
Abstract
Urothelial carcinoma is a tumor type featuring pronounced intertumoral heterogeneity and a high mutational and epigenetic load. The two major histopathological urothelial carcinoma types – the non-muscle-invasive and muscle-invasive urothelial carcinoma – markedly differ in terms of their respective typical mutational profiles and also by their probable cells of origin, that is, a urothelial basal cell for muscle-invasive carcinomas and a urothelial intermediate cell for at least a large part of non-muscle-invasive carcinomas. Both non-muscle-invasive and muscle-invasive urothelial carcinomas can be further classified into discrete intrinsic subtypes based on their typical transcriptomic profiles. Urothelial carcinogenesis shows a number of parallels to a urothelial regenerative response. Both of these processes seem to be dominated by specific stem cell populations. In the last years, the nature and location of urothelial stem cell(s) have been subject to many controversies, which now seem to be settled down, favoring the existence of a largely single urothelial stem cell type located among basal cells. Basal cell markers have also been amply used to identify urothelial carcinoma stem cells, especially in muscle-invasive disease, but they proved useful even in some non-muscle-invasive tumors. Analyses on molecular nature of urothelial carcinoma stem cells performed till now point to their great heterogeneity, both during the tumor development and upon intertumoral comparison, sexual dimorphism providing a special example of the latter. Moreover, urothelial cancer stem cells are endowed with intrinsic plasticity, whereby they can modulate their stemness in relation to other tumor-related traits, especially motility and invasiveness. Such transitional modulations suggest underlying epigenetic mechanisms and, even within this context, inter- and intratumoral heterogeneity becomes apparent. Multiple molecular aspects of urothelial cancer stem cell biology markedly influence therapeutic response, implying their knowledge as a prerequisite to improved therapies of this disease. At the same time, the notion of urothelial cancer stem cell heterogeneity implies that this therapeutic benefit would be most probably and most efficiently achieved within the context of individualized antitumor therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Urothelium
- Urothelial stem cells
- Urothelial regenerative response
- Lineage-tracing
- Lineage-depletion
- Urothelial carcinoma
- Urothelial carcinoma heterogeneity
- Non-muscle-invasive bladder cancer
- Muscle-invasive bladder cancer
- Intrinsic subtypes
- Urothelial carcinoma sexual dimorphism
- Urothelial carcinoma stem cells
- Stemness signaling pathway
- Sonic hedgehog
- Wnt/β-catenin pathway
- SOX-2
- STAT-3
- COX-2
- YAP-1
- Epithelial-mesenchymal transition
- Epigenetic plasticity
- DNMT-1
8.1 Cellular Heterogeneity Within Normal Urothelium
Urothelium represents a specialized pseudostratified epithelium that lines the urogenital ductal system from the ureters, through the bladder, to the proximal urethra. Its major task is to provide a practically impermeable blood-urine barrier. As is true for any other tissue, this specialized function is provided by terminally differentiated cells—superficial or umbrella cells lining the urothelial lumen. They are very large (50–100 μm), postmitotic, frequently bi- or multinucleated cells, and the blood-urine barrier function is mostly attributable to the function of highly specialized proteins—uroplakins. Assembling into a specific structure on the apical pole of umbrella cells called AUM (asymmetric unit membrane) plaques, uroplakins create an efficient barrier against water, solutes, and toxins that concentrate in the urine. In addition, adjacent superficial cells are connected by tight-junction proteins, limiting the possibility of pericellular transport. Last but not least, umbrella cells are not just a static barrier, but respond to changing bladder tonus by undergoing the regulated processes of endocytosis and exocytosis of discoidal fusiform-shaped vesicles (DFVs).
Beneath the superficial layer is one or several layers of intermediate cells, which are significantly smaller than umbrella cells (about 20 μm in diameter), sitting on a single layer of small (5–10 μm in diameter) cuboidal basal cells directly in contact with the basement membrane. Some of the intermediate cells are also in direct contact with the basement membrane, hence the notion of urothelium as transitional epithelium. Due to these very different size relations, relative representations of these three urothelial cell types differ dramatically, basal cells being the by far most prevalent cell type (about 90%). The histological analysis of whole mount adult mouse urothelium revealed that a single superficial cell could span the area of about 40 intermediate and basal cells (reviewed in Balsara and Li 2017).
What function do intermediate and basal cells serve? A traditional view has been to regard different urothelial cell types within the context of gradual differentiation. Accordingly, basal cells would include adult stem cells, intermediate cells would constitute transit-amplifying (TA) precursor cells, and umbrella cells correspond to terminally differentiated cells (Hatina and Schulz 2012). This tentative hierarchical model proved to be rather difficult to test experimentally, for several reasons. First, the small size and the resulting very high numbers of basal cells immediately arouse the question as to which of them actually correspond to stem cells; it is barely thinkable that 90% of cells in a tissue can represent stem cells. Urothelial stem cells are thus expected to constitute just a minority of basal cells, most of the basal cells corresponding likely to TA-precursor cells. Indeed, basal cells greatly differ in terms of their relative proliferation activity, a minority of cells being quiescent over a long period of time while the majority proliferating. This became apparent when the entire population of urothelial cells were forced to incorporate a specifically modified nucleotide (bromodeoxyuridine [BrdU]) and then observed for a long time. Over 90% of basal cells diluted BrdU gradually as they divided, while only about 9% preserved it because of their quiescence. Apparently, these minor LRC (label-retaining cells) are good candidates for urothelial stem cells (Kurzrock et al. 2008).
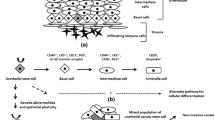
The second problem is the unusually slow physiological turnover of adult urothelium; the umbrella cells are surprisingly long-lived, their lifespan under physiological conditions reaching about 200 days or even a year. To experimentally study the lineage succession in urothelium, researchers frequently resort to studying either prenatal urothelial development or pathophysiological regenerative processes. Indeed, despite the largely quiescent state under physiological conditions, urothelium is able to mount a surprisingly rapid regenerative response to a variety of injuries. Experiments on lineage tracing and lineage depletion, performed largely in mice exposed to various types of urothelial damage, can help decipher an intrinsic cellular hierarchy. As explained in detail in other chapters of this treatise (Chap. 1), specific loci and associated gene regulatory sequences are used to drive expression of tamoxifen-inducible Cre-recombinase (CreERT2), in order to indelibly label a certain population of cells with a marker protein, lineage succession being then derived from a gradual inheritance of this marker protein expression by cells issued from this original recombined cell population. Alternatively, Cre-recombinase can unleash the expression of the diphtheria toxin receptor gene, and, if treated with the diphtheria toxin, the targeted Cre-recombinase expressing cells are specifically eliminated (Blanpain 2013). Implicit in using the lineage tracing or lineage depletion strategies is of course a thorough knowledge of genes and proteins whose expression is restricted to specific urothelial cell populations (Fig. 8.1). Such genes and proteins can thus serve as specific lineage markers and – by the same token – as specific drivers of CreERT2 inducible recombinase in a restricted and well-defined population of urothelial cells. Lineage-tracing experiments in mouse urothelium experienced an interesting evolution, which, for a certain period of time and in fact until recently, lead to two opposing theories on the identity of adult urothelial stem cells.
One landmark study used Sonic hedgehog (Shh) as the CreERT2 driver gene and uropathogenic Escherichia coli infection as a type of urothelium damage to induce a regenerative response. This model helped to discover a remarkable epithelial-mesenchymal reciprocal interaction. Uropathogenic E. coli infection leads to a rapid exfoliation of umbrella and most intermediate cells. The remaining basal cells respond by the secretion of the signaling molecule Sonic hedgehog, which signals to the lamina-propria and localized fibroblasts. They are activated and signal back to the urothelium with mitogenic (Wnt 2, Wnt 4, Fgf 16) and differentiation-inducing (Bmp-4 and -5) factors; the model assumes that the former have a shorter range of action, thus primarily inducing basal cell proliferation, whereas the latter can signal to a longer distance, thus reaching newly generated intermediate cells and inducing their differentiation (Shin et al. 2014b). Using Shh as Cre-ERT2 driver in lineage depletion experiments then led to slow urothelial degeneration over a period of 8–10 months, again reflecting the slow cell turnover of normal urothelium (Shin et al. 2014a). Finally, urothelium failure and the resulting death of manipulated mice ensued, apparently because urothelial stem cells have been eliminated. This model thus seems to corroborate the traditional view of urothelial differentiation, placing urothelial stem cells among the basal cells, gradually differentiating into intermediate cells and – eventually – into umbrella cells.
As elegant as this model may appear, it was not universally accepted. The major objection was that Shh was not a basal cell-specific marker, but its expression was shared by intermediate cells (Fig. 8.1). Using Krt 5 as a Cre-ERT2 driver gene instead seemed to yield quite a different picture (Gandhi et al. 2013). In response to a chemical damage induced by cyclophosphamide treatment applied once, twice or three times in succession, the authors did not evidence any succession of the labeled basal—intermediate—umbrella cells. But when uroplakin 2A served as a CreERT2 driver, vertical labeled units of intermediate and umbrella cells were observed. These results led the authors to form an alternative hypothesis placing two independent and autonomous cell populations, each replenished by different stem cells, into the adult urothelium. Accordingly, basal cells are one of the autonomous specific urothelial cell populations, with its own stem cells, and the intermediate cells include an independent stem cell population that self-renews and differentiates into umbrella cells. The study evidenced also an epithelial-mesenchymal crosstalk: suburothelial fibroblasts expressed enzymatic machinery to synthetize retinoic acid, which diffused to the urothelium and promoted the differentiation of umbrella cells (Gandhi et al. 2013).

The expression of CK5, CD44, and P-cadherin is uniform among all basal cells, unlike of CK14, which is expressed by a relatively small fraction of them. The expression of CK14 and CK17 may become ubiquitous throughout the entire urothelium, however, and this pattern signals squamous transdifferentiation. Source: (Wang et al. 2017; Balsara and Li 2017)
This conceptual quarrel about the identity of urothelial stem cell(s) seems now to have settled down, favoring the first model. Another experiment used the more restrictive basal stem cell marker Krt 14 as a CreERT2 driver and five rounds of cyclophosphamide treatment. Vertical labeled units spanned the entire thickness of urothelium, evidencing that basal layer stem cells can differentiate into umbrella cells. In vitro explant culture experiments on Krt 14–CreERT2-driven lineage depletion corroborated the exclusive requirement of Krt 14 expressing cells for urothelial regenerative activity (Papafotiou et al. 2016).
Why did these results differ? Part of the answer might lie in technical reasons – such as different mouse strains or a subtle difference in tamoxifen application – which can influence the effectiveness of Cre-mediated recombination and be one source of result variability between different studies. Indeed, two independent groups (Papafotiou et al. 2016; Schäfer et al. 2017) repeated the lineage tracing with Krt 5-CreERT2 as a driver and evidenced what the first analysis (Gandhi et al. 2013) failed to prove: the full-thickness urothelium differentiating units.
Other reasons may have a likely biological background. First and foremost, the different studies used different types of urothelial injury, resulting in different degrees and durations of the urothelial damage. Research on other organ systems (see Chap. 4) helped to discover two important phenomena: That not every regenerative response needs to resort to the activation of respective stem cells, and that the mobilization and proliferation of transit-amplifier precursors is a preferred way to mend relatively shorter and/or milder tissue damage. Indeed, the experiments that evidenced the basally located urothelial stem cells used either prolonged chemical damage, resulting in exfoliation of upper urothelial cell layers, or full-thickness urothelial injury induced surgically. In addition, different kinds of urothelial injuries vary in terms of the degree of activation of an inflammatory response (Wang et al. 2017), which might be one factor possibly affecting stem cells as well.
Finally, the results obtained by analyzing human urothelium, based on either a common pattern of X-chromosome inactivation or shared mitochondrial mutations (Gaisa et al. 2011), also point to the linear differentiation model, with urothelial stem cells localized among basal cells and intermediate cells serving as TA-precursors, eventually differentiating into umbrella cells.
As shown above, the knowledge of the cell population dynamics of adult urothelium has been improving recently. The same is not entirely true of embryonic and fetal urothelium, where our knowledge is still more scarce. Together with hindgut, urothelium starts to develop from endoderm around the middle of the mouse intrauterine development (i.e., from the embryonic day 10 [E10]), the whole process being regulated by the master stemness transcription factor p63 (Pignon et al. 2013). The adult hierarchical organization, with the CK 5-uniformly positive basal layer and CK 14-positive fraction of it, becomes evident only towards the end of gestation, from E16.5 (Gandhi et al. 2013; Paraskevopoulou et al. 2016). Interestingly, the CK 14-positive fraction is much higher in late embryonic and early postnatal phases, accounting for 20% and 30% of total urothelial cells, respectively, and sharply declining during further postnatal life (reaching 3.5% at 8 weeks) and adulthood (0.9% at 1 year of age) (Paraskevopoulou et al. 2016), a dynamics well reminiscent of a process of tissue aging, that is, a gradual decrease in stem cell abundance and competence. Until the appearance of the adult pattern of urothelial hierarchical organization, there are several unique stem cell populations specific for discrete stages of embryonic and fetal urothelial development. Well characterized are P-cells, which feature the unique marker profile Foxa2+ Upk+ p63+ Shh+ CK5−. Abundant between E11 and E13, at E14 P-cells are succeeded by I-cells, characterized by the typical marker profile Foxa2− Upk+ p63+ Shh+ CK5−. It comes as little surprise that suburothelial-mesenchyme—derived retinoic acid is also involved in the differentiation of embryonic and fetal urothelium, as revealed by the expansion of P-cells upon the urothelial-specific expression of a dominant negative mutation in the retinoic acid receptor alpha gene and their persistence until E14 (Gandhi et al. 2013). This means that adult regenerative response and embryonic and fetal development are governed by partially overlapping molecular machineries.
There is one more – perhaps even more flagrant – example of this overlap between the embryonic and regenerative urothelium biology. Between the E14 and immediate postnatal period (i.e., with a certain overlap between the P-cell– and I-cell—dominated urothelium and adult urothelial cell hierarchy), basal and intermediate cells strongly express the developmental transcription factor Sox-9. Afterwards, its expression becomes undetectable, only in order to be reactivated in regenerating urothelium. The underlying signaling pathway is the EGFR-triggered MAPK-pathway (Ras-Raf-ERK). Two signaling events – probably acting successively – activate it in basal urothelial cells. First, urine itself contains sufficient concentrations of epidermal growth factor (EGF) and other members of this growth factor family to activate EGFR. The point is that EGFR is basally located and umbrella cells in intact urothelium provide a sufficient barrier precluding any access of urine-borne growth factors to their basally located receptors. The immediate consequence of a urothelial injury is the extensive exfoliation of the umbrella cell layer—and therefore also of this signaling barrier. Second, promptly after the injury, the remaining urothelial cells activate the autocrine expression of several members of the EGF family (especially amphiregulin, HB-EGF, epiregulin, and epigen), further intensifying the regenerative response (Ling et al. 2011). Interestingly in this respect, phosphorylated ERK has been found as a specific molecular feature of stem cells in non-muscle-invasive papillary urothelial carcinomas (Hepburn et al. 2012); what needs to be explored is whether SOX-9 reactivation also follows and if yes, whether SOX-9 directly participated in cancer stem cell biology.
All the cases of urothelial regeneration discussed by now could resort to preexisting stem or potentially also progenitor cells that were either spared by the injury (caused by uropathogenic E. coli infections or various types of chemical injuries) or were available from the neighboring areas of uninjured urothelium (surgical injury). Can urothelium regenerate if urothelial stem or progenitor cells are directly damaged? A Shh-Cre- driven knockout of the gene coding for DNA-methyltransferase 1 seems to be such a case. So the answer is yes, it can, by employing a very unusual regeneration mechanism (Joseph et al. 2018). The Shh-directed Dnmt1 knockout results in global DNA hypomethylation, which triggers DNA damage, immediately followed by the activation of the DNA damage response pathway, eventually leading to apoptosis induction. The resulting acellular gaps along the basement membrane are filled by cells originating from the Wolffian duct, which subsequently transdifferentiate into fully competent urothelial cells; tissue recombination experiments showed that embryonic mesenchyme is crucial in this process. Wolffian ducts differentiate into seminal ducts, epididymis, and vas deferens in males; in females, they regress shortly before birth. Consequently, this unusual type of urothelial regeneration can happen in both sexes practically during the whole embryonic and fetal period. Unfortunately, the Shh-Cre- Dnmt1 knockout mice die shortly after birth, for reasons unrelated to the urogenital system. This precludes any direct analysis (even that of its existence) of this special urothelial regeneration mechanism (at least in males) during postnatal life. Therefore, any link to a somewhat similar human proliferative benign disease of the bladder known as nephrogenic adenoma of the bladder (Pavlidakey et al. 2010) could not be experimentally analyzed by now. In conclusion, depending on the type, extent, and timing of urothelial injury, stem and/or progenitor cells initiating a regenerative response can be quite heterogeneous.
8.2 Urothelial Carcinoma
Urothelial carcinoma represents a frequent malignancy, with a remarkable difference in terms of the incidence between the sexes, ranking as fourth most common cancer in men and 11th in women, with worldwide annual incidence exceeding 430,000 cases and mortality exceeding 160,000 cases (Antoni et al. 2017). For decades, it has been well established that urothelial carcinoma is a very heterogeneous disease, with the main histopathological discriminant being provided by the invasion into the muscle layer of the bladder. About 75% of the cases are diagnosed as non-muscle-invasive bladder cancer (NMIBC), either without any invasive properties registered at diagnosis (stage pTa) or with the invasion limited to lamina propria only (stage pT1). The remaining 25% are diagnosed as muscle-invasive bladder cancer (MIBC), whose depth of invasion and extent of spread are the major determinants of clinical staging (pT2–pT4). As many as 80% of NMIBC tumors behave clinically quite indolently, but many of them have a tendency to recur, necessitating their long-term surveillance by regular cystoscopies. Not only does such a surveillance procedure cause a substantial discomfort to patients, but also repeated cystoscopies are costly: Considering a total cost from diagnosis to death, bladder cancer is the most expensive cancer type. Ten to fifteen percent of originally NMIBCs ultimately progress to MIBCs, and the most important clinical parameter to forecast this dismal behavior is tumor grade. Several grading systems have been conceived over years, the most recent being the 2004 WHO grading system. It differentiates between low- and high-grade tumors based on architectural and cytologic atypia, and, in addition to them, introduces a histopathological diagnosis of the papillary urothelial neoplasm of low malignant potential (PUNLMP); in fact, the papillary growth pattern prevails among NMIBCs. The most telling clinical variable for MIBC is, as mentioned above, the disease stage. The overall 5-year survival rate is close to 50% for all stages, but only about 5% for metastatic disease. Most MIBCs, however, do not develop from progressing NMIBCs but rather along a separate pathway with a particular high-grade superficial neoplasia termed carcinoma in situ (pTis, also abbreviated as CIS) as a specific precursor lesion (Kamat et al. 2016).
Not only are NMIBC and MIBC clinically and pathologically different, they also develop – according to the current consensus – along different molecular and cellular pathways, a concept known as dual-track carcinogenesis. By the molecular pathway, we mainly understand genetic and epigenetic landscape—and indeed, mutational and epigenetic changes specific to either urothelial carcinoma type have been described. Most NMIBCs rely on the mutational activation of mitogenic signaling, especially the MAPK pathway. The major mutational hit is the type 3 receptor for fibroblast growth factors (FGFR3), mutations resulting mostly in its ligand-independent activation, which might be accompanied by overexpression. Alternatively, downstream signaling molecules are changed, like Ras (activation) or Notch (inactivation); the latter has a rather specific role in bladder cancer, activating ERK-phosphatases and thereby acting as a tumor suppressor. Further mitogenic changes have been detected, like cyclin D and E2F3 gene amplifications, the latter usually in high-grade tumors. Genome stability seems to be preserved, and p53 mutations are rare and confined to high-grade tumors as well. Their frequency is much higher in MIBC, complemented by pRb and PTEN mutations and genomic instability. Interestingly, among the most frequently mutated genes in both NMIBC and MIBC are those coding for chromatin-modifying enzymes that act epigenetically, with a surprising specificity for either tumor type. Inactivating mutations in the gene coding for H3K27 demethylase KDM6A are common in NMIBC whereas inactivating mutations in H3K4 methyltransferase MLL2 are quite common in MIBC; interestingly, both mutations seem to be mutually exclusive, suggesting common biological consequence(s). Indeed, since H3K27me3 is a repressive histone mark and H3K4me3 is an activating histone mark, both mutations could have a similar overall effect, namely that of preserving a generally silenced chromatin state (Choi et al. 2017).
The lineage-tracing and lineage-depletion experiments described in the previous section enabled us to define an additional important difference between NMIBC and MIBC, namely in terms of their respective cells of origin. The same transgenic mouse combinations as those discussed above were also exposed to urothelial-specific chemical carcinogen N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN) over extended periods of time. In the Shh-CreERT2 lineage-traced mice, BBN treatment led to the tumorigenic transformation of Shh-expressing cells into muscle-invasive carcinomas preceded by carcinoma-in-situ—like lesions, a process entirely prevented by their lineage depletion (Shin et al. 2014b). As noted above, since Shh lacks an exquisite specificity for basal urothelial cells, including stem cells, the definitive conclusion on the cell-type-specific origin of MIBC was here somewhat problematic. The same conclusion was independently drawn after a prolonged BBN treatment of Krt 5-CreERT2 lineage labeled mice, nevertheless. In contrast, Upk2-CreERT2 lineage labeled cells transformed exclusively into papillary NMIBC (Van Batavia et al. 2014). Consequently, the dual-track carcinogenic pathway outlined above should be extended to include also the different respective cells of origin for either type of urothelial carcinomas.
Interestingly, if lineage tracing was activated by tamoxifen treatment after CIS lesions have formed, a heterogeneous cellular pattern became apparent; obviously, CIS preserved the capacity of limited differentiation in form of Shh− progeny of Shh+ precursor cells. Strikingly, only the Shh+ cells presented tumor-initiating capacity, a crucial functional property of cancer stem cells. Apparently, Shh expression is crucial for early invasive carcinoma development.
It has been independently suggested that BBN treatment might initiate a pseudoregenerative response, Shhhigh cells of CIS lesions might thus have a selective advantage to the surrounding normal urothelium (Shin et al. 2014b). Interestingly in this regard, the explant outgrowth of CK14+ cells is uniquely dependent on β-catenin (Papafotiou et al. 2016), which is a downstream signal resulting from Shh expression within the context of urothelial regenerative response (Shin et al. 2011). As a result of this possible selective advantage, Shh+ CIS-cells can spread within a (histologically normal) urothelium, occupying its large areas. Surprisingly at the first look, if the lineage-tracing onset (i.e., tamoxifen application) was further delayed, up to the invasive carcinoma stage, the Shh-induced signal was completely lost. As advantageous as the Shh expression might be at the CIS stage, it appears to be disadvantageous at the invasive carcinoma stage, and hence the selection pressure to downregulate the Shh expression later during invasive carcinoma development (Shin et al. 2014b).
What could be the biological basis of this process? As specified above, later part of the urothelial regenerative response is mediated by differentiation-inducing factors secreted by suburothelial mesenchymal stromal cells under the influence of a Shh signal. These factors, especially Bmp-4 and -5, act as a break to tumor progression (Shin et al. 2014a, b). In addition, a targeted overexpression of the constitutively active gain-of-function mutant allele of β-catenin in urothelial basal cells resulted in low-grade papillary carcinomas (Lin et al. 2013), apparently violating the above-formulated rule of cells of origin specific to different carcinoma types. Plausibly, as beneficial as β-catenin might be for proliferation and intraurothelial spread, it might act inhibitory with regard to invasion. There presently is no consensus, however, about the exact impact of β-catenin activation in urothelial carcinogenesis. The above conclusion is thus entirely hypothetical, although some additional indices appear to support it, especially regarding the sexual dimorphism of urothelial carcinoma (see below). From another point of view, the dynamics of Shh expression could provide a clear example of heterogeneity of urothelial cancer stem cells in the course of tumor progression: In CIS, cancer stem cells feature Shh expression, while in invasive carcinomas, different markers and/or drivers characterize them.
A crucial question is whether these results of sophisticated experiments in mice are relevant for human urothelial carcinoma. First, SHH expression has been reported to be really downregulated in human MIBC, supporting the above outlined carcinogenic sequence (Shin et al. 2014b). Second, a specific bioinformatic tool has been constructed, combining stem cell-specific expression signatures of human invasive carcinoma stem cells, umbrella-basal cell-specific transcriptomic profile, NMIBC- and MIBC-specific mutations, and a variable factor of common vs distinct cell of origin. This tool was confronted with publicly available datasets including both NMIBC and MIBC cases. As a result, a better discriminating power has been achieved if distinct cells of origin were assumed, largely corroborating the mouse studies described above. Interestingly, NMIBC showed a rather perfect concordance with an expression signature characterizing expression changes in induced pluripotent stem cells, whereas, not surprisingly, MIBC-derived CSC-specific expression signatures as well as the umbrella-basal cell-specific transcriptome, were successful in identifying MIBC samples (Dancik et al. 2014). We could plausibly interpret these findings as indicating that cancer stemness is in MIBC, by and large, “inherited” from normal urothelial basal cells, while in NMIBC cancer stem cells seem to be generated de novo by dedifferentiation process—yet another clear demonstration of CSC heterogeneity in urothelial carcinoma.
8.3 Intrinsic Subtypes of Urothelial Carcinoma
Both NMIBC and MIBC are very heterogeneous groups—clinically, histopathologically, and molecularly. The clinical differences mostly concern an intrinsic propensity to progress on the one hand and a therapeutic response on the other. The difference in terms of a progression tendency is most dramatically expressed in NMIBCs, only a fraction of which showing a capacity to progress into muscle-invasive disease. Within both NMIBCs and MIBCs, individual tumors markedly differ also regarding their therapeutic responses. There are cases with an excellent sensitivity to Bacillus Calmette-Guérin (BCG) immunotherapy, the standard of care after the transurethral resection of NMIBC, as well as to cisplatin-based combination chemotherapy, a main systemic therapeutic option for MIBC. There are, however, primary refractory tumors that fail to respond at all, and there are initially sensitive tumors that develop resistance during treatment (Kamat et al. 2016).
Histopathologically, especially MIBC can present a lot of variants. The most common is squamous differentiation, which may in fact refer to the differentiation plasticity of normal urothelium, where squamous metaplasia of the trigon region of the bladder is quite common; interestingly, such squamous metaplasia may aggravate upon vitamin A deficiency. Up to 60% of MIBC cases may show focal squamous changes. The consensus is to diagnose these tumors as urothelial transitional carcinoma with squamous differentiation, and to reserve the diagnosis of squamous cell carcinoma to those rare cases in which squamous histopathology extends across the entire tumor (below 5% of cases). Likewise, focal glandular differentiation is also not entirely seldom (about 6% of MIBC cases), while pure adenocarcinoma is much rarer. There are a handful of additional histopathological variants (micropapillary, plasmocytoid, nested), all of which representing a diagnostic rarity (Chan and McConkey 2015; Kamat et al. 2016).
Assessing the underlying biology of urothelial carcinoma heterogeneity has only been possible with the advent of methods for characterizing complex transcriptomes. Landmark studies, for example, on diffuse large B-cell lymphoma and breast carcinoma, revealed that histopathologically uniform tumors can be classified into subtypes that clinically behave as distinct disease entities. Strikingly, differential expression of genes implicated in the normal differentiation of original cell types – that is, B-lymphocytes and mammary epithelial cells, respectively – turned out as a major discriminator, which allowed to identify processes of stemness and differentiation as crucial for intrinsic tumor subtypes (McConkey et al. 2015). Indeed, early studies in urothelial carcinoma transcriptome clearly demonstrated that NMIBC and MIBC are molecularly distinct disorders, and – notably within the context of this book – that transcriptomes of low- and high-grade urothelial carcinomas are very different. Several groups have independently approached the issue of MIBC intrinsic molecular subtypes, their results differing in terms of both the number of subtypes recognized and their nomenclature (Choi et al. 2017); within the context of this chapter, we will adopt the classification provided by The University of Texas M.D. Anderson Cancer Center. They identified two major subtypes, basal and luminal (Choi et al. 2014). Interestingly, this classification is remarkably reminiscent of (and also biologically substantiated, far beyond the issue of nomenclature) breast cancer molecular subtypes (Damrauer et al. 2014). Basal MIBCs are enriched for basal urothelial cell markers (CD44, P-cadherin, CK5, and CK14), EGFR, and squamous markers (CK6A, CK6B, CK6C, and CK16). Bioinformatic analysis of transcription factors revealed that p63, STAT-3, HIF-1, and NF-κB were involved. A fraction of basal MIBCs showed also upregulation of proteins responsible for epithelial-mesenchymal transition (EMT—Twist 1/2, Snai2, Zeb2, and vimentin). In contrast, luminal MIBC transcriptome was enriched for CK20 (a marker of umbrella cells, but also of CIS), CD24, FOXA1 (transcriptional activator of uroplakin genes), FGFR3, PPAR-γ, and GATA-3, the last two being the major transcription factors implicated. Luminal MIBC cases were also enriched for FGFR3 activating mutations, suggesting that they could correspond to progressed NMIBC. The analogous analysis of NMIBC resulted in the description of three subtypes (Hedegaard et al. 2016), termed subtype 1, 2, and 3. Subtype 2 bears the highest risk of progression. This subtype featured upregulated expression of late cell cycle genes, a feature that also turned out to be shared with luminal MIBC. The expression of CK14 was moderately increased, and subtype-2 tumors were also enriched for a CIS signature. Very interestingly, the expression of other “notorious” basal cell markers (CD44 and SHH) was rather downregulated in subtype-2 NMIBC tumors, whereas a distinct set of (bladder) cancer stem cell genes – including aldehyde dehydrogenases, CD133, nestin, and CD90 – were clearly upregulated, as was the expression of EMT factors. The expression pattern of urothelial basal and stemness genes among the intrinsic subtypes thus provides another glimpse on the heterogeneity of urothelial cancer stem cells.
Finally, a third subtype of MIBC has been revealed, called p53-like (Choi et al. 2014), after its characteristic expression profile reminiscent of wild-type p53 response genes. p53 mutations were, nevertheless, as common in p53-like tumors as in the two other subtypes, making the mechanism of specific upregulation of p53-downstream genes in this subtype rather mysterious. Notably, p53-like tumors show chemoresistance, and even initially chemosensitive basal or luminal tumors may switch to the p53-like subtype when manifesting acquired resistance. Besides p53-downstream genes, p53-like tumor-specific gene expression profile includes genes suggesting an enhanced activity of stromal cells, especially carcinoma-associated fibroblasts (CAFs). Adding to the issue of the stem-cell heterogeneity, this subtype of tumors may thus correspond to “acquired stemness” due to epithelial-mesenchymal interaction, as issue well known from the biology of normal urothelial cells and of the urothelial regenerative response (see above).
Of course, the question surfaces as to whether these subtypes are stable, and if they are, then to what extent: Are they really intrinsic, or can they change during tumor progression? A quite frequent switch to the p53-like subtype in the wake of chemotherapy could provide an example of the latter possibility. Another example was published recently. Most CIS cases could be classified as the luminal subtype, and they frequently progress to the basal subtype (Barth et al. 2018). Interestingly, as described above, this “subtype switch” coincides with a “stemness switch,” from Shh-dependent to Shh-independent. The question whether this is a mere coincidence or there is a mechanistic relationship between both these switches remains open.
8.4 Urothelial Carcinoma Stem Cells and Their Heterogeneity
As follows from the above discussion, urothelial carcinoma could be regarded as a stem cell disease. A link between a regenerative response – another stem-cell–dominated phenomenon – and urothelial carcinoma has been recognized a long time ago (reviewed in Hatina and Schulz 2012). As heterogeneous as urothelial carcinoma—considered as a clinical disease—clearly is, its stem cells would be as well. We can approach this heterogeneity of cancer stem cells at several levels, of which the first one may well regard their origin. As suggested above, the urothelial carcinoma stem cells might be of dual origin. In muscle-invasive tumors, which – according to the current consensus – originate from basally located urothelial stem cells, cancer stem cells are probably directly derived from normal stem cells; a lot of stemness mechanisms can thus passively pass from their cell of origin. On the other hand, superficial papillary carcinomas are believed to derive from intermediate cells. They practically always develop, however, a morphological architecture quite reminiscent of normal urothelium, including a basal layer. This similarity extends from morphological resemblance to common antigenic determinants, as evidenced by early experiments from 1980s (Dotsikas et al. 1987). This might be taken to challenge the notion of intermediate cells being the universal cell of origin for these tumors. However, if we embrace this notion of the universal cell-of-origin, the only thinkable explanation for the existence of basal-like cells in NMIBC would be that the papillary carcinoma basal layer regenerates by dedifferentiation, probably under a crucial microenvironmental influence. Therefore, paradoxically, urothelial carcinoma stem cells might appear, at least at the level of expression of certain basal cell-specific proteins, more homogeneous than they really are (see below).
It follows that basal cell markers have been always considered as good candidates to approach urothelial carcinoma stem cells. CD44 was the first (Chan et al. 2009), and it turned out to be applicable for only a fraction of tumors, but this fraction, in which CD44+ cells marked serially tumorigenic (=cancer stem) cells included a single pTa tumor as well, corroborating the conclusion above. The problem with CD44 might be that its expression might become uncoupled from stemness in a large proportion of NMIBC cases, where it can become ubiquitous, or the opposite may be true and it can be entirely lost, as is rather frequent in MIBC (Sugino et al. 1996). Anyway, concentrating on those tumors, where CD44 marks probable urothelial carcinoma stem cells, a pronounced heterogeneity as to the molecular mechanism of stemness preservation has been noticed. About 5% of cancer samples had activated β-catenin in their CD44+ cells, 20% expressed nuclear Bmi-1, 40% activated nuclear STAT-3, and 80% GLI-1, a transcription factor downstream of SHH. It is clear, however, that CD44+ urothelial CSCs represent just one subset. Independently, another basal stem cell marker, 67 KDa laminin receptor (67LR) has been used (He et al. 2009) and comparison of specific gene expression profiles identified between CD44+ and CD44− cells on one hand and 67LRbright and 67LRdim cells on the other hand clearly showed that these represent different stem cell subsets (Dancik et al. 2014), adding to the molecular heterogeneity discovered within CD44+ cells.
With the discovery and characterization of the intrinsic subtypes of both NMIBC and MIBC (see above), another level of stem-cell heterogeneity became apparent. Strikingly, the defined intrinsic subtypes feature widespread expression (i.e., so abundant that it becomes characteristic for bulk tumors, not just in cancer stem cells after a specific enrichment procedures) of certain stemness genes. Basal MIBCs thus show the abundant expression of basal stem cell markers (CD44, P-cadherin, CK5, and CK14) (Choi et al. 2014). If we accept the current view that a large proportion of luminal tumors derive from cluster 2 NMIBCs, similarly abundant expression of a completely different stemness genes (ALDH1A1, ALDH1A2, CD133, nestin, and CD90) (Hedegaard et al. 2016) becomes manifest. Does the widespread expression of these specific groups of stemness genes in the specific intrinsic subtypes result from stem cell expansion? Or does it rather reflect the uncoupling of expression of those genes from (still rare) urothelial cancer stem cells?
A special case of a stemness mechanism might be provided by CIS. As described above, CIS stem cells seem to be uniquely dependent on the Shh signaling. Could this explain a high proportion of CD44+ urothelial cancer stem cells relying on active GLI-factors? At best for a minor part probably, as CIS lesions were not among the tumors from which CD44+-urothelial CSCs were isolated and molecularly analyzed; we can perhaps only admit the simultaneous existence of CIS and a full-blown carcinoma, clinically not too rare a situation (McConkey et al. 2010). Consequently, alternative mechanisms explaining the activation of the GLI-transcription factor should be provided. For example, it has been shown that the Hedgehog pathway can be activated by chronic arsenic exposure, a well-known urothelial carcinoma carcinogen (Fei et al. 2010). The activation of nuclear STAT-3 might directly result from a stromal influence. It has been reported that urothelial cancer stem cells (in this case defined as CD14+ cells) are able to actively recruit myeloid cells and promote their differentiation into tumor-associated macrophages (TAMs) (Cheah et al. 2015). TAMs signal back to cancer cells by multiple mechanisms, including the secretion of inflammatory cytokines like Interleukin-6, a known activator of STAT-3 and a factor of adverse clinical prognosis in bladder cancer (Chen et al. 2013). Even arsenic, again, could be a signal to stimulate IL-6 secretion (Luo et al. 2013). Another possible source of a signal potentially leading to STAT-3 activation could be EGFR, which is overexpressed in basal-subtype MIBC (Choi et al. 2014).
A special example of microenvironmental promotion of urothelial carcinoma stem cells can be seen with certain primary chemoresistant tumors (Kurtova et al. 2015). The chief source of the stemness-promoting signals in this case, however, is not the stromal cells but the very urothelial carcinoma cells, somewhat reminiscent of the EGF-mediated regenerative response discussed above. Well-preserved cell hierarchy plays an essential mechanistic role. Cancer stem cells are endowed with specific mechanisms of self-protection, some of which have been also experimentally exploited to purify such cells, for example, the side population assay targeting the multidrug resistance efflux pumps or Aldefluor assay® targeting aldehyde dehydrogenases (Hatina et al. 2018). Consequently, as an immediate effect of chemotherapy, CSCs survive and non-CSCs begin to die. This death of non-CSCs is accompanied by the activation of a specific gene expression program, the wound response signature. One of the prominent genes activated is PTGS2, coding for cyclooxygenase-2 (COX-2), the enzyme catalyzing the synthesis of prostaglandin E2. PGE2 is consequently released from the dying cells, and it signals back to the surviving CSCs, stimulating their rapid entry into cell cycle. A malicious corollary is that the dying cells are immediately replenished by the progeny of surviving and proliferating stem cells, an elegant variation of the regenerative response discussed above, misused by the cancer to keep the cancer cell population largely constant; the tumor thus behaves as primarily refractory. One of the magics of this discovery is that the pharmacological inhibition of this specific cancer regenerative response is feasible: Celecoxib is an FDA-approved drug to inhibit COX-2, and in experiment it has been able to essentially thwart this repopulation-mediated chemoresistance, thus calling for expedite translational and clinical development (Kurtova et al. 2015).
Additional mechanisms of stemness preservation and promotion within the context of chemotherapy have been described. One attractive candidate is the Hippo-pathway downstream transcription factor YAP-1. In fact, YAP-1 has been found activated (i.e., nuclear) in a great proportion of chemoresistant urothelial carcinomas (Ciamporcero et al. 2016), and it promotes cancer stemness by directly activating SOX-2 (Ooki et al. 2017); indeed, SOX-2 has been independently described as a specific factor responsible for stemness in Aldefluor-bright cells of invasive urothelial carcinoma cell lines (Ferreira-Teixeira et al. 2015). Interestingly, the COX-2–PGE2 signaling seems to independently activate SOX-2 as well, via a let-7-miRNA-HMGA2 pathway, well characterized, for example, in sarcomas (Hatina et al. 2019): the PGE2 signal leads to the downregulation of the let-7 miRNA gene, resulting in the derepression of HMGA2 mRNA and the subsequent activation of the SOX-2 gene. The COX-2-PGE2 and YAP signaling pathways are connected by an intricate feedback loop, nonetheless. On the one hand, the PTGS2 gene coding for COX-2 is the YAP-1 downstream gene. On the other hand, YAP-1 is characterized by a pronounced antiapoptotic effect—which apparently could, on its own, mediate chemoresistance (Ciamporcero et al. 2016)—but, at the same time, this apoptosis inhibition limits the PGE2 signal (PGE2 is only released from dying cells) (Ooki et al. 2017). To make the things even a little bit more complicated, the COX-2-PGE2 signaling seems to independently activate STAT-3, too (Liu et al. 2016), and both COX-2-PGE2 signals and SOX-2 seem to be also activated by arsenic (Ooki et al. 2018). The molecular biology of urothelial carcinoma stem cells is thus coined by two recurrent themes: heterogeneity and signaling convergence.
A very special case of urothelial cancer and CSC heterogeneity is provided by the sexual dimorphism of the disease. Urothelial carcinoma is about four times more common in men than in women, yet the mortality is comparable, implying that women tend to have more aggressive disease (Kamat et al. 2016). A special biological mechanism has been recently proposed to explain these differences, providing another variation on the theme of signaling pathways convergence. The key has been provided by several mouse models that could reproduce various aspects of this sexual dimorphism. One of them was based on urothelial-specific knockout of the Foxa1 gene; FOXA1 is a transcription factor essential for urothelial differentiation, participating in the transcriptional activation of uroplakin genes, whose loss and/or mutation portend an unfavorable prognosis to bladder cancer patients (Reddy et al. 2015). Interestingly, a close relative, FOXA2 – whose expression is typical for P-cells during embryonic urothelial development (see above) and which is no more detectable in adult urothelium – is occasionally reactivated in urothelial carcinoma, a phenomenon that could be tentatively called “FOXA-switch.” Foxa1 urothelial-specific deletion resulted in umbrella cell damage, which could initiate a regenerative response, leading to different outcomes in male and female mice: basal cell hyperplasia in the former and keratinizing squamous metaplasia in the latter. Gene expression profiling and subsequent bioinformatic analysis to reveal upstream regulatory factors pertinent for the sex-specific divergent lesions yielded β-catenin as a master gene regulatory factor for male-specific basal cell hyperplasia and p63 for female-specific squamous metaplasia (Reddy et al. 2015). Both could be regarded as stemness regulators, providing an unexpected and unique example of stem-cell heterogeneity in urothelial precancerous lesions. Recall that both of them are involved in various aspects of urothelial development and regenerative response. Strikingly, the urothelial basal cell-specific expression of a constitutively active form of β-catenin resulted in hyperplasia followed by the development of low-grade papillary tumors, with a strong male-specific predilection (Lin et al. 2013). Finally, androgen-activated androgen receptor (AR) has been found as a transcription factor closely cooperating and directly physically interacting with β-catenin (Li et al. 2013; Lin et al. 2013). Plausibly, the constant AR-mediated moderate activation of β-catenin in males could lead to an increased overall frequency of urothelial hyperplasia, some of the hyperplastic lesions transforming to papillary carcinoma. This could explain the more frequent but at the same time less aggressive disease in males. Androgen-deprivation therapy could thus constitute a biologically substantiated therapeutic option to reduce recurrence rates in male bladder cancer, as recently proposed (Izumi et al. 2014).
Returning to the urothelial-specific Foxa1 knockout mice, they could be quite instrumental in another aspect of urothelial (cancer) stemness analysis. We have noted that high molecular weight cytokeratins (especially CK5 and CK14) could be regarded either as stem cell markers or as squamous-metaplasia markers (see above the legend to Fig. 8.1). Sexually dimorphic phenotypes of Foxa1 urothelial-specific deletion illustrate these two aspects brilliantly. In male mice, CK14 expression is strictly restricted to basal cells of hyperplastic lesions, implying CK14 as a stem cell marker. In sharp contrast, in female lesions CK14 is expressed throughout the entire thickness of the transformed urothelium, implying CK14 as a marker of squamous metaplasia in this case (Reddy et al. 2015).
8.5 Urothelial Cancer Stem Cell Plasticity
The issue of cancer stem cell heterogeneity becomes even more complicated if we invoke the issue of cancer cell plasticity. As discussed above, cancer stem cells are intimately linked to a particular cancer type and its stage of progression—recall the unique biology of CIS-stem cells, which directly depend on the Sonic hedgehog signal. Moving further along the cancer progression pathway, epithelial-to-mesenchymal transition (EMT) becomes the dominant theme in cancer cell plasticity. EMT represents an embryonic developmental program, reactivated, for example, as part of wound healing process as well as during cancer invasion and metastatic dissemination. Polarized stationary epithelial cells transform into motile mesenchymal-like cells that disengage from the (primary) tumor cell mass, invade the surrounding tissue structure, eventually reaching a blood or lymphatic vessel, enter it (intravasation), and leave it again at a secondary site (extravasation). A complex gene expression program dominates EMT, governed by a group of transcription factors, whose prominent role is to repress the CDH1 gene coding for the principal epithelial cell adhesion molecule E-cadherin: Twist-1 and -2, Zeb-1 and -2, Slug, Snail or Prrx1. The activated genes include vimentin and especially genes underlying single-cell motility and invasiveness. There is an elegant negative feedback loop mediated by the cluster of miRNA200: These miRNAs directly target Zeb-EMT-factors, thus promoting epithelial phenotype, and the Zeb represses the expression of these miRNA genes, thus keeping EMT running. A similar effect to miRNA200 has miRNA205, except that miRNA205 does not take part in this feedback regulation. Most carcinoma metastases display overly epithelial phenotypes, however, implying an opposite transformation process (mesenchymal-to-epithelial transition, MET) once a disseminated tumor cell reaches the secondary site and is about to resume growth to establish a clinically important metastasis (Brabletz 2012).
A crucial question within the context of this chapter is: What happens with cancer stem cells along this EMT-MET cascade? During the last decade, very controversial results have been obtained (Hatina 2012). Originally, Twist-1–activated EMT has been reported to directly connect with cancer stemness in breast cancer, thus promoting these two crucial cancer phenomena at once. This mechanism seemed to be valid also for other tumor types, supporting the earlier formulated concept of migrating cancer stem cells (Mani et al. 2008; Lehmann et al. 2016). More recently, an exact opposite has been found for Prrx1-dominated EMT in breast cancer, too, where stemness (measured, like in the Twist-1 experiment above, by sphere formation, self-renewal capacity, and the typical breast cancer stem cell marker profile CD44high CD24low) appeared antithetical to EMT and invasion (Ocaña et al. 2012). A great compatibility with the latter model has been convincingly demonstrated in urological tumor cell lines (both prostate and urothelial carcinoma) (Celià-Terrassa et al. 2012). Notably, there seems to be an intrinsic molecular connection between pluripotency genes and E-cadherin, and moreover, there also seems to be an intrinsic molecular antagonism between EMT-factors (Snail, Twist, and Zeb) and stemness. Invasive and motile cells produced only slowly growing and metastasis incompetent tumors whereas inoculating epithelial cell variants of the same cell lines resulted in an efficient metastatic spread. Likewise, knockdown of E-cadherin or of pluripotency factors resulted in a dramatic increase in motility and invasiveness, with a simultaneous decrease or loss of self-renewal and metastatic competence. The same was true upon overexpression of EMT-factors, with the simultaneous repression of both E-cadherin and pluripotency genes. Knockdown of EMT-factors, as expected, activated both epithelial character and stemness, both contributing to increased metastatic competence.
This study could certainly be reproached in various ways. First, stemness genes focused on involved pluripotency factors characterized in embryonic stem cells and induced pluripotent stem cells (OCT-4, SOX-2, Nanog, KLF-4 and -9, LIN28A). However, the analysis of molecular mechanisms of stemness of CD44+ urothelial carcinoma stem cells failed to provide evidence of a major role of these pluripotency factors (Chan et al. 2009), and only SOX-2 was later identified as a factor underlying a fraction of invasive urothelial CSCs (Ferreira-Teixeira et al. 2015), see above. On the other hand, the analysis of a clinical impact of expression of p63, a typical MIBC stemness gene (represented by a specific isoform ΔNp63) largely corroborated this concept: Expression of p63 significantly positively correlated with E-cadherin expression and significantly negatively correlated with Zeb-1 and -2 EMT-factors. Importantly, tumor samples with high p63/E-cadherin expression had especially quick lethal outcomes (median overall survival of 8 months, compared with 27 months in the low p63 expression group) (Choi et al. 2012).
Another reproach could be that the results on mirror-image-like dichotomy between motility and invasion, on the one hand, and stemness and metastatic competence, on the other hand, were arrived at by largely using cell lines that were stably genetically manipulated and in which either phenotype (epithelial metastatic or motile, invasive, and poorly metastatic) has been hard-wired into the genome by the genetic manipulation implemented. As we introduced above, however, the EMT and MET processes are largely plastic in real tumors, that is, the changes are only temporary and, by inference, predominantly epigenetically determined or microenvironmentally induced. Indeed, the authors showed that coculture of both “extreme” and genetically fixed prostate cancer cell sublines (a motile and invasive subline with suppressed stemness, and an epithelial metastatic subline with high stemness and low motility, respectively) induced in the latter a transitional EMT, allowing for temporary motility at the expense of decreased stemness, which could be resumed once the cells reached the secondary site.
The above model could indeed be quite realistic. By a serendipity, we established a rather similar experimental model for urothelial carcinoma as well, with a derivative RT-112 cell line (Fig. 8.2). Strikingly, our model does not rely on any genetic manipulation at all, so we can deliberately switch between the epithelial and EMT-like phenotypes, just by modifying cell density. This switch can proceed rapidly (5–7 days), implying that the cells have to be permanently simultaneously pre-programmed to enter either direction according to the momentary signals. Importantly, it seems that there is no black-and-white picture in the relationship between stemness and EMT, implying that both epithelial and EMT states may preserve stemness, but with different underlying molecular mechanisms. Indeed, one of the characteristic features of the basal MIBC intrinsic subtype is the simultaneous overexpression of both stemness and EMT markers. At the level of gene regulation, a peculiar phenomenon explaining this type of rapid plasticity is a poised chromatin conformation, at the same moment bearing repressive and activating chromatin marks at the crucial loci (Chaffer et al. 2013).

Density-dependent plasticity of RT-112-derivative cell line. Notice the mixed profile of stemness-related traits and stemness markers in both culture conditions
8.6 Epigenetic Regulation of Urothelial Carcinoma Stem Cells
We have already had the privilege to meet an epigenetic mechanism that hits urothelial stem cells really at the heart: According to all indices, the knockout of the gene coding for DNA methyltransferase 1 in the urothelium is able to target stem cells in the developing urothelium, and therefore a very special regenerative response must be resorted to, namely recruiting and reprogramming (stem) cells from the Wolffian duct (Joseph et al. 2018), see above. This particular epigenetic regulation seems also to be valid for CSCs. Indeed, DNMT1 has been published as a negative prognostic marker for urothelial carcinoma and its knockdown in two urothelial carcinoma cell lines resulted in a similar effect to that described for the embryonic urothelium, namely the activation of the DNA damage response pathway (Wu et al. 2011). There is also another plausible mechanistic explanation for the involvement of DNMT1 in cancer stemness. An analysis performed in breast cancer confirmed a crucial causal role of DNMT1 for normal mammary and breast cancer stem cells. In addition, the transcription factor ISL-1, acting as a differentiation regulator in multiple cell lineages, has been identified as a crucial mediator, whose gene promoter had to be hypermethylated in order to preserve stemness (Pathania et al. 2015). Importantly, ISL-1 is also essentially involved in urogenital development, its mutations predisposing to classic bladder exstrophy (Zhang et al. 2017). Moreover, the hypermethylation of ISL-1 portends a significant adverse prognosis for NMIBC, predicting both recurrence and progression (Kitchen et al. 2015). These observations make the mechanism discovered for breast cancer stem cells well worth testing in urothelial carcinoma.
As previously mentioned, urothelial carcinoma is also remarkable for its high frequency of mutations in chromatin modifiers, particularly KDM6A in NMIBC and MLL2 in MIBC, both mutations promoting a repressive chromatin conformation and acting thus in the same direction as does DNMT1. While these mutational changes are per definition common to all cancer cells, a specific stem cell epigenetic mechanism has been discovered recently. Another chromatin-modifying enzyme, leading to a repressive chromatin conformation and acting as H3K9 methyltransferase – KMT1A – has been found specifically overexpressed in urothelial carcinoma stem cells. Its immediate target in promoting cancer stemness is the promoter of the gene coding for the transcription factor GATA-3, which itself acts here as the direct transcriptional repressor of the STAT-3. By repressing GATA-3 (via grafting the H3K9me3 repressive chromatin marks on its promoter), KMT1A thus indirectly activates STAT-3 expression and promotes cancer stemness (Yang et al. 2017). Recall that GATA-3 belongs to transcription factors identified as master regulators of the luminal MIBC subtype, whereas STAT-3 enjoys a similar position for basal MIBC. The just described mechanism could thus underlie the luminal-to-basal switch during urothelial cancer progression. Moreover, if we accept the direct succession of luminal MIBC from cluster 2 NMIBC (see above) that seems to be dominated by an entirely different set of stemness genes (ALDH1A1, ALDH1A2, CD133, nestin, and CD90 – Hedegaard et al. 2016), see above, then we could reconstruct the urothelial carcinoma progression pathway in terms of a stemness switch. Plausibly, in luminal MIBC, the stemness molecular mechanisms are largely inherited from cluster 2 NMIBC, and the basal stemness molecular mechanisms are actively repressed by GATA-3. Luminal-to-basal switch would then be carried out epigenetically, via the KMT1A histone methyltransferase. At present, however, this scenario remains a hypothesis.
To make even this aspect a little more complicated, a completely inverse mechanism incorporating certain biological aspects of CSCs has been discovered very recently (Puig et al. 2018). Due to their quiescence, a small proportion of cancer cells across all tumor types behave as label-retaining cells. Classic in detecting stem cells, this approach was also applied for localizing urothelial stem cells among basal cells as well, as discussed above (Kurzrock et al. 2008). In cancer, these label-retaining cells or otherwise called slow-cycling cancer cells display a very specific gene expression profile. It combines, among the activated genes, certain stemness genes (especially for pluripotency factors) and genes coding for disparate detoxification and chemoresistance mechanisms and, among the repressed genes, the major part of cell cycle progression genes and DNA-replication genes, as well as genes responsible for energy metabolism. A crucial regulator of this specific phenotype is TET-2, catalyzing 5-methylcytosine oxidation to 5-hydroxymethylcytosine. This enzymatic reaction ushers the DNA-demethylation sequence, thereby acting in a completely opposite way to DNA-methyltransferases. Indeed, the level of 5-hmC portends a very adverse prognostic significance, especially for chemotherapy-treated patients. Although the survival analysis has been performed for colorectal carcinoma patients, quite likely it is also relevant for urothelial carcinoma, as bladder cancer represents a cancer type with the third highest proportion of high-5-hmC cases.
Obviously, the label-retaining cells, due to both their quiescence and a plethora of chemoresistance mechanisms expressed, cannot be eliminated by chemotherapy. These are exactly the cells that initiate cancer relapse, even years after an apparent cure, a picture well-known across all tumor types. Up to this point, label-retaining cells behave just like typical cancer stem cells. Thus, many results in the scientific literature describing the behavior of CSCs upon chemotherapy almost certainly actually addressed, at least in part, these cells. There is, nevertheless, one tremendous difference between true cancer stem cells on one hand and label-retaining or slow-cycling cancer cells on the other hand. The latter namely lack a self-renewal capacity. Slow-cycling cells yielded slow-cycling progeny with the same frequency as did rapidly proliferating cells (Puig et al. 2018). Slow cycling is thus an operational category, co-opting certain stemness traits and characterized by an extreme plasticity. Discovering their underlying biological mechanism opens an avenue for their pharmacological inhibition, with a potentially tremendous therapeutic impact.
References
Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F (2017) Bladder cancer incidence and mortality: a global overview and recent trends. Eur Urol 71(1):96–108
Balsara ZR, Li X (2017) Sleeping beauty: awakening urothelium from its slumber. Am J Physiol Renal Physiol 312(4):F732–F743
Barth I, Schneider U, Grimm T, Karl A, Horst D, Gaisa NT, Knüchel R, Garczyk S (2018) Progression of urothelial carcinoma in situ of the urinary bladder: a switch from luminal to basal phenotype and related therapeutic implications. Virchows Arch 472(5):749–758
Blanpain C (2013) Tracing the cellular origin of cancer. Nat Cell Biol 15(2):126–134
Brabletz T (2012) EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell 22(6):699–701
Celià-Terrassa T, Meca-Cortés O, Mateo F, Martínez de Paz A, Rubio N, Arnal-Estapé A, Ell BJ, Bermudo R, Díaz A, Guerra-Rebollo M, Lozano JJ, Estarás C, Ulloa C, Álvarez-Simón D, Milà J, Vilella R, Paciucci R, Martínez-Balbás M, de Herreros AG, Gomis RR, Kang Y, Blanco J, Fernández PL, Thomson TM (2012) Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Investig 122(5):1849–1868
Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA, Weinberg RA (2013) Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 154(1):61–74
Chan KS, McConkey DJ (2015) Cancer stem cells and intrinsic subtypes in bladder cancer. In: Bladder cancer: diagnosis and clinical management. Wiley-Blackwell, Hoboken, pp 342–352
Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J, Chang HY, van de Rijn M, Shortliffe L, Weissman IL (2009) Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A 106(33):14016–14021
Cheah MT, Chen JY, Sahoo D, Contreras-Trujillo H, Volkmer AK, Scheeren FA, Volkmer J-P, Weissman IL (2015) CD14-expressing cancer cells establish the inflammatory and proliferative tumor microenvironment in bladder cancer. Proc Natl Acad Sci U S A 112(15):4725–4730
Chen M-F, Lin P-Y, Wu C-F, Chen W-C, Wu C-T (2013) IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS One 8(4):e61901
Choi W, Shah JB, Tran M, Svatek R, Marquis L, Lee I-L, Yu D, Adam L, Wen S, Shen Y, Dinney C, McConkey DJ, Siefker-Radtke A (2012) p63 expression defines a lethal subset of muscle-invasive bladder cancers. PLoS One 7(1):e30206
Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee I-L, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, Siefker-Radtke A, Czerniak B, Dinney CPN, McConkey DJ (2014) Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell 25(2):152–165
Choi W, Ochoa A, McConkey DJ, Aine M, Höglund M, Kim WY, Real FX, Kiltie AE, Milsom I, Dyrskjøt L, Lerner SP (2017) Genetic alterations in the molecular subtypes of bladder cancer: illustration in the cancer genome atlas dataset. Eur Urol 72(3):354–365
Ciamporcero E, Shen H, Ramakrishnan S, Yu Ku S, Chintala S, Shen L, Adelaiye R, Miles KM, Ullio C, Pizzimenti S, Daga M, Azabdaftari G, Attwood K, Johnson C, Zhang J, Barrera G, Pili R (2016) YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene 35(12):1541–1553
Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, Yeh JJ, Milowsky MI, Iyer G, Parker JS, Kim WY (2014) Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc Natl Acad Sci U S A 111(8):3110–3115
Dancik GM, Owens CR, Iczkowski KA, Theodorescu D (2014) A cell of origin gene signature indicates human bladder cancer has distinct cellular progenitors. Stem Cells 32(4):974–982
Dotsikas G, Konowalchuk T, Major PP, Kovac PE, Ward GK, Stewart SS, Price GB, Elhilali MM, Mackillop WJ (1987) Cellular heterogeneity in normal and neoplastic human urothelium: a study using murine monoclonal antibodies. Br J Cancer 56(4):439–444
Fei DL, Li H, Kozul CD, Black KE, Singh S, Gosse JA, DiRenzo J, Martin KA, Wang B, Hamilton JW, Karagas MR, Robbins DJ (2010) Activation of hedgehog signaling by the environmental toxicant arsenic may contribute to the etiology of arsenic-induced tumors. Cancer Res 70(5):1981–1988
Ferreira-Teixeira M, Parada B, Rodrigues-Santos P, Alves V, Ramalho JS, Caramelo F, Sousa V, Reis F, Gomes CM (2015) Functional and molecular characterization of cancer stem-like cells in bladder cancer: a potential signature for muscle-invasive tumors. Oncotarget 6(34):36185–36201
Gaisa NT, Graham TA, McDonald SAC, Cañadillas-Lopez S, Poulsom R, Heidenreich A, Jakse G, Tadrous PJ, Knuechel R, Wright NA (2011) The human urothelium consists of multiple clonal units, each maintained by a stem cell. J Pathol 225(2):163–171
Gandhi D, Molotkov A, Batourina E, Schneider K, Dan H, Reiley M, Laufer E, Metzger D, Liang F, Liao Y, Sun T-T, Aronow B, Rosen R, Mauney J, Adam R, Rosselot C, Van Batavia J, McMahon A, McMahon J, Guo J-J, Mendelsohn C (2013) Retinoid signaling in progenitors controls specification and regeneration of the urothelium. Dev Cell 26(5):469–482
Hatina J (2012) The dynamics of cancer stem cells. Neoplasma 59(6):700–707
Hatina J, Schulz WA (2012) Stem cells in the biology of normal urothelium and urothelial carcinoma. Neoplasma 59(6):728–736
Hatina J, Parmar HS, Kripnerova M, Hepburn A, Heer R (2018) Urothelial carcinoma stem cells: current concepts, controversies, and methods. Methods Mol Biol 1655:121–136
Hatina J, Kripnerova M, Pesta M, Kuncova J, Sana J, Slaby O, Rodríguez R (2019) Sarcoma stem cell heterogeneity. In: Birbrair A (ed), Stem cells heterogeneity—novel concepts. Springer, New York. https://doi.org/10.1007/978-3-030-11096-3_7
He X, Marchionni L, Hansel DE, Yu W, Sood A, Yang J, Parmigiani G, Matsui W, Berman DM (2009) Differentiation of a highly tumorigenic basal cell compartment in urothelial carcinoma. Stem Cells 27(7):1487–1495
Hedegaard J, Lamy P, Nordentoft I, Algaba F, Høyer S, Ulhøi BP, Vang S, Reinert T, Hermann GG, Mogensen K, Thomsen MBH, Nielsen MM, Marquez M, Segersten U, Aine M, Höglund M, Birkenkamp-Demtröder K, Fristrup N, Borre M, Hartmann A, Stöhr R, Wach S, Keck B, Seitz AK, Nawroth R, Maurer T, Tulic C, Simic T, Junker K, Horstmann M, Harving N, Petersen AC, Calle ML, Steyerberg EW, Beukers W, van Kessel KEM, Jensen JB, Pedersen JS, Malmström P-U, Malats N, Real FX, Zwarthoff EC, Ørntoft TF, Dyrskjøt L (2016) Comprehensive transcriptional analysis of early stage urothelial carcinoma. Cancer Cell 30(1):27–42
Hepburn AC, Veeratterapillay R, Williamson SC, El-Sherif A, Sahay N, Thomas HD, Mantilla A, Pickard RS, Robson CN, Heer R (2012) Side population in human non-muscle invasive bladder cancer enriches for cancer stem cells that are maintained by MAPK signalling. PLoS One 7(11):e50690
Izumi K, Taguri M, Miyamoto H, Hara Y, Kishida T, Chiba K, Murai T, Hirai K, Suzuki K, Fujinami K, Ueki T, Udagawa K, Kitami K, Moriyama M, Miyoshi Y, Tsuchiya F, Ikeda I, Kobayashi K, Sato M, Morita S, Noguchi K, Uemura H (2014) Androgen deprivation therapy prevents bladder cancer recurrence. Oncotarget 5(24):12665–12674
Joseph DB, Chandrashekar AS, Abler LL, Chu L-F, Thomson JA, Mendelsohn C, Vezina CM (2018) In vivo replacement of damaged bladder urothelium by Wolffian duct epithelial cells. Proc Natl Acad Sci U S A 115(33):8394–8399
Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmström P-U, Choi W, Guo CC, Lotan Y, Kassouf W (2016) Bladder cancer. Lancet 388(10061):2796–2810
Kitchen MO, Bryan RT, Haworth KE, Emes RD, Luscombe C, Gommersall L, Cheng KK, Zeegers MP, James ND, Devall AJ, Fryer AA, Farrell WE (2015) Methylation of HOXA9 and ISL1 predicts patient outcome in high-grade non-invasive bladder cancer. PLoS One 10(9):e0137003
Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, Chan KS (2015) Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517(7533):209–213
Kurzrock EA, Lieu DK, Degraffenried LA, Chan CW, Isseroff RR (2008) Label-retaining cells of the bladder: candidate urothelial stem cells. Am J Physiol Renal Physiol 294(6):F1415–F1421
Lehmann W, Mossmann D, Kleemann J, Mock K, Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP, Brabletz T (2016) ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat Commun 7:10498
Li Y, Zheng Y, Izumi K, Ishiguro H, Ye B, Li F, Miyamoto H (2013) Androgen activates β-catenin signaling in bladder cancer cells. Endocr Relat Cancer 20(3):293–304
Lin C, Yin Y, Stemler K, Humphrey P, Kibel AS, Mysorekar IU, Ma L (2013) Constitutive β-catenin activation induces male-specific tumorigenesis in the bladder urothelium. Cancer Res 73(19):5914–5925
Ling S, Chang X, Schultz L, Lee TK, Chaux A, Marchionni L, Netto GJ, Sidransky D, Berman DM (2011) An EGFR-ERK-SOX9 signaling cascade links urothelial development and regeneration to cancer. Cancer Res 71(11):3812–3821
Liu Q, Yuan W, Tong D, Liu G, Lan W, Zhang D, Xiao H, Zhang Y, Huang Z, Yang J, Zhang J, Jiang J (2016) Metformin represses bladder cancer progression by inhibiting stem cell repopulation via COX2/PGE2/STAT3 axis. Oncotarget 7(19):28235–28246
Luo F, Xu Y, Ling M, Zhao Y, Xu W, Liang X, Jiang R, Wang B, Bian Q, Liu Q (2013) Arsenite evokes IL-6 secretion, autocrine regulation of STAT3 signaling, and miR-21 expression, processes involved in the EMT and malignant transformation of human bronchial epithelial cells. Toxicol Appl Pharmacol 273(1):27–34
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133(4):704–715
McConkey DJ, Lee S, Choi W, Tran M, Majewski T, Lee S, Siefker-Radtke A, Dinney C, Czerniak B (2010) Molecular genetics of bladder cancer: emerging mechanisms of tumor initiation and progression. Urol Oncol 28(4):429–440
McConkey DJ, Choi W, Dinney CPN (2015) Genetic subtypes of invasive bladder cancer. Curr Opin Urol 25(5):449–458
Ocaña OH, Córcoles R, Fabra Á, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA (2012) Metastatic colonization requires the repression of the epithelial-Mesenchymal transition inducer Prrx1. Cancer Cell 22(6):709–724
Ooki A, Del Carmen Rodriguez Pena M, Marchionni L, Dinalankara W, Begum A, Hahn NM, Van den Bussche CJ, Rasheed ZA, Mao S, Netto GJ, Sidransky D, Hoque MO (2017) YAP1 and COX2 coordinately regulate urothelial cancer stem-like cells. Cancer Res 78(1):168–181
Ooki A, Begum A, Marchionni L, VandenBussche CJ, Mao S, Kates M, Hoque MO (2018) Arsenic promotes the COX2/PGE2-SOX2 axis to increase the malignant stemness properties of urothelial cells. Int J Cancer 143(1):113–126
Papafotiou G, Paraskevopoulou V, Vasilaki E, Kanaki Z, Paschalidis N, Klinakis A (2016) KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat Commun 7:11914
Paraskevopoulou V, Papafotiou G, Klinakis A (2016) KRT14 marks bladder progenitors. Cell Cycle 15(23):3161–3162
Pathania R, Ramachandran S, Elangovan S, Padia R, Yang P, Cinghu S, Veeranan-Karmegam R, Arjunan P, Gnana-Prakasam JP, Sadanand F, Pei L, Chang C-S, Choi J-H, Shi H, Manicassamy S, Prasad PD, Sharma S, Ganapathy V, Jothi R, Thangaraju M (2015) DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun 6:6910
Pavlidakey PG, MacLennan GT, Goldman HB (2010) Nephrogenic adenoma of the bladder. J Urol 184(6):2535–2536
Pignon J-C, Grisanzio C, Geng Y, Song J, Shivdasani RA, Signoretti S (2013) p63-expressing cells are the stem cells of developing prostate, bladder, and colorectal epithelia. Proc Natl Acad Sci U S A 110(20):8105–8110
Puig I, Tenbaum SP, Chicote I, Arqués O, Martínez-Quintanilla J, Cuesta-Borrás E, Ramírez L, Gonzalo P, Soto A, Aguilar S, Eguizabal C, Caratù G, Prat A, Argilés G, Landolfi S, Casanovas O, Serra V, Villanueva A, Arroyo AG, Terracciano L, Nuciforo P, Seoane J, Recio JA, Vivancos A, Dienstmann R, Tabernero J, Palmer HG (2018) TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J Clin Investig 128(9):3887–3905
Reddy OL, Cates JM, Gellert LL, Crist HS, Yang Z, Yamashita H, Taylor JA, Smith JA, Chang SS, Cookson MS, You C, Barocas DA, Grabowska MM, Ye F, Wu X-R, Yi Y, Matusik RJ, Kaestner KH, Clark PE, DeGraff DJ (2015) Loss of FOXA1 drives sexually dimorphic changes in urothelial differentiation and is an independent predictor of poor prognosis in bladder cancer. Am J Pathol 185(5):1385–1395
Schäfer F-M, Algarrahi K, Savarino A, Yang X, Seager C, Franck D, Costa K, Liu S, Logvinenko T, Adam R, Mauney JR (2017) Mode of surgical injury influences the source of urothelial progenitors during bladder defect repair. Stem Cell Rep 9(6):2005–2017
Shin K, Lee J, Guo N, Kim J, Lim A, Qu L, Mysorekar IU, Beachy PA (2011) Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 472(7341):110–114
Shin K, Lim A, Odegaard JI, Honeycutt JD, Kawano S, Hsieh MH, Beachy PA (2014a) Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat Cell Biol 16(5):469–478
Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, Liao JC, Beachy PA (2014b) Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell 26(4):521–533
Sugino T, Gorham H, Yoshida K, Bolodeoku J, Nargund V, Cranston D, Goodison S, Tarin D (1996) Progressive loss of CD44 gene expression in invasive bladder cancer. Am J Pathol 149(3):873–882
Van Batavia J, Yamany T, Molotkov A, Dan H, Mansukhani M, Batourina E, Schneider K, Oyon D, Dunlop M, Wu X-R, Cordon-Cardo C, Mendelsohn C (2014) Bladder cancers arise from distinct urothelial sub-populations. Nat Cell Biol 16(10):982–991. 1–5
Wang C, Ross WT, Mysorekar IU (2017) Urothelial generation and regeneration in development, injury, and cancer. Dev Dyn 246(4):336–343
Wu C-T, Wu C-F, Lu C-H, Lin C-C, Chen W-C, Lin P-Y, Chen M-F (2011) Expression and function role of DNA methyltransferase 1 in human bladder cancer. Cancer 117(22):5221–5233
Yang Z, He L, Lin K, Zhang Y, Deng A, Liang Y, Li C, Wen T (2017) The KMT1A-GATA3-STAT3 circuit is a novel self-renewal signaling of human bladder cancer stem cells. Clin Cancer Res 23(21):6673–6685
Zhang R, Knapp M, Suzuki K, Kajioka D, Schmidt JM, Winkler J, Yilmaz Ö, Pleschka M, Cao J, Kockum CC, Barker G, Holmdahl G, Beaman G, Keene D, Woolf AS, Cervellione RM, Cheng W, Wilkins S, Gearhart JP, Sirchia F, Di Grazia M, Ebert A-K, Rösch W, Ellinger J, Jenetzky E, Zwink N, Feitz WF, Marcelis C, Schumacher J, Martinón-Torres F, Hibberd ML, Khor CC, Heilmann-Heimbach S, Barth S, Boyadjiev SA, Brusco A, Ludwig M, Newman W, Nordenskjöld A, Yamada G, Odermatt B, Reutter H (2017) ISL1 is a major susceptibility gene for classic bladder exstrophy and a regulator of urinary tract development. Sci Rep 7:42170
Acknowledgments
The original studies cited are supported by the Charles University Research Fund (Progres Q39), National Sustainability Program I (NPUI) Nr. LO1503 provided by the Ministry of Education, Youth and Sports of the Czech Republic (M.Kr., M.Ko, and J.K) and by Charles University Specific Student Research Projects Nr. 260394/2017, Nr. 260393/2017 (J.H.,M.K., M.P., J.K.)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kripnerova, M. et al. (2019). Urothelial Cancer Stem Cell Heterogeneity. In: Birbrair, A. (eds) Stem Cells Heterogeneity in Cancer. Advances in Experimental Medicine and Biology, vol 1139. Springer, Cham. https://doi.org/10.1007/978-3-030-14366-4_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-14366-4_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14365-7
Online ISBN: 978-3-030-14366-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)