
Abstract
RNA is abundantly modified by a range of covalent modifications, collectively termed the epitranscriptome. Of these modifications, N6-methyladenosine (m6A) is the most prevalent internal chemical tag in eukaryotic mRNA. Being cotranscriptionally deposited, it regulates almost all aspects of mRNA’s lifetime including maturation into mRNA, stability, distribution and protein translation. While m6A is likely present in all developing and adult mammalian tissues, here we highlight its distribution and reported functions in the mammalian brain. Additionally, we describe its potential to act as an encoding mechanism for activity- and experience-dependent adaptation and memory-formation. Such alterations may be positive when adjusting to outer challenges or negative when involved in maladaptive processes of the brain such as in the development of psychopathologies.
Consequently, studying this layer of gene expression control in the brain, alongside posttranslational regulation of proteins and epigenetics may inform us as to the molecular mechanisms underlying normal and pathological behaviors. Unfortunately, measuring m6A levels, patterns and especially dynamics still poses a major technological challenge especially in such a complicated organ as the brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Epitranscriptome
- N6-methyladenosine
- m6A
- Methyltransferase
- Demethylase
- m6A reader
- Brain functions
- Psychiatric disorders
1 Introduction
Over 100 covalent base modifications have been found in all domains of life including prokaryotes and eukaryotes but also archaea and viruses. They appear on almost all types of RNA including mRNA, tRNA, rRNA and snRNA (Boccaletto et al. 2018). Although many of these modifications and their potential to post-transcriptionally regulate gene expression have been known since the 1960s and 1970s, the field—now known as epitranscriptomics—attracted little attention until recent technological developments. In mammals, the most diverse RNA species regarding modified nucleotides are tRNA and rRNA, while only a very limited set of modifications is present on mRNA. The most abundant internal modification in mammalian mRNA is N6-methyladenosine, abbreviated to m6A, but many others exist including N1-methyladenosine, abbreviated to m1A (Dominissini et al. 2016; Li et al. 2016), pseudouridine Ψ (Carlile et al. 2014; Schwartz et al. 2014a; Li et al. 2015), 5-methylcytosine m5C (Dubin and Taylor 1975; Squires et al. 2012), and A-to-I editing (Levanon et al. 2004; Li et al. 2009).
2 m6A mRNA Methylation
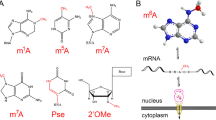
The biochemistry and cellular regulation of m6A has been described before in great detail here (Jia 2016) and elsewhere (e.g., Zhao et al. 2017). Internal mRNA m6A usually occurs in a fairly defined consensus motif DRm6ACH (with D = A, U or G; R = G or C; and H = A, U, C) (Wei et al. 1975; Schibler and Perry 1977). m6A is preferentially localized to the 3′UTR near the stop codon and in the 5′UTR of mRNAs, and to some degree, in the coding sequence (exon), the transcription start site (TSS) and in long internal exons (Dominissini et al. 2012; Meyer et al. 2012) (Fig. 1).

The m6A regulatory system including writers, erasers and readers; distribution of m6A on mRNA; and m6A reader proteins (adapted from Engel and Chen 2018)
Adenosine methylation at the N6-position, in contrast to e.g., N1-methylation in m1A, does not impair the Watson-Crick pairing with U but works via creating binding motifs increasing the accessibility for RNA-binding proteins (RBP; Liu et al. 2015, 2017), and modulating the mRNA secondary structure (Roost et al. 2015; Liu et al. 2015; Spitale et al. 2015).
The existence of both writer and eraser networks adding and removing m6A (as described in the following) has been widely accepted to indicate that m6A methylation is highly dynamic and a readily reversible system (Fig. 1).
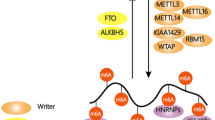
2.1 Writing m6A: The Methyltransferase Complex
A multiprotein methyltransferase complex transfers a methyl group from the donor substrate, S-adenosyl methionine, to the target RNA adenosines creating methylated adenosine (Bokar et al. 1994, 1997). This complex consists of two subunits with catalytic MT-A70 domains occurring in a heterodimer: METTL3 and METTL14 (Liu et al. 2014). Additional complex components include WTAP (Liu et al. 2014; Ping et al. 2014), KIAA1429 (Schwartz et al. 2014b), RBM15/B (Patil et al. 2016), and others, which enable target tethering and specificity as well as establishing the distinct nuclear localization pattern of the complex. Deposition of m6A likely occurs co-transcriptionally, i.e., on nascent pre-mRNA that is still tethered to genomic DNA (Slobodin et al. 2017; Ke et al. 2017). These latter studies however argue for rather static levels of m6A, i.e., conclude that once hnRNA has been released from the chromatin, m6A can only be removed by demethylation or mRNA decay. Posttranslational regulation of the methyltransferase proteins e.g., by phosphorylation has been described but may not necessarily regulate methylation activity per se (Schöller et al. 2018). Finally, conflicting roles for certain methyltransferase complex components aside from methylating nascent hnRNA or even those occurring in the cytoplasm have also been described but may be restricted to very special circumstances (Chen et al. 2015; Alarcón et al. 2015a; Lin et al. 2016).
2.2 Erasing m6A: FTO and ALKBH5
There are two known m6A demethylating enzymes enabling a potentially reversible and thus fully dynamic regulation of m6A: FTO (Jia et al. 2011) and ALKBH5 (Zheng et al. 2013). Interestingly, both were reported to have distinct subcellular and tissue distributions and thus potentially encode target- and tissue-specific regulation of m6A (Gerken et al. 2007; Vujovic et al. 2013; Zheng et al. 2013; Hess et al. 2013). Many reports even indicate that cellular regulation of e.g., FTO expression and activity is regulated thus enabling active regulation of m6A levels. However, recent results have dampened the original excitement over dynamic demethylation of m6A including the observation that FTO may preferentially demethylate a closely related and often co-detected modification, N6,2′-O-dimethyladenosine m6Am, in vitro and in vivo (Schwartz et al. 2014b; Linder et al. 2015; Mauer et al. 2017; Engel et al. 2018). These data suggest that reversibility of m6A may be less extensive than originally thought especially within physiological systems (Mauer et al. 2017; Mauer and Jaffrey 2018). Similar conclusions may be drawn from the fact that all known full mouse knockouts for FTO and ALKBH5 are, in contrast to all knockouts of the methyltransferases, viable after birth (Fischer et al. 2009; Zheng et al. 2013). In favor of active demethylation, localization of the enzymes and stoichiometry of m6A and m6Am, may allow FTO to target m6A in significant amounts in vivo with more recent data showing demethylation of all three mRNA methylated adenosines, m6A, m6Am, m1A, by FTO (Wei et al. 2018).
2.3 Readers of m6A
Given the wide abundance of m6A and even more diverse cellular functions of m6A (detailed below), a large part of functional specificity has to be achieved by the diverse range of m6A-interacting RBPs, the so called m6A readers. The most important family of m6A-readers consists of the YTH-domain-containing proteins, which bind directly to m6A. Currently known mammalian members of this family are YTHDF1-3 and YTHDC1-2. They have been assigned very diverse, often contradictory, yet sometimes cooperative cellular functions including promotion and inhibition of translation and decay (recently reviewed e.g., in Roundtree et al. 2017; Patil et al. 2018). The very diverse functions of the different YTH-family members may be regulated by several factors including cellular sub-localization, target-specificity and posttranslational regulation of the readers. Next to YTH-domain readers, m6A has also been reported to directly recruit eIF3 leading to a promotion of cap-independent translation (Meyer et al. 2015). Other proteins lacking a YTH domain, e.g., the hnRNP proteins HNRNPA2B1, HNRNPC, and HNRNPG (Liu et al. 2015, 2017; Alarcón et al. 2015a), may bind m6A instead via m6A-specific structural features. Finally, additional classes of direct binder proteins without a YTH-domain or m6A-specific structural features may exist including IGF2BP1–3 (Huang et al. 2018) and FMRP (Arguello et al. 2017; Edupuganti et al. 2017).
2.4 Cellular Functions of m6A
m6A cellular functions include the regulation of all stages of mRNA’s lifetime and thus establish a layer of secondary gene expression regulation (recently reviewed e.g., in Roundtree et al. 2017; Patil et al. 2018). Starting at the very beginning of mRNA’s life, m6A has been described to regulate the maturation of pre-mRNA into mature mRNA including 5′ capping, 3′ polyadenylation, splicing, nuclear processing and nuclear export of mRNAs. Thereby, m6A catalyzes differential splicing (Liu et al. 2015; Xiao et al. 2016; Ke et al. 2017) and differential polyA site usage (Ke et al. 2015; Molinie et al. 2016). m6A further promotes and also inhibits mRNA translation depending on the respective mRNA-m6A target and bound m6A-reader (Wang et al. 2015; Zhou et al. 2015; Meyer et al. 2015; Li et al. 2017a; Shi et al. 2017). Indicating the end of mRNA lifetime, methylation generally appears to accelerate mRNA decay (Wang et al. 2014) yet even this relationship is likely more complex than seen on first sight. m6A recognized by other effector proteins beyond the classical YTH-domain readers may have completely different effects on mRNA via third effector proteins. This includes the described interactions with ELAV-like RNA binding protein 1 (ELAV1/HuR) (Wang et al. 2014), intersections with miRNA biogenesis (Chen et al. 2015; Alarcón et al. 2015b), and interactions with the Toll-like receptor (TLR) family protein members TLR3 and TLR7 (Karikó et al. 2005).
3 m6A Distribution and Function in the Brain
The brain is one of the most complexly structured and regulated mammalian organs both during development and in adulthood. The adult brain especially, is a unique organ regarding gene expression regulation as it not only consists largely of postmitotic cells with very limited regeneration- and repair-capacity, but also because of the huge diversity and specialization of those cells. Thus, a mechanism of secondary gene expression regulation such as RNA methylation may be especially crucial in such a system.
Several studies have begun to uncover the functional significance of m6A regulation in the central nervous system (CNS) and its role in normal brain physiology during all stages of life from development to adulthood and encoding cellular plasticity in the adult brain. Thereby, m6A is abundant in the brain during all developmental stages with increasing levels during development (Meyer et al. 2012). In the adult brain, m6A is likely found in all brain structures of the CNS but also in the peripheral nervous system (PNS) (Weng et al. 2018). Region-specific methylation levels and patterns have been reported (Chang et al. 2017; Engel et al. 2018).
3.1 m6A in Brain Development
Knockout of the m6A methyltransferases in embryonic stem cells leads to embryonic lethality in all known cases, usually together with severe nervous system malformation (Fukusumi et al. 2008; Geula et al. 2015). In line, m6A has been described to be essential for mammalian cortical neurogenesis (Yoon et al. 2017). Loss of m6A in murine neural progenitor cells by removal of either METTL3 or METTL14 leads to prolonged cell cycle progression and delayed neuronal differentiation via suppression of neuronal lineage markers, thus, extending the cortical neurogenesis widely into postnatal stages (Yoon et al. 2017). Similarly, conditional knockout of Mettl3 using the prenatally expressing Nestin-Cre causes severe developmental defects both in cortical and cerebellar regions (Wang et al. 2018). Nestin-Cre Mettl3 conditional knockout mice, i.e., those with a knockout in prenatal brain cells, display cerebellar hypoplasia caused by drastically enhanced apoptosis of newborn cerebellar granule cells in the external granular layer leading to severe motoric deficits and death within the first 3 weeks after birth (Wang et al. 2018). Loss of FTO or FTO function in both mice and human leads to postnatal growth retardation, including microcephaly and increased postnatal lethality (Boissel et al. 2009; Gao et al. 2010). Interestingly, full knockut of the reader genes Ythdf1 or Ythdf2 does not lead to any gross brain development abnormalities, lethality or motor deficits (Ivanova et al. 2017; Shi et al. 2018).
m6A-profiling of human fetal forebrains and human brain organoids has revealed a conserved and unique m6A landscape similar to that in mouse embryonic forebrains (Yoon et al. 2017). In general, m6A has been described to be somewhat evolutionary conserved (Ma et al. 2017). Additional to stem cells in the developing brain, m6A is crucial for at least one of the two populations of neural stem cells (NSCs) remaining in the adult brain, the stem cells in the subgranular zone of the dentate gyrus (Li et al. 2017b). FTO loss in these cells reduces NSC proliferation and neuronal differentiation, reminiscent but not fully equal to the embryonic neurogenesis defect seen in METTL3 and METTL14 knockout mice. Finally, m6A modified RNAs also play a key role in brain cancer (Zhang et al. 2017; Cui et al. 2017).
3.2 m6A in the Adult Brain
Several detailed maps of m6A in the adult mammalian brain are available (Meyer et al. 2012; Hess et al. 2013; Chang et al. 2017; Merkurjev et al. 2018; Engel et al. 2018), reporting a total of approximately 10,000–20,000 m6A sites. Additionally, CNS RNA methylation has also been characterized in Drosophila melanogaster (Lence et al. 2016). In stark contrast to the deleterious effect of methyltransferases in the developing brain, loss of FTO, METTL3, or METTL14 in postnatal neurons only, e.g., via a conditional knockout using Camk2a-Cre driver lines, does not cause any major brain morphological changes or increase apoptosis of cells (Koranda et al. 2018; Engel et al. 2018; Zhang et al. 2018). The consequence of a loss of these enzymes in cells other than neurons, e.g., astrocytes, has not been investigated yet.
3.3 Sorting mRNAs in Complex Neurons by m6A?
m6A is involved in several mechanisms of regulating translocation of mRNA, including nuclear export (Zheng et al. 2013; Fustin et al. 2013) and sorting of mRNA into specific cytoplasmic aggregates like P-bodies and stress granules (Wang et al. 2015; Anders et al. 2018). Neurons are built more complexly than most mammalian cells, including higher polarization, higher fragmentation into specialized components like axons and dendrites, and a higher number of complex cell-to-cell connections. These cell-to-cell connections are highly regulated in the brain including changes of signal transmission efficacy and interactions of neurons with other cell types. Many such cellular changes are often realized via changes of gene expression control including compartmentalized regulation of protein translation e.g., at the synapse (Holt and Schuman 2013). Consequently, it has been speculated that m6A-modification of mRNA may regulate spatial sorting and compartmentalized protein translation control within neurons into axons, pre-synaptic nerve terminals, dendrites, and dendritic spines (Fig. 2). While a final demonstration of such a mechanism is still lacking, enrichment of synaptic and neuronal projection gene ontology terms has been reported repeatedly during the recent efforts to map m6A in the brain (Hess et al. 2013; Widagdo et al. 2016; Yoon et al. 2017; Merkurjev et al. 2018; Engel et al. 2018). Further, m6A has been reported to be localized to axons and to regulate axonal growth via local translation via GAP-43 (Yu et al. 2018) and several components of the m6A-machinery have been observed to be synaptically located including writers and erasers, classically considered to be nuclear proteins (Yu et al. 2018; Merkurjev et al. 2018). Consequently, synapses and neuronal somas have been reported to harbor their own specific epitranscriptome (Merkurjev et al. 2018).

Functions of m6A in the brain (adapted from Engel and Chen 2018)
3.4 FMRP
A potential key player involved in such a mechanism of localized translation regulation in neurons may be the fragile X mental retardation protein (FMRP). FMRP is a neuronal RNA-binding protein known for its role in metabotropic glutamate receptor (mGluR)-dependent signaling and synaptic plasticity (Waung and Huber 2009). It is found in neuronal RNA transport granules and regulates dendritic localization of RNAs. FMRP inhibits RNA local transcript translation including that occuring at the synapse (Holt and Schuman 2013). Overlap of the m6A consensus motif with the one of FMRP and high co-occurrence of FMRP binding sites in m6A modified target sites was observed early on (Anderson et al. 2016; Chang et al. 2017; Engel et al. 2018). Finally, actual binding of FMRP to m6A was shown recently (Arguello et al. 2017; Edupuganti et al. 2017). Competitive binding of m6A between FMRP and YTHDF1 and YTHDF2 was speculated upon as a potential mechanism of m6A-FMRP action (Edupuganti et al. 2017; Zhang et al. 2018).
3.5 Activity Dependent Regulation
A special property of neurons is their constant regulation via electrical and chemical signaling leading to amongst others activity-dependent regulation of gene expression. This enables the critical adaptiveness of the brain to outer and inner stimuli via short- and long-term alterations in gene expression, neuronal morphology, connectivity and ultimately regulation and behavior.
Similarly to m6A being involved in the basic cellular stress response (Dominissini et al. 2012; Zhou et al. 2015; Meyer et al. 2015; Xiang et al. 2017), it has also been described to be involved in cellular processes triggered upon neuronal signaling and activity-dependent regulation (Fig. 2). This includes both regulation in vitro, e.g., in primary neuronal cultures after KCl-induced neuronal depolarization (Widagdo et al. 2016), and in vivo in the adult brain after challenges in several brain regions, e.g., after fear conditioning (Widagdo et al. 2016; Walters et al. 2017; Zhang et al. 2018), stressful challenges (Engel et al. 2018), and also in the PNS after nerve injury (Weng et al. 2018). Levels of activity-dependent regulation include changes within the m6A machinery, altered global and target-mRNA specific m6A levels, and differential translation of downstream-effectors like immediate early genes (IEGs). In line with this, IEG function is widely impaired in Mettl3 knockout primary cortical neurons after fear conditioning (Zhang et al. 2018).
Furthermore, while activity-dependent gene expression changes for m6A enzymes and readers have been reported, the relation of dynamically regulated mRNA abundancy of e.g., Mettl3 or Fto to the respective active protein levels and cellular consequences of regulated m6A are still mostly unclear. This includes the very limited current knowledge on how the different m6A-enzymes and readers are regulated at the protein level in response to activity, including their subcellular localization, activity, and target specificity. Investigating the posttranslational regulation of the m6A-enzymes and readers, as for example shown or suggested for METTL3 and FTO via SUMOylation and ubiquitination (Tai et al. 2017; Zhu et al. 2018; Du et al. 2018) may provide valuable insight.
3.6 Brain Function, Electrophysiology and Behavior
Consistent with the concept of mRNA methylation being involved in the regulation of gene expression after neuronal activity, alterations of the m6A system via m6A enzyme knockouts were found to change neuronal electrophysiological properties. For example, long term potentiation (LTP) in the hippocampus was decreased after hippocampal knockout of Mettl3 (Zhang et al. 2018) (but observe conflicting data Engel et al. 2018), Fto (Engel et al. 2018), and Ythdf1 (Shi et al. 2018), while deletion of Mettl14 in the striatum led to increased neuronal excitability and reduced spike frequency adaptation (Koranda et al. 2018).
While the lack of m6A enzymes during development usually leads to severe developmental defects, enzyme deletion or depletion specifically in the adult brain causes only limited behavioral phenotypes, usually excluding effects on motor skills, movement properties, or anxiety-like behavior. Adult brain m6A-manipulation, reported to date, leads fairly specifically to memory impairment: Conditional knockout of Mettl3 in the hippocampus or forebrain excitatory neurons enhances cue-related memory consolidation after fear-conditioning and in the Morris Water Maze (MWM; Engel et al. 2018; Zhang et al. 2018), while full knockout of Ythdf1 reduces memory consolidation in both of these tests (Shi et al. 2018). Additionally, m6A deficiency via Mettl14 deletion in striatal neurons impairs learning and performance (Koranda et al. 2018). Conversely, knockout or knockdown of Fto in the prefrontal cortex or hippocampus enhances consolidation of cue- and or context-related fear memory (Wang et al. 2015; Widagdo et al. 2016; Walters et al. 2017) while impairing spatial learning and memory of mice in MWM and eight-arm maze test (Li et al. 2017b). Knockout of Fto also attenuates the response in cocaine-induced locomotion (Hess et al. 2013).
Together these studies show that although m6A seems to be much less crucial in the adult brain than during development, it is potentially important for specific brain functions. In line with m6A roles as a secondary mechanism of gene expression regulation, it may therefore be especially important for brain functions that require activity-dependent gene expression regulation, like memory formation.
4 m6A: Possible Implications for Psychiatric Disorders
Obesity and type-2 diabetes have been repeatedly associated with genetic polymorphisms in the first intron of the human FTO gene (Dina et al. 2007; Scuteri et al. 2007; Frayling et al. 2007), although the variant reported on may actually not affect the FTO locus itself but rather neighboring genes (Smemo et al. 2014; Stratigopoulos et al. 2014; Claussnitzer et al. 2015). Consequently, the physiological roles of FTO in the context of energy metabolism and expenditure and food intake have been extensively investigated but led to mixed results and as such the mechanisms remain unknown (Hess and Brüning 2014). Beyond such metabolic functions, the FTO variant has also been associated with several psychiatric disorders including Major Depressive Disorder (MDD) (Samaan et al. 2013; Milaneschi et al. 2014), Alzheimer’s Disease (AD) (Profenno et al. 2010; Keller et al. 2011; Reitz et al. 2012) and Attention Deficit Hyperactivity Disorder (ADHD) (Choudhry et al. 2013). It has further been indicated in memory processing capabilities in humans genome-wide association studies (Ho et al. 2010; Benedict et al. 2011; Keller et al. 2011). Additionally to FTO, a polymorphism related to ALKBH5 has been reported to be associated to MDD in a relatively small candidate gene association study (Du et al. 2015). However, recent more powered genetic association studies and meta-analyses did not report any association of gene variants close to any of the m6A-machinary genes to various psychiatric disorders, including MDD, Post-traumatic stress disorder, ADHD, and AD (Demontis et al. 2017*; Martin et al. 2017*; Purves et al. 2017*; Meier et al. 2018*; Duncan et al. 2018; Wray et al. 2018; Coleman et al. 2018*; Jansen et al. 2019, * not yet peer-reviewed preprints).
Beyond classic genetic association studies, increasing evidence suggests that dysregulation and maladaptation of transcriptional fine-tuning is central to the etiology of psychiatric disorders more so than monogenetic causes (Nestler et al. 2016). In agreement, our recent study comparing m6A of MDD patients and healthy controls in blood and derived cells found hardly any differences between the two groups except for after stimulation of stress-response signaling pathways (Engel et al. 2018). While these measurements were performed only in blood or blood derived cells, peripheral DNA methylation signatures related to neurobiological phenotypes may have some, albeit limited, similarity to central signatures (Davies et al. 2012; Farré et al. 2015; Hannon et al. 2015). Taken together, this indicates that the m6A-system may well be involved in the etiology of psychiatric disorders via changed regulation of gene expression, especially considering the encoding of activity-related gene expression regulation, and thus may lead to long-term changes contributing to psychiatric disorders.
5 Technological Challenges of Measuring m6A in the Brain
Technical approaches to detect m6A and challenges associated to them have been discussed in great detail here (Jia 2016) and elsewhere before (e.g., Schwartz and Motorin 2017; Helm and Motorin 2017; Schaefer et al. 2017). While the field of RNA modifications, especially m6A, has seen an incredible increase in attention over the last 10 years, studying those modifications still poses a major challenge given the limited availability of appropriate molecular tools and methods. This is especially true when aiming to quantify m6A dynamics. In the following section, we will summarize some of the current challenges related to the methods most used to detect m6A in the brain in vivo.
5.1 Global Detection Techniques
Two-dimensional thin layer chromatography (2-D TLC) and high-performance liquid chromatography coupled with triple-quadrupole tandem mass spectrometry (LC-MS/MS) were two of the earliest developed methods to detect and quantify modified nucleotides in RNA. Both require mRNA to be digested into single nucleotides first, which are then separated and detected based on their physico-chemical properties. This enables specific discrimination between different modifications, e.g., m1A and m6A. Stable isotope labelling approaches were developed to optimize these techniques but are mostly not applicable for analyzing intact mice or human organs (Popova and Williamson 2014; Kellner et al. 2014; Paulines and Limbach 2017). Both TLC and LC-MS/MS require comparably large amounts of input material and are usually performed on total RNA or mRNA preparations, and thus report an average global methylation signal derived across all nucleotides with a loss of sequence and target context. These techniques are thus greatly suited to detect major changes in levels of different modifications e.g., after knockout of enzymes or in some cases of experimental manipulations (Engel et al. 2018), but may fail to detect more subtle changes as often expected after stimulation of the adult brain.
Two additional techniques to measure global m6A-levels are antibody-based enzyme-linked colorimetric methods like Dot Blots and ELISAs. As with TLC and LC-MS/MS, these methods are commonly performed on total RNA or mRNA preparations but without previous digestion of the RNA. In contrast with the earlier described methods, they allow for low time-consuming and high throughput screening of samples and thus may be an entry point for characterizing m6A regulation in vivo. However, beyond being only able to report large and global differences in methylation, these methods also potentially suffer from antibody-associated issues (discussed below).
In contrast, a related method not suffering from the problems arising from global averaging of m6A modification is the SCARLET method (site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography, Liu et al. 2013) which quantifies nucleotides at one specific location with TLC. However, SCARLET also requires very high amounts of input material and radioactive labeling and thus is not readably applicable to study m6A dynamics in vivo.
5.2 m6A-Seq
The development of mapping m6A in a transcriptome-wide approach revolutionized the field in 2012 (m6A-Seq: Dominissini et al. 2012; meRIP-Seq: Meyer et al. 2012). Both, essentially equal, methods are based on affinity purification of fragmented mRNA with m6A-specific antibodies followed by random primed cDNA library generation, adaptor ligation and high throughput small read sequencing. Antibody-based affinity purification is needed because m6A, in contrast to other modifications as for example m1A (Dominissini et al. 2016), does not stop the most common reverse transcription enzymes or lead to base misincorporation. This may be overcome by using specific m6A-sensitive polymerases (Harcourt et al. 2013) or engineered reverse transcriptase with increased misincorporation in the opposite strand (Aschenbrenner et al. 2018), although both methods are not yet commonly established. This feature of m6A also limits the current availability of PCR based techniques.
While m6A-Seq enables a high throughput transcriptome-wide description of m6A, it comes with many significant problems: First of all, it is dependent on the antibody used with potential differences between the used antibodies and even affinity-differences across different batches of the same antibody. Antibodies may also detect related modifications like m6Am (Linder et al. 2015). Click chemistry protocols may improve antibody specificity (Hartstock et al. 2018) but are not yet implemented or tested in the majority of protocols. So far, only a limited number of antibodies have been used, leading to somewhat different signatures (Zeng et al. 2018) but a comprehensive study comparing their detection patterns is not available yet. Furthermore, the binding properties of the antibodies may be influenced by the adjacent nucleotide sequence and RNA secondary structures. Antibodies also may suffer from substantial background signal with sequence-selective capture of certain unmodified fragments (Schwartz et al. 2013). Moreover, all of these biases of the sequence read distributions are mixed and amplified by the structural biases inherent to RNA-Seq itself. The resulting bias-mixture can only be partially remedied by the typical correlation to input RNA-Seq common in m6A-Seq.
Secondly, the original m6A-Seq protocols required substantial quantities of input material (several micrograms of purified mRNA). Recent low input protocols overcome this problem by using library kits optimized for ultra-low input RNA-Seq (Zeng et al. 2018). These low-input protocols may however introduce even more additional biases. Superficial comparisons of data received with such low-input protocols have already showed significant differences from the respective classical m6A-Seq data set but this needs to be further investigated (Zeng et al. 2018).
Thirdly, classic m6A-Seq protocols do not detect the modified nucleotide itself but a pile-up of fragments called m6A-peaks which should, in principle, harbor the m6A site in its center. Such peaks, by design, are around 200 nt long (when using 100 nt RNA fragments) but often experiments result in even bigger peaks likely due to several m6A-sites close by. The m6A-sites appearing in clusters (Ke et al. 2015; Linder et al. 2015) and their potential dynamics will thus be partially lost in m6A-Seq. Further, due to commonly employed method of cDNA synthesis via random hexamer primers, m6A-peaks will not include 5′ ends of the mRNA if methylated, although 5′ mRNA m6A and m6Am methylation may still be inferred from a peak located at the 5′ UTR.
Aiming to improve these protocols, several protocols have been recently developed to enable a nucleotide-specific transcriptome-wide mapping of m6A (m6A-CLIP: Ke et al. 2015; miCLIP: Linder et al. 2015). These methods use UV-crosslinking of an m6A-antibody with mRNA-fragment leading to predictable mutation and truncation patterns in the cDNA strand during reverse-transcription that can be detected later in the sequencing data. Besides being prone to sequence and structural biases again, and the difficulty to map due to short fragments, the resulting data is often more noisy and less consistent than m6A-Seq data with many more replicates needed for consistent mapping and unclear quantitative potential.
In the future, detection of m6A via directly sequencing the RNA in its native form, e.g., while pulling through a nanopore, may overcome these challenges but just begins to be established for a wider audience (Liu et al. 2019).
5.3 Quantification of m6A-Seq
Most bioinformatics pipelines to analyze m6A-Seq data rely on comparisons of the enrichment of m6A-immunoprecipitated fragments over standard RNA-Seq signal, employing cutoffs to defined peak ranges and minimum-occurrence across several replicates to call the presence or absence of a peak. As a result, analysis results are heavily dependent on the bioinformatics algorithm used and seemingly arbitrary cutoffs chosen by the respective scientist with no consensus yet of best practices. Increasing the number of replicates used may often help to filter out sites that are spuriously detected aiming to increase robustness and reproducibility of the achieved maps. More dedicated analytical approaches are continuously being developed to better identify modified sites, more effectively integrate background levels and better filter out noise.
While binary maps of peaks and non-peaks can then be compared between two conditions, most likely in a physiological regulation of m6A, for example upon stimulation in the adult brain, quantitative regulation of m6A may be much more likely. While none of the m6A-Seq protocols have been originally developed for quantitative comparison of methylation states, methods to compare m6A-Seq signatures across conditions were proposed soon after the original methods description and are actively developed (Meng et al. 2014; Cui et al. 2015; Liu et al. 2016). For the special case of the mostly postmitotic adult brain, given enough replicates and a mild enough manipulation, underlying changes in mRNA expression may be assumed to be negligible, thus removing the need for extensive normalization to background levels.
Notably, a calibrated pulldown procedure called LAIC-Seq was establish recently as a tool to provide quantitative estimates of methylation stoichiometry (Molinie et al. 2016). However, this protocol uses immunoprecipitation of full-length mRNA rather than fragments with the m6A-antibody, thus averages m6A content across the entire transcript and likely suffers from extensive noise due to structural biases. Similar methods have been developed using qPCR instead of RNA-Seq to measure the abundance of m6A-containing (full-length) mRNA and m6A- calibrator spike-ins to allow for immunoprecipitation efficiency correction (Engel et al. 2018).
The perhaps biggest challenge to quantifying m6A in the (adult) brain is the lack of techniques to profile m6A in different brain cell subpopulations, leaving all currently available m6A-Seq data to be an average m6A signal from a widespread cell mixture. Employing knockout animals with conditional removal of m6A-related genes from distinct cell types may provide some insight about cell-specific m6A-signatures. Today, not even a comprehensive comparison of, for example, m6A-signatures in neuron versus astrocytes, the two major brain cell types, is available. The use of neuronal cultures to detect neuron-specific signals was employed before (Widagdo et al. 2016), but may carry limited significance due to neurons normally being highly embedded and structured in vivo.
5.4 Target Manipulation of m6A-Sites
Almost no reports are available describing the effects of manipulating m6A at a single target site (except Kane and Beemon 1987; Schwartz et al. 2013). To date, functional significance of RNA methylation has usually been shown by correlation, or broad manipulation of m6A levels by removal m6A enzymes. As a consequence, significance and cellular function of site-specific m6A is mostly unknown so far, although such manipulation should be easy to achieve using recent CRISPR/Cas9 techniques. One explanation may be that methyltransferases might set compensatory methylation at adjacent sites due to remaining m6A-site context when only removing the target site (Narayan et al. 1994; observe contrary results in Schwartz et al. 2013).
Rather than genetic manipulation of a target site, using CRISPR/Cas9 technology to recruit epitranscriptomic modulators to mRNA, potentially even in a temporally- and cell-type controlled manner, may provide much more insight into the cellular functions of specific m6A-sites (O’Connell et al. 2014).
6 Conclusions
The field of mRNA adenosine methylation has experienced a recent sudden take off, fueled by the transcriptome-wide mapping of m6A and many functional follow-up studies. While the core enzymes and reader protein have been well described, more and more details are added each year highlighting the function of m6A in adapting and fine-tuning gene expression especially after environmental stimulation and, in the brain, neuronal activity. Nevertheless, many questions about the regulatory mechanisms, complex interplay of RNA modifications, and the cellular consequences of RNA methylation remain unanswered. Brute force powered functional studies, based on removing enzymes from whole brain areas, have provided some valuable early insight into potential roles of m6A in the brain, but the actual functions of m6A in the brain and related pathology are still open. Methodological advancements to measure stimulated and activity-related m6A-changes in a time- and cell-specific manner as well as functional assays focusing on specific m6A sites may in the future answer the question of the actual significance of m6A in the brain.
References
Alarcón CR, Goodarzi H, Lee H et al (2015a) HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell 162:1299–1308. https://doi.org/10.1016/j.cell.2015.08.011
Alarcón CR, Lee H, Goodarzi H et al (2015b) N6-methyladenosine marks primary microRNAs for processing. Nature 519:482–485. https://doi.org/10.1038/nature14281
Anders M, Chelysheva I, Goebel I et al (2018) Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Sci Alliance 1:e201800113. https://doi.org/10.26508/lsa.201800113
Anderson BR, Chopra P, Suhl JA et al (2016) Identification of consensus binding sites clarifies FMRP binding determinants. Nucleic Acids Res 44:6649–6659. https://doi.org/10.1093/nar/gkw593
Arguello AE, DeLiberto AN, Kleiner RE (2017) RNA chemical proteomics reveals the N6-methyladenosine (m6A)-regulated protein-RNA interactome. J Am Chem Soc 139:17249–17252. https://doi.org/10.1021/jacs.7b09213
Aschenbrenner J, Werner S, Marchand V et al (2018) Engineering of a DNA polymerase for direct m6A sequencing. Angew Chem Int Ed 57:417–421. https://doi.org/10.1002/anie.201710209
Benedict C, Jacobsson JA, Rönnemaa E, Sällman-Almén M, Brooks S, Schultes B, Fredriksson R, Lannfelt L, Kilander L, Schiöth HB (2011) The fat mass and obesity gene is linked to reduced verbal fluency in overweight and obese elderly men. Neurobiol Aging 32(6):1159.e1–1159.e5. https://doi.org/10.1016/j.neurobiolaging.2011.02.006
Boccaletto P, Machnicka MA, Purta E et al (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46:D303–D307. https://doi.org/10.1093/nar/gkx1030
Boissel S, Reish O, Proulx K et al (2009) Loss-of-function mutation in the dioxygenase-encoding FTO gene causes severe growth retardation and multiple malformations. Am J Hum Genet 85:106–111. https://doi.org/10.1016/j.ajhg.2009.06.002
Bokar JA, Rath-Shambaugh ME, Ludwiczak R et al (1994) Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem 269:17697–17704
Bokar JA, Shambaugh ME, Polayes D et al (1997) Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3:1233–1247
Carlile TM, Rojas-Duran MF, Zinshteyn B et al (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515:143–146. https://doi.org/10.1038/nature13802
Chang M, Lv H, Zhang W et al (2017) Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol 7:170166. https://doi.org/10.1098/rsob.170166
Chen T, Hao Y-J, Zhang Y et al (2015) m6A RNA methylation is regulated by MicroRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16:289–301. https://doi.org/10.1016/j.stem.2015.01.016
Choudhry Z, Sengupta SM, Grizenko N et al (2013) Association between obesity-related gene FTO and ADHD. Obesity (Silver Spring) 21:E738–E744. https://doi.org/10.1002/oby.20444
Claussnitzer M, Dankel SN, Kim K-H et al (2015) FTO obesity variant circuitry and adipocyte browning in humans. New Engl J Med 373:895–907. https://doi.org/10.1056/NEJMoa1502214
Coleman JRI, Peyrot WJ, Purves KL et al (2018) Genome-wide gene-environment analyses of major depressive disorder and reported lifetime traumatic experiences in UK Biobank. bioRxiv. https://doi.org/10.1101/247353
Cui X, Zhang L, Meng J et al (2015) MeTDiff: a novel differential RNA methylation analysis for MeRIP-Seq data. IEEE/ACM Trans Comput Biol Bioinform 15(2):526–534. https://doi.org/10.1109/TCBB.2015.2403355
Cui Q, Shi H, Ye P et al (2017) m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep 18:2622–2634. https://doi.org/10.1016/j.celrep.2017.02.059
Davies MN, Volta M, Pidsley R et al (2012) Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 13:R43. https://doi.org/10.1186/gb-2012-13-6-r43
Demontis D, Walters RK, Martin J et al (2017) Discovery of the first genome-wide significant risk loci for ADHD. bioRxiv. https://doi.org/10.1101/145581
Dina C, Meyre D, Gallina S et al (2007) Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet 39:724–726. https://doi.org/10.1038/ng2048
Dominissini D, Moshitch-Moshkovitz S, Schwartz S et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206. https://doi.org/10.1038/nature11112
Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S et al (2016) The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530:441–446. https://doi.org/10.1038/nature16998
Du T, Rao S, Wu L et al (2015) An association study of the m6A genes with major depressive disorder in Chinese Han population. J Affect Disord 183:279–286. https://doi.org/10.1016/j.jad.2015.05.025
Du Y, Hou G, Zhang H et al (2018) SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res 46:5195–5208. https://doi.org/10.1093/nar/gky156
Dubin DT, Taylor RH (1975) The methylation state of poly A-containing messenger RNA from cultured hamster cells. Nucleic Acids Res 2:1653–1668
Duncan LE, Ratanatharathorn A, Aiello AE et al (2018) Largest GWAS of PTSD (N=20 070) yields genetic overlap with schizophrenia and sex differences in heritability. Mol Psychiatry 23:666–673. https://doi.org/10.1038/mp.2017.77
Edupuganti RR, Geiger S, Lindeboom RGH et al (2017) N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24:870–878. https://doi.org/10.1038/nsmb.3462
Engel M, Chen A (2018) The emerging role of mRNA methylation in normal and pathological behavior. Genes Brain Behav 17:e12428. https://doi.org/10.1111/gbb.12428
Engel M, Eggert C, Kaplick PM et al (2018) The role of m6A/m-RNA methylation in stress response regulation. Neuron 99:389–403.e9. https://doi.org/10.1016/j.neuron.2018.07.009
Farré P, Jones MJ, Meaney MJ et al (2015) Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 8:19. https://doi.org/10.1186/s13072-015-0011-y
Fischer J, Koch L, Emmerling C et al (2009) Inactivation of the Fto gene protects from obesity. Nature 458:894–898. https://doi.org/10.1038/nature07848
Frayling TM, Timpson NJ, Weedon MN et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894. https://doi.org/10.1126/science.1141634
Fukusumi Y, Naruse C, Asano M (2008) Wtap is required for differentiation of endoderm and mesoderm in the mouse embryo. Dev Dyn 237:618–629. https://doi.org/10.1002/dvdy.21444
Fustin J-M, Doi M, Yamaguchi Y et al (2013) RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155:793–806. https://doi.org/10.1016/j.cell.2013.10.026
Gao X, Shin Y-H, Li M et al (2010) The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS One 5:e14005. https://doi.org/10.1371/journal.pone.0014005
Gerken T, Girard CA, Tung Y-CL et al (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318:1469–1472. https://doi.org/10.1126/science.1151710
Geula S, Moshitch-Moshkovitz S, Dominissini D et al (2015) m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347:1002–1006. https://doi.org/10.1126/science.1261417
Hannon E, Lunnon K, Schalkwyk L, Mill J (2015) Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics 10:1024–1032. https://doi.org/10.1080/15592294.2015.1100786
Harcourt EM, Ehrenschwender T, Batista PJ et al (2013) Identification of a selective polymerase enables detection of N6-methyladenosine in RNA. J Am Chem Soc 135:19079–19082. https://doi.org/10.1021/ja4105792
Hartstock K, Nilges BS, Ovcharenko A et al (2018) Enzymatic or in vivo installation of propargyl groups in combination with click chemistry for the enrichment and detection of methyltransferase target sites in RNA. Angew Chem Int Ed 57:6342–6346. https://doi.org/10.1002/anie.201800188
Helm M, Motorin Y (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18:275–291. https://doi.org/10.1038/nrg.2016.169
Hess ME, Brüning JC (2014) The fat mass and obesity-associated (FTO) gene: obesity and beyond? Biochim Biophys Acta 1842:2039–2047. https://doi.org/10.1016/j.bbadis.2014.01.017
Hess ME, Hess S, Meyer KD et al (2013) The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 16:1042–1048. https://doi.org/10.1038/nn.3449
Ho AJ, Stein JL, Hua X, Lee S, Hibar DP, Leow AD, Dinov ID, Toga AW, Saykin AJ, Shen L, Foroud T, Pankratz N, Huentelman MJ, Craig DW, Gerber JD, Allen AN, Corneveaux JJ, Stephan DA, DeCarli CS, DeChairo BM, Potkin SG, Jack CR Jr, Weiner MW, Raji CA, Lopez OL, Becker JT, Carmichael OT, Thompson PM, Alzheimer’s Disease Neuroimaging Initiative (2010) A commonly carried allele of the obesity-related FTO gene is associated with reduced brain volume in the healthy elderly. Proc Natl Acad Sci U S A 107(18):8404–8409. https://doi.org/10.1073/pnas.0910878107
Holt CE, Schuman EM (2013) The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80:648–657. https://doi.org/10.1016/j.neuron.2013.10.036
Huang H, Weng H, Sun W et al (2018) Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20:285–295. https://doi.org/10.1038/s41556-018-0045-z
Ivanova I, Much C, Di Giacomo M et al (2017) The RNA m6A reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell 67:1059–1067.e4. https://doi.org/10.1016/j.molcel.2017.08.003
Jansen IE, Savage JE, Watanabe K et al (2019) Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. https://doi.org/10.1038/s41588-018-0311-9
Jia G (2016) RNA modification N6-methyladenosine in post-transcriptional regulation. In: Jurga S, Erdmann VA, Barciszewski J (eds) Modified nucleic acids in biology and medicine. Springer, Cham, pp 131–145
Jia G, Fu Y, Zhao X et al (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–887. https://doi.org/10.1038/nchembio.687
Kane SE, Beemon K (1987) Inhibition of methylation at two internal N6-methyladenosine sites caused by GAC to GAU mutations. J Biol Chem 262:3422–3427
Karikó K, Buckstein M, Ni H, Weissman D (2005) Suppression of RNA recognition by toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175. https://doi.org/10.1016/j.immuni.2005.06.008
Ke S, Alemu EA, Mertens C et al (2015) A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev 29:2037–2053. https://doi.org/10.1101/gad.269415.115
Ke S, Pandya-Jones A, Saito Y et al (2017) m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31:990–1006. https://doi.org/10.1101/gad.301036.117
Keller L, Xu W, Wang H-X et al (2011) The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzheimers Dis 23:461–469. https://doi.org/10.3233/JAD-2010-101068
Kellner S, Ochel A, Thüring K et al (2014) Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res 42:e142. https://doi.org/10.1093/nar/gku733
Koranda JL, Dore L, Shi H et al (2018) Mettl14 is essential for epitranscriptomic regulation of striatal function and learning. Neuron 99:283–292. https://doi.org/10.1016/j.neuron.2018.06.007
Lence T, Akhtar J, Bayer M et al (2016) m6A modulates neuronal functions and sex determination in Drosophila. Nature 540:242–247. https://doi.org/10.1038/nature20568
Levanon EY, Eisenberg E, Yelin R et al (2004) Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat Biotechnol 22:1001–1005. https://doi.org/10.1038/nbt996
Li JB, Levanon EY, Yoon J-K et al (2009) Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 324:1210–1213. https://doi.org/10.1126/science.1170995
Li X, Zhu P, Ma S et al (2015) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 11:592–597. https://doi.org/10.1038/nchembio.1836
Li X, Xiong X, Wang K et al (2016) Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol 12:311–316. https://doi.org/10.1038/nchembio.2040
Li A, Chen Y-S, Ping X-L et al (2017a) Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res 27:444–447. https://doi.org/10.1038/cr.2017.10
Li L, Zang L, Zhang F et al (2017b) Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet 26:2398–2411. https://doi.org/10.1093/hmg/ddx128
Lin S, Choe J, Du P et al (2016) The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62:335–345. https://doi.org/10.1016/j.molcel.2016.03.021
Linder B, Grozhik AV, Olarerin-George AO et al (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12:767–772. https://doi.org/10.1038/nmeth.3453
Liu N, Parisien M, Dai Q et al (2013) Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding. RNA 19:1848–1856. https://doi.org/10.1261/rna.041178.113
Liu J, Yue Y, Han D et al (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10:93–95. https://doi.org/10.1038/nchembio.1432
Liu N, Dai Q, Zheng G et al (2015) N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560–564. https://doi.org/10.1038/nature14234
Liu L, Zhang S-W, Gao F et al (2016) DRME: count-based differential RNA methylation analysis at small sample size scenario. Anal Biochem 499:15–23. https://doi.org/10.1016/j.ab.2016.01.014
Liu N, Zhou KI, Parisien M et al (2017) N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res 45:6051–6063. https://doi.org/10.1093/nar/gkx141
Liu H, Begik O, Lucas MC et al (2019) Accurate detection of m6A RNA modifications in native RNA sequences. bioRxiv 525741. https://doi.org/10.1101/525741
Ma L, Zhao B, Chen K et al (2017) Evolution of transcript modification by N6-methyladenosine in primates. Genome Res. https://doi.org/10.1101/gr.212563.116
Martin J, Walters RK, Demontis D et al (2017) A genetic investigation of sex bias in the prevalence of attention deficit hyperactivity disorder. bioRxiv 154088. https://doi.org/10.1101/154088
Mauer J, Jaffrey SR (2018) FTO, m6Am, and the hypothesis of reversible epitranscriptomic mRNA modifications. FEBS Lett 592:2012–2022. https://doi.org/10.1002/1873-3468.13092
Mauer J, Luo X, Blanjoie A et al (2017) Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541:371–375. https://doi.org/10.1038/nature21022
Meier S, Trontti K, Als TD et al (2018) Genome-wide association study of anxiety and stress-related disorders in the iPSYCH Cohort. bioRxiv 263855. https://doi.org/10.1101/263855
Meng J, Lu Z, Liu H et al (2014) A protocol for RNA methylation differential analysis with MeRIP-Seq data and exomePeak R/bioconductor package. Methods 69:274–281. https://doi.org/10.1016/j.ymeth.2014.06.008
Merkurjev D, Hong W-T, Iida K et al (2018) Synaptic N 6-methyladenosine (m6A) epitranscriptome reveals functional partitioning of localized transcripts. Nat Neurosci 1. https://doi.org/10.1038/s41593-018-0173-6
Meyer KD, Saletore Y, Zumbo P et al (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646. https://doi.org/10.1016/j.cell.2012.05.003
Meyer KD, Patil DP, Zhou J et al (2015) 5′ UTR m6A promotes cap-independent translation. Cell 163:999–1010. https://doi.org/10.1016/j.cell.2015.10.012
Milaneschi Y, Lamers F, Mbarek H et al (2014) The effect of FTO rs9939609 on major depression differs across MDD subtypes. Mol Psychiatry 19:960–962. https://doi.org/10.1038/mp.2014.4
Molinie B, Wang J, Lim KS et al (2016) m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome. Nat Methods. https://doi.org/10.1038/nmeth.3898
Narayan P, Ludwiczak RL, Goodwin EC, Rottman FM (1994) Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res 22:419–426. https://doi.org/10.1093/nar/22.3.419
Nestler EJ, Peña CJ, Kundakovic M et al (2016) Epigenetic basis of mental illness. Neuroscientist 22:447–463. https://doi.org/10.1177/1073858415608147
O’Connell MR, Oakes BL, Sternberg SH et al (2014) Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516:263–266. https://doi.org/10.1038/nature13769
Patil DP, Chen C-K, Pickering BF et al (2016) m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373. https://doi.org/10.1038/nature19342
Patil DP, Pickering BF, Jaffrey SR (2018) Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol 28:113–127. https://doi.org/10.1016/j.tcb.2017.10.001
Paulines M, Limbach P (2017) Comparative analysis of ribonucleic acid digests (CARD) by mass spectrometry. In: Lusser A (ed) RNA methylation. Springer, New York, pp 19–32
Ping X-L, Sun B-F, Wang L et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24:177–189. https://doi.org/10.1038/cr.2014.3
Popova AM, Williamson JR (2014) Quantitative analysis of rRNA modifications using stable isotope labeling and mass spectrometry. J Am Chem Soc 136:2058–2069. https://doi.org/10.1021/ja412084b
Profenno LA, Porsteinsson AP, Faraone SV (2010) Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 67:505–512. https://doi.org/10.1016/j.biopsych.2009.02.013
Purves KL, Coleman JRI, Rayner C et al (2017) The common genetic architecture of anxiety disorders. bioRxiv 203844. https://doi.org/10.1101/203844
Reitz C, Tosto G, Mayeux R et al (2012) Genetic variants in the Fat and Obesity Associated (FTO) gene and risk of Alzheimer’s disease. PLoS One 7:e50354. https://doi.org/10.1371/journal.pone.0050354
Roost C, Lynch SR, Batista PJ et al (2015) Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. https://doi.org/10.1021/ja513080v
Roundtree IA, Evans ME, Pan T, He C (2017) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200. https://doi.org/10.1016/j.cell.2017.05.045
Samaan Z, Anand SS, Anand S et al (2013) The protective effect of the obesity-associated rs9939609 A variant in fat mass- and obesity-associated gene on depression. Mol Psychiatry 18:1281–1286. https://doi.org/10.1038/mp.2012.160
Schaefer M, Kapoor U, Jantsch MF (2017) Understanding RNA modifications: the promises and technological bottlenecks of the ‘epitranscriptome’. Open Biol 7. https://doi.org/10.1098/rsob.170077
Schibler U, Perry RP (1977) The 5′-termini of heterogeneous nuclear RNA: a comparison among molecules of different sizes and ages. Nucleic Acids Res 4:4133–4149
Schöller E, Weichmann F, Treiber T et al (2018) Interactions, localization, and phosphorylation of the m6A generating METTL3-METTL14-WTAP complex. RNA 24:499–512. https://doi.org/10.1261/rna.064063.117
Schwartz S, Motorin Y (2017) Next-generation sequencing technologies for detection of modified nucleotides in RNAs. RNA Biol 14:1124–1137. https://doi.org/10.1080/15476286.2016.1251543
Schwartz S, Agarwala SD, Mumbach MR et al (2013) High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155:1409–1421. https://doi.org/10.1016/j.cell.2013.10.047
Schwartz S, Bernstein DA, Mumbach MR et al (2014a) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159:148–162. https://doi.org/10.1016/j.cell.2014.08.028
Schwartz S, Mumbach MR, Jovanovic M et al (2014b) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8:284–296. https://doi.org/10.1016/j.celrep.2014.05.048
Scuteri A, Sanna S, Chen W-M et al (2007) Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet 3:e115. https://doi.org/10.1371/journal.pgen.0030115
Shi H, Wang X, Lu Z et al (2017) YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res 27:315–328. https://doi.org/10.1038/cr.2017.15
Shi H, Zhang X, Weng Y-L et al (2018) m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563:249–253. https://doi.org/10.1038/s41586-018-0666-1
Slobodin B, Han R, Calderone V et al (2017) Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 169:326–337.e12. https://doi.org/10.1016/j.cell.2017.03.031
Smemo S, Tena JJ, Kim K-H et al (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507:371–375. https://doi.org/10.1038/nature13138
Spitale RC, Flynn RA, Zhang QC et al (2015) Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519:486–490. https://doi.org/10.1038/nature14263
Squires JE, Patel HR, Nousch M et al (2012) Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res 40:5023–5033. https://doi.org/10.1093/nar/gks144
Stratigopoulos G, Martin Carli JF, O’Day DR et al (2014) Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the FTO locus, causes increased adiposity in mice. Cell Metab 19:767–779. https://doi.org/10.1016/j.cmet.2014.04.009
Tai H, Wang X, Zhou J et al (2017) Protein kinase Cβ activates fat mass and obesity-associated protein by influencing its ubiquitin/proteasome degradation. FASEB J 31:4396–4406. https://doi.org/10.1096/fj.201601159RR
Vujovic P, Stamenkovic S, Jasnic N et al (2013) Fasting induced cytoplasmic Fto expression in some neurons of rat hypothalamus. PLoS One 8:e63694. https://doi.org/10.1371/journal.pone.0063694
Walters BJ, Mercaldo V, Gillon CJ et al (2017) The role of The RNA demethylase FTO (fat mass and obesity-associated) and mRNA methylation in hippocampal memory formation. Neuropsychopharmacology 42:1502–1510. https://doi.org/10.1038/npp.2017.31
Wang Y, Li Y, Toth JI et al (2014) N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16:191–198. https://doi.org/10.1038/ncb2902
Wang X, Zhao BS, Roundtree IA et al (2015) N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161:1388–1399. https://doi.org/10.1016/j.cell.2015.05.014
Wang C-X, Cui G-S, Liu X et al (2018) METTL3-mediated m6A modification is required for cerebellar development. PLoS Biol 16:e2004880. https://doi.org/10.1371/journal.pbio.2004880
Waung MW, Huber KM (2009) Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr Opin Neurobiol 19:319–326. https://doi.org/10.1016/j.conb.2009.03.011
Wei C-M, Gershowitz A, Moss B (1975) N6, O2′-dimethyladenosine a novel methylated ribonucleoside next to the 5′ terminal of animal cell and virus mRNAs. Nature 257:251–253. https://doi.org/10.1038/257251a0
Wei J, Liu F, Lu Z et al (2018) Differential m6A, m6Am, and m1A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell 71:973–985.e5. https://doi.org/10.1016/j.molcel.2018.08.011
Weng Y-L, Wang X, An R et al (2018) Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron 97:313–325.e6. https://doi.org/10.1016/j.neuron.2017.12.036
Widagdo J, Zhao Q-Y, Kempen M-J et al (2016) Experience-dependent accumulation of N6-methyladenosine in the prefrontal cortex is associated with memory processes in mice. J Neurosci 36:6771–6777. https://doi.org/10.1523/JNEUROSCI.4053-15.2016
Wray NR, Ripke S, Mattheisen M et al (2018) Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50:668. https://doi.org/10.1038/s41588-018-0090-3
Xiang Y, Laurent B, Hsu C-H et al (2017) RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543:573–576. https://doi.org/10.1038/nature21671
Xiao W, Adhikari S, Dahal U et al (2016) Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 61:507–519. https://doi.org/10.1016/j.molcel.2016.01.012
Yoon K-J, Ringeling FR, Vissers C, et al (2017) Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 0: doi: https://doi.org/10.1016/j.cell.2017.09.003
Yu J, Chen M, Huang H et al (2018) Dynamic m6A modification regulates local translation of mRNA in axons. Nucleic Acids Res 46:1412–1423. https://doi.org/10.1093/nar/gkx1182
Zeng Y, Wang S, Gao S et al (2018) Refined RIP-seq protocol for epitranscriptome analysis with low input materials. PLoS Biol 16:e2006092. https://doi.org/10.1371/journal.pbio.2006092
Zhang S, Zhao BS, Zhou A et al (2017) m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31:591–606.e6. https://doi.org/10.1016/j.ccell.2017.02.013
Zhang Z, Wang M, Xie D et al (2018) METTL3-mediated N 6-methyladenosine mRNA modification enhances long-term memory consolidation. Cell Res 28:1050. https://doi.org/10.1038/s41422-018-0092-9
Zhao BS, Roundtree IA, He C (2017) Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 18:31–42. https://doi.org/10.1038/nrm.2016.132
Zheng G, Dahl JA, Niu Y et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49:18–29. https://doi.org/10.1016/j.molcel.2012.10.015
Zhou J, Wan J, Gao X et al (2015) Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526:591–594. https://doi.org/10.1038/nature15377
Zhu T, Yong XLH, Xia D et al (2018) Ubiquitination regulates the proteasomal degradation and nuclear translocation of the fat mass and obesity-associated (FTO) protein. J Mol Biol 430:363–371. https://doi.org/10.1016/j.jmb.2017.12.003
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Engel, M., Chen, A. (2019). m6A mRNA Methylation in the Mammalian Brain: Distribution, Function and Implications for Brain Functions. In: Jurga, S., Barciszewski, J. (eds) The DNA, RNA, and Histone Methylomes. RNA Technologies. Springer, Cham. https://doi.org/10.1007/978-3-030-14792-1_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-14792-1_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14791-4
Online ISBN: 978-3-030-14792-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)