
Abstract
Of ~7000 gene products generated by an average cell, 2–3% of them are accounted for by the Hsp90 family of proteins. Mammals have two members of Hsp90, Hsp90α and Hsp90β, with which they respond to environmental stress, especially during tissue ischemia. Hsp90β fulfills the role of an important intracellular chaperone, whereas Hsp90α is dispensable inside the cell. Instead, Hsp90α gets secreted and the extracellular Hsp90α protects surrounding cells from hypoxia-induced cell death and promotes cell migration, during wound healing. The cell surface receptor, LRP-1, binds and mediates extracellular Hsp90α signaling by activating Akt kinases independently of Hsp90α’s intrinsic ATPase. Topical application of recombinant Hsp90α promotes healing of acute, diabetic and burn wounds. Tumors take advantage of these protective functions of both intracellular and extracellular Hsp90 family proteins to cope with the constant paucity of oxygen and nutrients within the tumor microenvironment. Unlike intracellular Hsp90β which is equally critical to the survival of both tumor and normal cells, secreted Hsp90α is non-essential for maintaining physiological homeostasis. In contrast, tumor cells constitutively secrete Hsp90α and use secreted Hsp90α to prevent tumor cell death under hypoxic conditions and to promote tumor cell invasion and metastasis. Monoclonal antibodies that selectively target tumor-secreted Hsp90α may prove more effective and less toxic than those that target the ATPase of the intracellular Hsp90β.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The 90-kDa heat-shock protein (Hsp90) was first reported half a century ago and described as an intracellular protein whose cellular levels increases in response to heat (Ritossa 1996). In the decades that followed, Hsp90 was further characterized as an evolutionarily-conserved and ATPase-driven chaperone molecule that plays an important role in the homeostasis of a wide range of living organisms under both physiological and pathophysiological conditions. In vertebrate cells, Hsp90 is a family of constitutively expressed proteins that protect hundreds of intracellular client proteins from possible damage or deterioration secondary to extracellular stress (Young et al. 2001; Whitesell and Lindquist 2005). Many cancer cells express levels of Hsp90 family proteins that are two to tenfold higher than their normal cell counterparts (Isaacs et al. 2003; Banerji 2009), and even amongst cancer cell lines which express relatively normal levels of Hsp90 protein, the Hsp90 in the cancer cells was reported “more active” than the Hsp90 expressed in the overexpressing cancer cells (Kamal et al. 2003). Interestingly, for long time the Hsp90 family proteins were not sought out as a target for cancer therapeutics. The reason behind this previous lack of investigation is that the HSP90AA1 and HSP90AB1 genes, which code for Hsp90α and Hsp90β, respectively, do not show any of the conventional traits of oncogenes like gene translocation, amplification, and oncogenic mutations. Regardless, the status of Hsp90 began to change in early 1990s, largely because of considerable new challenges emerging in the field of cancer therapeutics. Many cancer drugs designed to target an individual oncogene product or pathway began to encounter a common barrier, drug resistance. Cancer drug resistance can manifest from additional mutations in the same gene or by activation of a totally independent pathway in the same cancer cells (Neckers and Neckers 2002; Workman 2004; Workman et al. 2007). Therefore, it became desirable to produce drugs that could not only target a specific oncogenic mutation, but also inhibit multiple key signaling pathways that support the hallmarks of cancer pathogenesis (Hanahan and Weinberg 2011). Since Hsp90’s client proteins include many gene products that play critical roles in the regulation of a cell’s life-cycle (such as ERKB2, MET, RAF, AKT, BCR-ABL, CDK4, and HIF-1α) and Hsp90 serves as a “nodal protein” during multi-molecular complex formation for transformation (McClellan et al. 2007), it became widely accepted that inhibition of the Hsp90’s intrinsic ATPase could simultaneously shut down multiple growth-controlling signaling pathways in cancer cells and, therefore, decrease the genetic plasticity and development of a drug resistance (Workman 2004). Since 1999, many ATPase inhibitors entered clinical trials as potential cancer therapeutics, but few have obtained approval for use in humans.
Two independent studies made the discovery that Hsp90, and especially Hsp90α, are secreted by cells involved in tumor progression (Eustace and Jay 2004) and wound healing (Li et al. 2007), thereby unearthing a potentially new realm of function for this ancient family of intracellular proteins. Over the following decade, tremendous progress has been made in elucidating the functions of secreted Hsp90, its mechanism of action, and its therapeutic value using various preclinical models. Results of those studies in both wound healing and oncology have been summarized in several excellent review articles and book chapters dated up to 2013 (Tsutsumi and Neckers 2007; McCready et al. 2010; Li et al. 2012, 2013; Sidera et al. 2004; Hance et al. 2014). Therefore, in this chapter, we will focus on the more recent, novel progress that has occurred over the past several years, namely the delineation of the distinct roles of Hsp90α and Hsp90β, the stress-mediated regulation of Hsp90α secretion via exosomes, and the latest therapeutic innovations in wound healing and tumor progression. We apologize for the exclusion of many earlier, excellent quality research articles from our references that we obligated to omit in order to comply with the space restrictions.
1.1 HSP90α Versus HSP90β
Vertebrates have two distinct Hsp90 genes that encode Hsp90α and Hsp90β with a shared 86% identity in amino acid sequences. In addition, two organelle-residing isoforms, Grp94 (a 94-kDa protein in the lumen of the endoplasmic reticulum) and TRAP1 (a 75-kDa protein within mitochondria), are related to the Hsp90 proteins. Voss et al. reported that knockout of Hsp90β caused a primary defect in the embryonic allantois (a sac-like structure involved in nutrition and excretion), resulting in loss of liquid waste collection and gas exchange functions and consequent embryonic lethality at day 9.0/9.5 in mice (Voss et al. 2000). This finding demonstrated that Hsp90β is an essential chaperone during embryonic development. In addition, it suggested two possible relationships between Hsp90α and Hsp90β: (1) the role of Hsp90β is distinct and cannot be replaced by Hsp90α or (2) Hsp90β and Hsp90α work cooperatively to reach a minimum threshold level of activity required for successful chaperone functions to ensue, and therefore, reduction of either Hsp90β or Hsp90α would cause the observed defect. However, the reported phenotypes of Hsp90α-knockout mice appeared to challenge the second possibility. Picard’s group generated a mouse with an insertion in intron-10 of the Hsp90α gene using gene trapping techniques. This insertion was expected to produce a truncated Hsp90α protein lacking 36 amino acids in the C-terminal domain, however, the truncated protein was not detectable (likely due to compromised stability of the mutant protein). This outcome resulted in an unexpected Hsp90α-knockout mouse model, as reported (Grad et al. 2010). Udono’s group used a similar approach to generate conditional Hsp90α-knockout mice by floxing exons 9 and 10 in the Hsp90α gene (Imai et al. 2011). In contrast to the Hsp90β-knockout mice, lack of Hsp90α had little effect on embryonic mouse development, although these authors observed (1) lack of sperm in male mice due to an apparently higher rate of cell apoptosis in the testes and (2) a defect in translocation of extracellular antigens across endosomal membranes into the cytosol. Taken together, one may conclude that the two Hsp90 family members play unequal roles during development, in which the support provided by the ATPase-driven chaperone function of Hsp90β, not Hsp90α, is essential.
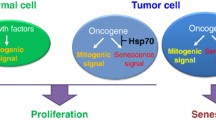
Hsp90α and Hsp90β have also shown distinct functions and mechanisms of action at cellular levels in vitro. Unmethylated CpG oligodeoxynucleotides (CpG ODNs) activate immune responses in a TLR9-dependent manner. Kuo et al. reported that only Hsp90β, and not Hsp90α, responded to CpG-B ODN stimulation and demonstrated a protective effect on serum starvation- and staurosporine- induced apoptosis in mouse macrophages and dendritic cells, although no RNAi experiments were carried out to explicitly address the role of Hsp90α (Kuo et al. 2007). Similarly, Chatterjee et al. reported that Hsp90β plays a more important role in control of multiple myeloma cell survival (Chatterjee et al. 2007). More convincingly, Jayaprakash and colleagues recently discovered a novel mechanism by which human dermal fibroblasts respond to environmental ischemia, in which Hsp90α and Hsp90β have distinct, non-interchangeable functions. They showed that within the ischemic environment of wounds, Hsp90α and Hsp90β work in conjunction to promote cell motility during wound healing. Under the stress, Hsp90β acts as a chaperone by binding to the cytoplasmic tail of the LDL Receptor-Related Protein-1 (LRP-1) and stabilizes the receptor at the cell surface. Hsp90α, on the other hand, is secreted into the extracellular space, where it signals through the Hsp90β-stabilized LRP-1 receptor to promote cell motility, leading to wound closure (Jayaprakash et al. 2015). Differences in the function of these two proteins have also been confirmed by Zou and colleagues during their work with the breast cancer cell line, MDA-MB-231. In their study, CRISPR/Case9 knockout of Hsp90α nullified the tumor cells’ ability to migrate, invade, and metastasize, but did not affect cell survival or growth. In contrast, knocking out Hsp90β resulted in tumor cell death. Intriguingly, extracellular supplementation with recombinant Hsp90α protein, but not Hsp90β, restored the tumorigenicity of the Hsp90α-knockout cells (Zou et al. 2016). These studies indicate that, unlike Hsp90β whose chaperone function is of equal importance in both tumor and normal cells, extracellular Hsp90α is non-essential for normal cells to maintain homeostasis but is a critical factor in the tumor microenvironment that promotes invasion and metastasis. The take home message is illustrated in Fig. 15.1 and summarized here: (1) The window for inhibitors to do more harm to cancer cells than normal cells may not be as wide as originally hoped. In theory, membrane permeable ATPase inhibitors, such as 17-AAG, could penetrate cells and cell organelles to prevent all Hsp90 isoforms from acting as chaperones in both normal and cancer cells. However, when this occurs, inhibition of Hsp90β or Grp94 is toxic to both cancer and normal cells in patients. (2) Isoforms and locations of distinct Hsp90 family members should be taken into consideration when designing potential therapeutics. Some of these isoforms are even secreted into the extracellular environment by certain cancer cells, especially those with constitutively elevated HIF-1α (hypoxia-inducible factor-1alpha) expression (detected in ~50% of all invasive human tumors). Unlike intracellular Hsp90β, secreted Hsp90α no longer depends upon an ATPase to function as (i) a survival factor for tumor cells under hypoxic conditions (Dong et al. 2015) and (ii) as a pro-motility factor promoting invasion (Cheng et al. 2008). Indeed, selective inhibition of secreted Hsp90α, which is nonessential for normal cells, could be an alternative therapeutic approach for certain cancers (Li et al. 2012, 2013). For the past decade, several studies have utilized this alternative approach to bypass targeting of intracellular Hsp90β, namely to selectively target the extracellular, tumor-secreted Hsp90α to inhibit tumor progression. In fact, the results from animal models employing this approach are encouraging (Stellas et al. 2007; Tsutsumi et al. 2008; Song and Lou 2010; Zou et al. 2016). This new approach remains to be tested in human trials.

Targeting tumor-secreted Hsp90α may be more effective and less toxic. Figure illustrates the consequences of inhibition or knockdown of Hsp90 family members depending on their intracellular or extracellular location and their cell of origin. ATPase inhibitors would indiscriminately inhibit all intracellular Hsp90 functions, however, inhibition of tumor-secreted Hsp90α would selectively target tumor progression pathways
1.2 Environmental Stress or Cellular Oncogene-Triggered, Exosome-Mediated Secretion of HSP90α
Yu and colleagues reported that γ irradiation induced exosome-mediated secretion of Hsp90β, but not Hsp90α, in a p53-dependent fashion, prompting proposal of the “DNA damage > p53 > Hsp90β secretion” pathway (Yu et al. 2006). Cheng et al. showed that in primary human keratinocytes, TGFα-induced Hsp90α membrane translocation and secretion were sensitive to inhibitors of exosome protein trafficking, but not to inhibitors of the conventional ER/Golgi protein trafficking pathway (Cheng et al. 2008). Li and colleagues showed that hypoxia (1% O2) induces Hsp90α secretion via HIF-1α and that blockade of the secreted Hsp90α functionality via neutralizing antibodies completely inhibits hypoxia-induced cell motility (Li et al. 2007). The identification of HIF-1α as a key upstream regulator of Hsp90α secretion also has an important implication for cancer pathogenesis. Hypoxia is a known micro-environmental stress that is connected to the growth, invasion, and metastasis of many solid tumors (Simon and Keith 2008). Under chronic hypoxic conditions, cancer cells are forced to adapt alternative and self-supporting mechanisms via HIF-1α for continued survival and expansion. It is likely why overexpression of HIF-1α has occurred in approximately 40% of the tumors in humans (Semenza 2007). Accordingly, surface expression and/or secretion of Hsp90α should become constitutive in those HIF-1α-overexpressing tumors. To support this notion, many tumor cell lines have been reported to secrete Hsp90α. Kuroita and colleagues reported purification of Hsp90α from the conditioned media of human hybridoma SH-76 cells (Kuroita et al. 1992). Eustace et al. reported presence of Hsp90α, but not Hsp90β, in the conditioned media of HT-1080 tumor cells (Eustace et al. 2004). Wang et al. reported secretion of Hsp90α by MCF-7 human breast cells (Wang et al. 2009). Suzuki and Kulkarni found Hsp90β secreted by MG63 osteosarcoma cells (Suzuki and Kulkarni 2010). Chen and colleagues reported secretion of Hsp90α by a colorectal cancer cell line, HCT-8 (Chen et al. 2010). Work by Tsutsumi and colleagues implied secretion of Hsp90α occurred in a variety of tumor cell lines (Tsutsumi et al. 2008). Finally, several recent studies from our laboratory demonstrated that seven breast cancer cell line constitutively secrete Hsp90α (Sahu et al. 2012; Dong et al. 2015; Zou et al. 2016). In these cells, HIF-1α plays a critical role in regulation of Hsp90α secretion, although the signaling steps between HIF-1α and the secretory machinery remain unknown.
What is known, however, is that Hsp90 proteins do not have the signal peptide (SP) required for secretion out of cells via the classical ER/Golgi protein secretory pathway. An alternative secretory pathway for molecules without SPs is mediated by secreted extracellular vesicles (EVs). EVs have several synonyms that are widely used in independent studies including “microvesicles”, “ectosomes”, “microparticles” and “exosomes” (Yáñez-Mó et al. 2015; Raposo and Stoorvogel 2013). Secretion of EVs by normal cells under stress is an evolutionarily conserved phenomenon observed in almost all cell types. Biologic origin and size variation account for the main distinctions among different kinds of EVs. Exosomes belong to a subtype of EVs with defined diameters between 30 and 150 nm and are derived from intraluminal vesicles (ILVs) within intracellular multivesicular bodies (MVB) (Harding et al. 1983; Pan et al. 1985; Johnstone et al. 1987). Nonetheless, due to technical limitations in purifying distinct EV populations, the term “exosome”, as currently used, refers to a population of EVs of varying sizes but with the majority being between 30 and 150 nm in diameter, instead of a single subtype of EV with a clearly defined population with respect to size and origin of production (Tkach and Théry 2016). In contrast to normal cells which secrete exosomes secondary to extracellular environmental stress, tumor cells constitutively secrete exosomes in a process driven by intracellular oncogenes (Kucharzewska and Belting 2013). In addition to proteins and peptides, secreted exosomes also contain other cargo molecules including DNA, mRNA, miRNA, and lipids. Consequently, exosomes are the most complex extracellular signaling entity identified for cell-to-cell communication to date (Théry et al. 2002; Witwer et al. 2013; Colombo et al. 2014). This new and increasingly recognized mechanism of intercellular communication has been demonstrated to play critical roles in host immune responses (Nolte et al. 2012), tissue repair (Yuana et al. 2013; Yáñez-Mó et al. 2015) and tumor invasion and metastasis (Kucharzewska and Belting 2013; Hoshino et al. 2015; Zhang et al. 2015). Three lines of evidence for exosome-mediated secretion of Hsp90α currently exist. First, using two chemical inhibitors, brefeldin A (BFA), which selectively blocks the classical ER/Golgi protein secretory pathway, and dimethyl amiloride (DMA), which blocks the exosome protein secretory pathway, several groups have shown that DMA selectively inhibits membrane translocation and secretion of Hsp90α or Hsp90β in various cell types (reviewed in Li et al. 2012). In contrast, BFA resulted in little inhibition of either Hsp90 protein secretion (Cheng et al. 2008). Second, Clayton and colleagues detected the presence of Hsp90α, among other proteins, during proteomic analysis of isolated exosomes from B-cell conditioned media (Clayton et al. 2005). Third, Yu et al. presented electron microscopic evidence of the presence of Hsp90 co-localized with exosomes (Yu et al. 2006). A recent study by Gou and colleagues demonstrated that the majority of tumor-secreted Hsp90α is present in the 100,000 g ultracentrifugation fraction of the cells’ conditioned media, together with exosome markers including flotillin, CD9, CD81, and Cd63 (Guo et al. 2017).
1.3 Regulation of HSP90α Secretion in Normal and Tumor Cells
While it is clear that environmental stress cues trigger release of exosomes in normal cells and intrinsic oncogenic signal(s) result in constitutive exosome secretion in many tumor cells (Raposo and Stoorvogel 2013), the mechanism by which the stress signals regulate exosome secretion remains elusive. In fact, there has been limited progress in this field until recently. First, the Rab27 small GTPases, Rab27a and Rab27b, were reported to regulate both exosome biogenesis and secretion. Rab27a and Rab27b regulate distinct steps of multi-vesicular endosome (MVE) docking to plasma membranes and exosome biogenesis; Rab27a regulates MVE breakdown and Rab27b regulates MVE distribution, formation, and secretion in various types of cells (Ostrowski et al. 2010; Zheng et al. 2013). However, Rab27a was also reported to regulate secretion of MMP9 and growth factors through the conventional ER/Golgi pathway (Kucharzewska and Belting 2013; Bobrie et al. 2012). Since the mechanisms of these two trafficking systems are completely unrelated, these observations generate more questions than answers. An interesting study by Sinha and colleagues showed that knockdown or overexpression of cortactin in cancer cells resulted in a respective decrease or increase in exosome secretion, without altering exosome cargo content. These authors proposed that cortactin promotes exosome secretion by binding to Arp2/3 and stabilizing cortical actin-rich MVE docking sites (Sinha et al. 2016). Our laboratory has studied stress-regulated and exosome-mediated secretion of Hsp90α during wound healing and tumor invasion. Our approach was to first understand how environmental stress signals trigger the secretion of Hsp90α, an exosome cargo molecule, which will garner insight into the larger question of how stress regulates exosome secretion. By taking advantage of a unique property of human keratinocytes in response to transforming growth factor-alpha (TGFα) and epidermal growth factor (EGF) in culture, we have recently identified a critical signaling molecule that links microenvironmental cues to exosome secretion. We found that, while both TGFα and EGF are known to utilize the same cell surface EGF receptor for transmembrane signaling and the same, previously undistinguishable, intracellular signaling networks for regulation of gene expression, we surprisingly found that only TGFα triggers secretion of Hsp90α. By comparing activations of 43 intracellular signaling molecules/pathways in the same cells in response to TGFα or EGF stimulation, we identified PRAS40 as a TGFα-specific downstream target. More importantly, activated PRAS40 acts not only as a regulator of TGFα-triggered and exosome-mediated secretion of Hsp90α, but rather as a common regulator of distinct microenvironmental and oncogenic signal-triggered exosome secretion in both normal and tumor cell types (Guo et al. 2017). PRAS40 is the first regulator identified for stress-induced exosome secretion.
The proline-rich Akt substrate of 40 kDa (PRAS40) was initially identified as a direct substrate of Akt kinase and a binding partner for the 14-3-3 scaffolding molecule (Kovacina et al. 2003). Most studies focused on PRAS40’s role in insulin, as well as NGF and PDGF, signaling to the mTOR (mammalian target of rapamycin) pathway (specifically mTORC1), which regulates cell metabolism, protein synthesis, and cell growth (Saito et al. 2004; Shimaya et al. 2004; Vander Haar et al. 2007; Sancak et al. 2010; Fonseca et al. 2007; Oshiro et al. 2007; Thedieck et al. 2007; Wang et al. 2007). In growth-arrested cells, PRAS40 was reported to bind, via the raptor subunit, to mTORC1 and inhibit mTOR kinase activity. Insulin stimulation activates Akt kinase mainly via threonine (Thr)-308 phosphorylation. The activated Akt kinase in turn phosphorylates PRAS40 on Thr-246. Thr-246-phosphorylated PRAS40 dissociates from mTORC1, resulting in activation of mTORC1, and (re-) associates with 14-3-3 (Vander Haar et al. 2007; Oshiro et al. 2007; Sancak et al. 2007). In addition to Akt, increased PIM1 kinase activity also correlated with increased PRAS40 phosphorylation. Activated mTORC1 phosphorylates PRAS40 at Ser-183, Ser-212 and Ser-221 (Wang et al. 2008; Zhang et al. 2009). Despite these reports, others showed that PRAS40 is not a common regulator of mTOR activation in response to extracellular signaling (Thedieck et al. 2007; Fonseca et al. 2008). Gou et al. demonstrated that stress signals activate Akt via phosphorylation at Thr-308. The Thr-308-phosphorylated Akt in turn activates PRAS40 via Thr-246 phosphorylation. More interestingly, Thr-246-phosphorylated PRAS40, even in the absence of stress, is both necessary and sufficient to cause exosome secretion, without affecting the ER/Golgi pathway. These findings are schematically depicted in Fig. 15.2.

A schematic representation of stress-triggered exosome secretion through PRAS40. Extracellular stress cues like cytokines, hypoxia, and H2O2 activate Akt kinase, which in turn phosphorylates PRAS40 at Thr-246. Activated PRAS40 communicates with a currently unknown intermediate(s), leading to exosome secretion. Secreted exosomes contain large quantities of DNA, RNA, and protein molecules, enabling much more efficient cell-to-cell communication than a single secreted molecule, like a peptide hormone, would alone. PRAS40 is the first linker identified between stress and exosome secretion. (Taken from Guo et al. 2017 with permission)
Identification of PRAS40 as a linker protein not only paves the way for understanding how stress regulates exosome secretion under pathophysiological conditions, but also provides direct support for previous studies that show PRAS40 has also a stimulating role in both normal and cancer cells. In those studies, PRAS40 (i) prevents stress-induced normal and tumor cell apoptosis and (ii) supports tumor progression in vitro and in vivo (Saito et al. 2004; Madhunapantula et al. 2007; Kazi and Lang 2010; Huang et al. 2012; Havel et al. 2015). Similar findings were also made in normal cells. Yu et al. reported that elevated PRAS40 levels protect motor neurons from spinal cord injury-induced cell death (Yu et al. 2008). Shin et al. showed that overexpression of PRAS40 prevents ischemic insults in the brain and oxidative stress-induced brain cell death (Shin et al. 2016). All these findings cannot be explained by the reported role of PRAS40 as an inhibitor of the mTOR pathway and leave the question of how PRAS40 exerts these “positive” functions unanswered. Our finding that Thr-246-phosphorylated PRAS40 regulates exosome secretion provides a possible mechanism for how PRAS40 protects cells from apoptosis and supports tumor progression – via secreted exosomes and their cargo protein, Hsp90α.
1.4 Secreted HSP90α Is Not Enclosed Inside Exosomes
There have been several lines of experimental support suggesting that secreted Hsp90α is not enclosed within the interior of exosomes. First, Eustace et al. reported that DMAG-N-oxide, a cell-impermeable version of geldanamycin that targets Hsp90’s ATPase, blocks tumor cell invasion (Eustace et al. 2004). Tsutsumi and colleagues reported similar findings with respect to in vitro cancer cell invasion and lung colonization by melanoma cells in mice (Tsutsumi et al. 2008). The results of these studies also implied that the Hsp90α’s intrinsic ATPase is still required for the extracellular functions of secreted Hsp90α. Second, Cheng and colleagues reported that the pro-motility activity of secreted Hsp90α from cell-conditioned medium could be completely blocked by neutralizing antibodies (Cheng et al. 2008). Furthermore, a specific anti-Hsp90α neutralizing monoclonal antibody, 1G6-D7, blocks the pro-motility and pro-invasion activities of isolated exosomes in vitro (Guo et al. 2017) and in vivo (Zou et al. 2016). Finally, RAP (LRP-1-associated protein), which competitively inhibits ligand binding and signaling through the extracellular domain of LRP-1 (the receptor for secreted Hsp90α), blocks enhanced cell motility by isolated exosomes (Guo et al. unpublished). The action of exosome-associated Hsp90α does not appear to be due to exosome rupturing or a slow release of cargo molecules, since the high stability of exosomes is widely reported. Nonetheless, these observations raised several questions. How does secreted Hsp90α anchor to the lipid membrane of exosomes? Does membrane anchoring occur before or after exosome secretion? If the membrane anchoring occurs intracellularly, how could exosomes diffuse through the plasma membrane with a highly hydrophilic polypeptide on the surface? If the anchoring occurs after exosomes are secreted into the extracellular environment, potential mechanisms are limited to speculation. All things considered, more research, especially work utilizing imaging and biochemistry approaches, needs to be done to gain insights into these puzzles.
1.5 Extracellular HSP90 Is Not a Chaperone Protein and Does Not Require Intrinsic ATPase or Dimerization to Function
One of the central debates in Hsp90 research was whether secreted Hsp90α still acts in an ATPase-dependent manner. Recent studies have provided several lines of convincing evidence that extracellular Hsp90α is no longer an ATPase-driven chaperone and instead acts as a novel class of signaling proteins that directly binds their receptors to triggers cellular responses. Taking advantage of the previous report that the Hsp90α-wt, the Hsp90α-E47D mutant, and the Hsp90α-E47A and Hsp90α-D93N mutants have 100%, 50%, and undetectable ATPase activity, respectively (Young et al. 2001), Cheng et al. made comparisons among the recombinant proteins encoded by these Hsp90α genes assessing their abilities to stimulate cell migration, a main function of secreted Hsp90α. They reported that all the ATPase mutant proteins retained similar degrees of pro-motility activity compared to Hsp90α-wt (Cheng et al. 2008). These authors then used sequential deletion mutagenesis to narrow down the pro-motility domain to a 115-amino acid fragment, called F-5 (aa-236 to aa-350), between the linker region (LR) and the middle (M) domain of the human Hsp90α. F-5, alone, was found to promote skin cell migration in vitro under serum-free conditions and wound healing in vivo as effectively as full-length Hsp90α-wt (Cheng et al. 2011). These findings demonstrate that the N-terminal ATPase domain and the C-terminal dimer-forming and co-factor-binding domain are dispensable with regard to extracellular Hsp90α’s ability to promote cell migration.
The next mystery elucidated was the identity of the molecular entity which determines the extracellular functions of secreted Hsp90α. Zou and colleagues took a clever approach by comparing amino acid substitutions between Hsp90α and Hsp90β (the former has extracellular functions and the latter does not). The authors first utilized the deletion mutagenesis approach to further narrow down the functional fragment from F-5 fragment to a 27-amino acid peptide fragment, called F-8. They then identified 8 amino acids within Hsp90α’s F-8 peptide that are substituted by variant amino acid residues within the corresponding F-8 peptide from Hsp90β. Sequential site-directed mutagenesis allowed them to identify two evolutionarily conserved lysine residues, lys-270 and lys-277, in the Hsp90α subfamily that determine extracellular Hsp90α functions. The Hsp90β subfamily lacks the dual lysine motif and extracellular functions. Substitution of lys-270 and lys-277 in Hsp90α with the two corresponding gly-262 and thr-269 from Hsp90β completely nullified the extracellular functions of Hsp90α. The reverse is also true. Substitutions of gly-262 and thr-269 in Hsp90β with lysines converted Hsp90β to an Hsp90α-like protein, at least in vitro. Intriguingly, these authors found that the dual lysine motif is conserved evolutionarily in all Hsp90α subfamily members and, similarly, the gly-262 and thr-269 are conserved in the Hsp90β subfamily proteins (Zou et al. 2016). A schematic representation of the structure and functional component requirements for intracellular Hsp90 and secreted Hsp90α is shown in Fig. 15.3.

Hsp90β is an intracellular chaperone, whereas Hsp90α is an extracellular factor. Hsp90β gene knockout is embryonic lethal in mice and causes cell death in culture dish. Absence of functional Hsp90α has little impact on mouse development and causes certain tumor cells to selectively lose their tumorigenicity. The action of intracellular Hsp90β is driven by an intrinsic ATPase, whereas action of secreted Hsp90α is determined by the dual lysine (Lys-270 and Lys-277) motif within the F-5 region. These lysine residues are present in all Hsp90α family members, but they are replaced by glycine-262 and Thr-269 in the Hsp90β family
1.6 Two Biological Functions of Extracellular HSP90α
Conventional wisdom dictates that locally released growth factors in an injured tissue constitute the principal driving force initiating wound healing (Werner and Grose 2003; Grose and Werner 2004). Under this assumption, growth factors are responsible for both inducing wound closure by promoting the lateral migration of epidermal keratinocytes and for remodeling and revascularizing the neodermis of the wounded space by inducing the inward migration of dermal fibroblasts and microvascular endothelial cells. Since the first report of a clinical trial using EGF to stimulate wound healing in 1989 (Brown et al. 1989), more than a dozen trials utilizing growth factors to promote wound healing have been conducted. However, only the human recombinant protein, platelet-derived growth factor-BB (PDGF-BB), has received US Food and Drug Administration (FDA) approval only for the treatment of diabetic limb ulcers (Regranex™/becaplermin gel 0.01%, Ortho-McNeil Pharmaceutical, Raritan, NJ). Following its FDA approval in 1997, multicenter and randomized clinical evaluations showed an overall ~15% improvement in wound closure by becaplermin gel at 100 μg PDGF-BB/g vesicle, a dose already more than a thousand-fold higher than the physiological range of PDGF-BB in human circulation (Steed 1995; Wieman et al. 1998; LeGrand 1998; Mandracchia et al. 2001; Embil and Nagai 2002). In 2008, the FDA added a black box warning to becaplermin gel due to an increased risk of cancer mortality in patients who required extensive treatments (≥3 tubes). This side-effect may not be altogether surprising, since autocrine release of PDGF-BB (c-sis), or its viral form (v-sis), at concentrations as low as 15–30 ng/ml can trigger cell transformation (Bejcek et al. 1992).
Li and colleagues concluded that the primary driving force behind skin wound closure does not originate from growth factors in circulation and subsequently began to search for novel factors secreted from the keratinocytes at the wound edge under stressful conditions like hypoxia and paucity of nutrients. Protein purification with the sensitive individual cell-based pro-motility assay, the colloidal gold motility assay, allowed them to identify Hsp90α as a possible candidate (Li et al. 2007; Cheng et al. 2008). Using purified recombinant Hsp90α protein, two important biological functions of extracellular Hsp90α in both in vitro cell culture and in vivo animal models were established: (1) promotion of cell-survival under stress and (2) stimulation of cell motility for tissue repair. First, using hypoxia as an in vitro model and burn wounds as an in vivo model, cells were observed to secrete Hsp90α to protect themselves from hypoxia- and heat- induced apoptosis (Dong et al. 2015; Bhatia et al. 2016). This finding is logically consistent with observed cellular behaviors in both physiological wound healing and cancer pathogenesis. During wound healing, cells in the hypoxic environment of the wound edge must remain viable, at least temporarily, prior to engaging in the repair process. During tumor growth, most solid tumors outgrow the nearest blood vessels and, therefore, are constantly challenged with a severely hypoxic microenvironment and possible cell death. To remain viable under these conditions, almost 50% of solid tumors have managed to maintain a level of constitutively expressed HIF-1α. As previously mentioned, hypoxia and HIF-1α are presently the best-characterized upstream stress cues that trigger Hsp90α secretion in both normal and tumor cells (Li et al. 2007; Woodley et al. 2009; Sahu et al. 2012). In addition to protecting cells from hypoxia-induced apoptosis, the second important biological function of secreted Hsp90α protein is its role as a novel class of motogen that promotes cell motility to “escape” the hazard microenvironment. Recombinant Hsp90α promotes cell migration, but not proliferation, of a wide range of cell types in the total absence of serum factors or any other exogenously supplemented large molecules (Cheng et al. 2008, 2011). Since migrating cells do not proliferate and proliferating cells cannot do motility at the same time, Hsp90α being a motogen, nut not mitogen, makes a perfect sense. Both the pro-survival and pro-motility functions of extracellular Hsp90α utilize a common signaling transduction pathway. Briefly, Hsp90α binds to subdomain II in the extracellular portion of the low-density lipoprotein receptor-related protein-1 (LRP-1). The NPVY, but not NPTY, motif in the intracellular tail of LRP-1 in turn connects Hsp90α signaling to serine-473 (but not threonine-308) phosphorylation in Akt kinases, leading to enhanced cell migration. Individual knockdown in cell culture or knockout of Akt1, Akt2, or Akt3 gene in mice demonstrates the importance of Akt1 and Ak2 in extracellular Hsp90α signaling and control of cell motility both in vitro and in vivo. Most convincingly, these two biological functions of secreted Hsp90α, promotion cell survival and cell motility, have been verified in wound healing models. Li’s group showed that topical application of recombinant Hsp90α protein strongly stimulates wound closure by promoting keratinocyte migration-driven re-epithelialization in mouse and pig wounds (Cheng et al. 2011; O’Brien et al. 2015). The pro-survival effects of extracellular Hsp90α were assessed using a burn wound healing model which undergoes a unique pathological process called “secondary burn wound progression”, in which the wound expands horizontally and vertically from the initial site of trauma. If left untreated, the cells in the expanded areas soon die of necrosis, apoptosis, or both, due to ischemia, infection, and accumulation of toxic metabolites. Bhatia et al. showed that topical application of recombinant Hsp90α dramatically reduced the degree of secondary burn wound progression by preventing heat-induced apoptosis of the surrounding cells (Bhatia et al. 2016). The secondary burn wound progression, surrounding cell death and prevention of apoptosis by topically applied Hsp90α protein in a pig wound model are as shown in Fig. 15.4. Identification of these two functions of extracellular Hsp90α provides direct support for independent studies from many laboratories (reviewed by Li et al. 2012, 2013).

Extracellular Hsp90α treatment prevents burn-induced cell apoptosis, but not necrosis. (a) “Secondary burn wound progression” from day 0 to day 4 following initial injury burn (120 °C, 30 s); (b) Human Hsp90α F-5 peptide treatment (panel c) prevents burn-induced cell apoptosis (brown stain) in cells surrounding the burn wound compared to no treatment (panel b); (c) F-5 treatment (panel c) demonstrates little rescuing effect on burn-induced cell necrosis (brown stain) compared to no treatment (panel b). (Taken and modified from Bhatia et al. with permission)
1.7 Keratinocyte-Secreted HSP90α During Wound Closure
The 1-year long skin wound healing process is divided into (1) an inflammatory phase, (2) a proliferation phase and (3) a maturation phase. Following a brief period of inflammation in response to skin injury, the proliferation phase begins and lasts a few weeks. During this phase, granulation tissue takes form and serves as the pavement support for epidermal keratinocytes at the wound edge to attach and migrate, ultimately resulting in resurfacing of the open wound. Therefore, this phase is also known as the re-epithelialization or wound closure phase. After wound closure, the time-consuming maturation phase, which involves extracellular matrix remodeling and neovascularization in the dermis, can last months to a year to complete. Most previous studies have primarily focused on the initial weeks of the inflammation and re-epithelization/wound closure phases due to lack of reliable animal models that could allow for analysis of the year-long wound maturation process (Singer and Clark 1999; Gurtner et al. 2008; Sen et al. 2009). However, despite years of studies and investment, there are few topical or systemic medications which effectively promote skin wound closure.
Since it has been widely accepted that identification of a natural “driver gene” for a given pathological process is the foundation for successful therapeutic development, we hypothesized that the natural driver factor(s) for wound closure was still at large. Proof of HSP90AA1’s driver gene nature during wound closure could facilitate development of a new and effective treatment of wounds. Bhatia and colleagues found that when skin is injured, there is a massive increase of Hsp90α protein in the wound bed (Bhatia et al. 2016). Using a unique mouse model which expresses a carboxyl terminal deletion mutant, Hsp90α-Δ, to prevent the dimerization and chaperone functions of Hsp90α, but spares the extracellular F-5-supported pro-motility function (Imai et al. 2011), allowed these authors to specifically test the role of the non-chaperone functions of secreted Hsp90α in normal wound closure. They showed that the chaperone-defective Hsp90α-Δ mutant mice had similar wound closure rates when compared to the Hsp90α wild type mice. Topical application of recombinant Hsp90α-Δ mutant protein promoted wound closure as effectively as full-length Hsp90α wild type protein. Finally, selective functional inhibition of secreted Hsp90α-Δ protein, via monoclonal antibodies targeting the F-5 region, disrupted normal wound closure in both Hsp90α wild type and Hsp90α-Δ mice. Thus, this study provides evidence that non-chaperone, extracellular Hsp90α is a potential driver for normal wound closure. An earlier study by Song and Luo which used regular nude mouse model and neutralizing antibodies against Hsp90 reported similar findings (Song and Luo 2010). As previously mentioned, within the 732-amino acid polypeptide of human Hsp90α, the fully functional therapeutic entity which promotes wound closure is located within a 115-amino acid fragment, F-5, between the linker region and middle domain of Hsp90α (Cheng et al. 2011). Topical application of F-5 peptide strongly promotes traumatic (full-thickness, excisional) wound closure, burn wound closure, and diabetic wound closure in mouse and porcine models. In side-by-side comparisons, FDA-approved becaplermin gel showed either minimal effects on acute wound (traumatic and burn wounds) closure or a much weaker effect on chronic wound (diabetic wounds) closure (Cheng et al. 2011; O’Brien et al. 2015; Bhatia et al. 2016). The effect of F-5 on burn wounds is especially encouraging since it is the first topically applied peptide that prevents secondary burn wound progression, a significant barrier for developing burn therapeutics. Currently, F-5 has entered industrial development as a new topical treatment for skin wounds.
But what qualifies secreted Hsp90α a driver for wound closure, rather than growth factors? Studies from our laboratory have identified three unique properties of extracellular Hsp90α, which are absent from conventional growth factors. First, extracellular Hsp90 is a common pro-motility factor for all three types of human skin cells involved in wound healing. Following skin injury, keratinocytes migrate laterally to cover the open wound and dermal fibroblasts and microvascular endothelial cells migrate inwardly to remodel the damaged tissue and reestablish a blood supply. Ideally, a single factor-based wound-healing agent should recruit all three types of skin cells into the wound bed. Secreted Hsp90α is exactly such an agent; it promotes motility in all three major types of human skin cells because each type of skin cell expresses a compatible level of LRP-1 for Hsp90α to bind (Cheng et al. 2008). In contrast, PDGF-BB only acts on dermal fibroblasts and not on keratinocytes or dermal microvascular endothelial cells due to lack of both PDGFRα and PDGFRβ on the latter two cell types (Cheng et al. 2011). Second, conventional growth factors are unable to override the inhibition of cell migration and proliferation caused by TGFβ3, which is co-present in the wound bed and present at especially high levels in inflammatory wounds (Bandyopahdhay et al. 2006). In contrast, even in the presence of TGFβ3, extracellular Hsp90α remains equally effective in promoting migration of all three types of human skin cells (Cheng et al. 2011). Third, extracellular Hsp90α is able to promote healing in diabetic wounds. All forms of diabetes are characterized by chronic hyperglycemia, which is believed to be one of the major factors delaying wound closure in diabetic patients (Brownlee 2001). A reported mechanism of wound-healing impairment mediated by hyperglycemia is destabilization of HIF-1α protein, the key regulator of Hsp90α secretion in the wound (Catrina et al. 2004; Botusan et al. 2008). While hyperglycemia blocks hypoxia-induced and serum-stimulated human dermal fibroblast migration, extracellular Hsp90α not only enhances hypoxia-driven migration in normal glycemic conditions, but also “rescues” migration of cells cultured in hyperglycemic conditions. In this case, extracellular Hsp90α promotes diabetic wound closure possibly by bypassing HIF-1α down-regulation induced by hyperglycemic conditions and jumpstarting migration of cells that otherwise are unable to raise HIF-1α level to respond to the environmental hypoxic cue. Finally, the notion that extracellular Hsp90α is a motogen, and not a mitogen (i.e. it does not stimulate cell proliferation) also makes sense from a physiologic perspective. Keratinocyte migration occurs almost immediately following skin injury, whereas the inward migration of dermal cells is not detected until 4 days afterward (Singer and Clark 1999). During wound healing, cell migration precedes cell proliferation and when a cell is actively migrating, it cannot proliferate simultaneously. Then, when and where does cell proliferation take place to replenish the lost tissue in wounded skin? We speculate that, as the cells at the wound edge move toward the wound bed, they leave “empty space” between themselves and the cells behind them. The cells behind the migrating cell front subsequently begin to proliferate after losing contact inhibition with the travelling cells. The stimuli responsible for this cell proliferation are likely plasma growth factors which have diffused from surrounding unwounded blood vessels, where TGFβ levels are low or undetectable (Bandyopahdhay et al. 2006). Thus, the role of cell proliferation in wound closure is to refill the space generated by the migrating cell front. The role for secreted Hsp90α in wound closure is to achieve initial wound closure as quickly as possible to prevent infection, water loss, and impact of severe environmental stress. After initial wound closure, many other factors (including growth factors and TGFβ) will participate in the remaining time-consuming wound remodeling processes for up to 1 year.
1.8 Tumor Cells Secrete HSP90α to Gain Invasive and Metastatic Advantages
It is clinically relevant to determine which tumors secrete and utilize secreted Hsp90α to gain invasive and metastatic advantages. So far, the best-characterized upstream oncogene that triggers Hsp90α secretion is the HIF1A gene which encodes HIF-1α (Li et al. 2007; Sahu et al. 2012). This finding is significant, since HIF-1α overexpression is associated with increased patient mortality in approximately 40% of solid tumors in humans (Semenza 2007, 2012a). Using breast cancer as a model, Dales et al. carried out anti-HIF-1α immunohistochemical assays on the frozen sections of 745 breast cancer samples and found that elevated levels of HIF-1α expression correlated with poor prognoses, lower overall survival, and higher risk of metastasis among both node-negative and node-positive patients (Dales et al. 2005). By using HIF-1α expression as a marker, it was estimated that approximately 25–40% of all invasive breast cancer samples are hypoxic, suggesting that HIF-1α can be used as a broader marker for breast cancer (Dales et al. 2005; Semenza 2012b). Sahu et al. have shown that down-regulation of endogenous HIF-1α in the breast cancer cell lines, MDA-MB-231 and MDA-MB-468, completely blocked the constitutive secretion of Hsp90α, and that secretion could be rescued by re-introducing the genes encoding WT-HIF-1α and CA-HIF-1α (constitutively active), but not DN-HIF-1α (dominant-negative) (Sahu et al. 2012). These data establish that HIF-1α is a direct upstream regulator of Hsp90α secretion.
The important role of tumor-secreted Hsp90α has been demonstrated in tumor cell-derived xenograft models. Tsutsumi and colleagues showed that the membrane impermeable 17-AAG inhibitor, DMAG-N-oxide, blocked the action of tumor-secreted Hsp90 and decreased tumor cell lung colonization in nude mice (Tsutsumi et al. 2008). Stellas et al. reported that the anti-Hsp90α monoclonal antibody, 4C5, inhibited breast cancer cell “deposits” in the lungs of nude mice (Stellas et al. 2010). A similar observation was made with an independent monoclonal antibody by Song and colleague (Song and Luo 2010). Sahu et al. showed that down-regulation of the LRP-1 receptor in MDA-MB-231 cells dramatically reduced lung colonization of the cancer cells in nude mice (Sahu et al. 2012). However, these studies suffered from several technical limitations, among the most notable being uncertainty regarding the specificity of the antibodies and their untested effects on tumors that had already formed, similar to cancer patients who initially come to see doctors. A recent study by Zou and colleagues took a systematic approach to address these issues. First, they showed that CRISPR/Case9 knockout of Hsp90α nullified the tumor cells’ ability to migrate, invade, and metastasize without affecting cell survival or growth. As expected, knockout of Hsp90β, the critical intracellular chaperone, led to tumor cell death. Extracellular supplementation with recombinant Hsp90α, but not Hsp90β, protein fully recovered tumorigenicity of the Hsp90α-knockout cells. Sequential mutagenesis identified two evolutionarily conserved lysine residues, lys-270 and lys-277, in the Hsp90α subfamily that determine extracellular Hsp90α function. The Hsp90β subfamily lacks the dual lysine motif and extracellular function. Zou et al. then constructed a monoclonal antibody, 1G6-D7, that targets the dual lysine region of secreted Hsp90α. 1G6-D7 inhibited both de novo tumor formation and expansion of pre-existing tumors in mice. As schematically shown in Fig. 15.5, this study suggests an alternative therapeutic approach to targeting tumor-secreted Hsp90α, instead of intracellular Hsp90α or Hsp90β, in cancer therapy. When these findings (the number of HIF-1α-overexpressing tumors, the biological functions of tumor-secreted Hsp90α, and Hsp90α’s effects on in vivo tumor progression), are taken together, one can extrapolate that tumor-secreted Hsp90α plays an important role in HIF-1α-overexpressing malignancies, which are approximately 40% of all tumors in humans.

1G6-D7 inhibits expansion of pre-formed tumors in vivo. (a) Based on the predicted crystal structure of Hsp90α, lysine-270 and lysine-277 are located in the unstructured linker region (LR) between the NTD and MD domains. F-5 inhibitors, such as mAb 1G6-D7, targeting the dual lysine residues (enlarged box) block secreted Hsp90α-triggered tumorigenesis; (b) A representative experiment demonstrating the effects of mAb 1G6-D7 on tumor progression over 35 days in mice injected with MDA-MB-231 cells (5 × 106). On day 5, with an average tumour size of 60 mm3, either vehicle, control mouse IgG, or 1G6-D7 was injected via IV (5 mg/kg) and around the tumour site (125 μg/injection). Measurement of tumor volumes in live mice (n = 5) were recorded every 5 days. (Taken from Zou et al. 2016 with permission)
1.9 Is Targeting Tumor-Secreted HSP90α More Effective and Less Toxic Than Targeting the Intracellular HSP90β Chaperone When Treating Tumors?
In contrast to the critical chaperone function of intracellular Hsp90β in normal cell homeostasis, no physiological function has been reported for secreted Hsp90α. Instead, all studies over the past decade have clearly shown that secretion of Hsp90α by normal cells is an emergency response to environmental insults, such as inflammation, heat, oxidation, and hypoxia, among others. Furthermore, the functions of secreted Hsp90α no longer require its intrinsic ATPase, which is the target site for small molecule inhibitors and the principle cause of cytotoxicity in clinical trials. Therefore, the F-5 epitope of tumor-secreted Hsp90α may represent an excellent target for design of safer and more effective inhibitors for treatment of tumors in which HIF1A is the upstream driver gene. We propose that this new type of anti-cancer drug should (i) selectively inhibit tumor-secreted Hsp90α (and not intracellular Hsp90β) and (ii) specifically target the dual lysine residues within the F-5 region. At least in theory, drugs that bear both properties should achieve high efficacy and pose minimal toxicity to patients.
1.10 Wound Healing and Tumor Progression: Similar Mechanisms with Opposite Outcomes
Wound healing has fixed beginning and end points, whereas cancer, once initiated, may continue to progress until a patient’s demise. In his analytical article published in the New England Journal of Medicine, Dvotak outlined the similarities and distinctions between wound healing and tumor stroma generation. He suggested that tumor stroma formation is a subversion of the normal wound healing process. He called tumors the “wounds that do not heal” (Dvorak 1986). When considering design of drugs which promote healing or inhibit tumorigenesis, it is critical to remember that targeting molecules which affect one process, will likely affect the other. This is especially important to consider when discussing Hsp90α as a drug target. For instance, when formulating a topical application of F-5 peptide to promote wound healing, a drug developer would have to consider whether or not the peptide could gain access to peripheral circulation and travel to sites where an early-stage tumor is already in progress. Under these circumstance, F-5 may aid the tumor’s progression and accelerate invasion and metastasis. On the other hand, administration of F-5 inhibitors, such as monoclonal antibodies to block tumor-secreted Hsp90α activity, might interfere with the wound healing process and negatively impact patients with surgical or chronic wounds. For example, numerous studies have shown that patients with type II diabetes are more likely to die from cancer than non-diabetics. Therefore, if a diabetic patient who was simultaneously suffering from a malignancy and a foot ulcer was treated with inhibitors of F-5 by an oncologist to slow cancer progression, the administered inhibitor could interfere with the healing of the foot ulcer. The reverse is also true. If the diabetic ulcer was treated by a wound specialist with F-5 peptide to promote healing, the peptide may travel through blood circulation to the tumor site and aid tumor invasion and metastasis. It is critical to know when and why patients should receive these potential treatments.
1.11 Is Extracellular HSP90α a “General Repair Molecule”?
Studies over the past few years have clearly indicated the previously contended notion that Hsp90α is not a critical intracellular chaperone, but Hsp90β is. First, the absence of Hsp90α, or at least its intracellular chaperone form, has a limited impact on mouse development (Grad et al. 2010; Imai et al. 2011), whereas Hsp90β knockout is lethal to developing embryos (Voss et al. 2000). These findings indicate that Hsp90α is less important than Hsp90β for homeostasis during development. Similarly, at the cellular level, Zou et al. have recently shown that CRISPR/Case9 knockout of Hsp90β in the MDA-MB-231 human breast cancer cell line led to tumor cell death, whereas knockout of Hsp90α in the same cells only eliminated the cancerous properties of the cells (Zou et al. 2016). Second, at the signaling level, Jayaprakash and colleagues reported discovery of a novel ischemia-response mechanism by which the two Hsp90 isoforms, Hsp90α and Hsp90β, work together, but have distinct and non-exchangeable functions involving LRP- receptor signaling during wound healing. Under hypoxic conditions or in paucity of nutrients, Hsp90β, but not Hsp90α, binds to the cytoplasmic tail of LRP-1 and stabilizes the receptor at the cell surface. Hsp90α, but not Hsp90β, is then secreted by the same cells into the extracellular space, where it binds and signals through the LRP-1 receptor to promote cell motility, leading to wound closure (Jayaprakash et al. 2015). This study provides a mechanistic support for the outcomes of the genetic studies in mice.
Currently, the hypothesis that Hsp90α was designed by nature as a “general repair” molecule is limited to indirect support and speculation. To proceed forward, this hypothesis needs to be tested in multiple tissue injury animal models. However, there is a preexisting narrative suggesting a role of Hsp90α in healing that has been conserved through evolution. Due to its unusual abundance of 2–3% of the total cellular protein in all types of normal cells, Csermely et al. have long argued that evolution would not have tolerated such a “waste” if the function of Hsp90 had only been as an intracellular chaperone (Csermely et al. 1998). Instead, we can view stockpiles of Hsp90α all over our bodies as fire stations dispersed around a city. When tissue is injured, the pre-stored Hsp90α will guarantee a quick response to repair signals immediately released into the wound environment. Likewise, it would not make sense for a city to only start building fire stations after the fire already took place. While Csermely’s hypothesis is a bold one, our studies have provided three unique qualifications that secreted Hsp90α has, whereas growth factors do not, as previously described. Extracellular Hsp90α’s apparent superiority over growth factors in promotion of wound closure has been reflected in experiments in which topical application of recombinant Hsp90α protein dramatically shortened the time of diabetic mouse wound closure from 35 days to ~18 days, a ~17 day reduction in healing time compared with the 5 day reduction achieved via treatment with becaplermin gel (Cheng et al. 2011)
Cancer cells have an interesting, love-hate relationship with the TGFβ protein family, whose members are regarded as both tumor suppressors and tumor promoters (Bachman and Park 2005). The inhibitory effects of TGFβ on cancer progression are reflective of their hindering effects on wound healing. To bypass the tumor suppressing effects of TGFβ, some tumors mutate either their type II (TβRII) or type I (TβRI) TGFβ receptor, whereas other tumors achieve bypass of TGFβ inhibition by eliminating its downstream signaling molecule, Smad4, which forms complexes with activated Smad2/3 to regulate gene expression in the nucleus. These alterations in the TGFβ signaling pathway presumably result in cancer cells which are insensitive to the anti-proliferation and anti-migration signals of TGFβ. However, aside from these examples, the majority of cancers do not appear to harbor mutations in TGFβ signaling components. How then do most cancers continue to proliferate and migrate without mutation-mediated bypass of TGFβ’s inhibitory signals? If we put the importance of HIF-1α in tumor progression and the recent discovery of the “HIF-1 > Hsp90α secretion > LRP-1 > tumor cell invasion” axis into perspective, one can extrapolate a possible answer to this puzzle from the following facts: (1) Approximately 40% of all human tumors has constitutively elevated expression of HIF-1α, the critical subunit of the master transcription factor for tissue oxygen homeostasis (Semenza 2007); (2) HIF-1α is a central upstream activator of Hsp90α secretion (Li et al. 2007; Sahu et al. 2012); and (3) secreted Hsp90α is required for cancer cell invasion in vitro (Eustace et al. 2004) and tumor formation in vivo (Stellas et al. 2007; Tsutsumi et al. 2008; Song and Lou 2010; Zou et al. 2016). Therefore, it is conceivable that secretion of Hsp90α is an alternative strategy for cancer cells to bypass the anti-motility effects of TGFβ without mutating TGFβ signaling components.
2 Conclusions
Between 2006 and 2015, the FDA’s overall likelihood of approval (LOA) from Phase I clinical trials for all therapeutic candidates was 9.6%, with the highest LOA in hematology (26.1%) and the lowest LOA in oncology (5.1%). Two critical features attributed to the success of advancing cancer trials were (i) targeting driver genes of the diseases and (ii) employing human disease-relevant animal models during preclinical studies. Mammals have two isoforms of heat shock protein-90 (Hsp90α and Hsp90β) with which they respond to environmental stress, especially during tissue ischemia. Hsp90α is not a critical chaperone like Hsp90β inside the cells. Instead, Hsp90α is secreted into the extracellular space by cells under stress, such as tissue injury. Secreted Hsp90α signals through the LRP-1 receptor to protect cells from hypoxia-induced apoptosis and to promote cell motility during wound closure. Tumors take advantage of these “useful” functions of secreted Hsp90α to cope with the constant paucity of oxygen and nutrients within the tumor microenvironment. Therefore, Hsp90α appears to be designed by nature as a “general repair” molecule with an exceptional large stockpile throughout the body of mammals. This design guarantees a quick first response by avoiding the travel delays of obstructed and damaged blood vessel “highways” and arrives equipped to extinguish TGFβ and hypoxia-induced inhibition. We look forward to exploring these new questions together, alongside the scientific community, with optimism and excitement.
Abbreviations
- 17-AAG:
-
17-(Allylamino)-17-demethoxygeldanamycin
- F-5:
-
fragment-5
- HIF-1:
-
hypoxia-inducible factor-1
- hsp90:
-
heat shock protein-90
- LRP-1:
-
LDL receptor-related protein-1
- PDGF:
-
platelet-derived growth factor
- PRAS40:
-
proline-rich Akt substrate of 40 kDa
References
Bachman KE, Park BH (2005) Duel nature of TGF-β signaling: tumor suppressor vs. tumor promoter. Curr Opin Oncol 17:49–54
Bandyopadhyay B, Fan JF, Guan SX, Li Y, Fedesco M, Chen M, Woodley DT, Li W (2006) A “traffic control” role for TGFbeta3: orchestrating dermal and epidermal cell motility during wound healing. J Cell Biol 172:1093–1105
Banerji U (2009) Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res 15:9–14
Bejcek BE, Hoffman RM, Lipps D et al (1992) The v-sis oncogene product but not platelet-derived growth factor (PDGF) A homodimers activate PDGF alpha and beta receptors intracellularly and initiate cellular transformation. J Biol Chem 267:3289–3293
Bhatia A, O’Brien K, Chen M et al (2016) Dual therapeutic functions of F-5 fragment in burn wounds: preventing wound progression and promoting wound healing in pigs. Mol Ther Methods Clin Dev 3:16401–16411
Bobrie A, Krumeich S, Reyal F et al (2012) Rab27a supports exosome-dependent and-independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res 72:4920–4930
Botusan IR, Sunkari VG, Savu O et al (2008) Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci U S A 105:19426–19431
Brown GL, Nanney LB, Griffen J et al (1989) Enhancement of wound healing by topical treatment with epidermal growth factor. New Engl J Med 321:76–79
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820
Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L (2004) Hyperglycemia regulates hypoxia-inducible factor-1α protein stability and function. Diabetes 53:3226–3232
Chatterjee M, Jain S, Stühmer T, Andrulis M, Ungethüm U, Kuban RJ, Lorentz H, Bommert K, Topp M, Krämer D, Müller-Hermelink HK, Einsele H, Greiner A, Bargou RC (2007) STAT3 and MAPK signaling maintain overexpression of heat shock proteins 90alpha and beta in multiple myeloma cells, which critically contribute to tumor-cell survival. Blood 109:720–728
Chen JS, Hsu YM, Chen CC, Chen LL, Lee CC, Huang TS (2010) Secreted heat shock protein 90α induces colorectal cancer cell invasion through CD91/LRP-1 and NF-κB-mediated integrin αV expression. J Biol Chem 285:25458–25466
Cheng C, Fan J, Fedesco M et al (2008) Transforming growth factor alpha (TGFalpha)-stimulated secretion of HSP90alpha: using the receptor LRP-1/CD91 to promote human skin cell migration against a TGFbeta-rich environment during wound healing. Mol Cell Biol 28:3344–3358
Cheng C, Sahu D, Tsen F (2011) A fragment of secreted hsp90cl carries properties that enable it to accelerate effectively both acute and diabetic wound healing in mice. J Clin Invest 121:4348–4361
Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z (2005) Induction of heat shock proteins in B-cell exosomes. J Cell Sci 118:3631–3638
Colombo M, Raposo G, Théry C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Ann Rev Cell Dev Biol 30:255–289
Csermely P, Schnaider T, So C, Prohászka Z, Nardai G (1998) The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol Ther 79:129–168
Dales JP, Garcia S, Meunier-Carpentier S et al (2005) Overexpression of hypoxia-inducible factor HIF-1α predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int J Cancer 116:734–739
Dvorak HF (1986) Tumors: wounds that do not heal. New Engl J Med 315:1650–1659
Dong H, Zou M, Bhatia A, Jayaprakash P, Hofman F, Ying Q, Chen M, Woodley DT, Li W (2015) Breast Cancer MDA-MB-231 cells use secreted heat shock protein- 90alpha (Hsp90α) to survive a hostile hypoxic environment. Sci Rep 6:20605
Embil JM, Nagai MK (2002) Becaplermin: recombinant platelet derived growth factor, a new treatment for healing diabetic foot ulcers. Expert Opin Biol Ther 2:211–218
Eustace BK, Jay DG (2004) Extracellular roles for the molecular chaperone, hsp90. Cell Cycle 3:1096–1098
Eustace BK, Sakurai T, Stewart JK et al (2004) Functional proteomic screens reveal an essential extracellular role for hsp90α in cancer cell invasiveness. Nat Cell Biol 6:507–514
Fonseca BD, Smith EM, Lee VH-Y, MacKintosh C, Proud CG (2007) PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem 282:24514–24524
Fonseca BD, Lee VH-Y, Proud CG (2008) The binding of PRAS40 to 14-3-3 proteins is not required for activation of mTORC1 signalling by phorbol esters/ERK. Biochem J 411:141–149
Grad I, Cederroth CR, Walicki J et al (2010) The molecular chaperone Hsp90α is required for meiotic progression of spermatocytes beyond pachytene in the mouse. PLoS One 5:e15770
Grose R, Werner S (2004) Wound-healing studies in transgenic and knockout mice. Mol Biotechnol 28:147–166
Guo J, Jayaprakash P, Dan J et al (2017) PRAS40 connects microenvironmental stress signaling to exosome-mediated secretion. Mol Cell Biol 37:e00171–e00117
Gurtner GC, Werner S, Barrandon Y, Longaker MT (2008) Wound repair and regeneration. Nature 453:314–321
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Harding C, Heuser J, Stahl P (1983) Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol 97:329–339
Havel JJ, Li Z, Cheng D, Peng J, Fu H (2015) Nuclear PRAS40 couples the Akt/mTORC1 signaling axis to the RPL11-HDM2-p53 nucleolar stress response pathway. Oncogene 34:1487–1498
Hoshino A, Costa-Silva B, Shen T-L et al (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527:329–335
Huang L, Nakai Y, Kuwahara I, Matsumoto K (2012) PRAS40 is a functionally critical target for EWS repression in Ewing’s sarcoma. Cancer Res 72:1260–1269
Imai T, Kato Y, Kajiwara C et al (2011) Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. PNAS 108:16363–16368
Isaacs JS, Xu W, Neckers L (2003) Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 3:213–217
Jayaprakash P, Dong H, Zou M et al (2015) Hsp90α and Hsp90β together operate a hypoxia and nutrient paucity stress-response mechanism during wound healing. J Cell Sci 128:1475–1480
Hance MW, Nolan KD, Isaacs JS (2014, May 6) The double-edged sword: conserved functions of extracellular hsp90 in wound healing and cancer. Cancers (Basel) 6(2):1065–1097
Johnstone RM, Adam M, Hammond J, Orr L, Turbide C (1987) Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem 262:9412–9420
Kamal A, Thao L, Sensintaffar J et al (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425:407–410
Kazi AA, Lang CH (2010) PRAS40 regulates protein synthesis and cell cycle in C2C12 myoblasts. Mol Med 16:359–371
Kovacina KS, Park GY, Bae SS et al (2003) Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 278:10189–10194
Kucharzewska P, Belting M (2013) Emerging roles of extracellular vesicles in the adaptive response of tumour cells to microenvironmental stress. J Extracell Vesicles 5(2). https://doi.org/10.3402/jev.v2i0.20304
Kuo CC, Liang CM, Lai CY, Liang SM (2007) Involvement of heat shock protein (Hsp) 90β but not Hsp90α in antiapoptotic effect of CpG-B oligodeoxynucleotide. J Immunol 178:6100–6108
Kuroita T, Tachibana H, Ohashi H, Shirahata S, Murakami H (1992) Growth stimulating activity of heat shock protein 90α to lymphoid cell lines in serum-free medium. Cytotechnology 8:109–117
LeGrand EK (1998) Preclinical promise of becaplermin (rhPDGF-BB) in wound healing. Am J Surg 176:48S–54S
Li W, Li Y, Guan S et al (2007) Extracellular heat shock protein-90α: linking hypoxia to skin cell motility and wound healing. EMBO J 26:1221–1233
Li W, Sahu D, Tsen F (2012) Secreted heat shock protein-90 (Hsp90) in wound healing and cancer. Biochim Biophys Acta 1823:730–741
Li W, Tsen F, Sahu D, Bhatia A, Chen M, Multhoff G, Woodley DT (2013) Extracellular Hsp90 (eHsp90) as the actual target in clinical trials: intentionally or unintentionally. IRCMB 34:124–150
Madhunapantula SV, Sharma A, Robertson GP (2007) PRAS40 deregulates apoptosis in malignant melanoma. Cancer Res 67:3626–3636
Mandracchia VJ, Sanders SM, Frerichs JA (2001) The use of becaplermin (rhPDGF-BB) gel for chronic nonhealing ulcers. A retrospective analysis. Clin Podiatr Med Surg 18:189–209
McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J (2007) Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131:121–135
McCready J, Sims JD, Chan D, Jay DG (2010) Secretion of extracellular hsp90α via exosomes increases cancer cell motility: a role for plasminogen activation. BMC Cancer 10:294
Neckers L, Neckers K (2002) Heat-shock protein 90 inhibitors as novel cancer chemotherapeutic agents. Expert Opin Emerg Drugs 7:277–288
Nolte-’t Hoen EN, Buermans HP, Waasdorp M, Stoorvogel W, Wauben MH, ’t Hoen PA (2012) Deep sequencing of RNA from immune cell-derived vesicles uncovers the selective incorporation of small non-coding RNA biotypes with potential regulatory functions. Nucleic Acids Res 40:9272–9285
Oshiro N, Takahashi R, Yoshino K et al (2007) The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 282:20329–20339
O’Brien K, Bhatia A, Tsen F, Chen M, Wong AK, Woodley DT, Li W (2015) Identification of the critical therapeutic entity in secreted Hsp90α that promotes wound healing in newly re-standardized healthy and diabetic pig models. PLoS One 9:e113956
Ostrowski M, Carmo NB, Krumeich S et al (2010) Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat Cell Biol 12:19–30
Pan BT, Teng K, Wu C, Adam M, Johnstone RM (1985) Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J Cell Biol 101:942–948
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383
Ritossa F (1996) Discovery of the heat shock response. Cell Stress Chaperones 1:97–98
Sahu D, Zhao Z, Tsen F et al (2012) A potentially common peptide target in secreted heat shock protein-90α for hypoxia-inducible factor-1α–positive tumors. Mol Biol Cell 23:602–613
Saito A, Narasimhan P, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH (2004) Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci 24:1584–1593
Sancak Y, Thoreen CC, Peterson TR et al (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25:903–915
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303
Semenza GL (2007) Evaluation of HIF-1 inhibitors as anticancer agents. Drug Discov Today 12:853–859
Semenza GL (2012a) Hypoxia-inducible factors in physiology and medicine. Cell 148:399–408
Semenza GL (2012b) Molecular mechanisms mediating metastasis of hypoxic breast cancer cells. Trends Mol Med 18:534–543
Sen CK, Gordillo GM, Roy S et al (2009) Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 17:763–771
Shimaya A, Kovacina KS, Roth RA (2004) On the mechanism for neomycin reversal of wortmannin inhibition of insulin stimulation of glucose uptake. J Biol Chem 279:55277–55282
Shin MJ, Kim DW, Jo HS et al (2016) Tat-PRAS40 prevent hippocampal HT-22 cell death and oxidative stress induced animal brain ischemic insults. Free Radic Biol Med 97:250–262
Sidera K, Samiotaki M, Yfanti E, Panayotou G, Patsavoudi E (2004) Involvement of cell surface HSP90 in cell migration reveals a novel role in the developing nervous system. J Biol Chem 279:45379–45388
Simon MC, Keith B (2008) The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 9:285–296
Singer AJ, Clark RA (1999) Cutaneous wound healing. New Engl J Med 341:738–746
Sinha S, Hoshino D, Hong NH et al (2016) Cortactin promotes exosome secretion by controlling branched actin dynamics. J Cell Biol 214:197–213
Song X, Luo Y (2010) The regulatory mechanism of Hsp90alpha secretion from endothelial cells and its role in angiogenesis during wound healing. Biochem Biophys Res Commun. 16 398(1):111–117
Steed DL (1995) Clinical evaluation of recombinant human platelet–derived growth factor for the treatment of lower extremity diabetic ulcers. J Vasc Surg 21:71–81
Stellas D, Karameris A, Patsavoudi E (2007) Monoclonal antibody 4C5 immunostains human melanomas and inhibits melanoma cell invasion and metastasis. Clin Cancer Res 13:1831–1838
Stellas D, El Hamidieh A, Patsavoudi E (2010) Monoclonal antibody 4C5 prevents activation of MMP2 and MMP9 by disrupting their interaction with extracellular HSP90 and inhibits formation of metastatic breast cancer cell deposits. BMC Cell Biol 11:51
Suzuki S, Kulkarni AB (2010) Extracellular heat shock protein HSP90beta secreted by MG63 osteosarcoma cells inhibits activation of latent TGF-beta1. Biochem Biophys Res Commun 398:525–531
Thedieck K, Polak P, Kim ML et al (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2:e1217
Théry C, Zitvogel L, Amigorena S (2002) Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579
Tkach M, Théry C (2016) Communication by extracellular vesicles: where we are and where we need to go. Cell 164:1226–1232
Tsutsumi S, Neckers L (2007) Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci 98:1536–1539
Tsutsumi S, Scroggins B, Koga F et al (2008) A small molecule cell-impermeant Hsp90 antagonist inhibits tumor cell motility and invasion. Oncogene 27:2478–2487
Vander Haar E, Lee S, Bandhakavi S, Griffin TJ, Kim D-H (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9:316–323
Voss AK, Thomas T, Gruss P (2000) Mice lacking HSP90beta fail to develop a placental labyrinth. Development 127:1–11
Wang L, Harris TE, Roth RA, Lawrence JC (2007) PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem 282:20036–20044
Wang L, Harris TE, Lawrence JC (2008) Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem 283:15619–15627
Wang X, Song X, Zhuo W et al (2009) The regulatory mechanism of Hsp90α secretion and its function in tumor malignancy. Proc Natl Acad Sci 106:21288–21293
Werner S, Grose R (2003) Regulation of wound healing by growth factors and cytokines. Physiol Rev 83:835–870
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5:761–772
Wieman TJ, Smiell JM, Su Y (1998) Efficacy and safely of a topical gel formulation of recombinant human platelet-derived growth factor-BB (becaplermin) in patients with chronic neuropathic diabetic ulcers: a phase III randomized placebo-controlled double-blind study. Diabetes Care 21:822–827
Witwer KW, Buzas EI, Bemis LT et al (2013) Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles 2:20360
Woodley DT, Fan J, Cheng CF et al (2009) Participation of the lipoprotein receptor LRP1 in hypoxia-HSP90α autocrine signaling to promote keratinocyte migration. J Cell Sci 122:1495–1498
Workman P (2004) Altered states: selectively drugging the Hsp90 cancer chaperone. Trends Mol Med 10:47–51
Workman P, Burrows F, Neckers L, Rosen N (2007) Drugging the cancer chaperone HSP90. Ann N Y Acad Sci 1113:202–216
Yáñez-Mó M, Siljander PRM, Andreu Z et al (2015) Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4:27066
Young JC, Moarefi I, Hartl FU (2001) Hsp90. J Cell Biol 154:267–274
Yu X, Harris SL, Levine AJ (2006) The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res 66:4795–4801
Yu F, Narasimhan P, Saito A, Liu J, Chan PH (2008) Increased expression of a proline-rich Akt substrate (PRAS40) in human copper/zinc-superoxide dismutase transgenic rats protects motor neurons from death after spinal cord injury. J Cereb Blood Flow Metab 28:44–52
Yuana Y, Sturk A, Nieuwland R (2013) Extracellular vesicles in physiological and pathological conditions. Blood Rev 27:31–39
Zhang F, Beharry ZM, Harris TE et al (2009) PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells. Cancer Biol Ther 8:846–853
Zhang L, Zhang S, Yao J et al (2015) Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527:100–104
Zheng Y, Campbell EC, Lucocq J, Riches A, Powis SJ (2013) Monitoring the Rab27 associated exosome pathway using nanoparticle tracking analysis. Exp Cell Res 319:1706–1713
Zou M, Bhatia A, Dong H et al (2016) Evolutionarily conserved dual lysine motif determines the non-chaperone function of secreted Hsp90alpha in tumour progression. Oncogene 36:2160–2171
Acknowledgments
We thank many of our current and previous lab colleagues who made contributions to the work described in this review. We apologize if we failed to acknowledge all publications on secreted Hsp90, while this review was being written. This study was supported by NIH grants GM066193 and GM067100 (to W. L.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lincoln, V., Tang, X., Chen, M., Li, W. (2019). Extracellular HSP90α Versus Intracellular HSP90β in Wound Healing and Cancer. In: Asea, A., Kaur, P. (eds) Heat Shock Proteins in Signaling Pathways. Heat Shock Proteins, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-030-03952-3_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-03952-3_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03951-6
Online ISBN: 978-3-030-03952-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)