
Abstract
N 6-methyladenosine (m6A) is the most prevalent internal methylation in messenger RNA (mRNA). This biochemically reversible modification is deposited by m6A methyltransferases, removed by m6A demethylases and recognized by different RNA-binding proteins. Depending on the localization of m6A and its reader proteins, an array of cellular processes ranging from RNA maturation and export in nucleus, to degradation and translation in cytoplasm, can be affected and consequently lead to diverse cell fates. The essential role of m6A in normal tissue development as well as tumor progression has been revealed in the past few years, emphasizing an additional layer of gene expression regulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
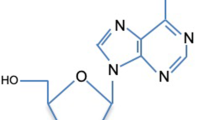
N 6-methyladenosine (m6A) modification (Fig. 1), the most abundant internal methylation on mRNA, has emerged as a key regulatory mark on messenger RNA (mRNA). Despite the demonstration of methylation of ribosomal RNA (rRNA) and transfer RNA (tRNA) in mammalian cells by 1970, mRNA methylation was not ascertained until mRNA could be purified with polyA selection. With the pursuit of cap structure of mRNA (or heterogeneous nuclear RNA, hnRNA), Perry and Rottman laboratories found that the internal regions of mRNA and hnRNA are frequently methylated by m6A in mammalian cells, with an estimated occurrence of ∼1–2 m6A per 1000 nucleotides (Desrosiers et al. 1974; Perry et al. 1975). Subsequent mutation and enzymatic footprinting assays by a number of groups revealed that m6A is deposited within a consensus sequence Pu(G>A)m6AC(A/C/U) (where Pu represents purine) (Schibler et al. 1977; Wei and Moss 1977; Kane and Beemon 1987). In mammalian cells, the majority of m6A modifications are catalyzed by a multicomponent methylation complex identified by Rottman laboratory (Bokar et al. 1994), which contains the core catalytic component METTL3 (methyltransferase like 3, initially called MT-A70) (Bokar et al. 1997). m6A was thought to be irreversible for decades until the groundbreaking discovery by Chuan He laboratory with their exciting report of the first m6A demethylase FTO (fat mass and obesity-associated protein) in mammalian cells (Jia et al. 2011). Another member of AlkB subfamily of the Fe(II)/2-oxoglutarate (2OG) dioxygenase superfamily, ALKBH5 (alkB homologue 5), was soon discovered to have a similar m6A demethylase activity (Zheng et al. 2013). These remarkable discoveries revived the interest from RNA research community and led to the functional characterization of m6A. Meanwhile, transcriptomic analysis afforded by high-throughput sequencing enables global mapping and measurement of m6A at levels of cells, tissues and species, with the use of an antibody that specifically recognizes m6A (Meyer et al. 2012; Dominissini et al. 2012; Schwartz et al. 2013; Batista et al. 2014; Wang et al. 2014b). These m6A-seq studies, ranging from yeast to mammalian cells, confirmed that m6A residues within the consensus sequences are scattered in coding sequences (CDS), 3′ untranslated regions (UTRs) and 5′ UTRs, and are more enriched in the last exon around stop codon, suggesting a highly conserved m6A deposition machinery. Pioneer studies by James E. Darnell and others (Sommer et al. 1978; Friderici et al. 1976) suggested a correlation between m6A and mRNA instability, which has been elucidated by He laboratory and others with the demonstration of YTHDF2 (YTH domain-containing family protein 2)-dependent degradation of a subset of mRNAs bearing m6A (Wang et al. 2014a; Du et al. 2016). Shortly after, m6A has been shown to participate in many critical aspects of RNA regulation that include alternative splicing, nuclear export and translation, which are carried out by a variety of RNA-binding proteins and are ultimately essential for many major biological processes, such as embryonic stem cell (ESC) differentiation, circadian clock, and spermatogenesis (Geula et al. 2015; Batista et al. 2014; Wang et al. 2014b; Fustin et al. 2013; Zheng et al. 2013).

The structures of mRNA methylation
2 m6A Sequencing Technologies Enable “Epitranscriptomic” Studies
The transcriptome-wide profile of m6A in eukaryotic cells was first accomplished by two m6A-seq (or MeRIP-seq) studies based on the immunoprecipitation with a specific anti-m6A antibody and high-throughput sequencing (Dominissini et al. 2012; Meyer et al. 2012). m6A-seq starts with purified RNAs that are subjected to polyA-selection or ribosomal RNA depletion, fragmentation, and immunoprecipitation followed by high-throughput sequencing. This straightforward method led to the identification of m6A-marked transcripts corresponding to more than 7000 human genes. When coupled with methyltransferase depletion to reduce the detection background, this approach can achieve a relatively high resolution (Schwartz et al. 2013). Given the limitation of resolution due to the cumulative signals from multiple m6A residues residing within the same ∼100–200 nucleotide fragments, the CLIP-based m6A-CLIP (Cross-Linking and Immunoprecipitation) and PA-m6A-seq (photocrosslinking-assisted m6A sequencing) were introduced to facilitate higher resolution. Both methods use UV irradiation to capture RNA-antibody interactions by creating a covalent crosslink between the antibody and its bound RNA. However, unlike the m6A-CLIP that allows the antibody to be crosslinked directly at m6A sites (Ke et al. 2015; Linder et al. 2015), PA-m6A-seq UV irradiation induces T-to-C transitions at 4-thiouridine residues in m6A nearby regions, which therefore requires the search of consensus sequence to infer the precise m6A sites (Chen et al. 2015). Consistent with the early observations regarding a biased m6A distribution in 3′ end of mRNA (Perry et al. 1975), nearly all m6A-seq studies have m6A peaks mapped enriched in 3′ UTRs and near stop codons. It should be noted that this antibody also binds to m6Am, a frequent modification adjacent to the cap of mRNA, which yields “false” m6A peaks within 500 nt from the transcription start site (TSS). Nonetheless, these peaks can be extracted from m6A-CLIP mapped sites by in silico analysis of their overlap with TSSs and their localization in a sequence context matching the core initiator motif (Mauer et al. 2017).
3 m6A Is Primarily Deposited by the METTL3–METTL14 Methyltransferase
The first cloned mRNA m6A methyltransferase METTL3 was identified from a partially purified Hela cell nuclear fraction referred to as MT-A, which contains the subunit that binds to S-adenosylmethionine (SAM), the catalytic co-factor for methyltransferase (Bokar et al. 1997; Bokar et al. 1994). METTL3 has a co-localization with nuclear speckles and a widespread expression pattern in human tissues (Bokar et al. 1997). A phylogenomic analysis revealed over 40 bacterial and eukaryotic proteins that share sequence similarity with the methyltransferase domain of human METTL3 (Bujnicki et al. 2002). METTL3 orthologs are conserved throughout all eukaryotes that include Saccharomyces cerevisiae (IME4), Drosophila melanogaster (IME4) and Arabidopsis Thaliana (MTA). In S. cerevisiae, m6A can be detected in sporulating cells at low levels that requires the catalytic activity of Ime4 (Inducer of Meiosis 4) (Clancy et al. 2002). In D. melanogaster, Ime4 mutants are viable but develop behavioural defects with a sex bias towards maleness due to reduced female-specific splicing of the Sex lethal (Sxl), a master switch gene that controls sex determination and dosage compensation (Haussmann et al. 2016; Lence et al. 2016). The METTL3 orthologs are essential for developing embryo to progress past the globular stage in A. thaliana (Zhong et al. 2008) and required for embryonic development in mammalian cells (Geula et al. 2015). During the course of evolution, METTL3-mediated m6A RNA modification turns to be more actively involved in critical biological processes. Noteworthy, m6A is also present in bacterial mRNAs that carry a unique consensus motif of GCCAU. With a distinct modification pattern from those in eukaryotes, these m6A-modified genes are associated with respiration, amino acids metabolism, stress response and small RNAs. Homologs of mammalian m6A methyltransferases have not been reported in bacteria, suggesting a distinct bacterial pathway of m6A modification (Deng et al. 2015).
The MT-A fraction most likely contains METTL14 (methyltransferase-like 14) as well, which partners with METTL3 to form a heterodimer (Liu et al. 2014; Wang et al. 2014b), since monomeric METTL3 is nearly inactive (Wang et al. 2016a; Scholler et al. 2018). Beyond the mammalian cells, METTL14 orthologs are present in A. thaliana (MTB/EMB1691) and D. melanogaster (CG7818). Structural analyses revealed that METTL14 has no catalytic ability, but instead, it provides an RNA-binding platform and stabilizes METTL3 conformation in the tight asymmetric heterodimer (Wang et al. 2016a, b; Sledz and Jinek 2016). A narrow groove lined with conserved positively charged residues that are contributed by the two methyltransferase domains (MTDs) in the heterodimeric METTL3-METTL14 complex, allows single-stranded RNA to fit in, so that the METTL3-METTL14 complex can methylate target adenosine in the consensus sequence of single-stranded RNAs, rather than long stable duplex structures (Wang et al. 2016a, b; Sledz and Jinek 2016; Narayan et al. 1994). In addition to the MTDs, the methyltransferase activity requires two CCCH-type zinc fingers of METTL3 and a domain of Arginine-Glycine-Glycine (RGG) repeats on the C-terminus of METTL14 (Scholler et al. 2018; Wang et al. 2016a) (Fig. 2). Moreover, METTL3-METTL14 can form heterotetramers through the METTL14 homodimer (Ruzicka et al. 2017; Liu et al. 2014), which may affect substrate recognition or catalytic activity.

Schematic domain structures of human mRNA m6A methyltransferases
Interestingly, whereas the METTL3-containing fraction (MT-A) from Hela cell nuclear extract displayed low methyltransferase activity, the addition of an METTL3-absent, methylation-deficient fraction (MT-B) fully restored the activity (Bokar et al. 1994, 1997). There are two possibilities that may be not mutually exclusive: MT-B releases METTL3-METTL14 from an inhibitory state of MT-A; or METTL3-METTL14 interacts with the cofactors in MT-B to create a highly active “Super Methylation Complex” (SMC). Although the exact composition of MT-B fraction (875 kDa) has yet to be defined, some lines of evidence suggest that METTL3-METTL14 may form a SMC through direct or indirect interactions with the cofactors. The evolutionally conserved SMC is consisted of METTL3-METTL14 core methylation subunits and some cofactors that at least include WTAP (Wilms tumour 1-associated protein), KIAA1429 (VIRMA, vir like m6A methyltransferase associated), RBM15/RBM15B (RNA binding motif protein 15/15B), HAKAI (E3 ubiquitin-protein ligase Hakai) and ZC3H13 (zinc finger CCCH domain-containing protein 13) (Schwartz et al. 2014; Horiuchi et al. 2013; Knuckles et al. 2018; Yue et al. 2018; Zhong et al. 2008). Whereas the core methylation subunits METTL3 and METTL14 tend to have comparable stoichiometry and remain a biochemically stable complex, the cofactors are less associated with the core subunits (Knuckles et al. 2018), which suggests that a highly active SMC may be short-lived to prevent hyperactivity of the methyltransferase, that methylation on different sites is tightly controlled by cofactors, and that heterogenous SMCs may exist in cells.
HAKAI, an E3-ubiquitin ligase that interacts with MTB in A. thaliana and WTAP complex through the RING finger domain, has been shown to affect m6A levels to some extent (Ruzicka et al. 2017; Yue et al. 2018; Horiuchi et al. 2013). WTAP has no methyltransferase capacity, but it directly binds to METTL3 and is required for efficient methylation in vivo (Ping et al. 2014; Liu et al. 2014; Zhong et al. 2008). WTAP orthologs that include FIP37 in A. thaliana, Fl(2)d in D. melanogaster and Mum2 in S. cerevisiae, are all METTL3 partners. In addition, GST-WTAP has been shown to bind to the 3′ UTR of Cyclin A2 mRNA directly in vitro (Horiuchi et al. 2006), suggesting its potential role in selectivity of m6A methylation.
On the other hand, the association between RBM15/RBM15B proteins and METTL3-METTL14 is WTAP-dependent. An RBM15/RBM15B-containing SMC is likely to be recruited to RBM15/RBM15B binding sites to methylate adjacent consensus motifs (Patil et al. 2016; Kan et al. 2017). Depletion of VIRMA, another WTAP-associated protein, reduces 3′ UTR methylation and seems to increase some 5′ UTR methylation as well (Yue et al. 2018). Likewise, ZC3H13 mainly affects 3′ UTR methylation and appears to bridge RBM15/RBM15B and WTAP through its C-terminal region (Wen et al. 2018; Knuckles et al. 2018). Interestingly, loss of Zc3h13 induces translocation of a great majority of WTAP, Virma, Hakai, Mettl3 and Mettl4 from nucleus to cytoplasm, where WTAP remains to be associated with Virma, Hakai, Mettl3 and Mettl4 (Wen et al. 2018). Since m6A modification in polyadenylated RNA has only been found to occur in the nucleus (Wen et al. 2018; Ke et al. 2017; Sommer et al. 1978), ZC3H13 with a related nuclear localization of the m6A processing machinery is essential for m6A methylation in vivo. Orthologs of VIRMA, RBM15/RBM15B, and ZC3H13 have also been found in D. melanogaster, suggesting an evolutionally conserved mechanism for m6A regulation. Then, the question is if there is any SMC component that can stimulate the intrinsic METTL3-METTL14 activity. Thus far, it is not clear whether these proteins influence m6A methylation directly or indirectly since these cofactors do not exist excusively in SMC—they exert cellular functions in other complexes as well. As revealed by genetic studies in Drosophila, ime4, mettl14, YT521-B mutants are homozygous viable, whereas fl(2)d (WTAP), Virilizer (VIRMA) and Nito (RBM15/RBM15B) mutants are homozygous lethal (Kan et al. 2017), which suggests that the participation of methyltransferase in a WTAP complex is not essentially required for the function of the latter, whose defects have broad effects on the gene expression beyond m6A.
Since METTL3-METTL14 complexes prepared from E. coli, insect cells and mammalian cells display similar activities in vitro, post-translational modifications are not expected to have a substantial impact on the methylation activity (Wang et al. 2016a). Indeed, although multiple phosphorylation sites have been identified in METTL3 and METTL14, none of them seems to affect methylation (Scholler et al. 2018). However, unlike phosphorylation, SUMOylation of METTL3 in the N-terminal region near the two CCCH motifs is suggested to repress its methyltransferase activity with an unknown mechanism (Du et al. 2018).
4 METTL16 Is an m6A Methyltransferase That Prefers Structured RNAs
METTL16, the mammalian ortholog of Escherichia coli methyltransferase for A1618 in the 23S ribosomal RNA, was reported to be an m6A methyltransferase for U6 snRNA A43 methylation and for ~20% m6As in either total RNA or polyadenylated RNA in human HEK293 cells (Pendleton et al. 2017; Warda et al. 2017). It is found that 82% of the METTL16-dependent m6A sites are in introns or spanned intron-exon boundaries (Pendleton et al. 2017), and 87% of the METTL16-bound pre-mRNA introns are constitutively spliced (Warda et al. 2017). Despite a far less methylation scope relative to the METTL3-METTL14 complex, homozygous Mettl16−/− knockout only allows mouse embryonic development until blastocyst stage (E3.5) and causes developmental arrest around the time of implantation (Mendel et al. 2018). Loss of Mettl16 leads to massive transcriptome dysregulation that includes reduced mRNA levels of the SAM synthetase Mat2a (Mendel et al. 2018), which encodes a catalytic subunit (α2) in the methionine adenosyltransferase isoenzyme MAT II. METTL16 introduces m6A at six vertebrate-conserved hairpin structures in the MAT2A 3′ UTR, wherein the binding of METTL16 at hp1 regulates the expression of a nuclear retained-intron isoform fated for nuclear decay. Different from hp1, m6As in the hp2–6 cluster promote degradation of MAT2A mRNA without affecting intron retention (Pendleton et al. 2017; Shima et al. 2017; Pendleton et al. 2018). When SAM level is low, METTL16 dwells longer on hp1, which promotes the splicing of an otherwise retained proximal upstream intron to facilitate MAT2A expression and to maintain SAM homeostasis. Of note, it is the binding of METTL16 rather than its methyltransferase activity that induces MAT2A splicing through the METTL16-VCR domains (Pendleton et al. 2017) (Fig. 2). Interestingly, MAT2A protein also associates with METTL16 in mammalian cells (Shima et al. 2017), suggesting an intimate involvement of METTL16 in the SAM-sensing feedback mechanism.
Apart from its interaction with U6 snRNA, METTL16 also associates with other non-coding RNAs, such as XIST (X inactive specific transcript) and MALAT1 (metastasis associated lung adenocarcinoma transcript 1) (Pendleton et al. 2017; Warda et al. 2017). MALAT1 contains a unique triple helix structure that is specifically recognized by METTL16 (Brown et al. 2016), suggesting that METTL16 may prefer structured RNAs. Structural analyses of METTL16 show that the RNA-binding groove comprising positively charged residues from the N-terminal module and the MTD allows RNA binding for subsequent methylation of the target adenosine, which is captured by a hydrophobic pocket (Ruszkowska et al. 2018; Doxtader et al. 2018; Mendel et al. 2018). The MTD contains a polypeptide loop that adopts an autoinhibitory conformation, characterized by a key lysine residue near the SAM binding site. Destabilizing mutations within this K-loop lead to a hyperactivity of METTL16 (Doxtader et al. 2018). Although a consensus sequence for METTL16 methylation is absent, the target adenosine must be unpaired and surrounded by stems. The transition region of the RNA stem-loop is a key region affecting catalysis, which possibly involves a disordered loop within the MTD that is not required for RNA binding (Doxtader et al. 2018; Mendel et al. 2018). Interestingly, the C-terminal region of METTL16 is required for an METTL16/MALAT1 RNA triple helix interaction with a stoichiometry of 2:1 (Ruszkowska et al. 2018), but it is unclear whether the two METTL16 molecules exist as monomers and homodimer in the complex.
5 m6A Methylation Occurs Before Splicing and Depends on Transcription
The early radioactive labeling experiments by J. E. Darnell laboratory suggested that (1) m6A deposition mainly occurs in exon regions of mRNA co-transcriptionally or shortly after transcription completion (Lavi et al. 1977; Sommer et al. 1978) and (2) methylation can occur before splicing (Chen-Kiang et al. 1979). These pioneer studies defined the m6A-formation step in the course of mRNA biogenesis and processing in steady-state cells, although it is unknown when m6A occurs in cells under stress conditions, such as heat shock and ultraviolet-induced DNA damage response (Zhou et al. 2015; Xiang et al. 2017). In agreement with these concepts, METTL3 can localize to transcription active sites in an RNA-dependent manner as marked by RNAP II (RNA polymerase II) co-localization (Haussmann et al. 2016). An association between METTL3 and RNAP II was observed when cells were treated with camptothecin, an topoisomerase I inhibitor (Slobodin et al. 2017). Moreover, upon transcription inhibition by actinomycin D, WTAP nulear localization is altered, which may partially explain the m6A dependence on transcription (Little et al. 2000). On the other hand, the intronic existence of m6A in eukaryotic cells (Carroll et al. 1990; Dominissini et al. 2012), albeit at a very low abundance, suggests that at least these mRNAs are methylated before spliced. The m6A-CLIP study from Robert B. Darnell laboratory showed that a majority of m6As are added to exons of nascent pre-mRNA (Ke et al. 2017). However, ~43–49% and ~29–34% of binding sites for METTL3/METTL14/WTAP identified by PAR-CLIP largely fall into intergenic regions and introns, respectively (Liu et al. 2014). Consistent with the PAR-CLIP study, ~57% of early m6A peaks around consensus motif or motifs sharing an SAG core arose within introns by bromouridine (BrU) pulse-chase of nascent RNAs (Louloupi et al. 2018). The discrepancies could be caused by different factors that include microRNAs (Alarcon et al. 2015) in these regions, distinct kinetics of transcription elongation, RNA processing, methylation and demethylation, among others.
Despite the establishment of m6A as a primarily co-transcriptional process, the mechanisms underlying the interplay of m6A deposition and transcription are unclear. Several intriguing questions of m6A formation are begging answers: (1) only selected consensus sequences are methylated; (2) methylation is nonstoichiometric, with most genes displaying less than 50% methylation levels; (3) methylation of a gene transcript may be changed in different conditions. While the selection of primary sequences may reflect a context effect of the RNA substrates which emphasizes the interaction with SMC components, the latter two facts are likely to be contributed by different transcription activators, co-factors or regulators. This conceptual model can be supported by several lines of evidence: (1) the recruitment or displacment of METTL3 at genomic regions can be induced by transcription activators in response to stimulus or development conditions; (2) an METTL3 accumulation in nuclear speckles where most proteins are not active in RNA processing suggests an inducible interaction between METTL3 and nascent RNAs that are synthesized in the chromatin-associated regions. This model also implies that histone modifications and DNA methylation may play indirect role in m6A formation (Zhang 2018).
For the METTL3-METTL14 complex, it is tempting to hypothesize that the sequential recruitment of m6A methylation cofactors to nascent transcripts following transcription activation nucleates a pre-methylation complex (PMC), which further assembles to an active SMC upon interaction with the METTL3-METTL14 core subunits (Zhang 2018). In this transcription → PMC (without core subunits) → SMC → m6A methylation pathway, RNA-binding proteins (RBPs) with different target sequence preferences, such as RBM15 (and possibly WTAP), first bind to nascent transcripts and seed the PMC. If the methylation core subunits join, a SMC will be formed, stabilized and able to modify nearby consensus sequences around RBP binding sites. In this methylation pathway, the PMC formation on a near consensus sequence and its transition to a SMC would be rate-limiting after transcription activation. It would appear therefore that various associations between RBPs and SMC components, or potential heterogenous SMCs under different conditions, cooperatively define the precise temporal and spatial m6A landscapes.
For METTL16 methyltransferase, the relationship between m6A decoration and transcription is currently unknown. The METTL16 substrate U6 snRNA is transcribed by RNA polymerase III. An early study showed that U6 A43 can be methylated by HeLa whole cell S-100 extract with a high efficiency (Shimba et al. 1995), suggesting transcription might not be essential to this type of methylation. Whether m6A methylation of RNA pol II transcribed genes by METTL16 depends on transcription warrants further study.
6 m6A Demethylation Occurs on Nascent Transcripts in Mammalian Cells
The first identified m6A demethylase FTO was initially shown to utilize α-ketoglutarate (α-KG)-and Fe(II) to demethylate 3-methylthymine (3-meT) and 3-methyluracil (3-meU) in single-stranded DNA and RNA, respectively, with low in vitro activities (Jia et al. 2008; Gerken et al. 2007). Later, FTO was found to demethylate ~8–20% cellular m6As in mammalian cells (Wei et al. 2018a; Jia et al. 2011), with N 6-hydroxymethyladenosine (hm6A) and N 6-formyladenosine (f6A) as intermediate products during the demethylation (Fu et al. 2013). Soon after, the target spectrum of FTO in vivo was further extended to include the N 6, 2′-O-dimethyladenosine (m6Am) adjacent to the N 7-methylguanosine (m7G) cap in mRNA (Mauer et al. 2017) (Fig. 1), N 1-methyladenosine (m1A) in tRNA, m6A in U6 RNA and m6Am in several snRNAs (Wei et al. 2018a). Notably, whereas the demethylase activity of FTO on cap m6Am is higher relative to internal m6A, (Wei et al. 2018a; Mauer et al. 2017), the cellular levels of m6Am in polyadenylated RNA are only ~1/10–1/25 of that of m6A (Wei et al. 2018a). Upon demethylation, the m6Am of m7Gpppm6Am is converted to 2′-O-methyladenosine (Am) via an N 6-hydroxymethyl intermediate (m7Gppphm6Am). The consequence of demethylation was thought to reduce RNA stability (Mauer et al. 2017). However, a more careful estimation of demethylation effect on the transcripts containing m6Am without any m6A suggests no statistically noticeable increase of RNA levels upon FTO knockdown in HEK293T cells (Wei et al. 2018a). Considering the lack of m6Am in Arabidopsis and Drosophila polyadenylated RNA, whether the reversible modification near cap plays any important role in gene expression remains an open question. The other demethylase ALKBH5 has been reported to demethylate an estimated ~9–30% of total m6As in human cells (Zheng et al. 2013; Zhang et al. 2017). Both FTO and ALKBH5 belong to the mammalian AlkB family of Fe(II)- and α-ketoglutarate-dependent dioxygenases that is consisted of nine AlkB homologs (ALKBH1-8, FTO). FTO and ALKBH5 are not essential for embryonic development, but each possesses non-redundant functions in specific tissues. Fto is widely expressed in adult and fetal tissues, with expression highest in the brain. Fto−/− mice show postnatal growth retardation with a significant reduction in adipose tissue and lean body mass (Fischer et al. 2009). Alkbh5 is also widely expressed, with most abundant expression in the testes where it is essential for spermatogenesis and male fertility (Zheng et al. 2013). The characterization of ALKBH10B and ALKBH9B, two Arabidopsis ALKBH5 orthologs with an m6A demethylase activity on single-stranded RNA, indicates that demethylation occurs not solely in mammalian cells. Inactivation of ALKBH10B demethylase activity destabilizes mRNA transcripts of FT, SPL3, and SPL9, delays flowering, and represses vegetative growth (Duan et al. 2017). ALKBH9B was identified as an interactive protein with coat protein (CP) of alfalfa mosaic virus (AMV) by an yeast two-hybrid screen. ALKBH9B positively regulates AMV infection and influences m6A levels in the AMV genome. In contrast, cucumber mosaic virus (CMV) CP fails to interact with ALKBH9B. Moreover, m6A-modified viral RNAs of CMV differ from those of AMV in that neither the methylation levels nor viral infection is affected by ALKBH9B, suggesting a selectivity for substrate demethylation (Martinez-Perez et al. 2017). Interestingly, ALKBH9B exhibits a partial colocalization with siRNA bodies and P bodies in cytoplasm, thus raising the possibility of cytoplasmic demethylation in A. thaliana.
Because the mammalian demethylases were found to mainly localize in nucleus, demethylation in mammalian cells was thought to be a nuclear event in normal conditions (Jia et al. 2011; Zheng et al. 2013; Zhang et al. 2017). However, FTO is also found in the cytoplasm of some cell lines (Wei et al. 2018a). HEK293T cells expressing a cytoplasm-retained FTO mutant for lack of a nuclear localization signal at N-terminal region show reduced m6Am but not m6A in polyadenylated RNA. A more detailed analysis on nuclear and cytoplasmic fractions suggested that the FTO-mediated demethylation of m6A is prominent in the nucleus, whereas m6Am is more affected in the cytoplasm of HEK293T cells. This observation contrasts with the finding that FTO knockdown in some acute myeloid leukemia (AML) cell lines leads to increased m6A levels in both nucleus and cytoplasm, although the m6Am levels only increase in the cytoplasm (Wei et al. 2018a). These findings unambiguously favor nucleus as the primary site for FTO-mediated m6A demethylation and also point out that FTO-mediated demethylation of m6Am mainly occurs in cytoplasm. The concomitant m6A change in cytoplasm could be caused by RNA export and turnover of the m6A-methylated fraction.
Since m6A of mammalian cell mRNA is added and removed in nucleus, an interesting question is whether demethylation takes place on mature mRNA or nascent transcripts. Based on the subcellular fractionation experiments, Zhang et al. (2017) measured the levels of ALKBH5 protein and FOXM1 (Forkhead box M1) transcripts in three subcellular fractions: a cytoplasmic fraction, a soluble nuclear fraction (nucleoplasmic) and an insoluble fraction that contains chromatin and associated ternary transcription complexes (RNAPII with attached nascent transcripts). It is found that, in glioblastoma cells, FOXM1 nascent transcripts are exclusively present in the insoluble fraction, where there is also a large amount of ALKBH5 proteins. The depletion of ALKBH5 increases m6A levels on FOXM1 nascent transcripts and mature mRNAs equally. The native RNA immunoprecipitation with nuclear ALKBH5 confirmed its interaction with FOXM1 nascent rather than mature transcripts even though the latter is readily detected in nucleus, indicating that nascent transcripts attached to the chromatin are the nuclear substrates for ALKBH5 (Zhang et al. 2017). Similar subcellular fractionation experiments carried out by R.B. Darnell laboratory were further coupled with m6A-CLIP to map and quantify each m6A in RNAs from the three fractions (Ke et al. 2017). In HeLa cells, about 10% of the m6As in pre-mRNA exons disappear when these RNAs are released from chromatin, whereas >99% of nucleoplasmic m6As remain unchanged when they become cytoplasmic (Ke et al. 2017). These two studies pointed out that, in steady state, m6A demethylation takes place in the nascent transcripts associated with chromatin. However, where demethylation occurs in response to stress is not known (Xiang et al. 2017; Zhou et al. 2018). Since the demethylase FTO is active in cytoplasm where m6A demethylation rarely happens, it is tempting to speculate an unidentified factor that directs demethylases to nascent transcripts and perhaps stimulates their activities as well. The possible candidates include protein modifications, long non-coding RNAs and other trans and cis factors, depending on what is achieved from certain contexts. A relevant example that has been illustrated above, is the specific interaction of ALKBH9B and AMV CP. Similarly, viral infection induces a critical acetylation of the nuclear DEAD-box helicase member DDX46 at Lys470 that allows ALKBH5 recruitment via DDX46’s DEAD helicase domain to demethylate m6A-modified antiviral transcripts, such as MAVS, TRAF3 and TRAF6, and therefore to enforce their retention in the macrophage nucleus and to reduce their translation (Zheng et al. 2017) (the molecular mechanism of nuclear retention will be discussed later). Long non-coding RNAs with sequence complementary to pre-mRNA have also been suggested to enhance ALKBH5-mediated demethylation on specific nascent transcripts (Zhang et al. 2017).
In contrast to the preferential exonic deposition of m6A as reported by R. B. Darnell laboratory, two studies addressing FLAG-FTO targets by CLIP showed the preferential binding of FTO in introns relative to exons (Bartosovic et al. 2017; Wei et al. 2018a). It should be mentioned that the m6A consensus motif is not enriched in FTO binding sequences and that the FTO binding sites appear not to overlap with m6A sites very well (Bartosovic et al. 2017). Nevertheless, it agrees with the reports that show a large fraction of early m6As and METTL3-METTL14 binding sites in introns (Liu et al. 2014; Louloupi et al. 2018). An explanation would be these intronic m6As are extremely short-lived due to frequent demethylation. In fact, this is not entirely impossible because upon UV stress, m6A can be efficiently added by METTL3 and removed by FTO both within a few minutes (Xiang et al. 2017). Future studies should provide direct evidence to prove the emergence of a large group of intronic m6As in cells depleted of any m6A demethylase activity, if the undetectable intronic m6As are indeed because of rapid demethylation.
7 Nuclear m6A Regulates Alternative Splicing and Export
Since m6A methylation occurs before splicing completion, m6A was conceived to participate in the regulation of RNA processing a few decades ago (Stoltzfus and Dane 1982; Camper et al. 1984). However, embryonic stem cells depleted of Mettl3 have all m6A-containing constitutive exons spliced normally, with only a minor change of alternative splicing events, which suggests no common obligatory role of m6A in splicing (Ke et al. 2017). It is of importance to be noted that any change of alternative splicing by depletion of methyltransferase or demethylase cannot be simply interpreted as a direct effect of m6A, because some small non-coding RNAs involved in splicing are possibly regulated by m6A or m6Am and, as exemplified by METTL16, methylation activity is not required for its regulation of MAT2A intron retention. A direct contribution of m6A to alternative splicing is that m6A destabilizes the hairpin structure to increase the accessibility of its surrounding single-stranded RNA binding motifs to RNA binding proteins, such as heterogeneous nuclear ribonucleoprotein C (HNRNPC) and HNRNPG, which modulate the alternative splicing of some gene transcripts (Liu et al. 2015, 2017). In the case of HNRNPC, the presence of methylation in an m6A-containing stem loop enhances the HNRNPC interaction with the opposing poly-U strand that is otherwise buried within their local RNA structures. The majority of these m6A-switches are found in introns and depend on METTL3-METTL14 mediated methylation. Knockdown of HNRNPC or METTL3-METTL14 resulted in differentially expressed splice variants associated with 221 m6A-switch-containing genes in HEK293T cells (Liu et al. 2015). Similarly, m6A-switches also modulate the binding affinity of HNRNPG to RNA (Liu et al. 2017). On the other hand, the cofactors of methyltransferase with additional roles in splicing may provide a connection with m6A, such as WTAP, which facilitates alternative splicing (Ortega et al. 2003). Likewise, YT521-B, the ortholog of human m6A binding protein YTHDC1, regulates alternative splicing of Sxl in D. melanogaster (Haussmann et al. 2016; Lence et al. 2016). Moreover, Alkbh5 knockout in the mouse testes leads to several hundred of shorter 3′ UTR transcripts during spermatogenesis (Tang et al. 2018), likely due to alternative splicing or alternative polyA (APA) usage in the last exon (Ke et al. 2015). APA could be partially mediated by VIRMA, an SMC component that is associated with polyadenylation cleavage factors CPSF5 (cleavage and polyadenylation specificity factor subunit 5) and CPSF6 in an RNA-dependent manner (Yue et al. 2018). Depletion of VIRMA or METTL3 leads to 3′ UTR lengthening of a few hundred of genes in HeLa cells, indicating a potential role of m6A in the selection of proximal polyadenylation sites (Yue et al. 2018). Again, despite the correlation, unresolved is if the m6A within 3′ UTR per se regulates APA, or vice versa.
After splicing is completed, RNA export may also be influenced by m6A. A delayed appearance of mRNA in the HeLa cell cytoplasm caused by methyltransferase inhibitors was reported by Rottman laboratory based on radioactive labeling (Camper et al. 1984). Subsequent studies showed that a delayed cytoplasmic appearance of bPRL mRNA was accompanied with nuclear accumulation of precursor RNA (Carroll et al. 1990). Similarly, the application of methylation inhibitors leads to a prolonged nuclear retention of two key clock genes, PER2 (period circadian clock 2) and ARNTL (aryl hydrocarbon receptor nuclear translocator like), and thereby an altered schedule of gene expression that results in an elongated circadian period (Fustin et al. 2013). On the other hand, RNA export is enhanced by ALKBH5 knockdown (Zheng et al. 2013), which may be mediated by YTHDC1, the only nuclear exclusive YTH domain family member. Despite its role in D. melanogaster, YTHDC1 mainly binds to exons (Xu et al. 2014) and has very minor effect on splicing in Hela cells (Roundtree et al. 2017b). Depletion of YTHDC1 induces a nuclear accumulation of mature m6A-marked mRNAs and a decrease in their cytoplasmic appearance. The nuclear export function of YTHDC1 is mediated by an RNA-independent interaction between the C-terminal YTHDC1 and SRSF3 (serine/arginine-rich splicing factor 3), which interacts with the canonical mRNA export receptor NXF1 (nuclear RNA export factor 1) (Roundtree et al. 2017b).
8 Cytoplasmic m6A Regulates mRNA Stability and Translation Efficiency
Before the advance of the “omics” era, biochemical studies detected a positive correlation between m6A decoration and mRNA decay (Friderici et al. 1976; Sommer et al. 1978). These radioactive labeling experiments provided the first evidence of the functional relevance of m6A. The principle of m6A-mediated post-transcriptional gene regulation is that RBPs recruited or repelled by m6As modify the fate of mRNA, such as decay and translation, depending on the function of RBP per se or through the association with a traditional pathway. Among these RBPs, the proteins harboring a YTH domain are described as the m6A “readers” that specifically recognize and regulate m6A-decorated transcripts (Dominissini et al. 2012; Wang et al. 2014a). The mammalian YTH domain proteins that have been largely characterized by He laboratory include the cytoplasmic YTHDF1, YTHDF2, YTHDF3, YTHDC2 and the nuclear YTHDC1, all of which employ their conserved aromatic cage to recognize m6A (Xu et al. 2014; Zhu et al. 2014). With the list of “reader” proteins extended by other RBPs that are absent of a YTH domain, a critical regulatory network has emerged that impacts multiple aspects of mRNA homeostasis, which cannot be achieved by the primary sequence alone.
8.1 m6A and mRNA Decay
The first confirmed m6A reader-mediated effect on gene expression arose from the mechanistic studies of YTHDF2 (Wang et al. 2014a). YTHDF2 accelerates mRNA deadenylation and degradation, with the C-terminal YTH domain specifically recognizing m6A-methylated RNA and the N-terminal region interacting with the CCR4–NOT deadenylase complex through CNOT1 (CCR4–NOT transcription complex subunit 1) (Wang et al. 2014a; Du et al. 2016). In early life of zebrafish embryos, the depletion of Ythdf2 stabilizes m6A-modified maternal mRNAs and delays maternal-to-zygotic transition (Zhao et al. 2017). Likewise, Ythdf2 is maternally required for oocyte maturation (Ivanova et al. 2017). Moreover, Ythdf2 depletion leads to a disturbed m6A-dependent degradation of neural development-related mRNA targets, and impaired proliferation and differentiation capabilities of neural stem/progenitor cells, consequently leading to defective neurogenesis. Homozygous Ythdf2 −/− mice in C57BL/6 background are embryonic lethal, with the majority of Ythdf2 −/− embryos lost between E14.5 and E18.5. Semi-lethality was observed for the Ythdf2 +/− mice, with one third of the surviving mice having malfunctioning eyes (Li et al. 2018). It is not surprising that depletion of m6A methyltransferase, accompanied with a failure of m6A-induced decay of methylated transcripts, would give rise to similar or more severe phenotypes. Indeed, a loss of m6A methyltransferase exhibits significant impacts on mouse ESC differentiation and preimplantation development (Batista et al. 2014; Wang et al. 2014b; Geula et al. 2015). Mettl3 knockout mice display more severe early embryo lethality phenotypes than Ythdf2 knockout, with E5.5-E7.5 Mettl3 −/− embryos having abnormal characteristics (Geula et al. 2015), indicating additional mechanisms for m6A-mediated effects as discussed later. Interestingly, depending on the dominance of already-expressed genes in different cell states to be stabilized, the loss of methyltransferase in murine ESCs can lead to opposite cell fates, i.e., an improved self-renewal in naїve state but a defective cell regeneration in primed state (Wang et al. 2014b; Geula et al. 2015; Batista et al. 2014). In general, mRNAs that encode key proteins with regulatory functions tend to have more m6As with faster turnover (Wang et al. 2014b; Ke et al. 2017). A proposed model describes that m6A induces rapid clearance of a group of mRNAs encoding key regulators, such as transcription factors, in a coordinated manner during the transition of cell states (Roundtree et al. 2017a). Additionally, m6A may destabilize some transcripts by repelling RBPs without a YTH domain, such as G3BP1 (G3BP stress granule assembly factor 1) and HuR (ELAV-like protein 1) (Edupuganti et al. 2017; Wang et al. 2014b).
Not only m6A methyltransferase and its cofactors are evolutionally conserved, the “readers” coevolve from yeast to mammals as well (Schwartz et al. 2013). A phylogenetic analysis of 297 YTH-containing proteins from 32 representative species suggests that 57 of them belong to the DC group and 240 to the DF group (Scutenaire et al. 2018). Eleven of the 13 members of the Arabidopsis YTH protein family, namely ECT1-11 for Evolutionarily Conserved C Terminus, fall into the YTHDF clade. One of the two YTHDC-type proteins possesses three highly conserved N-terminal zinc fingers in addition to the YTH domain and is a member of the plant polyadenylation complex (CPSF30; AT1G30460). ECT2 is required for trichome branching and involved in the redundant control of timing of leaf formation with ECT3 (Arribas-Hernandez et al. 2018). Surprisingly, by formaldehyde-assisted crosslinking of ECT2 and associated RNA in A. thaliana, G. Jia laboratory found that the majority of ECT2-binding sites are located within the 3′ UTR of mRNA containing a unique URUAY motif (R=G>A, Y=U>A, where the majority [over 90%] UGUAY)(Wei et al. 2018b). ECT2 appears to bind methylated UGUAA and methylated GGACU with similar affinities (Wei et al. 2018b), suggesting the sequence bias may reflect the relative abundance of methylated motifs in the cells, and in line with this, Arabidopsis nuclear extracts exhibit higher methylation activity for UGUA relative to GGACU. However, the RRACH is more significantly enriched than UGUAY in overall m6A peaks, and interestingly, UGUAY is identical to a specific UGUAACA methylation signature near the start codon (Luo et al. 2014). The ECT2 binding motifs also share sequence similarity with far upstream elements that regulate polyadenylation, likely lending evidence of a potential regulatory role in RNA processing (Wei et al. 2018b). However, ECT2 mostly accumulates in the cytoplasm with a diffuse pattern or aggregates in stress granules upon heat stress (Scutenaire et al. 2018; Arribas-Hernandez et al. 2018), which suggests its function on cytoplasmic RNAs. This is partially evidenced by ECT2-dependent stabilization of some RNA transcripts associated with trichome morphogenesis (Wei et al. 2018b).
ECT2 is not the first reader that was found to stabilize m6A-marked transcripts. In mammalian cells, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs; including IGF2BP1/2/3) were reported to enhance RNA stability and translation under normal and stress conditions through the K homology domains that recognize m6A residues (Huang et al. 2018). Moreover, depletion of FTO appears to increase the levels of some m6A-methylated transcripts (Wei et al. 2018a). Therefore, despite the m6A-mediated destabilization of a majority of transcripts, a subgroup of transcripts become more stable, which diverts the fates of mRNAs for dynamic physiological roles.
8.2 m6A and mRNA Translation
In bacterial translation systems, m6A within coding regions acts as a barrier to tRNA accommodation during translation elongation (Choi et al. 2016). However, m6A was found to associate with enhanced translation efficiency of dihydrofolate reductase transcripts (Tuck et al. 1999). Recent studies support the positive role of m6A in translation through several mechanisms. First, the cap-dependent translation can be enhanced by the m6A-bound YTHDF1 through recruitment of eIF3 (translation initiation factor complex 3) (Wang et al. 2015). Interestingly, YTHDF1 was found to associate with G3BP1 in an RNA-dependent manner (Wang et al. 2015). Although genetic deletion of YTHDF1 allows mice to develop normally at least to four months of age, these mice show learning and memory defects, impaired hippocampal synaptic transmission and long-term potentiation due to the loss of YTHDF1-dependent expedited translation of m6A-methylated neuronal mRNAs in response to neuronal stimulation in the hippocampus (Shi et al. 2018). Moreover, YTHDF3 also promotes translation through interaction with YTHDF1 (Shi et al. 2017). Surprisingly, cytoplasmic METTL3 enhances translation of mRNAs bearing m6As that are close to stop codon independently of its methyltransferase activity or YTHDF1. In cytoplasm, in addition to a direct METTL3-m6A interaction, the N-terminal region of METTL3 binds eIF3h (translation initiation factor 3 subunit h) to facilitate mRNA looping, thus promoting translation of a large subset of oncogenic mRNAs, such as BRD4 (bromodomain-containing protein 4) in human primary lung tumors (Lin et al. 2016; Choe et al. 2018). Second, m6As within 5′ UTR can be recognized by eIF3, which recruits the 43S complex and initiates translation in a cap- and internal ribosome entry site (IRES)-independent manner (Meyer et al. 2015; Zhou et al. 2015). ABCF1 (ATP-binding cassette sub-family F member 1) and YTHDF3 appear to contribute to this type of m6A-facilitated mRNA translation to some extent (Coots et al. 2017). Other RBPs implicated in translation include YTHDC2 that increases translation efficiency (Hsu et al. 2017), and FMR1 (fragile X mental retardation 1) (Edupuganti et al. 2017), a polyribosome-associated neuronal RNA-binding protein associated with Fragile X syndrome that represses translation by stalling ribosome translocation.
Elucidating m6A function at different conditions is central for unraveling the role of m6A in more complex processes. Upon heat shock, YTHDF2 translocates into nucleus and binds to the m6A within 5′ UTR, which blocks FTO-mediated demethylation and hence enhances translation of stress-induced transcripts (Zhou et al. 2015). During integrated stress response, the 5′ UTR-located m6A controls ribosome scanning and start codon selection, while demethylation promotes the reinitiation of ATF4 (activating transcription factor 4) translation (Zhou et al. 2018). These complex m6A-mediated pathways of translational regulation undoubtedly add new dimensions to the dynamic effects of m6A in relation to gene expression.
9 m6A Methyltransferases and Demethylases Play Important Roles in Cancer
The comprehensive functional analyses for m6A regulators in cancer were initiated by an FTO study in leukemia and an ALKBH5 study in glioblastoma (Li et al. 2017; Zhang et al. 2017), and have been extended by a plethora of studies focused on the dysregulation of m6A methylation and associated proteins. FTO is highly expressed in several subtypes of AMLs, including those that carry t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA translocation, FLT3 gene internal tandem duplication (ITD) mutations and/or NPM1 mutation. FTO demethylates 3′ or/and 5′ UTR-located m6As on the transcripts of ASB2 (ankyrin repeat and SOCS box containing 2) and RARA (retinoic acid receptor alpha), which encode key regulators in all-trans-retinoic acid (ATRA)-induced differentiation of leukemia cells. It therefore enhances ASB2 and RARA mRNA decay and induces downregulation of these two genes. The oncogenic role of FTO in leukemic cell transformation and leukemogenes was confirmed by a series of in vitro and in vivo assays (Li et al. 2017). Interestingly, the oncogenic activity of FTO is inhibited by 2-hydroxyglutarate (R-2HG), an oncometabolite competitive inhibitor of α-KG-dependent dioxygenases that is produced by the cancer-associated mutant IDH1/2 (isocitrate dehydrogenase 1/2) (Su et al. 2018). In leukemic cells that highly express FTO with a “medium” level of MYC, FTO affects the stability of MYC transcripts by changing m6A levels at MYC 5′ UTR and CDS regions, which confers R-2HG sensitivity in these cells. However, hyperactivation of MYC-associated pathways renders leukemic cells resistant to R-2HG. On the other hand, in glioblastoma multiforme (GBM), ALKBH5 is highly expressed in the patient-derived glioma stem cells (GSCs) and informs poor survival for GBM patients (Zhang et al. 2017). ALKBH5 depletion impaires GSC growth in vitro and in vivo due to the loss expression of FOXM1, an essential transcription factor for GSC self-renewal and proliferation. ALKBH5 binds to and demethylates the 3′ UTR of FOXM1 nascent transcripts, thereby enhancing FOXM1 mRNA expression through interaction with HuR, an abundant nuclear RBP that promotes pre-mRNA expression. The demethylation seems to be facilitated by a long noncoding RNA transcribed antisense to the FOXM1, which harbors an overlapping region with FOXM1 3′ UTR.
Interestingly, although these demethylases behave as oncogenes in these cancers, the methyltransferase complex does not simply play the opposite role, i.e., METTL3-METTL14 being oncogenic in AMLs (Barbieri et al. 2017; Vu et al. 2017; Weng et al. 2018). Studies from different laboratories suggest that METTL3-METTL14 complex may employ multiple mechanisms to activate oncogenic pathways that at least include some key transcription factors, such as MYC (Barbieri et al. 2017; Vu et al. 2017; Weng et al. 2018). Starting with a catalytic activity domain-focused Cas9-induced indel mutation screen in mouse primary leukemia cells, Barbieri et al. (Barbieri et al. 2017) confirmed the requirement of METTL3, METTL14 and METTL16 for cell growth in ten human AML cell lines. They also found that METTL3 and METTL14 can be crosslinked to TSSs of distinct groups of coding genes that are decorated with bimodal trimethylation of lysine 4 on histone H3 (H3K4me3) marks by chromatin immunoprecipitation assays. The promoter-bound METTL3, likely recruited by CEBPZ (CCAAT enhancer binding protein zeta), enhances translation of target genes without affecting mRNA levels, such as the transcription factors SP1 and SP2, which activate the transcription of MYC oncogene. Similarly, Vu et al. (2017) reported METTL3 as an essential gene to maintain the undifferentiated state and survival of hematopoietic stem/progenitor cells (HSPCs) and myeloid leukemia cell lines. METTL3 depletion reduces translational efficiency and causes decreased levels of MYC, BCL2 and PTEN (phosphatase and tensin homolog) protein expression in AML cells. Weng et al. (2017) showed that METTL14 is expressed at high levels in HSPCs and AML cells carrying t(11q23), t(15;17), or t(8;21) and is downregulated during myeloid differentiation. METTL14 sustains the development and maintenance of AMLs and self-renewal of leukemia stem/initiation cells through promoting stability and translation of MYB and MYC mRNAs. METTL14 itself is transcriptionally suppressed by the transcription factor SPI1. In addition, mutations of methyltransferases have also been implicated in tumor development. The hotspot R298P mutation of METTL14 in endometrial tumors is correlated with reductions in m6A methylation, and promotes proliferation and tumorigenicity of endometrial cancer cells through activation of the AKT pathway (Liu et al. 2018).
The connections of m6A and tumor development are not limited to aforementioned types of tumor. A growing body of studies have indicated that multiple types of cancer take advantage of the tunable regulatory mechanisms to survive and propagate, through mutations or aberrant expression of components of the m6A regulatory network (e.g., writers, erasers and readers).
10 Conclusion and Future Perspective
mRNA m6A methylation was discovered over 40 years ago. However, the attempts from RNA biologists to understand the function and regulation of this abundant modification at molecular, cellular and tissue levels have just started in very recent years, because of the important biological functions of reversible RNA methylation revealed by a few groups (Zheng et al. 2013; Li et al. 2017; Zhang et al. 2017) and the advance of new technologies afforded by high-throughput sequencing. A current view of m6A on mRNA is that it is deposited, removed, sensed and perhaps regulated by several conserved machineries from fly to human, and that m6A plays distinct roles in life when incorporated into diverse biological processes. In Fig. 3, a simplified model of METTL3-METTL14-mediated m6A deposition is hypothesized—the recruitment of a Pre-Methylation Complex (PMC) with subsequent assembly to a Super Methylation Complex (SMC) allows efficient m6A methylation at consensus sequence adjacent to the RBP binding sites. The intracellular and extracellular signals that trigger transcription allow m6A methylation for specific genes, while the selection of consensus motif to be modified is decided by the PMC (Fig. 3a). The signals may also regulate PMC component expression, modification, or localization, etc., and as a result, m6A distribution. On the other hand, the “free” methyltransferase core subunits have intrinsic ability to bind and catalyze single-stranded RNA with low activity, likely giving rise to abortive methylation in the absence of PMC or low methylation “noise” in cells (Fig. 3b). Therefore, the sequential recruitment of SMC components ensures accurate m6A deposition in a large number of transcripts. After methylation, m6A demethylation occurs on a subset of nascent transcripts in mammalian cells, which is regulated by a poorly understood mechanism. Depending on the functions of the RBPs that display higher or lower binding affinities to m6A-marked transcripts, a wide range of events throughout RNA life are influenced by m6As that include RNA processing, localization, decay and translation, which altogether modulate gene expression for dynamic physiological functions. Elucidating how m6A affects gene expression not only aids our understanding of essential physiology but also enables novel therapeutic approaches for human diseases.

A hypothetical model of METTL3-METTL14-dependent m6A deposition. The overlaps of proteins indicate confirmed or proposed interactions
References
Alarcon CR, Lee H, Goodarzi H et al (2015) N6-methyladenosine marks primary microRNAs for processing. Nature 519:482–485
Arribas-Hernandez L, Bressendorff S, Hansen MH et al (2018) An m(6)A-YTH module controls developmental timing and morphogenesis in arabidopsis. Plant Cell 30:952–967
Barbieri I, Tzelepis K, Pandolfini L et al (2017) Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552:126–131
Bartosovic M, Molares HC, Gregorova P et al (2017) N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3′-end processing. Nucleic Acids Res 45:11356–11370
Batista PJ, Molinie B, Wang J et al (2014) m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15:707–719
Bokar JA, Rath-Shambaugh ME, Ludwiczak R et al (1994) Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem 269:17697–17704
Bokar JA, Shambaugh ME, Polayes D et al (1997) Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3:1233–1247
Brown JA, Kinzig CG, DeGregorio SJ et al (2016) Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc Natl Acad Sci USA 113:14013–14018
Bujnicki JM, Feder M, Radlinska M et al (2002) Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol 55:431–444
Camper SA, Albers RJ, Coward JK et al (1984) Effect of undermethylation on mRNA cytoplasmic appearance and half-life. Mol Cell Biol 4:538–543
Carroll SM, Narayan P, Rottman FM (1990) N6-methyladenosine residues in an intron-specific region of prolactin pre-mRNA. Mol Cell Biol 10:4456–4465
Chen K, Lu Z, Wang X et al (2015) High-resolution N(6)-methyladenosine (m(6)A) map using photo-crosslinking-assisted m(6)A sequencing. Angew Chem Int Ed Engl 54:1587–1590
Chen-Kiang S, Nevins JR, Darnell JE Jr (1979) N-6-methyl-adenosine in adenovirus type 2 nuclear RNA is conserved in the formation of messenger RNA. J Mol Biol 135:733–752
Choe J, Lin S, Zhang W et al (2018) mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561:556–560
Choi J, Ieong KW, Demirci H et al (2016) N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol 23:110–115
Clancy MJ, Shambaugh ME, Timpte CS et al (2002) Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res 30:4509–4518
Coots RA, Liu XM, Mao Y et al (2017) m(6)A facilitates eIF4F-independent mRNA translation. Mol Cell 68:504–514 e7
Deng X, Chen K, Luo GZ et al (2015) Widespread occurrence of N6-methyladenosine in bacterial mRNA. Nucleic Acids Res 43:6557–6567
Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA 71:3971–3975
Dominissini D, Moshitch-Moshkovitz S, Schwartz S et al (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201–206
Doxtader KA, Wang P, Scarborough AM et al (2018) Structural basis for regulation of METTL16, an S-adenosylmethionine homeostasis factor. Mol Cell 71:1001–1011.e4
Du H, Zhao Y, He J et al (2016) YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun 7:12626
Du Y, Hou G, Zhang H et al (2018) SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res 46:5195–5208
Duan HC, Wei LH, Zhang C et al (2017) ALKBH10B is an RNA N(6)-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 29:2995–3011
Edupuganti RR, Geiger S, Lindeboom RGH et al (2017) N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24:870–878
Fischer J, Koch L, Emmerling C et al (2009) Inactivation of the Fto gene protects from obesity. Nature 458:894–898
Friderici K, Kaehler M, Rottman F (1976) Kinetics of Novikoff cytoplasmic messenger RNA methylation. Biochemistry 15:5234–5241
Fu Y, Jia G, Pang X et al (2013) FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat Commun 4:1798
Fustin JM, Doi M, Yamaguchi Y et al (2013) RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155:793–806
Gerken T, Girard CA, Tung YC et al (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318:1469–1472
Geula S, Moshitch-Moshkovitz S, Dominissini D et al (2015) Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347:1002–1006
Haussmann IU, Bodi Z, Sanchez-Moran E et al (2016) m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540:301–304
Horiuchi K, Umetani M, Minami T et al (2006) Wilms’ tumor 1-associating protein regulates G2/M transition through stabilization of cyclin A2 mRNA. Proc Natl Acad Sci USA 103:17278–17283
Horiuchi K, Kawamura T, Iwanari H et al (2013) Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem 288:33292–33302
Hsu PJ, Zhu Y, Ma H et al (2017) Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res 27:1115–1127
Huang H, Weng H, Sun W et al (2018) Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20:285–295
Ivanova I, Much C, Di Giacomo M et al (2017) The RNA m(6)A Reader YTHDF2 is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell 67:1059–1067 e4
Jia G, Yang CG, Yang S et al (2008) Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett 582:3313–3319
Jia G, Fu Y, Zhao X et al (2011) N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7:885–887
Kan L, Grozhik AV, Vedanayagam J et al (2017) The m(6)A pathway facilitates sex determination in Drosophila. Nat Commun 8:15737
Kane SE, Beemon K (1987) Inhibition of methylation at two internal N6-methyladenosine sites caused by GAC to GAU mutations. J Biol Chem 262:3422–3427
Ke S, Alemu EA, Mertens C et al (2015) A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev 29:2037–2053
Ke S, Pandya-Jones A, Saito Y et al (2017) m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31:990–1006
Knuckles P, Lence T, Haussmann IU et al (2018) Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev 32:415–429
Lavi U, Fernandez-Munoz R, Darnell JE Jr (1977) Content of N-6 methyl adenylic acid in heterogeneous nuclear and messenger RNA of HeLa cells. Nucleic Acids Res 4:63–69
Lence T, Akhtar J, Bayer M et al (2016) m(6)A modulates neuronal functions and sex determination in Drosophila. Nature 540:242–247
Li Z, Weng H, Su R et al (2017) FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell 31:127–141
Li M, Zhao X, Wang W et al (2018) Ythdf2-mediated m(6)A mRNA clearance modulates neural development in mice. Genome Biol 19:69
Lin S, Choe J, Du P et al (2016) The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62:335–345
Linder B, Grozhik AV, Olarerin-George AO et al (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12:767–772
Little NA, Hastie ND, Davies RC (2000) Identification of WTAP, a novel Wilms’ tumour 1-associating protein. Hum Mol Genet 9:2231–2239
Liu J, Yue Y, Han D et al (2014) A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10:93–95
Liu N, Dai Q, Zheng G et al (2015) N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518:560–564
Liu N, Zhou KI, Parisien M et al (2017) N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res 45:6051–6063
Liu J, Eckert MA, Harada BT et al (2018) m(6)A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol 20:1074–1083
Louloupi A, Ntini E, Conrad T et al (2018) Transient N-6-methyladenosine transcriptome sequencing reveals a regulatory role of m6A in splicing efficiency. Cell Rep 23:3429–3437
Luo GZ, MacQueen A, Zheng G et al (2014) Unique features of the m6A methylome in Arabidopsis thaliana. Nat Commun 5:5630
Martinez-Perez M, Aparicio F, Lopez-Gresa MP et al (2017) Arabidopsis m(6)A demethylase activity modulates viral infection of a plant virus and the m(6)A abundance in its genomic RNAs. Proc Natl Acad Sci USA 114:10755–10760
Mauer J, Luo X, Blanjoie A et al (2017) Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature 541:371–375
Mendel M, Chen KM, Homolka D et al (2018) Methylation of structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol Cell 71:986–1000. e11
Meyer KD, Saletore Y, Zumbo P et al (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149:1635–1646
Meyer KD, Patil DP, Zhou J et al (2015) 5′ UTR m(6)A promotes cap-independent translation. Cell 163:999–1010
Narayan P, Ludwiczak RL, Goodwin EC et al (1994) Context effects on N6-adenosine methylation sites in prolactin mRNA. Nucleic Acids Res 22:419–426
Ortega A, Niksic M, Bachi A et al (2003) Biochemical function of female-lethal (2)D/Wilms’ tumor suppressor-1-associated proteins in alternative pre-mRNA splicing. J Biol Chem 278:3040–3047
Patil DP, Chen CK, Pickering BF et al (2016) m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537:369–373
Pendleton KE, Chen B, Liu K et al (2017) The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169:824–835.e14
Pendleton KE, Park SK, Hunter OV et al (2018) Balance between MAT2A intron detention and splicing is determined cotranscriptionally. RNA 24:778–786
Perry RP, Kelley DE, Friderici K et al (1975) The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5′ terminus. Cell 4:387–394
Ping XL, Sun BF, Wang L et al (2014) Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24:177–189
Roundtree IA, Evans ME, Pan T et al (2017a) Dynamic RNA modifications in gene expression regulation. Cell 169:1187–1200
Roundtree IA, Luo GZ, Zhang Z et al (2017b) YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6:e31311
Ruszkowska A, Ruszkowski M, Dauter Z et al (2018) Structural insights into the RNA methyltransferase domain of METTL16. Sci Rep 8:5311
Ruzicka K, Zhang M, Campilho A et al (2017) Identification of factors required for m(6) A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol 215:157–172
Schibler U, Kelley DE, Perry RP (1977) Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J Mol Biol 115:695–714
Scholler E, Weichmann F, Treiber T et al (2018) Interactions, localization, and phosphorylation of the m(6)A generating METTL3-METTL14-WTAP complex. RNA 24:499–512
Schwartz S, Agarwala SD, Mumbach MR et al (2013) High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155:1409–1421
Schwartz S, Mumbach MR, Jovanovic M et al (2014) Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8:284–296
Scutenaire J, Deragon JM, Jean V et al (2018) The YTH domain protein ECT2 is an m(6)A reader required for normal trichome branching in arabidopsis. Plant Cell 30:986–1005
Shi H, Wang X, Lu Z et al (2017) YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 27:315–328
Shi H, Zhang X, Weng YL et al (2018) m(6)A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563:249–253
Shima H, Matsumoto M, Ishigami Y et al (2017) S-Adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep 21:3354–3363
Shimba S, Bokar JA, Rottman F et al (1995) Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res 23:2421–2426
Sledz P, Jinek M (2016) Structural insights into the molecular mechanism of the m(6)A writer complex. Elife 5:e18434
Slobodin B, Han R, Calderone V et al (2017) Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell 169:326–337.e12
Sommer S, Lavi U, and Darnell J E, Jr. (1978) The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J Mol Biol 124:487-499
Stoltzfus CM, Dane RW (1982) Accumulation of spliced avian retrovirus mRNA is inhibited in S-adenosylmethionine-depleted chicken embryo fibroblasts. J Virol 42:918–931
Su R, Dong L, Li C et al (2018) R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172:90–105 e23
Tang C, Klukovich R, Peng H et al (2018) ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci USA 115:E325–EE33
Tuck MT, Wiehl PE, Pan T (1999) Inhibition of 6-methyladenine formation decreases the translation efficiency of dihydrofolate reductase transcripts. Int J Biochem Cell Biol 31:837–851
Vu LP, Pickering BF, Cheng Y et al (2017) The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23:1369–1376
Wang X, Lu Z, Gomez A et al (2014a) N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505:117–120
Wang Y, Li Y, Toth JI et al (2014b) N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16:191–198
Wang X, Zhao BS, Roundtree IA et al (2015) N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161:1388–1399
Wang P, Doxtader KA, Nam Y (2016a) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63:306–317
Wang X, Feng J, Xue Y et al (2016b) Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534:575–578
Warda AS, Kretschmer J, Hackert P et al (2017) Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep 18:2004–2014
Wei CM, Moss B (1977) Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry 16:1672–1676
Wei J, Liu F, Lu Z et al (2018a) Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell 71(973–985):e5
Wei LH, Song P, Wang Y et al (2018b) The m(6)A Reader ECT2 controls trichome morphology by affecting mRNA stability in arabidopsis. Plant Cell 30:968–985
Wen J, Lv R, Ma H et al (2018) Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell 69:1028–1038.e6
Weng H, Huang H, Wu H et al (2017) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22:191–205
Weng H, Huang H, Wu H et al (2018) METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22:191–205 e9
Xiang Y, Laurent B, Hsu CH et al (2017) RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543:573–576
Xu C, Wang X, Liu K et al (2014) Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 10:927–929
Yue Y, Liu J, Cui X et al (2018) VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 4:10
Zhang S (2018) Mechanism of N(6)-methyladenosine modification and its emerging role in cancer. Pharmacol Ther 189:173–183
Zhang S, Zhao BS, Zhou A et al (2017) m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31:591–606.e6
Zhao BS, Wang X, Beadell AV et al (2017) m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Zheng G, Dahl JA, Niu Y et al (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49:18–29
Zheng Q, Hou J, Zhou Y et al (2017) The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat Immunol 18:1094–1103
Zhong S, Li H, Bodi Z et al (2008) MTA is an arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20:1278–1288
Zhou J, Wan J, Gao X et al (2015) Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526:591–594
Zhou J, Wan J, Shu XE et al (2018) N(6)-methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell 69:636–647 e7
Zhu T, Roundtree IA, Wang P et al (2014) Crystal structure of the YTH domain of YTHDF2 reveals mechanism for recognition of N6-methyladenosine. Cell Res 24:1493–1496
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zhang, S. (2019). The Role of mRNA m6A in Regulation of Gene Expression. In: Jurga, S., Barciszewski, J. (eds) The DNA, RNA, and Histone Methylomes. RNA Technologies. Springer, Cham. https://doi.org/10.1007/978-3-030-14792-1_14
Download citation
DOI: https://doi.org/10.1007/978-3-030-14792-1_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14791-4
Online ISBN: 978-3-030-14792-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)