
Abstract
The heat shock proteins (Hsp), the family of molecular chaperons, are key proteins in protein folding and maturation. The client proteins of Hsp are critical in number of biological processes including cellular proliferation, differentiation, survival, metastasis, invasion, and angiogenesis. Thus, Hsp family becomes one of the desirable targets for cancer treatment. It has been demonstrated that Hsp overexpress in multiple myeloma and linked in poor prognosis and relapse. This chapter describes about the Hsp and their possible link with the pathogenesis of multiple myeloma. It addresses the advancement and challenges in the development of Hsp inhibitors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Heat Shock Proteins (Hsp) are the family member of highly conserved proteins (Wu et al. 2017). In addition to their most studied role in protein folding, Hsp are also involved in intracellular trafficking, signaling pathways, and immune responses. These functions enable Hsp to play critical roles in the regulation of protein homeostasis, cell survival, development and differentiation. Based on the molecular weight, mammalian Hsp have been classified into five families: Hsp100, Hsp90, Hsp70, Hsp60, and small Hsp such as Hsp27. These proteins expressed differentially in the cells depending on the cell conditions, though often times these proteins are constitutively expressed. These proteins can be found in different cellular compartments, for example, Hsp90 is produced in the nucleus and cytoplasm, while Hsp60 is produced in the mitochondria (Whitley et al. 1999). Based on the studies it has been illustrated that each family members are designated for specific function for example, Hsp90 family found to be involved in steroid receptor complex formation, while Hsp70 plays an important role in protein synthesis process including folding, and Hsp60 provides protein stability. Along with the co-chaperones, Hsp90 protein forms a huge complex, which interacts with variety of proteins involved in various biological processes such as proliferation, survival, angiogenesis, cell cycle, invasion, and metastasis. Importantly, it has been reported that a complex of Hsp90 is required for activation of transcription factors (e.g. p53, NF-κB, HIF-1α), MAPK kinases (e.g. RAF-1), receptor tyrosine kinases (e.g. erbB) (Zagouri et al. 2012). Hsp70 interacts with a variety of proteins irrespective of their conformational stages, for example it may bind to unfolded, and natively folded proteins. Hsp70 protects the cells against cellular stresses, lethal injuries, and apoptosis, thus, promotes the cell survival. In cancer cells, it regulates intrinsic and extrinsic apoptosis by modulating caspase-independent apoptotic pathways (Kumar et al. 2016). Because of the critical role of Hsp70 in cancer cell survival and proliferation, mammoth efforts have been made to develop Hsp70 inhibitors for cancers mostly other than multiple myeloma (Kumar et al. 2016).
The small Hsp are conserved among species. This family of Hsp play very important role in autophagy, degradation of proteasomes, development, differentiation, and stress tolerance. In addition, small Hsp are anti-apoptotic in nature. The mutations in these proteins result in number of pathological conditions such as myopathies, neuropathies and cataract. It has been demonstrated that small Hsp bind with copper and suppress the generation of reactive oxygen species (Madamanchi et al. 2001). This property of small Hsp is implicated in copper homeostasis and neurodegenerative disorders, such as Alzheimer, Parkinson (Bakthisaran et al. 2015). All together small Hsp have both beneficial and detrimental effects on human health.
In most of the cancers, Hsp are found to be overexpressed, which is linked with the proliferation, survival, invasion, and metastasis (Lianos et al. 2015; Teng et al. 2012). Intracellularly, Hsp stabilize a number of oncogenic proteins, and facilitate the interaction of various signaling pathways associated with cancer. Because of their role in regulation and stabilization of a number of oncogenic proteins, the expression of Hsp is associated with poor prognosis of cancers including gastric, liver, prostate, osteosarcomas, breast, endometrial, uterine cervical, and bladder carcinomas. Hsp27 regulates the stability, nuclear shuttling and transcriptional activity of androgen receptor that are involved in prostate cancer (Azad et al. 2015; Cordonnier et al. 2015; Teng et al. 2012; Zagouri et al. 2012). Altogether, Hsp thus prove to be an excellent anti-cancer targets (Ciocca and Calderwood 2005; Zhang et al. 2014).
Multiple myeloma, is a plasma cell neoplasm. The hallmark of multiple myeloma is the production of monoclonal proteins and bone loss (Palumbo and Anderson 2011; Rajkumar and Kumar 2016). In last one decade the treatment options for multiple myeloma evolved beyond the expectations and now 5-year survival expectancy is more than 50%, which is remarkable. Nonetheless, almost every patient relapse after successful initial treatment, thus there is always a demand for novel drug development. Because Hsp regulate a number of key proteins which are integral in the pathogenesis of multiple myeloma, it has become attractive therapeutic target. Along these lines, number of Hsp inhibitors have been developed and shown promise (Born et al. 2013; Duus et al. 2006; Richardson et al. 2011c). Importantly, studies have demonstrated that these inhibitors possess the potential to inhibit drug resistance. How Hsp inhibitors control drug resistance is not fully understood, though a number of mechanisms have been proposed, for instance, inhibitor of Hsp27 induces the release of mitochondrial protein, Smac, and inhibits dexamethasone resistance in multiple myeloma cells (Chauhan et al. 2003).
In this chapter, we briefly discussed the roles of Hsp family members and current advancement in the development of Hsp inhibitors, particularly in multiple myeloma. Moreover, the strategies and challenges in targeting Hsp90 in particular is discussed in detail.
2 Role of Heat Shock Proteins in Cancer Cell Signaling
Hsp proteins play critical role in cancer cell survival (Calderwood and Gong 2016), because they interact with a variety of client proteins involved in regulation of cell cycle and inhibition of apoptosis (Chatterjee and Burns 2017). Thus, the importance of Hsp become more relevant in cancer cells as compared to their normal counterpart, because malignant cells require more energy in order to survive in challenging hostile environment. Consequently, the high levels of Hsp are reported in variety of tumors including solid as well as hematological cancers including multiple myeloma (Ciocca and Calderwood 2005; Flandrin et al. 2008; Saluja and Dudeja 2008; Zhang et al. 2014).
2.1 Hsp90
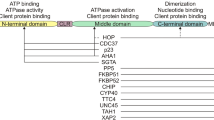
The monomer Hsp90 contains three highly conserved domains: amino terminal domain (NTD), middle domain (MD), and carboxy terminal domain (CTD). Each domain is unique in function, for example, NTD mediates the binding of ATP, and MD facilitates the binding of Hsp90 to client proteins, and hydrolyzes ATP, while CTD mediates the dimerization of Hsp90, which is essential for its function (Fig. 12.1) (Ciglia et al. 2014; Colombo et al. 2008; Hawle et al. 2006; Nemoto et al. 1995; Ratzke et al. 2010). Both NTD and MD are connected by a long, flexible, charged linker. Monomer Hsp90 also contains peculiar motif at CTD site called MEEVD motif. The MEEVD motif facilitates the binding of Hsp90 to specific co-chaperons those contain TPR domains (Zuehlke and Johnson 2010). The ATP free Hsp90 exist in V-shaped open conformation. ATP binding induces conformation changes, which leads to a closed and active conformation. The overexpression Hsp90 has been observed in a variety of malignancies including multiple myeloma (Zhang et al. 2014). It has been demonstrated that proteins integral in cell cycle, kinases, transcription factors, survival and anti-apoptotic proteins are the client of Hsp90 (Wayne et al. 2011). Thus Hsp90’s client proteins regulate number of key processes linked with “hallmarks of cancer” (Chatterjee and Burns 2017; Miyata et al. 2013).

Schematic presentation of Hsp90 protein structure. The homodimer of Hsp90 contains a N terminal domain (NTD), connecting linker region (CR), middle domain (MD), and a C terminal domain (CTD). Each domain has a specific function, for example NTD is responsible for ATPase and co-chaperone binding, MTD binds to client protein, and facilitates co-chaperon binding, and CTD is instrumental in dimerization. Targeting specific domains has developed various small molecule inhibitors. The inhibitors highlighted in red color have been identified in other than multiple myeloma
2.2 Hsp70
The chaperon Hsp70 family facilitates protein folding process of client proteins (Saibil 2013). The overexpression of Hsp70 is linked with poor prognosis and aggressiveness of cancers (Lee et al. 2013; Murphy 2013). The HSP70’s client proteins play key role in anti-apoptosis, senescence, and autophagy (Mayer and Bukau 2005). It has been reported that Hsp70 is anti-apoptotic in nature and promotes the cancer cell survival and linked with drug resistance in a variety of cancers including multiple myeloma (Nimmanapalli et al. 2008; Reikvam et al. 2013; Yang et al. 2012). Number of mechanisms have been proposed regarding the role of Hsp70 in the regulation of apoptosis. Hsp70 protects the degradation of Bcl-2, suppresses Bax translocation to the mitochondria, and inhibits the recruitment of caspase 9 to apoptosome by binding to Apaf-1 (Li et al. 2000; Rerole et al. 2011). Overall, these studies demonstrate that targeting of Hsp70 is required in order to induce apoptosis and treatment of cancers including multiple myeloma (Bailey et al. 2015; Zhang et al. 2014).
2.3 Hsp27
Hsp27 is overexpressed in a variety of cancers including multiple myeloma, breast, colorectal, ovarian, and prostate that is correlated with poor prognosis, chemo-resistance, and tumor aggressiveness (Chauhan et al. 2003; Langdon et al. 1995; Voll et al. 2014; Wei et al. 2011; Yu et al. 2010; Zhang et al. 2014, 2016). Not only Hsp27 is critical in tumor survival, it is also instrumental in actin dynamics, cell migration and invasion. Studies have demonstrated that knocking down of Hsp27 leads to suppression of bone metastasis in a breast tumor model (Gibert et al. 2012). Elevated levels of Hsp27 transcripts have been observed in multiple myeloma cells (Zhang et al. 2014). Using oligonucleotide array, Chauhan et al. (2004) showed that Hsp27 mRNA is highly expressed in Dexamethasone-resistant multiple myeloma cells versus Dexamethasone -sensitive multiple myeloma cells, which further supports its role in chemo resistance.
3 Molecular Structural Sites to Target Hsp90
Hsp90 proteins are evolutionarily conserved molecular chaperons accounting for almost 1–2% of total protein under normal condition (non-stressful) and 5–6% under stressful condition (Usmani and Chiosis 2011). The protein Hsp90, regulated by more than ten genes, is mainly an intracellular non-secretary localized in the cytoplasm, endoplasmic reticulum, mitochondria and small amount in nucleoplasm (Chen et al. 2005; ProteinAtlas 2018). However, Hsp90 are also secreted and found in the extracellular environment of cancer cells to some extent with role in metastasis (Wong and Jay 2016). Considering intracellular abundance of Hsp90, small molecules and peptides may be ideal drug tools to target Hsp90 than the large molecules. Indeed, over a decade, a several synthetic molecules have been developed to inhibit Hsp90 and some also made to the clinical trials although none is yet approved by Food and Drug Administration (FDA). The possible drug-able sites on Hsp90 can be predicted by understanding the roles of different sites of Hsp90. As mentioned earlier the Hsp90 constitutes: NTD, connecting flexible linker region, MD, and CTD (Fig. 12.1) (Richardson et al. 2011b). The NTD is the site for nucleotide (ATP/ADP) binding and interaction with co-chaperons. It is the site where ADP-ATP exchange and hydrolysis of ATP to ADP occurs. The binding of ATP makes the conformational changes in the Hsp90 molecule which is also coordinated by co-chaperons such as Hip, Hop, Cdc37, p23, immunophilins, and Aha1 which bind at NTD, MD or CTD. These co-chaperons regulate the interaction of Hsp90 with nucleotides and substrate proteins (a.k.a. client proteins). Most of the client proteins bind to the MD during “immature” phase of the complex – just before ATP hydrolysis – and eventually becomes “mature” complex upon ATP hydrolysis (Sidera and Patsavoudi 2014). The set of required co-chaperons may differ with the client proteins which emphasizes the significance of the role of co-chaperons in Hsp90 activity. Hence, all three sites on Hsp90 play crucial role in the function of Hsp90 and therefore all three sites are plausible drug-able sites. However, each site poses challenges and benefits to target with the therapeutic. This basic understanding of mechanics of Hsp90 hints some possible targeting strategies: (1) Targeting ATP-binding site on Hsp90, (2) Targeting interaction of co-chaperons with Hsp90, and (3) Targeting interaction of Hsp90 with its client proteins.
3.1 Targeting ATP-Binding Site on Hsp90 (NTD and CTD)
Geldanamycin, radiciol and their derivatives bind to NTD and interfere with binding of ATP to NTD. However, it is important to note that geldanamycin and radiciol exhibit two different modes of binding within ATP-binding pocket. Although these drugs initially looked promising, they failed in clinical trials either due to toxicity or not having enough therapeutic effects, lowering the risk-benefit ratio. This might be due to ubiquitous nature of ATP binding sites on more than 500 kinases present in humans and hence more possibilities of off-target effects. However, this does not mean more specific ATP-competitive inhibitors cannot be developed. By rational drug design more selective inhibitors targeting cancer cells can be developed. The rational for development of cancer cell specific Hsp90 inhibitors is that cancer cell-derived Hsp90 shows 100-fold higher binding affinity to the inhibitors than Hsp90 from normal cells (Kamal et al. 2003). Moreover, two types of Hsp90 exists in cancer cells: major housekeeping Hsp90 which have low affinity for inhibitors and another type of stressed Hsp90 in complex with multi-chaperones with high affinity conformation (Moulick et al. 2011). The small molecule PU-H71, which was a result of rational drug design, showed binding specificity for stressed cancer specific Hsp90-oncoprotein complex in chronic myeloid leukemia cells (Bcr-Abl-Hsp90), melanoma cells (B-Raf-Hsp90) and breast cancer cells (Her3-Hsp90 and Raf1-Hsp90). Per http://www.ClinicalTrials.gov, as on April, 2018, PU-H71 is being studied (“recruiting” status) in clinical trials as a combination therapy for breast cancer (with Nab-Paclitaxel) and myelofibrosis (with rituximab). Additionally, Phase 1 clinical trial for PU-H71 alone in patients with advanced malignancies shows status as “active but not recruiting”, while another clinical trial in patients with solid tumors and low-grade Hodgkin’s lymphoma has been terminated. Phase 1 clinical trial data indicated that PU-H71 was well tolerated with some tumor regression in cervical squamous cell carcinoma (SCC; 22.6%), triple negative breast cancer (8.3%), ER positive breast cancer (25.6%), penile SCC (20.8%), and marginal zone lymphoma (20.6%) (Gerecitano et al. 2015). However, its effect in multiple myeloma is not yet clear. In this trial, 5 patients out of 40 exhibited dose limiting toxicities such as mucositis, liver enzyme elevation (ALT and AST), nausea/vomiting, intolerable myalgia, anemia, and intolerable headache (Gerecitano et al. 2015).
CTD of Hsp90 has another putative ATP-binding site which may be involved in allosteric regulation of conformational change of Hsp90 during activity. Novobiocin was the first small molecule inhibitor to be identified as inhibitor of Hsp90 at CTD (Marcu et al. 2000). Eventually cisplatin, epigallocatechin-3-gallate (a green tea component), and taxol were also identified as CTD inhibitors (Donnelly and Blagg 2008). A novel CTD inhibitor, KU675, has shown to be effective in vitro in prostate cancer cell lines. KU675 selectively inhibited HSP90α (Kd = 191 μM) over HSP90β (Kd = 726 μM) (Liu et al. 2015). However, its activity in multiple myeloma cells is not known. One probable advantage with CTD-binding Hsp90 inhibitors is that they do not induce stress response in cells and hence less chances of developing resistance.
3.2 Targeting Interaction of Co-chaperons with Hsp90
Binding of co-chaperons is essential for the activity of Hsp90. The list of Hsp90 co-chaperons includes Aha1/Ch1 (stimulates Hsp90 ATPase), p23 (involved in maturation of client proteins at later stage), Cdc37 (involved in protein kinase folding), Cns1 (uncertain role), Unc45 (involved in myosin assembly), PP5 (protein phosphatase), Tom70 (mitochondrial preprotein import), Chip (ubiquitin ligase), and HOP (connects Hsp70 and Hsp90, and transfers client proteins) (Caplan 2003). Inhibition of Hsp90 at NTD has shown to induce heat shock response by upregulating Hsp70 and hence conferring resistance to Hsp90 inhibitors. In view of that, targeting interaction of co-chaperon with Hsp90 seems to be a viable alternate strategy to make Hsp90 dysfunctional. Novobiocin which was initially discovered to bind ATP binding site at CTD, also interferes with binding of p23 and Hsc70 with Hsp90 and hence its activity. The co-chaperon p23 helps in maturation of the client proteins. Hence, in absence of maturation, the client proteins will undergo proteasome-dependent degradation. Similarly, celastrol and gedunin were also identified to inactivate p23 and substrate maturation (Chadli et al. 2010; Patwardhan et al. 2013). Before identifying celastrol and gedunin as p23 inhibitors, these agents were identified as inhibitors of co-chaperon Cdc37 during gene-expression based target pathway identification analysis (Hieronymus et al. 2006). Mechanistic studies revealed that celastrol induces large conformational change in N-terminal ‘kinase-binding’ domain and middle ‘Hsp90 (N-terminal)-binding domain’ of Cdc37 resulting in disruption of Cdc37-Hsp90 complex formation and destabilization of numerous kinase client proteins (Sreeramulu et al. 2009). However, although optimistic, efficacy of these inhibitors need yet to be investigated in multiple myeloma cancer cell types. Another important co-chaperon Aha1 binds to NTD and MD of Hsp90 and stimulates ATPase activity at those sites. siRNA mediated selective inhibition of Aha1 decreased activity of C-Raf, and levels of Mek1/2 and Erk1/2 in colon cancer cells (Holmes et al. 2008). Recently identified small molecule HAM-1 binds to NTD of Hsp90 overlapping with Aha1 interacting site (Stiegler et al. 2017). Thus HAM-1 interferes with transient interaction of Aha1 and NTD of Hsp90, without dissociating Hsp90-Aha1 complex, but affects catalytic activity of Hsp90 and activity of Hsp90-Aha1 dependent client proteins in yeast.
3.3 Targeting Interaction of Hsp90 with Its Client Proteins
The main function of Hsp90 is to ensure proper folding of client proteins for their proper activity. Hence, targeting the interaction of Hsp90 and their substrate becomes a logical strategy to inhibit function of Hsp90. A considerable body of evidence from structural and functional analysis indicates that middle segment of Hsp90 plays a crucial role in binding of Hsp90 with the client proteins (Meyer et al. 2003). One of such client proteins is androgen receptor (AR). Targeting AR has been a promising strategy to treat prostate cancer. In vitro studies revealed that Hsp90 stabilizes AR in the inactive conformation and is responsible for the nuclear translocation and transcriptional activity of androgen bound-AR (Georget et al. 2002). Although these studies reveal necessity of Hsp90-AR interaction in prostate cancer, it is less doubtful that this mechanism will be applicable for other client proteins that are key to multiple myeloma. A topoisomerase-I inhibitor, camptothecin, has been shown to disrupt association between Hsp90 and AR, interferes in binding of androgen to AR leading to inhibition of cell growth in LNCaP prostate cancer cells (Liu et al. 2010). Another such example of Hsp90-client protein interaction is Hsp90-survivin. Survivin plays a key role in promoting cell proliferation and preventing apoptosis in cancer cells (Wheatley and McNeish 2005). Fortugno et al. reported that survivin proteins remain physically associated with Hsp90 in human cervical cancer HeLa cells (Fortugno et al. 2003). They further demonstrated that Hsp90 is required for stability of survivin and inhibition of Hsp90 by geldanamycin induced proteasome-dependent degradation of survivin. Plescia et al., creatively identified minimal amino acid sequence in survivin that can block Hsp90-survivin interaction by using synthetic peptidyl mimicry (Plescia et al. 2005). The resultant peptide from this sequence was named shepherdin. Shepherdin inhibited interaction of survivin and Hsp90 not by binding at the middle domain, but at N-terminal ATP binding site of Hsp90 which is slightly different than geldanamycin site in that shepherdin does not require geldanamycin-specific D93 site for its activity. Shepherdin selectively killed cancer cells (prostate carcinoma and HeLa cells) over normal cells indicating less probability of having off-target effects or overt toxicity. Moreover, shepherdin also showed promising anticancer effects in prostate and breast cancer mouse models. Altogether, these studies suggest that strategy to disrupt Hsp90-client protein interaction may well work in multiple myeloma as well.
4 Hsp90 Inhibitors
There are many Hsp90 inhibitors in the clinical trials, however none is approved by FDA or European Medicines Agency (EMA) so far for the treatment of multiple myeloma (Table 12.1). The Hsp90 inhibitors are being studied in clinical trials either alone or in combination with existing preferred therapeutics which predominantly include bortezomib and dexamethasone. However, the clinical trial results indicated that Hsp90 inhibitors are more effective in combination with standard multiple myeloma chemotherapy. The molecular mechanisms for its reasons are discussed in following section. The obvious reason for better efficacy in combination with bortezomib is by combined inhibition of Hsp90 and proteasomal degradation leads to increased accumulation of unfolded proteins and enhanced unfolded protein response (UPR) in cancer cells leading to activation of apoptotic pathways.
The 17-AAG was the first geldanamycin derivative to enter in the clinical trials. However, poor solubility, limited bioavailability, and overt toxicity limited its success. In these trials, 17-AAD was studied as a single agent with no/minimal efficacy in multiple myeloma patients. However, some adverse events like diarrhea, hepatotoxicity, gastrointestinal complaints, and nausea were observed in patients. However, combination of 17-AAG with bortezomib in Phase 1/2 and abbreviated Phase 2 clinical trials showed some promising response rate and diminished severity of bortezomib-induced peripheral neuropathy in multiple myeloma patients (Richardson et al. 2010, 2011a). A Hsp90 inhibitor, NVP-AU922, was studied in Phase 1/1B clinical trials either as a monotherapy or in combination with bortezomib for treatment of multiple myeloma. During the treatment, 12 out of 24 (50%) patients discontinued the NVP-AU922 treatment for various reasons while 8 out of remainder 12 showed adverse events without partial or complete response which may be due to compensatory upregulation of Hsp70, another chaperon like Hsp90. However, 66.6% patient showed disease stabilization. All the five patients enrolled in NVP-AU922 (50 mg/m2) plus bortezomib (1.3 mg/m2) combination therapy showed adverse events and 3 out of 5 showed dose limiting toxicities (Seggewiss-Bernhardt et al. 2015). Similar effect of disease stabilization was observed in multiple myeloma patients during Phase 1 clinical trial of PF-04929113, a Hsp90 inhibitor (Reddy et al. 2013). The treatment with PF-04929113 also showed some adverse events (Table 12.1). Both NVP-AU922 and PF-04929113 had shown significant anticancer activity in in vitro and in vivo models (Okawa et al. 2009; Stuhmer et al. 2008).
5 Challenges in Targeting Hsp90
It is intriguing to question that why none of the Hsp inhibitors have been yet approved by FDA in spite almost 20 such inhibitors have gone through clinical trials. The analysis of data published about Hsp90 inhibitors indicate two possible reasons for their failure: No better efficacy than existing treatment regime; and toxicity/adverse events. Clinical trials have revealed that inhibition of Hsp90 did not lead to a remarkable cure rather at the most it stabilized the disease (Ramanathan et al. 2007; Reddy et al. 2013; Seggewiss-Bernhardt et al. 2015). One of the main reasons for Hsp90 inhibitors for not achieving desirable efficacy in cancer patients is induction of heat shock response in cancer cells in response to Hsp90 inhibition and induction of other compensatory mechanisms. This compensatory phenomenon is not new to be observed in cancer cells. To adhere to the Darwin’s theory of evolution, cancer cells remodel themselves to survive the repeated attacks of chemotherapy. Normal eukaryotic cells (and cancer cells as well) have a huge incomprehensible network of cellular proteins and their signaling. Under normal conditions, there are some ancillary and “redundant” (not really!) pathways, the role of which may not be much appreciated in normal cells by the scientists. However, these “redundant” pathways drive the survival process in cancer cells when their “main” networks are disrupted by chemotherapy. The similar process has been indicated to be involved in targeting Hsp90. During clinical trials of Hsp90 inhibitors, expression of Hsp70 protein is monitored as pharmacodynamic biomarker for Hsp90-inhibitors and indeed, Hsp70 was found to be upregulated in the patients treated with Hsp90 inhibitor 17-AAG (Banerji et al. 2005; Goetz et al. 2005; Ramanathan et al. 2007; Solit et al. 2007). It is hypothesized from the molecular studies that the resistance to Hsp90 inhibition by 17-AAG and NVP-AUY922 may be a result of compensatory overexpression of Hsp70 and Hsp27 chaperons (McCollum et al. 2006; Seggewiss-Bernhardt et al. 2015). Hsp90 is a negative regulator of HSF-1 and inhibition of Hsp90 activates HSF-1 in homotrimer form. The activated HSF-1 in turn induces expression of chaperones such Hsp70 and Hsp27 and co-chaperons like Aha1 which have major pro-survival roles. Hsp70 binds to Bax (a proapoptotic protein) preventing it to localize in mitochondria and induce apoptosis (Guo et al. 2005). Considering anti-apoptotic role of Hsp70, no wonder that Hsp70 offers protection against neurodegenerative diseases and trauma by modulating inflammatory and apoptotic pathways (Magrane et al. 2004; Yenari 2002). This is considered as a plausible mechanism behind neuroprotective effect of tanespimycin, a Hsp90 inhibitor, in bortezomib-induced peripheral neuropathy (Argyriou et al. 2008; Cavaletti and Jakubowiak 2010; Flint et al. 2009; Richardson et al. 2011a). Clinically, other compensatory mechanisms conferring resistance to Hsp90 inhibitors needs to be shunted for their full effect. The other chaperones like Hsp70 and Hsp27 needs to be inhibited simultaneously either by another inhibitor or a single inhibitor with selective poly pharmacologic effect blocking these chaperones along with Hsp90 (McCollum et al. 2008). The siRNA mediated abrogation of Hsp70 along with Hsp90 inhibition has proved to be an effective strategy to overcome Hsp70 medicated resistance to 17-AAG treatment in acute myelogenous leukemia (AML) cells (Guo et al. 2005). Similarly, Hsp70 inhibition with VER155008 has also exhibited potentiation of Hsp90 inhibitor (17-AAG)-induced apoptosis in human colon cancer cells (Massey et al. 2010). The other small molecule inhibitors of Hsp70 such as A17, ADD70 and PES, have been developed although for non-multiple myeloma cancers (Jego et al. 2013). There is hope that these small molecule inhibitors of Hsp70 can be effective as a co-treatment with Hsp90 inhibitors in multiple myeloma as well. In 2015, Lu et al. had reported discovery of KU675, a selective small molecule inhibitor of Hsp90α (Kd = 191 μM) and Hsp70 (Kd = 76.3 μM) over Hsp90β (Kd = 726 μM), and anticancer activity in prostate cancer cells (Liu et al. 2015). It is intriguing to question whether this dual inhibitor of Hsp90 and Hsp70 will show activity in multiple myeloma cells. The natural flavonoid quercitin and synthetic small molecule inhibitor KNK437 have been found to inhibit Hsp70 expression at mRNA level mediated by HSF-1 inhibition in HeLa human cervical cancer, HL-60 AML or COLO 320DM human colon carcinoma cells (Guo et al. 2005; Nagai et al. 1995; Yokota et al. 2000). Moreover, HSF-1 induction in response to Hsp90 inhibition with 17-AAD also induces activity of co-chaperons like Aha1 which increases the phosphorylation of pro-survival kinases like Mek1/2 and Erk1/2 and activity of c-Raf in HT-29 colon cancer cells (Holmes et al. 2008). However, selective silencing of Aha1 along with 17-AAD in these studies sensitized HT-29 cells to 17-AAD treatment with two- to threefold increase in apoptosis. Although, these results need to be confirmed in multiple myeloma cells, these strongly indicate that combination therapy of Hsp90 inhibitors and co-chaperon inhibitors may be effective in developing such a therapeutic strategy for multiple myeloma. Actually, in 2017, Stiegler et al. identified small molecule HAM-1 as an inhibitor of Hsp90-Aha1 complex (Stiegler et al. 2017). Co-inhibition of HSF-1, which is considered as main cause of developing ‘heat shock response’-mediated resistance to Hsp90 inhibitors, may also seem an attractive strategy. But, as a word of caution, targeting HSF-1 with small molecule inhibitors may bring challenges of non-specific inhibition of HSF-1 in normal cells as well, which can lead to toxicity. This issue can be circumvented by synthesizing cancer specific antibody-drug conjugates which can be delivered specifically to tumors only and thus avoiding any untoward effects resulting from inhibition of HSF-1 in normal cells. The alternate strategy could be to inhibit the final product of these co-chaperones, like glutathione (GSH) of Hsp27, which is also responsible for conferring resistance to Hsp90 inhibitors (McCollum et al. 2006). The delivery of the drug to the inaccessible Hsp90 located in the mitochondria would be challenging, which may also confer some degree of resistance to Hsp90 inhibitors (Piper and Millson 2011).
Another challenge is to develop relatively safe Hsp90 inhibitors. Inherently, any therapeutic that binds to the ATP-binding pocket of kinases are promiscuous for the target with low selectivity. This results in increased toxicity and adverse events. This observation is supported by adverse events reported the clinical trials on Hsp90 inhibitors. These adverse events ranged from ocular toxicity (night blindness, photophobia, and visual impairment), diarrhea, nausea, prolonged QTc, thrombocytopenia, peripheral neuropathy, pneumonia, and hyponatremia. However, the ultimate decision for the development of such inhibitors targeting ATP-binding sites depends on continual efforts in improving their specificity and their risk-benefit ratio.
6 Conclusions
It has now been established that targeting one protein or signaling pathways would not be sufficient for multiple myeloma treatment. Therefore, targeting selective multiple proteins and signaling pathways concurrently is the future of cancer treatment, particularly multiple myeloma. Along these lines, combination therapies are now common practice in myeloma treatment. Furthermore, poly pharmacological agents have been also appreciated and got great amount of attention recently. It may be worthwhile approach to target chaperone proteins because these proteins regulate a number of key events in carcinogenesis. As Hsp are highly expressed in cancer cells as compared to their normal counterpart, it makes them an attractive target. Importantly, early results from combination clinical trial are encouraging. Furthermore, targeting several Hsp together may be a good option as it has been observed that simultaneous inhibition of Hsp90 and Hsp70 is more effective compared to single targeting, nonetheless, further studies are required to fully validate this observation (Cavanaugh et al. 2015). Despite the progress in Hsp inhibitor development there are several challenges that need to be addressed. First, the complete understanding of the mechanism is required, because Hsp exist in several isoforms and perform similar functions. Dissecting the roles of individual Hsp and the effect of combined inhibition of multiple Hsp is the key in order to develop an effective treatment strategy against myeloma. Second, the development of suitable Hsp inhibitors for the clinic remains a major challenge. Overall, much more effort is needed for targeting Hsp and exploiting this protein for better treatment in multiple myeloma.
Abbreviations
- ADP:
-
Adenosine diphosphate
- Aha1:
-
activator of HSP90 ATPase activity 1
- ALT:
-
alanine aminotransferase
- Apaf-1:
-
apoptotic peptidase activating factor 1
- AST:
-
aspartate aminotransferase
- ATP:
-
adenosine triphosphate
- Bax:
-
Bcl-2-associated X
- Bcl-2:
-
B-cell lymphoma-2
- Cdc37:
-
cell division cycle 37
- Chip:
-
carboxy terminus of Hsc70 interacting protein
- Cns1:
-
tetratricopeptide repeat domain 4
- CR:
-
connecting linker region
- CTD:
-
C terminal domain
- ErbB:
-
epidermal growth factor receptor
- ER:
-
Estrogen receptor
- Her3:
-
erb-b2 receptor tyrosine kinase 3
- HIF-1α:
-
hypoxia inducible factor 1 subunit alpha
- Hip:
-
Hsc70-interacting protein
- Hop:
-
Hsp70-Hsp90 organizing protein
- HSF-1:
-
heat shock transcription factor-1, Hsp, heat shock proteins
- MAPK:
-
Mitogen-activated protein kinase 1
- MEEVD:
-
Met-Glu-Glu-Val-Asp motif
- Mek1/2:
-
mitogen activated protein kinase kinase
- MD:
-
middle domain
- NF-κB:
-
nuclear factor kappa-light-chain- enhancer of activated B cells
- NTD:
-
N terminal domain
- PP5:
-
protein phosphatase 5
- p23:
-
prostaglandin E synthase 3
- Smac:
-
second Mitochondria-derived activator of caspases
- Tom70:
-
translocase of outer mitochondrial membrane 70
- TPR:
-
tetratricopeptide repeat domains
- Unc45:
-
unc-45 myosin chaperone B
References
Argyriou AA, Iconomou G, Kalofonos HP (2008) Bortezomib-induced peripheral neuropathy in multiple myeloma: a comprehensive review of the literature. Blood 112:1593–1599
Azad AA, Zoubeidi A, Gleave ME, Chi KN (2015) Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat Rev Urol 12:26–36
Bailey CK, Budina-Kolomets A, Murphy ME, Nefedova Y (2015) Efficacy of the HSP70 inhibitor PET-16 in multiple myeloma. Cancer Biol Ther 16:1422–1426
Bakthisaran R, Tangirala R, Rao CM (2015) Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta 1854:291–319
Banerji U, O’Donnell A, Scurr M et al (2005) Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J Clin Oncol 23:4152–4161
Born EJ, Hartman SV, Holstein SA (2013) Targeting HSP90 and monoclonal protein trafficking modulates the unfolded protein response, chaperone regulation and apoptosis in myeloma cells. Blood Cancer J 3:e167
Calderwood SK, Gong J (2016) Heat shock proteins promote cancer: it’s a protection racket. Trends Biochem Sci 41:311–323
Caplan AJ (2003) What is a co-chaperone? Cell Stress Chaperones 8:105–107
Cavaletti G, Jakubowiak AJ (2010) Peripheral neuropathy during bortezomib treatment of multiple myeloma: a review of recent studies. Leuk Lymphoma 51:1178–1187
Cavanaugh A, Juengst B, Sheridan K, Danella JF, Williams H (2015) Combined inhibition of heat shock proteins 90 and 70 leads to simultaneous degradation of the oncogenic signaling proteins involved in muscle invasive bladder cancer. Oncotarget 6:39821–39838
Chadli A, Felts SJ, Wang Q et al (2010) Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J Biol Chem 285:4224–4231
Chatterjee S, Burns TF (2017) Targeting heat shock proteins in cancer: a promising therapeutic approach. Int J Mol Sci 18:E1978
Chauhan D, Li G, Hideshima T et al (2003) Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood 102:3379–3386
Chauhan D, Li G, Auclair D et al (2004) 2-Methoxyestardiol and bortezomib/proteasome-inhibitor overcome dexamethasone-resistance in multiple myeloma cells by modulating heat shock Protein-27. Apoptosis 9:149–155
Chen B, Piel WH, Gui LM, Bruford E, Monteiro A (2005) The HSP90 family of genes in the human genome: insights into their divergence and evolution. Genomics 86:627–637
Ciglia E, Vergin J, Reimann S et al (2014) Resolving hot spots in the C-terminal dimerization domain that determine the stability of the molecular chaperone Hsp90. PLoS One 9:e96031
Ciocca DR, Calderwood SK (2005) Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10:86–103
Colombo G, Morra G, Meli M, Verkhivker G (2008) Understanding ligand-based modulation of the Hsp90 molecular chaperone dynamics at atomic resolution. Proc Natl Acad Sci U S A 105:7976–7981
Cordonnier T, Bishop JL, Shiota M et al (2015) Hsp27 regulates EGF/beta-catenin mediated epithelial to mesenchymal transition in prostate cancer. Int J Cancer 136:E496–E507
Donnelly A, Blagg BS (2008) Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem 15:2702–2717
Duus J, Bahar HI, Venkataraman G et al (2006) Analysis of expression of heat shock protein-90 (HSP90) and the effects of HSP90 inhibitor (17-AAG) in multiple myeloma. Leuk Lymphoma 47:1369–1378
Flandrin P, Guyotat D, Duval A et al (2008) Significance of heat-shock protein (HSP) 90 expression in acute myeloid leukemia cells. Cell Stress Chaperones 13:357–364
Flint OP, Kwagh J, Wang FY et al (2009) Tanespimycin prevents bortezomib toxicity and preserves neuronal morphology in primary rat dorsal root ganglion cultures. Blood 114:1112–1112
Fortugno P, Beltrami E, Plescia J et al (2003) Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A 100:13791–13796
Georget V, Terouanne B, Nicolas JC, Sultan C (2002) Mechanism of antiandrogen action: key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry 41:11824–11831
Gerecitano JF, Modi S, Rampal R et al. (2015) Phase I trial of the HSP-90 inhibitor PU-H71. J Clin Oncol 33:2537
Gibert B, Eckel B, Gonin V et al (2012) Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br J Cancer 107:63–70
Goetz MP, Toft D, Reid J et al (2005) Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J Clin Oncol 23:1078–1087
Guo F, Rocha K, Bali P et al (2005) Abrogation of heat shock protein 70 induction as a strategy, to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res 65:10536–10544
Hawle P, Siepmann M, Harst A, Siderius M, Reusch HP, Obermann WM (2006) The middle domain of Hsp90 acts as a discriminator between different types of client proteins. Mol Cell Biol 26:8385–8395
Hieronymus H, Lamb J, Ross KN et al (2006) Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10:321–330
Holmes JL, Sharp SY, Hobbs S, Workman P (2008) Silencing of HSP90 cochaperone AHA1 expression decreases client protein activation and increases cellular sensitivity to the HSP90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Cancer Res 68:1187–1196
Jego G, Hazoume A, Seigneuric R, Garrido C (2013) Targeting heat shock proteins in cancer. Cancer Lett 332:275–285
Jhaveri K, Taldone T, Modi S, Chiosis G (2012) Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 1823:742–755
Kamal A, Thao L, Sensintaffar J et al (2003) A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425:407–410
Kumar S, Stokes J 3rd, Singh UP et al (2016) Targeting Hsp70: a possible therapy for cancer. Cancer Lett 374:156–166
Langdon SP, Rabiasz GJ, Hirst GL et al (1995) Expression of the heat shock protein HSP27 in human ovarian cancer. Clin Cancer Res 1:1603–1609
Lee HW, Lee EH, Kim SH, Roh MS, Jung SB, Choi YC (2013) Heat shock protein 70 (HSP70) expression is associated with poor prognosis in intestinal type gastric cancer. Virchows Arch 463:489–495
Li CY, Lee JS, Ko YG, Kim JI, Seo JS (2000) Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem 275:25665–25671
Lianos GD, Alexiou GA, Mangano A et al (2015) The role of heat shock proteins in cancer. Cancer Lett 360:114–118
Liu S, Yuan Y, Okumura Y, Shinkai N, Yamauchi H (2010) Camptothecin disrupts androgen receptor signaling and suppresses prostate cancer cell growth. Biochem Biophys Res Commun 394:297–302
Liu W, Vielhauer GA, Holzbeierlein JM et al (2015) KU675, a concomitant heat-shock protein inhibitor of Hsp90 and Hsc70 that manifests isoform selectivity for Hsp90alpha in prostate Cancer cells. Mol Pharmacol 88:121–130
Madamanchi NR, Li S, Patterson C, Runge MS (2001) Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol 21:321–326
Magrane J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci 24:1700–1706
Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM (2000) The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem 275:37181–37186
Massey AJ, Williamson DS, Browne H et al (2010) A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol 66:535–545
Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62:670–684
McCollum AK, TenEyck CJ, Sauer BM, Toft DO, Erlichman C (2006) Up-regulation of heat shock protein 27 induces resistance to 17-allylamino-demethoxygeldanamycin through a glutathione-mediated mechanism. Cancer Res 66:10967–10975
McCollum AK, TenEyck CJ, Stensgard B et al (2008) P-glycoprotein-mediated resistance to Hsp90-directed therapy is eclipsed by the heat shock response. Cancer Res 68:7419–7427
Meyer P, Prodromou C, Hu B et al (2003) Structural and functional analysis of the middle segment of hsp90: implications for ATP hydrolysis and client protein and cochaperone interactions. Mol Cell 11:647–658
Miyata Y, Nakamoto H, Neckers L (2013) The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des 19:347–365
Moulick K, Ahn JH, Zong H et al (2011) Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90. Nat Chem Biol 7:818–826
Murphy ME (2013) The HSP70 family and cancer. Carcinogenesis 34:1181–1188
Nagai N, Nakai A, Nagata K (1995) Quercetin suppresses heat shock response by down regulation of HSF1. Biochem Biophys Res Commun 208:1099–1105
Nemoto T, Ohara-Nemoto Y, Ota M, Takagi T, Yokoyama K (1995) Mechanism of dimer formation of the 90-kDa heat-shock protein. Eur J Biochem 233:1–8
Nimmanapalli R, Gerbino E, Dalton WS, Gandhi V, Alsina M (2008) HSP70 inhibition reverses cell adhesion mediated and acquired drug resistance in multiple myeloma. Br J Haematol 142:551–561
Okawa Y, Hideshima T, Steed P et al (2009) SNX-2112, a selective Hsp90 inhibitor, potently inhibits tumor cell growth, angiogenesis, and osteoclastogenesis in multiple myeloma and other hematologic tumors by abrogating signaling via Akt and ERK. Blood 113:846–855
Palumbo A, Anderson K (2011) Multiple myeloma. N Engl J Med 364:1046–1060
Patwardhan CA, Fauq A, Peterson LB, Miller C, Blagg BSJ, Chadli A (2013) Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J Biol Chem 288:7313–7325
Piper PW, Millson SH (2011) Mechanisms of resistance to Hsp90 inhibitor drugs: a complex mosaic emerges. Pharmaceuticals (Basel) 4:1400–1422
Plescia J, Salz W, Xia F et al (2005) Rational design of shepherdin, a novel anticancer agent. Cancer Cell 7:457–468
ProteinAtlas (2018) The human protein atlas (https://www.proteinatlas.org/)
Rajkumar SV, Kumar S (2016) Multiple myeloma: diagnosis and treatment. Mayo Clin Proc 91:101–119
Ramanathan RK, Egorin MJ, Eiseman JL et al (2007) Phase I and pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin in adult patients with refractory advanced cancers. Clin Cancer Res 13:1769–1774
Ratzke C, Mickler M, Hellenkamp B, Buchner J, Hugel T (2010) Dynamics of heat shock protein 90 C-terminal dimerization is an important part of its conformational cycle. Proc Natl Acad Sci U S A 107:16101–16106
Reddy N, Voorhees PM, Houk BE, Brega N, Hinson JM, Jillela A (2013) Phase I trial of the HSP90 inhibitor PF-04929113 (SNX5422) in adult patients with recurrent, refractory hematologic malignancies. Cl Lymph Myelom Leuk 13:385–391
Reikvam H, Nepstad I, Sulen A, Gjertsen BT, Hatfield KJ, Bruserud O (2013) Increased antileukemic effects in human acute myeloid leukemia by combining HSP70 and HSP90 inhibitors. Expert Opin Investig Drugs 22:551–563
Rerole AL, Jego G, Garrido C (2011) Hsp70: anti-apoptotic and tumorigenic protein. Methods Mol Biol 787:205–230
Richardson PG, Badros AZ, Jagannath S et al (2010) Tanespimycin with bortezomib: activity in relapsed/refractory patients with multiple myeloma. Br J Haematol 150:428–437
Richardson PG, Chanan-Khan AA, Lonial S et al (2011a) Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Brit J Haematol 153:729–740
Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC (2011b) Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Brit J Haematol 152:367–379
Richardson PG, Mitsiades CS, Laubach JP, Lonial S, Chanan-Khan AA, Anderson KC (2011c) Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers. Br J Haematol 152:367–379
Saibil H (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 14:630–642
Saluja A, Dudeja V (2008) Heat shock proteins in pancreatic diseases. J Gastroenterol Hepatol 23(Suppl 1):S42–S45
Seggewiss-Bernhardt R, Bargou RC, Goh YT et al (2015) Phase 1/1B trial of the heat shock protein 90 inhibitor NVP-AUY922 as monotherapy or in combination with bortezomib in patients with relapsed or refractory multiple myeloma. Cancer 121:2185–2192
Sidera K, Patsavoudi E (2014) HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov 9:1–20
Siegel D, Jagannath S, Vesole DH et al (2011) A phase 1 study of IPI-504 (retaspimycin hydrochloride) in patients with relapsed or relapsed and refractory multiple myeloma. Leuk Lymphoma 52:2308–2315
Solit DB, Ivy SP, Kopil C et al (2007) Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. Clin Cancer Res 13:1775–1782
Sreeramulu S, Gande SL, Gobel M, Schwalbe H (2009) Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem Int Ed 48:5853–5855
Stiegler SC, Rubbelke M, Korotkov VS et al (2017) A chemical compound inhibiting the Aha1-Hsp90 chaperone complex. J Biol Chem 292:17073–17083
Stuhmer T, Zollinger A, Siegmund D et al (2008) Signalling profile and antitumour activity of the novel Hsp90 inhibitor NVP-AUY922 in multiple myeloma. Leukemia 22:1604–1612
Teng Y, Ngoka L, Mei Y, Lesoon L, Cowell JK (2012) HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J Biol Chem 287:10051–10059
Usmani SZ, Chiosis G (2011) HSP90 inhibitors as therapy for multiple myeloma. Clin Lymphoma Myeloma Leuk 11(Suppl 1):S77–S81
Voll EA, Ogden IM, Pavese JM et al (2014) Heat shock protein 27 regulates human prostate cancer cell motility and metastatic progression. Oncotarget 5:2648–2663
Wayne N, Mishra P, Bolon DN (2011) Hsp90 and client protein maturation. Methods Mol Biol 787:33–44
Wei L, Liu TT, Wang HH et al (2011) Hsp27 participates in the maintenance of breast cancer stem cells through regulation of epithelial-mesenchymal transition and nuclear factor-kappaB. Breast Cancer Res 13:R101
Wheatley SP, McNeish IA (2005) Survivin: a protein with dual roles in mitosis and apoptosis. Int Rev Cytol 247:35–88
Whitley D, Goldberg SP, Jordan WD (1999) Heat shock proteins: a review of the molecular chaperones. J Vasc Surg 29:748–751
Wong DS, Jay DG (2016) Emerging roles of extracellular Hsp90 in cancer. Adv Cancer Res 129:141–163
Wu J, Liu T, Rios Z, Mei Q, Lin X, Cao S (2017) Heat shock proteins and cancer. Trends Pharmacol Sci 38:226–256
Yang X, Wang J, Zhou Y, Wang Y, Wang S, Zhang W (2012) Hsp70 promotes chemoresistance by blocking Bax mitochondrial translocation in ovarian cancer cells. Cancer Lett 321:137–143
Yenari MA (2002) Heat shock proteins and neuroprotection. Adv Exp Med Biol 513:281–299
Yokota S, Kitahara M, Nagata K (2000) Benzylidene lactam compound, KNK437, a novel inhibitor of acquisition of thermotolerance and heat shock protein induction in human colon carcinoma cells. Cancer Res 60:2942–2948
Yu Z, Zhi J, Peng X, Zhong X, Xu A (2010) Clinical significance of HSP27 expression in colorectal cancer. Mol Med Rep 3:953–958
Zagouri F, Bournakis E, Koutsoukos K, Papadimitriou CA (2012) Heat shock protein 90 (hsp90) expression and breast cancer. Pharmaceuticals (Basel) 5:1008–1020
Zhang L, Fok JH, Davies FE (2014) Heat shock proteins in multiple myeloma. Oncotarget 5:1132–1148
Zhang Y, Tao X, Jin G et al (2016) A targetable molecular chaperone Hsp27 confers aggressiveness in hepatocellular carcinoma. Theranostics 6:558–570
Zuehlke A, Johnson JL (2010) Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93:211–217
Acknowledgements
Authors are thankful for the proofreading and editing services of Cooper Medical School of Rowan University.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kale, V.P., Phadtare, S., Amin, S.G., Pandey, M.K. (2019). Targeting Heat Shock Proteins in Multiple Myeloma. In: Asea, A., Kaur, P. (eds) Heat Shock Proteins in Signaling Pathways. Heat Shock Proteins, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-030-03952-3_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-03952-3_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03951-6
Online ISBN: 978-3-030-03952-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)