mastroeni D, Grover A, Delvaux E et al (2010) Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging 31:2025–2037
Matsumoto L, Takuma H, Tamaoka A et al (2010) CpG demethylation enhances alpha-synuclein expression and affects the pathogenesis of Parkinson’s disease. PLoS One 5:e15522
McGraw CM, Samaco RC, Zoghbi HY (2011) Adult neural function requires MeCP2. Science 333:186
McMillan CT, Russ J, Wood EM et al (2015) C9orf72 promoter hypermethylation is neuroprotective: neuroimaging and neuropathologic evidence. Neurology 84:1622–1630
Meadows JP, Guzman-Karlsson MC, Phillips S et al (2016) Dynamic DNA methylation regulates neuronal intrinsic membrane excitability. Sci Signal 9:ra83
Mehta P, Kaye W, Raymond J et al (2018) Prevalence of amyotrophic lateral sclerosis - United States, 2014. MMWR Morb Mortal Wkly Rep 67:216–218
Mellen M, Ayata P, Dewell S et al (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151:1417–1430
Messerschmidt DM, Knowles BB, Solter D (2014) DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev 28:812–828
Michel L (2018) Environmental factors in the development of multiple sclerosis. Rev Neurol (Paris) 174:372–377
Miller CA, Sweatt JD (2007) Covalent modification of DNA regulates memory formation. Neuron 53:857–869
Mink JW (1996) The basal ganglia: focused selection and inhibition of competing motor programs. Prog Neurobiol 50:381–425
Mo A, Mukamel EA, Davis FP et al (2015) Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86:1369–1384
Mollica PA, Reid JA, Ogle RC et al (2016) DNA methylation leads to DNA repair gene down-regulation and trinucleotide repeat expansion in patient-derived huntington disease cells. Am J Pathol 186:1967–1976
Morahan JM, Yu B, Trent RJ et al (2009) A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 10:418–429
Moutsianas L, Jostins L, Beecham AH et al (2015) Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet 47:1107–1113
Moyon S, Casaccia P (2017) DNA methylation in oligodendroglial cells during developmental myelination and in disease. Neurogenesis (Austin) 4:e1270381
Moyon S, Ma D, Huynh JL et al (2017) Efficient remyelination requires DNA methylation. eNeuro 4
Nan X, Ng HH, Johnson CA et al (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389
Neven KY, Piola M, Angelici L et al (2016) Repetitive element hypermethylation in multiple sclerosis patients. BMC Genet 17:84
Ng LJ, Cropley JE, Pickett HA et al (2009) Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res 37:1152–1159
Ng CW, Yildirim F, Yap YS et al (2013) Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci U S A 110:2354–2359
Nichol K, Pearson CE (2002) CpG methylation modifies the genetic stability of cloned repeat sequences. Genome Res 12:1246–1256
Norden DM, Godbout JP (2013) Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39:19–34
Oates N, Pamphlett R (2007) An epigenetic analysis of SOD1 and VEGF in ALS. Amyotroph Lateral Scler 8:83–86
Okano M, Xie S, Li E (1998) Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet 19:219–220
Okano M, Bell DW, Haber DA et al (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Orru M, Zanoveli JM, Quiroz C et al (2011) Functional changes in postsynaptic adenosine A(2A) receptors during early stages of a rat model of Huntington disease. Exp Neurol 232:76–80
Pandya JD, Royland JE, MacPhail RC et al (2016) Age- and brain region-specific differences in mitochondrial bioenergetics in Brown Norway rats. Neurobiol Aging 42:25–34
Park M, Min B, Jeon K et al (2017) Age-associated chromatin relaxation is enhanced in Huntington’s disease mice. Aging (Albany NY) 9:803–822
Penner MR, Parrish RR, Hoang LT et al (2016) Age-related changes in Egr1 transcription and DNA methylation within the hippocampus. Hippocampus 26:1008–1020
Pensold D, Zimmer G (2018) Single-cell transcriptomics reveals regulators of neuronal migration and maturation during brain development. J Exp Neurosci 12:1179069518760783
Pensold D, Symmank J, Hahn A et al (2017) The DNA methyltransferase 1 (DNMT1) controls the shape and dynamics of migrating POA-derived interneurons fated for the murine cerebral cortex. Cereb Cortex 27:5696–5714
Pezzi JC, de Bem CM, da Rocha TJ et al (2017) Association between DNA methyltransferase gene polymorphism and Parkinson’s disease. Neurosci Lett 639:146–150
Pihlstrom L, Berge V, Rengmark A et al (2015) Parkinson’s disease correlates with promoter methylation in the alpha-synuclein gene. Mov Disord 30:577–580
Pihlstrom L, Blauwendraat C, Cappelletti C et al (2018) A comprehensive analysis of SNCA-related genetic risk in sporadic parkinson disease. Ann Neurol 84:117–129
Poddar J, Pradhan M, Ganguly G et al (2018) Biochemical deficits and cognitive decline in brain aging: Intervention by dietary supplements. J Chem Neuroanat 95:70–80
Pohodich AE, Zoghbi HY (2015) Rett syndrome: disruption of epigenetic control of postnatal neurological functions. Hum Mol Genet 24:R10–R16
Pritchard CA, Cox DR, Myers RM (1989) Methylation at the Huntington disease-linked D4S95 locus. Am J Hum Genet 45:335–336
Reik W, Maher ER, Morrison PJ et al (1993) Age at onset in Huntington’s disease and methylation at D4S95. J Med Genet 30:185–188
Richter J, Appenzeller S, Ammerpohl O et al (2012) No evidence for differential methylation of alpha-synuclein in leukocyte DNA of Parkinson’s disease patients. Mov Disord 27:590–591
Rizvi S, Raza ST, Mahdi F (2014) Telomere length variations in aging and age-related diseases. Curr Aging Sci 7:161–167
Robertson KD, Uzvolgyi E, Liang G et al (1999) The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 27:2291–2298
Rodier F, Coppe JP, Patil CK et al (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11:973–979
Roos RA (2010) Huntington’s disease: a clinical review. Orphanet J Rare Dis 5:40
Roost MS, Slieker RC, Bialecka M et al (2017) DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat Commun 8:908
Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700
Rudenko A, Dawlaty MM, Seo J et al (2013) Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79:1109–1122
Ruzov A, Tsenkina Y, Serio A et al (2011) Lineage-specific distribution of high levels of genomic 5-hydroxymethylcytosine in mammalian development. Cell Res 21:1332–1342
Sadovnick AD, Baird PA (1988) The familial nature of multiple sclerosis: age-corrected empiric recurrence risks for children and siblings of patients. Neurology 38:990–991
Samkoff LM, Goodman AD (2011) Symptomatic management in multiple sclerosis. Neurol Clin 29:449–463
Samorajski T (1976) How the human brain responds to aging. J Am Geriatr Soc 24:4–11
Sanchez-Mut JV, Aso E, Panayotis N et al (2013) DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain 136:3018–3027
Sandler M (1972) L-dopa metabolism in man (with special reference to parkinsonism). Sci Basis Med Annu Rev:161–173
Sanosaka T, Imamura T, Hamazaki N et al (2017) DNA methylome analysis identifies transcription factor-based epigenomic signatures of multilineage competence in neural stem/progenitor cells. Cell Rep 20:2992–3003
Santiago M, Antunes C, Guedes M et al (2014) TET enzymes and DNA hydroxymethylation in neural development and function - how critical are they? Genomics 104:334–340
Sarraf SA, Stancheva I (2004) Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol Cell 15:595–605
Sasaki S, Maruyama S (1994) Synapse loss in anterior horn neurons in amyotrophic lateral sclerosis. Acta Neuropathol 88:222–227
Sawcer S, Ban M, Maranian M et al (2005) A high-density screen for linkage in multiple sclerosis. Am J Hum Genet 77:454–467
Scarpa S, Fuso A, D'Anselmi F et al (2003) Presenilin 1 gene silencing by S-adenosylmethionine: a treatment for Alzheimer disease? FEBS Lett 541:145–148
Scheltens P, Blennow K, Breteler MM et al (2016) Alzheimer’s disease. Lancet 388:505–517
Schiffer D, Cordera S, Cavalla P et al (1996) Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci 139(Suppl):27–33
Schmitt I, Kaut O, Khazneh H et al (2015) L-dopa increases alpha-synuclein DNA methylation in Parkinson’s disease patients in vivo and in vitro. Mov Disord 30:1794–1801
Schwendimann RN (2006) Treatment of symptoms in multiple sclerosis. Neurol Res 28:306–315
Sedelnikova OA, Horikawa I, Zimonjic DB et al (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6:168–170
Shatunov A, Mok K, Newhouse S et al (2010) Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol 9:986–994
Siegmund KD, Connor CM, Campan M et al (2007) DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2:e895
Sieradzan KA, Mann DM (2001) The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol Appl Neurobiol 27:1–21
Simon-Sanchez J, Schulte C, Bras JM et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Singer J, Roberts-Ems J, Riggs AD (1979) Methylation of mouse liver DNA studied by means of the restriction enzymes msp I and hpa II. Science 203:1019–1021
Smith KJ, McDonald WI (1999) The pathophysiology of multiple sclerosis: the mechanisms underlying the production of symptoms and the natural history of the disease. Philos Trans R Soc Lond Ser B Biol Sci 354:1649–1673
Smith AR, Smith RG, Condliffe D et al (2016) Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol Aging 47:35–40
Sokratous M, Dardiotis E, Bellou E et al (2018) CpG Island methylation patterns in relapsing-remitting multiple sclerosis. J Mol Neurosci 64:478–484
Song Y, Ding H, Yang J et al (2014) Pyrosequencing analysis of SNCA methylation levels in leukocytes from Parkinson’s disease patients. Neurosci Lett 569:85–88
Stewart KR, Veselovska L, Kelsey G (2016) Establishment and functions of DNA methylation in the germline. Epigenomics 8:1399–1413
Stroud H, Feng S, Morey Kinney S et al (2011) 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol 12:R54
Sun YM, Zhang YB, Wu ZY (2017) Huntington’s disease: relationship between phenotype and genotype. Mol Neurobiol 54:342–348
Sweatt JD (2016) Dynamic DNA methylation controls glutamate receptor trafficking and synaptic scaling. J Neurochem 137:312–330
Szulwach KE, Li X, Li Y et al (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 14:1607–1616
Szwagierczak A, Bultmann S, Schmidt CS et al (2010) Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res 38:e181
Tahiliani M, Koh KP, Shen Y et al (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324:930–935
Takizawa T, Nakashima K, Namihira M et al (2001) DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell 1:749–758
Tan YY, Wu L, Zhao ZB et al (2014) Methylation of alpha-synuclein and leucine-rich repeat kinase 2 in leukocyte DNA of Parkinson's disease patients. Parkinsonism Relat Disord 20:308–313
Tanzi RE (1999) A genetic dichotomy model for the inheritance of Alzheimer’s disease and common age-related disorders. J Clin Invest 104:1175–1179
Tanzi RE (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2
Tanzi RE, Kovacs DM, Kim TW et al (1996) The gene defects responsible for familial Alzheimer’s disease. Neurobiol Dis 3:159–168
Teter B, Rozovsky I, Krohn K et al (1996) Methylation of the glial fibrillary acidic protein gene shows novel biphasic changes during brain development. Glia 17:195–205
Theilmann JL, Robbins CA, Hayden MR (1989) Methylation at the D4S95 locus and predictive testing. Am J Hum Genet 45:477–479
Titova N, Qamar MA, Chaudhuri KR (2017) The nonmotor features of Parkinson’s disease. Int Rev Neurobiol 132:33–54
Trapp BD, Nave KA (2008) Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci 31:247–269
Trapp BD, Peterson J, Ransohoff RM et al (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285
Tremolizzo L, Messina P, Conti E et al (2014) Whole-blood global DNA methylation is increased in amyotrophic lateral sclerosis independently of age of onset. Amyotroph Lateral Scler Frontotemporal Degener 15:98–105
Turek-Plewa J, Jagodzinski PP (2005) The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett 10:631–647
Unnikrishnan A, Hadad N, Masser DR et al (2018) Revisiting the genomic hypomethylation hypothesis of aging. Ann N Y Acad Sci 1418:69–79
Uysal F, Ozturk S, Akkoyunlu G (2017) DNMT1, DNMT3A and DNMT3B proteins are differently expressed in mouse oocytes and early embryos. J Mol Histol 48:417–426
van Blitterswijk M, DeJesus-Hernandez M, Rademakers R (2012) How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Curr Opin Neurol 25:689–700
Van Dam D, De Deyn PP (2011) Animal models in the drug discovery pipeline for Alzheimer’s disease. Br J Pharmacol 164:1285–1300
van der Flier WM, Pijnenburg YA, Fox NC et al (2011) Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE varepsilon4 allele. Lancet Neurol 10:280–288
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509:439–446
Villar-Menendez I, Blanch M, Tyebji S et al (2013) Increased 5-methylcytosine and decreased 5-hydroxymethylcytosine levels are associated with reduced striatal A2AR levels in Huntington’s disease. NeuroMolecular Med 15:295–309
Waalwijk C, Flavell RA (1978) DNA methylation at a CCGG sequence in the large intron of the rabbit beta-globin gene: tissue-specific variations. Nucleic Acids Res 5:4631–4634
Wagner M, Steinbacher J, Kraus TF et al (2015) Age-dependent levels of 5-methyl-, 5-hydroxymethyl-, and 5-formylcytosine in human and mouse brain tissues. Angew Chem Int Ed Eng 54:12511–12514
Wakabayashi K, Takahashi H, Obata K et al (1992) Immunocytochemical localization of synaptic vesicle-specific protein in Lewy body-containing neurons in Parkinson’s disease. Neurosci Lett 138:237–240
Wakabayashi K, Hayashi S, Yoshimoto M et al (2000) NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol 99:14–20
Wang H (2018) Modeling neurological diseases with human brain organoids. Front Synaptic Neurosci 10:15
Wang F, Yang Y, Lin X et al (2013a) Genome-wide loss of 5-hmC is a novel epigenetic feature of Huntington’s disease. Hum Mol Genet 22:3641–3653
Wang Y, Wang X, Li R et al (2013b) A DNA methyltransferase inhibitor, 5-aza-2'-deoxycytidine, exacerbates neurotoxicity and upregulates Parkinson’s disease-related genes in dopaminergic neurons. CNS Neurosci Ther 19:183–190
Wasmuth JJ, Hewitt J, Smith B et al (1988) A highly polymorphic locus very tightly linked to the Huntington’s disease gene. Nature 332:734–736
Wei S, Hua HR, Chen QQ et al (2017) Dynamic changes in DNA demethylation in the tree shrew (Tupaia belangeri chinensis) brain during postnatal development and aging. Zool Res 38:96–102
Wen L, Li X, Yan L et al (2014) Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol 15:R49
West RL, Lee JM, Maroun LE (1995) Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci 6:141–146
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3
Wilson VL, Smith RA, Ma S et al (1987) Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem 262:9948–9951
Wu H, Coskun V, Tao J et al (2010) Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329:444–448
Xi Z, Yunusova Y, van Blitterswijk M et al (2014) Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology 83:1476–1478
Xie W, Barr CL, Kim A et al (2012) Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148:816–831
Yan S, Tu Z, Liu Z et al (2018) A huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington’s disease. Cell 173:989–1002, e1013
Yoder JA, Walsh CP, Bestor TH (1997) Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13:335–340
Young PE, Kum Jew S, Buckland ME et al (2017) Epigenetic differences between monozygotic twins discordant for amyotrophic lateral sclerosis (ALS) provide clues to disease pathogenesis. PLoS One 12:e0182638
Zadel M, Maver A, Kovanda A et al (2018) DNA methylation profiles in whole blood of Huntington’s disease patients. Front Neurol 9:655
Zhang RR, Cui QY, Murai K et al (2013) Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell 13:237–245
Zhang M, Xi Z, Ghani M et al (2016) Genetic and epigenetic study of ALS-discordant identical twins with double mutations in SOD1 and ARHGEF28. J Neurol Neurosurg Psychiatry 87:1268–1270
Zhao X, Ueba T, Christie BR et al (2003) Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A 100:6777–6782
Zhou D, Robertson KD (2016) Role of DNA methylation of genomic stability. In: Tollefsbol TO (ed) Translational epigenetics series. Elsevier, London
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bruggeman, E.C., Yao, B. (2019). DNA Methylation in Neuronal Development and Disease. In: Jurga, S., Barciszewski, J. (eds) The DNA, RNA, and Histone Methylomes. RNA Technologies. Springer, Cham. https://doi.org/10.1007/978-3-030-14792-1_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-14792-1_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14791-4
Online ISBN: 978-3-030-14792-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)