
Abstract
Bone marrow failure (BMF) syndromes can be classified into inherited and acquired. In addition to the classic clinical presentation of inadequate hematopoiesis (anemia, leukopenia, and thrombocytopenia), which can initially be isolated cytopenias and then progress to marrow aplasia, inherited bone marrow failure syndromes can present with characteristic non-hematologic clinical findings. Patients can be diagnosed shortly after birth or the diagnosis can be delayed into adulthood. BMF syndromes can be premalignant conditions that can progress into myelodysplastic syndrome and/or acute leukemia. Some patients are at higher risk of solid tumors. Early diagnosis, prompt management of complications (infections, bleeding, organ failures), and early referral for hematopoietic stem cell transplantation can be associated with improved outcome. In this chapter we will discuss the most common bone marrow failure syndromes, acquired idiopathic aplastic anemia, Fanconi anemia, Diamond-Blackfan anemia, dyskeratosis congenita, and Shwachman-Bodian-Diamond syndrome.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bone marrow failure
- Aplastic anemia
- Fanconi anemia
- Dyskeratosis congenita
- Diamond-Blackfan anemia
- Shwachman-Bodian-Diamond syndrome
Acquired Aplastic Anemia
Acquired aplastic anemia in children is a rare disorder (two per million children per year in North America and Europe [1]) associated with variable peripheral blood cytopenias and a hypocellular bone marrow. It can be broadly divided into mild to moderate (non-severe), severe, and very severe depending on the degree or severity of the peripheral blood cytopenias in the presence of a hypocellular marrow. Mild to moderate aplastic anemia is defined as marrow cellularity of <50% and any two of the following: absolute neutrophil count (ANC) <1500/μl, platelet count <100,000/μl, and absolute reticulocyte count (ARC) <40,000/μl [2]. Severe aplastic anemia (SAA) is associated with severe hypoplastic marrow (bone marrow cellularity <25%) and at least two of the following: ANC <500/μl, platelet count <20,000/μl, and absolute reticulocyte count <20,000/μl [3]. Very severe aplastic anemia (vSAA) is defined by fulfilling criteria for SAA plus ANC of <200/μl [4]. Mild to moderate aplastic anemia can be associated with spontaneous resolution or can progress to SAA [5]. Severe and very severe aplastic anemias (SAA) are life-threatening disorders. Major advances in diagnostic and therapeutic (including immunosuppressive therapy or IST) resulted in long-term survival for more than 90% of the cases [6]. Acquired aplastic anemia can be secondary to irradiation, drugs (e.g., chemotherapy, chloramphenicol, NSAIDs, and benzene), infections (EBV, CMV, HHV6, parvovirus B19, HIV, and hepatitis), pregnancy, nutritional deficiency (e.g., vitamin B12, folate and trace elements), and immune diseases (e.g., SLE) or “idiopathic” when no clear etiology can be identified. There is strong evidence that idiopathic acquired aplastic anemia is an immune destruction-mediated phenomena associated with an abnormal hematopoietic microenvironment [7,8,9]. The clinical signs and symptoms of aplastic anemia result from the pancytopenias. Spontaneous petechial skin rash, easy bruising, epistaxis, gum bleeding, or heavy menses in postmenarchal females are manifestations of thrombocytopenia. Anemia can present as fatigue, lack of energy, exercise intolerance, respiratory distress, and pallor. Infectious complications (e.g., fever, gingivitis, and skin abscesses) are a consequence of neutropenia. Organomegaly is not a typical clinical finding in aplastic anemia, and its presence should raise the suspicion of other diagnoses (infections and malignancies). Prompt diagnostic evaluation and early therapeutic intervention (IST or hematopoietic stem cell transplantation HSCT) are associated with improved outcomes [10]. Inherited bone marrow failure syndromes (IBMFS) and hypoplastic myelodysplastic syndrome (MDS) can have similar clinical presentation and must be distinguished from idiopathic acquired aplastic anemia. It is important to obtain a detailed history including exposure to medications and chemicals, comprehensive family history, and complete physical examination. Standard laboratory evaluations include complete blood count, reticulocyte count, peripheral smear, chemistries, unilateral bone marrow aspiration, and biopsy for histology and cytogenetics. Chromosomal breakage studies, telomeres’ length, and genetic testing may be helpful to rule out IBMFS. Testing for paroxysmal nocturnal hemoglobinuria (PNH) by flow cytometry [11] is also important as PNH can present with marrow failure and pancytopenia and can also be an early clonal evolution in SAA. Once the diagnosis is established, HLA typing of the patient and related family members should be sent. Despite improvement in survival for patients with SAA [12], infection and bleeding remain as major causes of morbidity and mortality [13, 14]. SAA patients are profoundly immunocompromised and at risk of life-threatening infections including fungal infections [15]. Prophylactic antimicrobials (fungal and Pneumocystis jiroveci prophylaxis) are recommended [16] especially during periods of prolonged leukopenia and IST. Antibacterial prophylaxis may be considered in patients with vSAA [16]. Fever and neutropenia require immediate medical attention and hospitalization as they can progress rapidly to septic shock. Cultures (blood and urine), imaging (e.g., CT scans or chest X-ray), and prompt initiation of broad-spectrum antimicrobials (including antifungal agents for prolonged fever) are critical and can be lifesaving. Treatment should be initiated without waiting for culture results. There are currently no data to support the routine use of growth factors (GCSF or GMCSF) in patients with SAA. It may shorten the duration of neutropenia but did not improve overall survival, and prolonged use of high doses may be associated with malignant transformation [17, 18]. On the other hand, one large meta-analysis showed no association between the use of growth factors and the progression to MDS, acute leukemia, or PNH [19]. Granulocyte transfusions may be considered in life-threatening infections and neutropenia knowing that there is limited data to support it [20]. Several studies reported a negative impact of the number of blood products transfused prior to HSCT on the outcome after HSCT [21], and a restrictive blood product transfusion policy should be applied in patients with SAA who are candidates for HSCT; that being said, this should not delay a transfusion when indicated. Patients with active bleeding should receive platelet transfusions. Prophylactic platelet transfusion in asymptomatic patients with platelet count of <10,000/μl is recommended [22]. Higher platelet transfusion threshold (<20,000/μl) is recommended in patients at risk of rapid platelet consumption (e.g., fever or patients receiving IST) or those with history of life-threatening bleeding (gastrointestinal or central nervous system bleeding) [16]. There is no data to support the use of non-HLA-matched apheresis platelets over pooled platelets in non-allosensitized patients [23]. HLA-matched single-donor apheresis platelets should be used if the patient is platelet refractory and HLA antibodies are positive [24]. The addition of the thrombopoietin receptor agonist eltrombopag to the standard IST regimen was associated with improved hematologic response after 6 months without increasing the risk of clonal evolution [25]. Red blood cell transfusion is recommended for patients with hemoglobin level <8 g/dl or those with symptomatic anemia, and a higher threshold may be indicated in patients with other comorbidities (cardiac and/or respiratory) [26]. Erythropoietin should be avoided due to the resulting delay in definitive SAA therapy . Transfusions from related family members should be avoided as this may increase the risk of HLA sensitization and graft rejection if a matched related donor (MRD) bone marrow transplant could be an option. Blood products should always be leukoreduced and irradiated [27]. CMV-negative blood products are not routinely recommended if universal leukodepletion is applied [28, 29]. Treatment options for patients with SAA are hematopoietic stem cell transplantation (HSCT) and IST. The backbone of IST is antithymocyte globulin (ATG) in combination with the calcineurin inhibitor cyclosporine. Steroids are used during ATG therapy to prevent serum sickness (fever, rash, bronchospasm, and elevated liver enzymes 2–3 weeks after starting ATG) followed by rapid taper. Prolonged steroid use in SAA is ineffective and can increase the risk of infectious complications and gastrointestinal hemorrhage in the setting of severe thrombocytopenia. Horse ATG was proven to be associated with higher rates of hematologic response (68%) at 6 months and superior overall survival (96% at 3 years) compared to rabbit ATG [13]. ATG can be associated with rare immediate severe allergic (anaphylactic) reaction that can be predicted by skin test followed by desensitization for those who are allergic [30]. Horse ATG should be infused over at least 4 h through a 0.2 to 1 micron inline filter. Strongly consider premedication with antipyretics, antihistamines, and/or corticosteroids to prevent reactions. Cyclosporine blood levels should be closely monitored to avoid toxicities (nephrotoxicity, hypertension, hypertensive encephalopathy, hepatotoxicity, electrolyte abnormalities, and opportunistic infections). The current recommendation of primary therapy is HSCT with a bone marrow graft for patients who have MRD [31, 32]. IST is usually the first line of therapy in patients who lack full HLA-matched related donor and is associated with hematologic recovery in 50–70% of cases with excellent long-term survival [33]. Matched unrelated donor (MUD) or unrelated cord blood HSCT should be offered to all patients who do not respond to IST within 3–6 months [34, 35]. MUD HSCT was reported to be superior to second course of IST [36]. If MUD is unavailable, a second course of IST may be considered. Recent data may suggest that first-line therapy with HSCT forms MUD or even haploidentical donor HSCT is comparable or even superior to IST [37].
Fanconi Anemia
Fanconi anemia (FA) is a genetic disease characterized by marrow failure, predisposition to malignancies, and congenital abnormalities. This disorder was first described by pediatrician Dr. Guido Fanconi in 1927 after describing a family of three brothers with aplastic anemia and developmental defects [38]. FA is the most common of the rare inherited bone marrow failure syndromes, with a heterozygous carrier frequency of 1 in 300, with at least 20 different gene mutations described to date, known as “complementation groups,” on which FANCA gene mutations are the most common. FA is a widely heterogeneous disease associated with multiple congenital malformations in different organ systems. Examples include the skin (hyper or hypopigmentation), musculoskeletal (thenar hypoplasia, absence or hypoplasia of the radius/thumbs, scoliosis, clubfoot), gastrointestinal (esophageal atresia, tracheoesophageal fistula, duodenal web, anal atresia), cardiopulmonary (ventricular septal defects, patent ductus arteriosus, pulmonary atresia), central nervous system (hydrocephalus, microcephaly), urogenital (undescended testes, micropenis, vaginal hypoplasia, urethral stenosis), renal (horseshoe kidney, hydronephrosis), ears (deafness, low-set ears), and endocrine (failure to thrive, short stature, glucose intolerance, infertility) [39]. FA is not classically associated with mental retardation. Patients with FA are often followed by multiple subspecialists since birth due to these congenital anomalies, which should raise suspicion for the diagnosis. However, up to 25% of patients will not display classic phenotypic features of the disease. A unifying feature is marked chromosomal instability and defects in DNA repair, for which increased breaks and radial formations in chromosomes are seen when cultured with diepoxybutane (DEB) or mitomycin C (MMC). This functional testing should be performed in a CLIA (Clinical Laboratory Improvement Amendments)-approved lab that has established expertise in performing this assay as a reference center. On occasion, patients’ blood lymphocytes may not exhibit the degree of hypersensitivity to DEB and MMC crosslinking agents that would be expected for someone with FA, known as revertant somatic mosaicism [40, 41]. This occurs when there is spontaneous functional correction of the gene mutation and can sometimes lead to a milder phenotype of marrow failure. However, this could then create false-negative results on chromosomal fragility testing. If there is strong suspicion for FA, a skin biopsy should be sent to perform DEB/MMC testing on fibroblasts [42]. Greater than 90% of all patients will develop marrow failure before the age of 40 years old, with most presenting within the first decade of life [43]. Patients with FA have increased risk of developing all forms of hematological malignancies, with the most common being acute myeloid leukemia (10–37%) and myelodysplastic syndrome (11–34%), which often presents as refractory cytopenias with multilineage dysplasia. Patients with hematological malignancies frequently present during teenage years or young adulthood [44]. Solid tumor risk is also high, with a 700-fold risk of squamous cell carcinomas compared to the general population. Solid tumors are rare in the FA pediatric population, with exception of those with FANCD1/BRCA2, FANCN/PALB2, and FANCJ/BRIP1 complementation groups. Chemotherapy regimens must be adjusted when treating hematological malignancies and solid tumors for concern of increased toxicity. Most patients will need to undergo bone marrow transplantation for marrow failure. The conditioning regimen must also be limited in doses of agents which cross-link DNA. Fatal sinusoidal obstruction syndrome (SOS) as well as severe mucositis can affect management in the critical care setting. X-rays and CT scans, while necessary, should be thoughtfully used to limit ionizing radiation exposures. Each FA patient’s underlying congenital defects must be considered when he or she becomes critically ill. For example, problems with tracheal stenosis could affect airway management; specific heart defects could impact fluid management. Patients who have urogenital concerns could be more prone to urinary tract infections and urosepsis when immunocompromised. Prior transfusion burden could impact iron overload, which can further increase the risk of SOS in FA.
Diamond-Blackfan Anemia
Diamond-Blackfan anemia (DBA) was initially described by Diamond and Blackfan in 1938 [45]. However, it was not until 25 years later that a full case series based on 25 years of experience on congenital hypoplastic anemia was published. This mainly described patients who had normocytic normochromic anemia, lacked erythroid precursors in the bone marrow, and had the absence of clinical leukopenia and/or thrombocytopenia [46]. Since then, despite the advances and efforts in understanding the etiology of the disease, the mainstay therapies remain unchanged. These therapies are packed red blood cells’ (PRBCs) transfusion, corticosteroids, and hematopoietic stem cell transplantation (HSCT), the last one reserved for severe non-remission cases. DBA usually presents early in life, even in utero, due to the early severity of anemia (hydrops fetalis). Therefore, the classic phenotype of a patient with DBA is short stature, thumb anomalies, and congenital heart disease. This is in the context of macrocytic normochromic anemia (not described by Blackfan and Diamond) and reticulocytopenia in patients before 1 year of age [47, 48]. Nonetheless, at least 50% of the patients have only one congenital anomaly, and in approximately 25% of the cases, patients may go unnoticed due to the lack of anomalies and present later in life as adolescents or young adults [48,49,50,51]. Bone marrow biopsy will confirm normocellular marrow with the lack of erythroid precursors [51]. The diagnosis is further confirmed through an elevated pre-transfusion serum adenosine deaminase (erythrocytes) with a sensitivity and specificity of 84% and 95%, respectively [52]. This can be confirmed with the ongoing increased knowledge of the pathophysiology of DBA, locating the biogenesis in the ribosomes in at least 50% of the cases, with the majority of them inherited in an autosomal dominant pattern with incomplete penetrance [53]. These ribosome gene mutations seem to activate p53 pathways leading to the apoptosis of progenitor cells [54,55,56,57]. Among approximately 20 different genes, mutations in the RPS19 gene can be detected in up to 25% of the cases of DBA. In addition, genes such as GATA1 and TSR2 (XLR) have been described [51]. Differential diagnoses are described in this chapter such as Fanconi anemia, Shwachman-Diamond syndrome, dyskeratosis congenita, or transient erythroblastopenia of childhood (transient red cell hypoplasia). Once the diagnosis is confirmed, urgent treatment with PRBCs is needed due to severe anemia. Then, corticosteroids should be started at a dose of 2 mg/Kg a day in patients older than 12 months [47]. Corticosteroids may achieve a goal of complete or partial remission in up to 80% of the cases; however eventually a subset of patients will become refractory; only 37% patients will have a sustained response without the need of PRBCs [47, 58]. Response is expected within 4 weeks after initiation of therapy. If patients continue to require transfusion(s), then they are considered to have refractory disease. All data from corticosteroid treatment is based on retrospective studies. Tapering should be slow, and the maintenance dose recommended to avoid toxicities should be less than 0.5 mg/kg/day. In addition, the use of steroids before 1 year of age is avoided due to the significant impact on growth and development. Patients on prolonged steroids warrant close monitoring for steroid-related complications (i.e., bone density, susceptibility to infections). If patients become refractory, then they may require chronic PRBCs transfusions with the goal of keeping the hemoglobin above 8 g/dl to lessen effects on growth and development [49, 59]. The major and most detrimental effect of chronic transfusions is the development of iron overload and the subsequent multiorgan involvement that could increase morbidity and mortality in the transfusion-dependent population. This should be promptly treated with iron chelation, and patient’s organs such as the heart and liver should be closely monitored for dysfunction due to iron overload.
In addition, patients with DBA are at increased risk of malignancies such as gastrointestinal carcinomas, myelodysplastic syndromes, and myeloid leukemia, and in rare occasions this could be the initial presentation [47, 60, 61]. HSCT is the only curative option for the hematological manifestations in patients with DBA who are transfusion dependent and steroid refractory or patients with other hematopoietic abnormalities such as MDS/leukemia. Chemotherapy and posttransplant complications require co-management with transplant physicians and a hematologist-oncologist [62].
Dyskeratosis Congenita
Dyskeratosis congenita (DKC) is a rare and highly variable phenotype condition that was initially described in 1906 in a dermatologic conference [63]. The principal triad of abnormal skin pigmentation, nail dystrophy, and mucosal leukoplakia characterizes the most severe phenotype [63,64,65]. In addition, patients with DKC are at higher risk of bone marrow failure, variable immunodeficiency (severe opportunistic infections), myelodysplastic syndrome/acute myeloid leukemia, solid tumors, and pulmonary fibrosis. Other nonclassical features may develop later in adulthood and include early gray hair, liver cirrhosis, emphysema, esophageal and urethral strictures, and head/neck squamous cell carcinoma, making the diagnosis more difficult and delayed [48]. The sole presence of pulmonary fibrosis and bone marrow failure in a patient has been found to be highly predictive of DKC [66].
The biological finding, however not unique to patients with DKC, is abnormally short telomere length, which encompasses the whole spectrum of telomere biology disorders [67, 68]. Telomeres are essential for maintaining chromosomal (DNA) stability [68]. The measurement can be performed clinically in peripheral lymphocytes via Flow-FISH [69, 70]. Lymphocytes’ telomere lengths below the first percentile have a sensitivity and specificity of 97% and 91%, respectively [70]. Genetic mutation identified in 50–60% of the cases can be helpful in confirming the diagnosis. The most common gene mutation is in the DKC1 gene, which is inherited in an X-linked recessive fashion, can be seen in up to 25% of all cases. However, up to 25% of patients with DKC have unknown genetic variants. Other genes include TERC, TERT (associated with adult-onset pulmonary fibrosis), RTEL1, and TINF2 among others (14 pathogenic known genes) that are inherited in autosomal dominant or recessive pattern [71]. The treatment utilized for patients with telomeropathies is based on using derivatives of androgens or synthetic steroids to improve telomere length [62]. Hematologic response is seen in up to 60–80% of the patients. However, common side effects include virilization, dyslipidemias, liver toxicity, splenic peliosis, bone fractures, and muscle cramps [72, 73]. Bone marrow failure in patients with DKC does not respond to immunosuppressive therapy [74]. The aforementioned treatments and complications describe clearly the complexity in managing patients with DKC. Moreover, these patients due to their liver and lung issues and their ongoing supportive therapies should be closely monitored for potential additional toxicities, especially medications that affect the DNA and/or are metabolized in the liver. HSCT is the only potential curative strategy for the patients who do not have a hematological response to medical treatment or for patients with severe bone marrow failure/immunodeficiency [75, 76]. Due to high sensitivity to DNA-disrupting medications, patients are conditioned with reduced intensity preparative regimens for their transplant. As described before, some of the phenotypes may be aggravated by the HSCT, and the complications warrant co-management with the bone marrow transplant physician and the respective specialists [59].
Schwachman-Bodian-Diamond Syndrome
First described by two different groups in 1964 for which the disease has its namesake [77, 78], Schwachman-Bodian-Diamond syndrome (SBDS ) is characterized by progressive marrow failure (most commonly with neutropenia), exocrine pancreatic insufficiency, acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS), and skeletal abnormalities. It is inherited in an autosomal recessive fashion with an incidence of 1 in 77,000 and a carrier frequency of 1 in 110 [79]. SBDS is a clinical diagnosis in which a genetic diagnosis can be made in approximately 90% of individuals who have biallelic mutations in the SBDS gene, which is located on chromosome 7 [80]. SBDS uniformly has an exocrine pancreatic defect associated with steatorrhea and often failure to thrive, similar to that of a patient with cystic fibrosis. Evaluation includes checking fat-soluble vitamin levels (vitamins A, D, E, K), low serum trypsinogen, elevated fecal fat levels, and MRI imaging of the pancreas demonstrating a small-sized pancreas with fatty replacement [81]. The marrow dysfunction has been characterized as neutropenia, although thrombocytopenia and anemia can also occur. One of the most concerning features of this disease is the risk of transformation to AML or MDS, with an estimated risk of 19% at 20 years and 36% at 30 years [82]. Other concerns for this disease include an association with endocrine abnormalities including insulin-dependent diabetes mellitus, growth hormone deficiency, hypothyroidism, cardiac issues including myocardial necrosis [83], and skeletal abnormalities typically at growth plates, with more than 50% of patients having metaphyseal dysostosis or rib cage abnormalities [84]. Patients are referred to hematopoietic stem cell transplant (HSCT) for progressive cytopenias, development of AML, or MDS with excessive blasts [85]. Congenital defects of the skeletal system and associated endocrine and cardiac dysfunction may create additional concerns in the posttransplant setting when considering fluid management, sepsis, and cardiorespiratory concerns.
Intensive Care Considerations
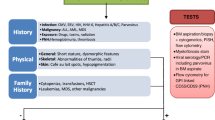
Patients with bone marrow failure syndromes (congenital and acquired) are at risk of life-threatening complications related to their primary disease or as a result of treatment (Table 7.1). Isolated cytopenias and pancytopenia can result in life-threatening hemorrhage, respiratory and/or cardiac failure, and sepsis. Chronic blood product transfusions is the mainstay of supportive treatment for most marrow failure syndromes which are associated with high risk of iron overload resulting in multiple organs’ damage/dysfunction (Table 7.1), risk of transmission of infectious diseases, alloimmunization, and transfusion reactions (including TRALI or transfusion-related acute lung injury). Inherited marrow failure syndromes can be associated with congenital malformations (craniofacial, thoracic cage, cardiac, renal, pulmonary among others) which can impact ICU care in regard to airway management, fluid balance, and risk of infectious complications.
References
Montane E, Ibanez L, Vidal X, et al. Epidemiology of aplastic anemia: a prospective multicenter study. Haematologica. 2008;93(4):518–23.
Howard SC, Naidu PE, Hu XJ, et al. Natural history of moderate aplastic anemia in children. Pediatr Blood Cancer. 2004;43(5):545–51.
Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45(3):355–63.
Bacigalupo A, Hows J, Gluckman E, et al. Bone marrow transplantation (BMT) versus immunosuppression for the treatment of severe aplastic anaemia (SAA): a report of the EBMT SAA working party. Br J Haematol. 1988;70(2):177–82.
Khatib Z, Wilimas J, Wang W. Outcome of moderate aplastic anemia in children. Am J Pediatr Hematol Oncol. 1994;16(1):80–5.
Hartung HD, Olson TS, Bessler M. Acquired aplastic anemia in children. Pediatr Clin N Am. 2013;60(6):1311–36.
Young NS, Scheinberg P, Calado RT. Aplastic anemia. Curr Opin Hematol. 2008;15(3):162–8.
Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–19.
Knospe WH, Crosby WH. Aplastic anaemia: a disorder of the bone-marrow sinusoidal microcirculation rather than stem-cell failure? Lancet. 1971;1(7688):20–2.
Locasciulli A, Oneto R, Bacigalupo A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 2007;92(1):11–8.
Sutherland DR, Kuek N, Davidson J, et al. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom. 2007;72(3):167–77.
Valdez JM, Scheinberg P, Nunez O, Wu CO, Young NS, Walsh TJ. Decreased infection-related mortality and improved survival in severe aplastic anemia in the past two decades. Clin Infect Dis. 2011;52(6):726–35.
Scheinberg P, Nunez O, Weinstein B, et al. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N Engl J Med. 2011;365(5):430–8.
Tichelli A, Schrezenmeier H, Socie G, et al. A randomized controlled study in patients with newly diagnosed severe aplastic anemia receiving antithymocyte globulin (ATG), cyclosporine, with or without G-CSF: a study of the SAA Working Party of the European Group for Blood and Marrow Transplantation. Blood. 2011;117(17):4434–41.
Weinberger M, Elattar I, Marshall D, et al. Patterns of infection in patients with aplastic anemia and the emergence of Aspergillus as a major cause of death. Medicine (Baltimore). 1992;71(1):24–43.
Hochsmann B, Moicean A, Risitano A, Ljungman P, Schrezenmeier H. Supportive care in severe and very severe aplastic anemia. Bone Marrow Transplant. 2013;48(2):168–73.
Ohara A, Kojima S, Hamajima N, et al. Myelodysplastic syndrome and acute myelogenous leukemia as a late clonal complication in children with acquired aplastic anemia. Blood. 1997;90(3):1009–13.
Jeng MR, Naidu PE, Rieman MD, et al. Granulocyte-macrophage colony stimulating factor and immunosuppression in the treatment of pediatric acquired severe aplastic anemia. Pediatr Blood Cancer. 2005;45(2):170–5.
Gurion R, Gafter-Gvili A, Paul M, et al. Hematopoietic growth factors in aplastic anemia patients treated with immunosuppressive therapy-systematic review and meta-analysis. Haematologica. 2009;94(5):712–9.
Quillen K, Wong E, Scheinberg P, et al. Granulocyte transfusions in severe aplastic anemia: an eleven-year experience. Haematologica. 2009;94(12):1661–8.
Champlin RE, Horowitz MM, van Bekkum DW, et al. Graft failure following bone marrow transplantation for severe aplastic anemia: risk factors and treatment results. Blood. 1989;73(2):606–13.
Marsh JC, Ball SE, Cavenagh J, et al. Guidelines for the diagnosis and management of aplastic anaemia. Br J Haematol. 2009;147(1):43–70.
Schrezenmeier H, Seifried E. Buffy-coat-derived pooled platelet concentrates and apheresis platelet concentrates: which product type should be preferred? Vox Sang. 2010;99(1):1–15.
Laundy GJ, Bradley BA, Rees BM, Younie M, Hows JM. Incidence and specificity of HLA antibodies in multitransfused patients with acquired aplastic anemia. Transfusion. 2004;44(6):814–25.
Townsley DM, Scheinberg P, Winkler T, et al. Eltrombopag added to standard immunosuppression for aplastic anemia. N Engl J Med. 2017;376(16):1540–50.
Welte M. Erythrocyte transfusion: update of the guidelines “therapy with blood components and plasma derivatives”. Anaesthesist. 2009;58(11):1150–8.
Marsh J, Socie G, Tichelli A, et al. Should irradiated blood products be given routinely to all patients with aplastic anaemia undergoing immunosuppressive therapy with antithymocyte globulin (ATG)? A survey from the European Group for Blood and Marrow Transplantation Severe Aplastic Anaemia Working Party. Br J Haematol. 2010;150(3):377–9.
Nichols WG, Price TH, Gooley T, Corey L, Boeckh M. Transfusion-transmitted cytomegalovirus infection after receipt of leukoreduced blood products. Blood. 2003;101(10):4195–200.
Vamvakas EC. Is white blood cell reduction equivalent to antibody screening in preventing transmission of cytomegalovirus by transfusion? A review of the literature and meta-analysis. Transfus Med Rev. 2005;19(3):181–99.
Bielory L, Wright R, Nienhuis AW, Young NS, Kaliner MA. Antithymocyte globulin hypersensitivity in bone marrow failure patients. JAMA. 1988;260(21):3164–7.
Yoshida N, Kobayashi R, Yabe H, et al. First-line treatment for severe aplastic anemia in children: bone marrow transplantation from a matched family donor versus immunosuppressive therapy. Haematologica. 2014;99(12):1784–91.
Bacigalupo A, Brand R, Oneto R, et al. Treatment of acquired severe aplastic anemia: bone marrow transplantation compared with immunosuppressive therapy--The European Group for Blood and Marrow Transplantation experience. Semin Hematol. 2000;37(1):69–80.
Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017;129(11):1428–36.
Samarasinghe S, Steward C, Hiwarkar P, et al. Excellent outcome of matched unrelated donor transplantation in paediatric aplastic anaemia following failure with immunosuppressive therapy: a United Kingdom multicentre retrospective experience. Br J Haematol. 2012;157(3):339–46.
Pagliuca S, Peffault de Latour R, Volt F, et al. Long-term outcomes of cord blood transplantation from an HLA-identical sibling for patients with bone marrow failure syndromes: a report from eurocord, cord blood committee and severe aplastic anemia Working Party of the European Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2017;23(11):1939–48.
Kosaka Y, Yagasaki H, Sano K, et al. Prospective multicenter trial comparing repeated immunosuppressive therapy with stem-cell transplantation from an alternative donor as second-line treatment for children with severe and very severe aplastic anemia. Blood. 2008;111(3):1054–9.
Cheng Y, Xu Z, Zhang Y, et al. First-line choice for severe aplastic anemia in children: transplantation from a haploidentical donor vs immunosuppressive therapy. Clin Transpl. 2017 PMID: 29297952
Fanconi G. Familiaere infantile pernizisaartige anaemie. Jahrbuch Kinderheild. 1927;117:257–80.
Triemstra J, Rhodes L, Waggoner DJ, Onel K. A review of Fanconi anemia for the practicing pediatrician. Pediatr Ann. 2015;44(10):444–5, 448, 450, 452
Soulier J, Leblanc T, Larghero J, et al. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood. 2005;105(3):1329–36.
Gregory JJ Jr, Wagner JE, Verlander PC, et al. Somatic mosaicism in Fanconi anemia: evidence of genotypic reversion in lymphohematopoietic stem cells. Proc Natl Acad Sci U S A. 2001;98(5):2532–7.
Fargo JH, Rochowski A, Giri N, Savage SA, Olson SB, Alter BP. Comparison of chromosome breakage in non-mosaic and mosaic patients with Fanconi anemia, relatives, and patients with other inherited bone marrow failure syndromes. Cytogenet Genome Res. 2014;144(1):15–27.
Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood. 2003;101(4):1249–56.
Peffault de Latour R, Soulier J. How I treat MDS and AML in Fanconi anemia. Blood. 2016;127(24):2971–9.
Diamond LK, Blackfan KD. Hypoplastic anemia. Am J Dis Child. 1938;56:464.
Diamond LK, Allen DM, Magill FB. Congenital (erythroid) hypoplastic anemia. A 25-year study. Am J Dis Child. 1961;102:403–15.
Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116(19):3715–23.
West AH, Churpek JE. Old and new tools in the clinical diagnosis of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2017;2017(1):79–87.
Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859–76.
Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med. 2014;46(6):353–63.
Clinton C, Gazda HT. Diamond-Blackfan Anemia. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews((R)). Seattle: University of Washington; 1993.
Fargo JH, Kratz CP, Giri N, et al. Erythrocyte adenosine deaminase: diagnostic value for Diamond-Blackfan anaemia. Br J Haematol. 2013;160(4):547–54.
Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115(16):3196–205.
McGowan KA, Li JZ, Park CY, et al. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40(8):963–70.
Fumagalli S, Di Cara A, Neb-Gulati A, et al. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009;11(4):501–8.
Dutt S, Narla A, Lin K, et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117(9):2567–76.
Keel SB, Doty RT, Yang Z, et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008;319(5864):825–8.
Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006;46(5):558–64.
Calado RT, Cle DV. Treatment of inherited bone marrow failure syndromes beyond transplantation. Hematology Am Soc Hematol Educ Program. 2017;2017(1):96–101.
Vlachos A, Rosenberg PS, Kang J, Atsidaftos E, Alter BP, Lipton JM. Myelodysplastic syndrome and gastrointestinal carcinomas characterize the cancer risk in Diamond Blackfan anemia: a report from the Diamond Blackfan anemia registry. Blood. 2016;128(122):333.
Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119(16):3815–9.
Alter BP. Inherited bone marrow failure syndromes: considerations pre- and posttransplant. Hematology Am Soc Hematol Educ Program. 2017;2017(1):88–95.
Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12(12):753–64.
Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol. 2000;110(4):768–79.
Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–6.
Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011;117(21):5607–11.
Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107(7):2680–5.
Savage SA, Alter BP. The role of telomere biology in bone marrow failure and other disorders. Mech Ageing Dev. 2008;129(1–2):35–47.
Baerlocher GM, Lansdorp PM. Telomere length measurements in leukocyte subsets by automated multicolor flow-FISH. Cytometry A. 2003;55(1):1–6.
Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110(5):1439–47.
Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97(3):353–9.
Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014;165(3):349–57.
Townsley DM, Dumitriu B, Young NS. Danazol treatment for telomere diseases. N Engl J Med. 2016;375(11):1095–6.
Al-Rahawan MM, Giri N, Alter BP. Intensive immunosuppression therapy for aplastic anemia associated with dyskeratosis congenita. Int J Hematol. 2006;83(3):275–6.
Dietz AC, Orchard PJ, Baker KS, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46(1):98–104.
Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013;19(8):1238–43.
Bodian M, Sheldon W, Lightwood R. Congenital hypoplasia of the exocrine pancreas. Acta Paediatr. 1964;53:282–93.
Shwachman H, Diamond LK, Oski FA, Khaw KT. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964;65:645–63.
Goobie S, Popovic M, Morrison J, et al. Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7. Am J Hum Genet. 2001;68(4):1048–54.
Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97–101.
Toiviainen-Salo S, Raade M, Durie PR, et al. Magnetic resonance imaging findings of the pancreas in patients with Shwachman-Diamond syndrome and mutations in the SBDS gene. J Pediatr. 2008;152(3):434–6.
Donadieu J, Leblanc T, Bader Meunier B, et al. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica. 2005;90(1):45–53.
Savilahti E, Rapola J. Frequent myocardial lesions in Shwachman's syndrome. Eight fatal cases among 16 Finnish patients. Acta Paediatr Scand. 1984;73(5):642–51.
Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009;23(2):233–48.
Dror Y, Donadieu J, Koglmeier J, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242:40–55.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer International Publishing
About this chapter

Cite this chapter
Khazal, S., Galvez Silva, J.R., Thakar, M., Margolis, D. (2019). Bone Marrow Failure. In: Duncan, C., Talano, JA., McArthur, J. (eds) Critical Care of the Pediatric Immunocompromised Hematology/Oncology Patient. Springer, Cham. https://doi.org/10.1007/978-3-030-01322-6_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-01322-6_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-01321-9
Online ISBN: 978-3-030-01322-6
eBook Packages: MedicineMedicine (R0)