
Abstract
Diffuse large B-cell lymphoma (DLBCL) is currently treated with immunochemotherapy based on the R-CHOP regimen (see Chap. 5). Depending on the clinical risk factors summarized by the International Prognostic Index (IPI) and some molecular characteristics of the tumor, about two thirds of patients are cured with standard first-line therapy, whereas one third is primarily refractory or relapse in the further course of disease (Friedberg JW, Hematol Am Soc Hematol Educ Program 2011:498–505, 2011). Thus, the addition of rituximab to CHOP chemotherapy has dramatically improved survival of patients with DLBCL and largely contributed to the development of monoclonal antibodies (mAbs) in DLBCL and more generally in cancer (Coiffier B et al., N Engl J Med 346:235–242, 2002). This first clinical success of immunotherapy led different companies to develop new mAbs targeting new surface molecules or being modified to elicit increased immune activity or to bring chemotherapy or a radioactive isotope closer to the tumor cells. Patients who relapse after R-containing first-line therapy (Gisselbrecht et al., J Clin Oncol 28(27):4184–4190, 2010) showed their dismal prognosis; the recently published SCHOLAR-1 study described an objective response rate of only 26% (complete response rate 7%) and a median overall survival of 6.3 months for patients with refractory DLBCL (Crump M et al., Blood 130:1800–1808, 2017). Taken together, the enormous difficulties in successfully managing patients suffering from refractory or relapsed DLBCL demonstrate that novel therapeutic approaches are urgently required. In contrast to other novel approaches discussed below, chimeric antigen receptor T cells (CAR-T) showed remarkable response rates and ongoing remissions in early clinical trials, even in heavily pretreated patients who had failed multiple salvage regimens. Thus, CAR T cells seem to become an important pillar in the therapeutic management of lymphoma even when immunochemotherapy, radiotherapy, and the use of small molecules fail.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
Diffuse large B-cell lymphoma (DLBCL) is currently treated with immunochemotherapy based on the R-CHOP regimen (see Chap. 5). Depending on the clinical risk factors summarized by the International Prognostic Index (IPI) and some molecular characteristics of the tumor, about two thirds of patients are cured with standard first-line therapy, whereas one third is primarily refractory or relapse in the further course of disease [1]. Thus, the addition of rituximab to CHOP chemotherapy has dramatically improved survival of patients with DLBCL and largely contributed to the development of monoclonal antibodies (mAbs) in DLBCL and more generally in cancer [2]. This first clinical success of immunotherapy led different companies to develop new mAbs targeting new surface molecules or being modified to elicit increased immune activity or to bring chemotherapy or a radioactive isotope closer to the tumor cells. Patients who relapse after R-containing first-line therapy [3] showed their dismal prognosis; the recently published SCHOLAR-1 study described an objective response rate of only 26% (complete response rate 7%) and a median overall survival of 6.3 months for patients with refractory DLBCL [4]. Taken together, the enormous difficulties in successfully managing patients suffering from refractory or relapsed DLBCL demonstrate that novel therapeutic approaches are urgently required. In contrast to other novel approaches discussed below, chimeric antigen receptor T cells (CAR-T) showed remarkable response rates and ongoing remissions in early clinical trials, even in heavily pretreated patients who had failed multiple salvage regimens. Thus, CAR T cells seem to become an important pillar in the therapeutic management of lymphoma even when immunochemotherapy, radiotherapy, and the use of small molecules fail.
2 Monoclonal Antibody Therapy
Monoclonal antibodies (mAbs) are undoubtedly one of the therapeutic revolutions of the last 10 years in oncology. Because of the absolute specificity of the antibody for its target, they perfectly illustrate the concept of targeted therapy highlighted at the end of the nineteenth century. Their success can be explained by the progress of biotechnologies made in the 1980s, allowing their humanization. Rituximab (MabThera®, Rituxan®) was the first “humanized” mAb marketed worldwide. The results obtained in non-Hodgkin’s lymphomas, especially in DLBCL, and the rapidity of its clinical development largely explain the enthusiasm for this class of drugs. The progress made in understanding the mechanisms of action of this antibody and its ability to interact with the immune system have consequences applicable to all monoclonal antibodies and have allowed the development of new format of mAbs and gave us way to better use these compounds. These successes explain why this antibody is a hope for patients and a model for doctors, scientists, and drug manufacturers.
Given the success of rituximab, various companies first developed monoclonal antibodies that recognize the different proteins expressed on the surface of the B lymphocyte. To date, no other monoclonal antibody targeting a protein expressed by DLBCL cells has been approved. Other strategies were then developed. All of them aim to improve the cellular cytotoxicity of the antibody either by modifying the Fc portion of the antibody itself, enabling it to better recruit the immune effector cells, or by directing one of the two Fab against a protein expressed by immune cells. In the first case, it is essentially the natural killer (NK) cells and the macrophages which are recruited by the optimized Fc portion allowing an improvement of the antibody-dependent cellular cytotoxicity (ADCC); in the other case, the bispecific antibody will recruit the T lymphocytes via CD3 allowing a T-cell killing.
More recently, the demonstration of the mechanisms controlling the immune response during tumorigenesis has made possible identification of key proteins (immune checkpoint) that can be targeted by monoclonal antibodies. It is still too early to say how this strategy will modify the management of DLBCL, but these antibodies could at least in combination contribute to improve long-term control of this disease.
2.1 Anti-CD20 Monoclonal Antibodies
In 1984, the second workshop of human leukocyte differentiation antigens identified 11 new clusters of differentiation (CD16 to CD26), including CD20, expressed by all B lymphocytes and B-cell lymphoma. Among the antibodies of this CD20 cluster, clone 1F5 (mouse IgG2a) was selected by Oncogen in Seattle and tested in four patients with lymphomas at the University of Washington [5]. The patient receiving the highest dose experienced a 90% reduction in tumor mass, but the remission lasted only 6 weeks. At that time, IDEC Pharmaceuticals developed a chimeric monoclonal antibody from the murine monoclonal anti-CD20 2B8 antibody (mu-2B8). This antibody was chimerized with the constant domains κ and γ1 (human IgG1 version or ch-C2B8) or κ and γ4 (human IgG4 version) [6]. Only the human IgG1 version was able to activate the complement and recruit the effectors of the immunity, proved to be lymphopenic in the macaque and was developed under the name of rituximab.
Rituximab, combined with chemotherapy, is now the gold standard for treatment of DLBCL [2, 3]. A better knowledge of its mechanisms of action allowed to understand the development of novel anti-CD20 antibodies and, in a broader sense, part of recent development in DLBCL immunotherapy.
2.1.1 Rituximab: Mechanisms of Action
Rituximab is a “bifunctional” molecule combining functions related to the recognition of the antigen (and, therefore, specific for the epitope) and functions related to the Fc portion (crystallizable fragment) common to all IgG1. The properties related to the Fc portion make it possible to distinguish the IgG1 monoclonal antibodies from the different isotype monoclonal antibodies (Table 16.1). Thus, IgG1 and IgG3 are the classes of IgG with the greatest capacity to recruit the immune system (effector cells and complement). In mice, however, IgG2a and, to a lesser extent, IgG2b have this property. This difference underlines the difficulties to interpret experiments using humanized monoclonal antibodies in the murine model. The Fc portion of the IgGs is also capable to bind to a receptor named FcRn (or Brambell factor) expressed by endothelial cells, epithelial cells, and syncytiotrophoblast cells. Interaction with this receptor ensures their transplacental or transepithelial passage and allows IgGs to escape the lysosomal degradation that accounts for the longer half-life of IgG compared to other immunoglobulins.
2.1.1.1 Mechanisms Related to Target Recognition
CD20 is the target antigen of rituximab, and the advent of this therapeutic antibody has led to important advances in the knowledge of this protein and its functions. CD20 is a transmembrane protein (Fig. 16.1) that has characteristics that make it a therapeutic target of choice [7]. Thus, CD20 is expressed by most B lymphocytes but is absent or poorly expressed by B-progenitors or plasma cells, thus maintaining immunoglobulin levels and peripheral lymphoid reconstitution after treatment. After binding to the antibody, CD20 is neither regulated nor released from the plasma surface. This is the extracellular domain, which carries the epitopes recognized by anti-CD20 antibodies. The homology with murine CD20 is 73% and is located essentially in the transmembrane regions. The extracellular domain of murine CD20 differs from that of human CD20 for 16 of the 43 amino acids explaining [8] the lack of rituximab binding to murine CD20.

Structure of human CD20. CD20 is a non-glycosylated protein with four transmembrane domains (Tetraspan). The extracellular domain carries the epitopes recognized by the various anti-CD20 antibodies. Alanine and proline located at positions 170 and 172 are important residues in the determination of the rituximab epitope. The sequence between residues 219 and 225 plays an important role in the migration of CD20 into lipid rafts
The function of CD20 has remained unknown for a long time, and its role as a calcium channel has been now demonstrated [9]. However, knockout mice for the gene coding for CD20 do not show phenotypic abnormalities [10], which could testify either to the minor role of CD20 in the physiology of the B lymphocyte or to a certain biological redundancy with other proteins. The use of antibodies directed against CD20 has long been the only method of understanding its function. Two types of properties reported initially to the recognition of two different epitopes could be identified: the first, found with rituximab, but also with other types of anti-CD20 (2H7, B1), leads to inhibitory signals inducing apoptosis and/or antiproliferative activity, whereas the 1F5 antibody activates cell proliferation. In reality, there is a wide variety of epitopes within the extracellular domains, although some residues are critical for antiproliferative activity (Fig. 16.1) [8]. The binding of the antibody to its target induces, under certain conditions, the migration of the antigen within lipid rafts present on the surface of the plasma membrane [11]. This movement is dependent on an amino acid sequence (219–225) located within the intracytoplasmic portion and not present in the mouse. This property allowed the identification of antibodies inducing (type I antibodies: rituximab, 2H7, etc.) or not (type II antibodies: B1, Ly1, etc.) this migration. The translocation of CD20 within these structures allows its colocalization with proteins ensuring the signal transduction. Only the antibodies inducing this migration are able to complement activation (see below) [12]. In vitro and in some cell models, rituximab induces apoptotic cell death. Several pathways for apoptosis activation have been described, in particular by the mitogen-activated protein kinase (MAPK), NFκΒ, protein kinase C (PKC), or ceramides or BCL-2. The activation of such pathways would explain the synergies observed with certain chemotherapeutic agents (fludarabine, cisplatin, anthracyclines).
2.1.1.2 Mechanisms Related to the Fc Portion
The ability of the Fc portion of IgG1 to interact with cellular immune effectors or complement confers on the set of monoclonal antibodies belonging to this class of common cytolytic properties which largely explain the therapeutic activity of these molecules.
2.1.1.2.1 Complement-Dependent Cell Lysis
Complement is an important actor in the eradication of malignant cells. The activation of the classical complement pathway by immunoglobulins (IgG1, IgG3, and IgM) requires the binding of the antibody to its target allowing the binding of the C1q protein to the Fc portion of the antibody. This binding will trigger a proteolytic cascade leading to the formation of a large amount of C3b allowing the formation of the membrane attack complex (MAC) and the destruction of the cell (complement-dependent cell lysis or CDC). It also allows the migration of cells from inflammation (via C3a and C5a) to the activation site and the opsonization of C3b on the target cell allowing its interaction with complement receptors (CR3 and CR4) expressed by immune cells (NK cells, monocytes, neutrophils). Thus, the complement constitutes a system allowing both the direct lysis of the target cell and the establishment of a cytolytic cellular response.
Many in vitro studies have shown that rituximab induces CDC on fresh lymphoma cells. The activation of complement by rituximab was perfectly shown in a syngeneic model of murine lymphoma expressing human CD20 (EL4-huCD20) [13]. In this model, the therapeutic activity of rituximab was not found with mice deficient for C1Q. In humans, administration of rituximab is accompanied by increased concentrations of complement degradation products (C3b/c, C4b/c) [14]. It has been observed in vitro that the complement activation may be different depending on the histological type of lymphoma. The role of the expression level of CD20 or proteins that negatively regulate complement (CD46, CD55, and CD59) on this activity has long been discussed. Recent results have clearly demonstrated that CDC is correlated with the expression level of CD20 by the target cell [15]. The ability of an anti-CD20 antibody to activate complement is also related to the recognized epitope and its ability to relocate CD20 within the lipid rafts. Thus, rituximab or 2H7 that induce effective redistribution (type I) results in CDC, while murine B1 or Ly1 antibodies do not induce CDC due to their inability to migrate CD20 within lipid rafts (type II).
2.1.1.2.2 Receptor-Dependent Cellular Lysis at the Fc Portion of the Antibody
The Fc portion of rituximab is capable to interact with receptors at the Fc portion of IgG or FcγRs (Fig. 16.2). By the recruitment of cells expressing these receptors (Table 16.2), immunoglobulins participate in the implementation of immune effector mechanisms such as antibody-dependent cell phagocytosis (ADCP) and ADCC.

Structure of the different receptors for the Fc portion of IgG immunoglobulins (FcγRs). FcγRIIb is the only inhibitory receptor due to the presence of an ITIM motif (immunoreceptor tyrosine-based inhibition motif) in its intracytoplasmic portion. The presence of an ITAM motif (immunoreceptor tyrosine-based activation motif) in the intracytoplasmic domain or within an associated accessory chain gives to the other FcγRs activating properties
The ability of rituximab to induce an ADDC or to mediate ADPC has been demonstrated in vitro on human lymphomatous lines, and the involvement of FcγRs has been shown in a mouse model [16]. The involvement of these receptors and particularly FcγRIIIa has been shown in humans. In fact, this receptor exhibits a nucleotide polymorphism leading to the substitution of the amino acid located at position 158. Thus, two variants of the receptor are possible, one with a valine at position 158 (FcγRIIIa-158 V) and the other with a phenylalanine (FcγRIIIa-158F). This substitution is accompanied by a modification of the affinity between the FcγRIIIa receptor and the Fc portion of the IgG1 [17]. The influence of this amino acid is not surprising since it is located in the site of interaction between these two proteins. A study including patients with follicular lymphoma showed that patients homozygous for the high-affinity receptor for the Fc fragment (FcγRIIIa-158 V) had a better clinical response to rituximab [18]. Since this receptor is expressed by monocytes and NK cells, essential actors of ADCC, this cytolytic mechanism is considered today as an important mode of action of rituximab. Above all, this work emphasized the importance of the interaction between FcγRs and the Fc portion of the antibody. The influence of FcγRIIIa-158 V/F polymorphism on rituximab response was however inconstantly found in DLBCL clinical trials. This could be related either to the lack of importance of ADCC in this histology or by the chemotherapy associated with rituximab which could reduce rituximab-mediated ADCC.
2.1.1.2.3 Anti-Lymphoma-Specific Immunity
There are a number of arguments for the initiation of specific anti-lymphoma immunity during treatment with rituximab that murine models seem to confirm [19]. Indeed, most antigen-presenting cells (dendritic cells, macrophages) express FcγRs whose role in the therapeutic activity of the mAbs has been demonstrated. Moreover, a number of clinical observations could account for this mechanism: delayed response to treatment and increase in the duration of response to reprocessing. The hypothesis is that the antibody-induced cytolytic mechanisms would induce the presentation of antigens specific for lymphoma by antigen-presenting cells leading to the establishment of a specific immune response. The confirmation of such a mechanism could lead to a modification of the conditions of rituximab use and would open up new ways to optimize its therapeutic activity.
2.1.2 Other Anti-CD20 Monoclonal Antibodies
2.1.2.1 Glyco-Modified Anti-CD20 Monoclonal Antibodies
The principle of these mAbs is to modify the Fc portion in order to obtain a better affinity for FcγRIIIa. These can be obtained either by modifying the oligosaccharide located between the two Fc arms of the antibody (obinutuzumab, ublituximab) or by mutating the region of the Fc portion involved in the interaction with FcγRIIIa (ocaratuzumab, PRO131921). To date, only obinutuzumab has been tested in clinical trials in DLBCL, and ocaratuzumab and PRO131921 development are stopped.
2.1.2.1.1 Obinutuzumab
Obinutuzumab is the first humanized glycoengineered IgG1 anti-CD20 mAb to be tested in clinical trials. Obinutuzumab has been humanized by grafting the complementarity-determining region sequences from the murine antibody B-ly1 into human VH and VL acceptor frameworks [20]. Obinutuzumab was expressed from Chinese hamster ovary (CHO) K1 cell lines engineered to constitutively overexpress the heavy and light chains of obinutuzumab. Those cell lines also express recombinant wild-type β-1,4-N-acetyl-glucosaminyltransferase III and wild-type Golgi α-mannosidase II leading to accumulation of antibody glycoforms containing bisected, non-fucosylated oligosaccharides attached to asparagine 297 in the Fc region.
Such modifications induce increased affinity of obinutuzumab to both FcγRIIIa-158 V and FcγRIIIa-158F compared with rituximab translating into an increased induction of ADCC relative to rituximab in vitro [20]. Obinutuzumab is a type II anti-CD20 mAbs and thus exhibits lower level of CDC in vitro. It differs from most anti-CD20 type I mAbs investigated (rituximab, ofatumumab, ublituximab, ocaratuzumab) (Table 16.3). Other characteristics of type II mAbs have been described differentiating these mAbs from type I antibodies: homotypic adhesion resulting in non-caspase-dependent direct cell death, half-maximal CD20 binding at saturating conditions, and less or no CD20 modulation. Except lack of CD20 modulation, all these in vitro properties have been described for obinutuzumab [21] and thus enhance direct cell death compared to rituximab [22]. Preclinical development of obinutuzumab elucidated such differences, leading to the proposal of a model of mechanism of action [20, 23]. Obinutuzumab conformational structure is different to that of rituximab. Firstly, obinutuzumab binds CD20 in a different overlapping epitope than rituximab and in a different orientation [24, 25]. In comparison with rituximab, obinutuzumab is rotated 90° around the Fab middle axis and tilts 70° toward the carboxy-terminus of the CD20 epitope. Moreover, the elbow angle between VH and CH1 is 30° wider. This characteristic could be related to amino acid substitution at position 11 substituting a leucine for a valine [20]. This results in a new spatial arrangement between CD20 and the antibody, and, unlike rituximab, obinutuzumab can bind its two Fab arms on the same CD20 tetramer. This difference in binding CD20 antigen (i.e., intra-CD20 tetramer for type II vs. inter-CD20 tetramer for type I) led authors to propose a dynamic model of interaction [24] explaining the majority of in vitro observations.
An obinutuzumab phase I/II study [26], including previously rituximab-treated DLBCL patients, demonstrated an overall response rate of 30%, which was not different from that obtained with rituximab [27]. Untreated DLBCL patients were randomized to receive eight cycles of either rituximab-CHOP21 or obinutuzumab-CHOP21 in the GOYA study [28]. No difference was found in terms of overall response rate or progression-free survival (PFS). Such results indicate that the improvement of FcγRIIIA-mediated mechanisms and/or enhancement of direct cytotoxicity does not translate into higher clinical activity in DLBCL, possibly due to low-level CDC observed with obinutuzumab. Final results of randomized phase III GAINED study including younger DLBCL patients receiving more intensive chemotherapy with obinutuzumab or rituximab are awaited soon.
2.1.2.1.2 Ublituximab
Ublituximab is a chimeric IgG1k produced by rat hybridoma YB2/0 cells [29] resulting in low percentage of fucosylated glycoforms. In vitro data demonstrated higher ADCC than rituximab but similar direct toxic effect or CDC. No trial testing of ublituximab in DLBCL is ongoing (Table 16.4).
2.1.3 Anti-CD20 Monoclonal Antibodies with Increased Affinity for CD20
Ofatumumab is the only representative of this anti-CD20 class. It was selected because its affinity for CD20 appeared to be greater than that observed with rituximab [30]. In fact, it is also a type I antibody, but its off-rate is decreased contributing to a greater aggregation of ofatumumab-CD20 complexes within the lipid rafts, a condition favorable to the recruitment of C1Q, leading to CDC activation. Thus, ofatumumab is characterized by a better CDC than rituximab, especially when antibody concentrations or CD20 expression is low [31, 32]. This property appears to be related to the recognition of a different CD20 epitope [31], ofatumumab, recognizing two epitope sites with which it interacts via strong bonds [33]. Unfortunately, ofatumumab failed to demonstrate any clinical advantage in relapse/refractory DLBCL compared to rituximab when associated with chemotherapy (Table 16.4) [34].
2.2 Bispecific Monoclonal Antibodies
Bispecific mAbs are able to bind two different targets simultaneously and might potentially induce more powerful antitumor response. Different formats of bispecific antibodies are developing. Blinatumomab is a single-chain protein comprising the antigen-binding domains of two different antibodies joined by a non-immunogenic linker that allows for the rotational flexibility to bind two different antigen epitopes on separate cells in close proximity. Blinatumomab contains binding regions for the B-cell lineage-specific antigen, CD19, as well as the invariant CD3ε subunit of the T-cell receptor (TCR) present on all T lymphocytes. Compared to full IgG antibodies (150 KDa), bispecific antibodies exhibit improved tissue distribution and better tumor penetration. Their bivalent nature confers them probably a prolonged target retention. All these characteristics may result in synergistic effect on tumor destruction. A phase II evaluated blinatumomab in 25 DLBCL patients [35]. Among 21 evaluable patients, the overall response after one cycle was 43%, including complete response in 19%, and three patients experienced late complete response in follow-up. Patients experienced grade 3 neurologic events with encephalopathy and aphasia (each 9%) and tremor, speech disorder, dizziness, somnolence, and disorientation (each 4%). Those neurological adverse events are presumably caused by release cytokines. Importantly, continuous infusion is required to ensure sustained effective serum concentration due to a very short half-time of less than 2 h related to the absence of Fc portion and renal excretion. Larger trials evaluating blinatumomab in relapse/refractory DLBCL are ongoing.
The progress of biotechnologies allows today to design new generation of bispecific monoclonal antibodies. Indeed, it is now possible to construct bispecific antibodies while preserving the properties of interest of the Fc portion. For example, the bispecific can retain the format of an IgG1, thus preserving a favorable pharmacokinetic profile of this isotype (in relation to its binding to FcRn) and mutating the CH2 region involved in the interaction with FcγR and C1Q, allowing to repeal any ADCC or CDC. Of course, the absence of such modifications of this region will make it possible to obtain a trifunctional antibody (e.g., CD16xCD3xCD20).
2.3 Monoclonal Antibodies Targeting Immune Cells
The emergence of treatments targeting immune checkpoints owes much to the advances in biotechnologies and the ability to produce mAbs directly targeting proteins involved in controlling the immune response. At the same time, considerable progress has been made since the 1970s to better understand the role of the immune response in the development of tumorigenesis. Today, and following the success of targeting immune checkpoints, monoclonal antibodies against proteins involved in immune control are an emerging treatment of DLBCL.
2.3.1 Monoclonal Antibodies Targeting Inhibitory Checkpoints
The concept of immune checkpoint emerged when we understood the role of immunological surveillance made by the immune system during tumor development. Thus, the tumor is able to regulate negatively the immune system allowing it to escape to its control. Immune evasion by cancers is accomplished through a variety of mechanisms, including upregulation of negative costimulatory molecules, such as PD1 and CTLA-4.
Three inhibitory checkpoints are currently targeted in DLBCL (Table 16.5).
2.3.1.1 Antigen-Presenting Cell (APC)/T-Cell Interactions: CTLA-4 Pathway
TCR stimulation by an antigenic peptide in the context of a major histocompatibility complex (MHC) molecule leads to CTLA-4 upregulation on the plasma membrane of the activated T cell where it outcompetes CD28 for B7 ligand binding, resulting in costimulation and downregulation of effector T-cell functions. CTLA-4 expression is mainly expressed by T cells in the lymph nodes where initial tumor antigen presentation is thought to occur. Anti-CTLA4 antibody ipilimumab has been tested in phase I trial [36]. Objective response rate was low (11%), but durable complete response suggested that ipilimumab may have additive and durable effect as combination therapy.
2.3.1.2 Tumor Cell/Activating T-Cell Interactions: PD1 Pathway
PD1 is expressed by activated T cells located in peripheral tissues, including those residing within the tumor. PD1 interacts with PD-L1, which is broadly expressed, and with PD-L2 expressed mainly on hematopoietic cells. The upregulation of PD-1, which occurs after tumor antigen recognition and its binding to PD-L1 or PD-L2 ligands, inhibits intracellular signaling pathways and blocks further T-cell activation. PD-L1 expression is usually induced by inflammatory signals, such as IFNγ produced by activated T cells attempting to execute an active antitumor response (adaptive resistance). In some tumor, PD-L1 expression could be induced through activation of the AKT pathway or gene amplifications, outside inflammatory signals (innate resistance). Primary mediastinal lymphomas exhibit gene fusions between MHC class II transactivator (CIITA) and PD-L1 or PD-L2 placing those genes under the transcriptional control of the CIITA promoter leading to expression of PD-L1 in around 40% of cases [37]. A subset of Epstein-Barr virus (EBV)-associated lymphomas also display gene amplification leading to PD-L1 and PD-L2 overexpression. Outside specific mechanisms leading to PD-L1 (over)expression, PD-L1 is expressed in around 30% of DLBCL [37]. In a phase II study including 66 DLBCL patients [38], three doses of pidilizumab were infused after autologous stem cell transplantation. The 16-month PFS was 70%, and among the 35 patients with measurable disease after autologous stem cell transplantation, the overall response rate after pidilizumab treatment was 51%.
2.3.1.3 Tumor Cell/Macrophage Interactions: CD47 Pathway
CD47 is now recognized as a “marker of self” and broadly expressed by tumor cells. Its ligand, signal regulatory protein alpha (SIRPα), is expressed by macrophage and, when it is engaged by CD47, decreases phagocytosis and cytotoxicity. CD47/SIRPα inhibits therefore the activation of macrophages and other myeloid cells against tumors and thereby acts as a myeloid-specific immune checkpoint. CD47 expression is increased in DLBCL, correlates with cells of origin, and confers worse clinical prognosis [39]. Blocking antibodies against CD47 enable phagocytosis of lymphoma cells by macrophages and synergize with rituximab in vitro. Combination therapy with anti-CD47 antibody and rituximab eliminates lymphoma in xenograft mouse model [39]. Phase I testing CD47/SIRPα-targeting therapeutics is ongoing.
In the future, characterization of multiple immune checkpoints will drive the further clinical development of other checkpoint inhibitors, such as TIM3, LAG-3, KIR, and VISTA.
2.3.2 Monoclonal Antibodies Targeting Costimulatory Receptors
Another attractive alternative is to use agonistic antibodies that target stimulatory molecules expressed by T cells; CD137 and CD40 are the most prominent of these molecules. Activation of T cells by antibodies may lead to critical adverse events warranting particular attention during clinical development.
CD137 (4-1BB) is a surface glycoprotein belonging to the tumor necrosis factor receptor family. CD137 is an inducible costimulatory molecule expressed on a variety of immune cells, including activated CD4+ and CD8+, T cells, NK cells, monocytes, and dendritic cells. Agonistic mAbs against this receptor have been shown to induce tumor-specific T-cell responses able to eradicate tumor cells in murine models [40]. In a syngeneic murine lymphoma model and in a xenotransplanted human lymphoma model, sequential administration of rituximab followed by anti-CD137 mAb has potent anti-lymphoma activity in vivo [41]. These results suggested that stimulation of CD137 could enhance NK cell killing by ADCC and thereby augment rituximab efficacy. A phase I trial evaluating utomilumab in combination with rituximab in patients with relapsed or refractory CD20-positive lymphomas demonstrated a significant antitumor efficacy and no dose-limiting toxicities. In addition, no patients discontinued treatment due to treatment-related adverse events [42].
CD40 is also a tumor necrosis factor receptor family member expressed on APC, B cells, and monocytes. In murine models, CD40 agonistic antibodies have shown exceptional therapeutic activity in the treatment of CD40-positive B-cell lymphomas with 80–100% of mice cured [43].
Despite some success achieved by mAbs targeting immune cells in DLBCL patients, there are still a number of patients who do not benefit from single-agent therapy. To enhance efficacy, synergistic combinations are now proposed including co-targeting of inhibitory checkpoint (anti-CTLA-4 and anti-PD1/PD-L1) or co-targeting of inhibitory checkpoint and costimulatory receptors (anti-PD1/PDL1 and anti-CD137).
3 CAR T-Cell Therapy
3.1 Design and Differences
CAR T cells are T cells expressing a chimeric antigen receptor (CAR) introduced in vitro by varying vector systems. The CAR consists of three major structural elements: an extracellular domain with an antibody-derived single-chain variable fragment (scFv), a spacer and linker domain, and finally an intracellular signaling domain like CD3ζ connected to important costimulatory domains like CD28 (KTE-C19) or CD137 (4-1BB) (CTL019, JCAR017) (Fig. 16.3) [44]. Most CAR T-cells are autologous, but allogeneic CAR T cells are also being developed. To overcome critical HLA barriers and to allow evading to host-mediated immunity and deliver anti-lymphoma effects without graft versus host disease (GVHD), a smart method is used to target the constant region of the T-cell receptor alpha chain (TRAC), thereby disrupting cell surface expression of TCRαβ [45]. Such allogenic CAR T cells are also named universal CAR T cells (UCAR-T) reflecting their ability to be effective in all patients without restriction to distinct HLA molecules.

Structure of chimeric antigen receptor. A chimeric antigen receptor (CAR) consists of an antibody-derived single-chain variable fragment (scFv) binding to the tumor antigen (e.g., CD19), a spacer and linker region, and an intracellular signaling domain activating the T cell
For treatment of B-cell malignancies, the scFv has been directed against the B-cell surface antigen CD19 representing an ideal therapeutic target since apart from virtually all B-cell lymphomas only normal B cells and follicular dendritic cells express CD19 [46]. CAR T cells directed against CD19 specifically bind to B cells, get activated by downstream signaling, and initiate a cytotoxic response and cytokine release. In contrast to normal T cells, which for different reasons are not completely understood (e.g., checkpoint inhibition, immunosuppressive tumor environment, downregulation of MHC-presented tumor antigens) and fail to induce effective tumor lysis, CAR T cells induce rigorous killing of lymphoma cells. The binding affinity as well as the exact epitope location the scFv binds to will finally determine the efficacy of the CAR system.
The off-tumor toxicity of CD19-binding CAR T cells is limited with the resulting B-cell aplasia being clinically well manageable. The lack of an in many respects ideal target like CD19 explains the difficulty to design appropriate CAR T cells for other hematological malignancies and especially for solid tumors although important progress is being made [47].
The so-called first-generation CAR T cells consisted of the scFv and the CD3ζ domain; clinical responses to these constructs were still limited [48]. By the adding costimulatory domains like CD28 or 4-1BB (second-generation CARs), a significantly higher antitumor activity as well as an increased persistence of CAR T cells could be achieved [49]. Interestingly, costimulation via CD28 was associated with strong activation, increased cytokine release, and enhanced tumor lysis, whereas costimulation using the 4-1BB domain is said to induce longer persistence of CAR T cells and more durable tumor control [48]. Further benefit of CARs with several costimulatory domains (third generation) was reported in preclinical settings although clinical experience with such molecules is limited. Overstimulation may not only decrease the tumor response but also induce severe side effects.
The transmembrane and extracellular spacer and linker domain is a pivotal structural element for the stability and functionality of the CAR optimizing the T cell to target cell engagement. The phenotype of the transduced T cells represents a further key point. More immature T cells with a phenotype of memory T cells show a significant better tumor control and a longer survival compared to more differentiated effector and effector memory T cells. In most clinical trials, investigators used unselected transduced T cells. Therefore, the importance of specifically selected T cells for the success of CAR T-cell therapy is not yet clear, and further investigation is highly warranted.
3.2 Practical Aspects
Autologous CAR T-cell manufacturing starts with leukapheresis from the patient’s blood. The apheresis product is transported to a central manufacturing facility where T cells are isolated from the collected blood cells. It is possible to enrich T-cell subsets such as CD4+, CD8+, CD25+, or CD62L+ T cells. The isolated T cells are expanded by T-cell activation via different systems using, e.g., beads coated with anti-CD3 and anti-CD28 antibodies. The expanded and activated T cells are finally transduced with the CAR system, most often using lentiviral or γ-retroviral vector systems. The final product is cryopreserved and shipped back to the study site. The time from leukapheresis to delivery of the ready-to-use CAR T-cell product may take between 14 and 40 days. This time period is relatively long and may necessitate administration of further chemo-immunotherapy to the patient in order to prevent massive tumor growth and further worsening of the patient’s performance status.
T-cell-depleting chemotherapy prior to CAR T-cell infusion is associated with an impairment of regulatory T cells, an increase of T-cell-activating cytokines like IL-15, and an activation of antigen-presenting cells [50, 51] and may contribute to the success of CAR T-cell therapy.
The limitation of autologous CAR T cells is that they need to be customized, whereas allogenic CAR T cells (UCART) could be produced independently of the patient and the tumor and can be taken off the shelf when needed.
3.3 CAR T Cells for Treatment of Relapsed and Refractory B-Cell Lymphoma
Clinically, three CD19 CAR T-cell constructs have been used for treatment of refractory B-cell lymphoma: CTL019 (tisagenlecleucel, Novartis Pharmaceuticals, Basel, Switzerland), KTE-C19 (axicabtagene ciloleucel (axi-cel), Kite Pharma, Santa Monica, USA) and JCAR017 (lisocabtagene maraleucel (liso-cel), Juno Therapeutics, Seattle, USA; Celgene, New Jersey, USA) (Table 16.6).
CTL019 uses the CD137 costimulatory domain and a lentiviral vector transduction. Since August 2017, CTL019 is approved by the FDA to treat children and young adult patients up to 25 years with refractory disease or in relapse of B-cell precursor acute lymphoblastic leukemia. KTE-C19 uses CD28 as costimulatory domain and was originally constructed at the National Cancer Institute [52]. In October 2017, the FDA approved KTE-C19 for adult patients with refractory or relapsed DLBCL after at least two different therapy lines. JCAR017 was developed by Juno Therapeutics and Celgene Corporation and is under investigation for treatment of relapsed and/or refractory DLBCL. In January 2018, Celgene announced the acquisition of Juno Therapeutics. Regulatory approval for JCAR017 in the USA is expected in 2019.
3.3.1 Clinical Experience with CAR T Cells
Based on the promising initial results at the NCI, the phase II multicenter study ZUMA-1 (NCT02348216) enrolled 111 patients with refractory DLBCL, primary mediastinal B-cell lymphoma, or transformed follicular lymphoma to be treated with KTE-C19 CAR T cells [53]. Refractory disease was defined as progressive or stable disease after the most recent therapy or relapse within 12 months after autologous transplantation. The patients received a conditioning regimen of fludarabine (30 mg/m2) and cyclophosphamide (500 mg/m2) on days −5 to −3 before administration of a target dose of two million of CAR T cells per kilogram body weight on day 0. The conditioning regimen as well as the minimum number of CAR T cells necessary to elicit reliable tumor responses had been examined in previous studies.
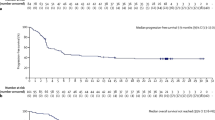
Among patients who received KTE-C19, the objective response rate was 82%, with complete responses in 54% of cases. With a median follow-up of 15.4 months, remissions were ongoing in 42% (including 40% complete response (CR) remissions). The overall survival rate at 18 months was 52%. Three patients died during treatment. The time from leukapheresis to final delivery of the CAR T-cell product took a median of only 17 days.
The major side effects developing after infusion of CAR T cells were myelosuppression, occurrence of the cytokine release syndrome (CRS), and neurologic side effects. 93% of patients suffered from CRS with 13% of patients experiencing CRS of grade 3 or higher. The symptoms resolved within a median of 8 days except for two patients who died due to hemophagocytic lymphohistiocytosis and cardiac arrest.
Severe neurologic events (grade 3 or higher) comprising encephalopathy, confusion, aphasia, or somnolence occurred in about one third of cases. Except for four events occurring in patients who eventually died, all neurologic symptoms resolved over a median time of 17 days after infusion. Altogether, 43% of patients needed tocilizumab (a humanized monoclonal antibody against the interleukin-6 receptor) and 27% received glucocorticoids to handle CRS and/or neurologic events. Interestingly, high numbers of CAR T cells in peripheral blood were correlated with clinical response and the occurrence of neurologic events but not with the severity of CRS.
The second CAR T-cell system CTL-019 was studied in the JULIET trial [54]. JULIET enrolled 147 patients with chemorefractory DLBCL after at least two different therapies. Patients had to be ineligible for or had to have failed to autologous transplantation. JULIET was an international study with participation of 27 centers in 10 different countries. The time from apheresis to delivery to the treating physician was 39 days. Ninety-nine patients received a single infusion of CTL019 CAR T cells at a median dose of 3.1 × 108 cells. In 16 of 48 cases, CAR T cells could not be infused because the patient had died prior to delivery. Like in the ZUMA trial, most patients received conditioning with fludarabine (25 mg/m2) and cyclophosphamide (250 mg/m2) for 3 days or bendamustine (90 mg/m2) for 2 days. Of the 81 patients infused, 40% achieved a CR which was maintained in 30% of cases after 3 and 6 months, respectively. The response rates were consistent across disease subgroups. Three patients died due to disease progression; no death was reported due to CTL09 infusion.
Looking at adverse events, CRS occurred in about two thirds of patients, and 15% of patients experienced grade 3 and 8% grade 4 toxicity. 15% of patients received tocilizumab in order to control CRS. Neurological events (of grade 3 or 4) occurred in 12% of cases. Infections of higher grade were described in about one of five patients.
In the TRANSCEND trial patients with refractory DLBCL, but also PMBL, follicular lymphoma of grade 3B and mantle cell lymphoma were enrolled and treated with JCAR017 [55]. All patients received fludarabine (30 mg/m2) and cyclophosphamide (300 mg/m2) for 3 days prior to CAR T-cell infusion. In contrast to the previously described trials, patients in TRANSCEND received transduced CD4-positive and CD8-positive CAR T cells in a predefined 1:1 ratio at different dose levels.
In the DLBCL cohort, 68 patients could be evaluated: 56% of patients achieved a CR, and almost 40% maintained CR at 3 and 6 months follow-up. Remarkably, the rate of CRS was rather low (30%) with only 1% of grade 4 toxicity. Severe neurotoxicity (grades 3–4) occurred in 14% of cases.
To summarize, clinical experience with second-generation CAR T cells used in order to treat refractory DLBCL and other aggressive B-cell malignancies appears very promising. The patients treated with CAR T cells were true poor-risk patients with dismal prognosis whatever salvage treatment would have been administered. The SCHOLAR-1 study attributed a median overall survival of 6 months to such patients [4]. In this poor prognostic setting, treatment with CAR T cells resulted in a CR rate of approximately 50%. Most of the complete remissions were durable although follow-up times are still limited. As an important caveat, it must be taken into account that only those patients surviving rather long periods of time without or with mild chemotherapy were put on CAR T-cell trials. The typical patient suffering from refractory or multiple relapsed aggressive B-cell lymphoma, however, will need immediate and aggressive therapy. It is difficult to understand that many patients on CAR T-cell trials treated with mild and atypical chemotherapy survived time periods up to 145 days before CAR T cells were infused.
The major acute toxicities of CAR T-cell infusion represent the cytokine release syndrome and neurologic events. CRS and neurologic events can be severe to life-threatening and need immediate therapy. Over time, management of CRS and neurologic side effects substantially improved. With the use of tocilizumab and glucocorticoids, both CRS and neurologic complications are mostly reversible, and severe adverse events seem to be observed less frequently.
3.4 Perspectives
CAR T cells hold great promise in clinical settings with very poor prognosis. To further optimize the potential of CAR T cells, the aim must be to investigate what kind of CAR T-cell system is most powerful for which malignancy and how to further boost its therapeutic power. Target selection is one crucial issue. CAR T cells for B-cell neoplasia so far address the antigen CD19. One new approach in order to avoid tumor cell escape is the development of bispecific CAR T cells directed against two antigens, like CD19 and CD20 (Fig. 16.4a) [56]. The infusion of a mixture of different CAR T cells and the creation of T cells with different CARs are possible approaches to prevent tumor escape.

Activation of bispecific CAR T cells. (a) Bispecific CAR T cells can be activated by two different antigens (e.g., CD19 and CD20) and minimize the risk of tumor escape by downregulation of surface proteins. (b) These CAR T cells express two different CARs: one connected to CD3ζ and the other to the costimulatory domain. Only when both CARs bind to their target (e.g., CD19 and CD20), the full T-cell activation is triggered
To further enhance tumor binding, CAR T cells expressing two different CARs directed against different antigens have been constructed. One CAR is linked to CD3ζ, and the other CAR is linked to the costimulatory domain: only when both antigens are bound the full CAR T-cell reaction is triggered (Fig. 16.4b) [57]. With an alternative approach, both antigens must be present on the target cell: a NOTCH receptor binds to the first antigen, gets cleaved within the cell membrane, and translocates into the nucleus to induce expression of the “effector CAR.” Only if the effector CAR binds to the second target, a cytotoxic response occurs [58, 59].
Even if CAR T cells succeed in recognizing the malignant cells, there are multiple inhibitory effects that may hinder efficient tumor lysis. One important player is PD1. Theoretically, the combination of CAR T cells with checkpoint inhibitors (PD1-inhibitors) may enhance antitumor effects. In mouse models, the CAR T-cell efficacy could indeed be boosted by the simultaneous use of PD1 inhibitors [59].
The combination of CAR T cells with small molecules also holds great potential. Pretreatment with ibrutinib may improve expansion of CAR T cells in vitro and in vivo. In mouse models of CLL, the combination of ibrutinib and CAR T cells led to enhanced tumor clearance and survival [60].
The most serious side effects of CAR T cells are the cytokine release syndrome and neurologic events. To better control potentially life-threatening side effects, researchers develop CAR T-cell systems which are transient or can be destroyed on demand. Introduction of suicide genes or transient transduction of CAR T cells via RNA electroporation represents examples on how to create CAR T cells that can be irreversibly destroyed if clinically necessary [61]. Of course, the irreversible depletion of CAR T cells will increase the risk of relapse. Therefore, the next logical step was the development of ON-OFF switch CAR T-cell systems. One established system is a chimeric CAR that dimerizes only in presence of a small molecule. Only the dimerized CAR transduces downstream signaling and hence activation of the T cells (Fig. 16.5) [62]. A further advancement is the development of switchable CAR T cells that are activated by binding to a peptide neo-epitope of a tumor antigen-specific Fab molecule. Not the CAR itself but the infused antibody binds to the tumor surface. In a second step, the CAR T cells bind to the antibody and get activated. This technique leads to a tunable control of CAR T-cell activity. Moreover, different tumor antigens can be addressed with the same CAR T cells by infusing a mixture of different antibodies (Fig. 16.6) [63]. Clinical trials will show to what extent these technologies can further improve the long-term response rates of CAR T cells.

ON-OFF switch CARs dimerize in the presence of a small molecule (ON). Only the dimerized CAR transduces the downstream signaling and hence activation of the T cell

A tumor antigen-specific Fab molecule binds to the tumor target (e.g., CD19), and the CAR is directed vs. the Fab molecule
References
Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematol Am Soc Hematol Educ Program. 2011;2011:498–505.
Coiffier B, Lepage E, Briere J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346(4):235–42.
Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184–90.
Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–8.
Press OW, Appelbaum F, Ledbetter JA, et al. Monoclonal antibody 1F5 (anti-CD20) serotherapy of human B cell lymphomas. Blood. 1987;69(2):584–91.
Reff ME, Carner K, Chambers KS, et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994;83(2):435–45.
Cartron G, Watier H, Golay J, Solal-Celigny P. From the bench to the bedside: ways to improve rituximab efficacy. Blood. 2004;104(9):2635–42.
Polyak MJ, Deans JP. Alanine-170 and proline-172 are critical determinants for extracellular CD20 epitopes; heterogeneity in the fine specificity of CD20 monoclonal antibodies is defined by additional requirements imposed by both amino acid sequence and quaternary structure. Blood. 2002;99(9):3256–62.
Li H, Ayer LM, Lytton J, Deans JP. Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem. 2003;278(43):42427–34.
O’Keefe TL, Williams GT, Davies SL, Neuberger MS. Mice carrying a CD20 gene disruption. Immunogenetics. 1998;48(2):125–32.
Polyak MJ, Tailor SH, Deans JP. Identification of a cytoplasmic region of CD20 required for its redistribution to a detergent-insoluble membrane compartment. J Immunol. 1998;161(7):3242–8.
Cragg MS, Morgan SM, Chan HT, et al. Complement-mediated lysis by anti-CD20 mAb correlates with segregation into lipid rafts. Blood. 2003;101(3):1045–52.
Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171(3):1581–7.
van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115(4):807–11.
van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin Cancer Res. 2006;12(13):4027–35.
Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443–6.
Dall'Ozzo S, Tartas S, Paintaud G, et al. Rituximab-dependent cytotoxicity by natural killer cells: influence of FCGR3A polymorphism on the concentration-effect relationship. Cancer Res. 2004;64(13):4664–9.
Cartron G, Dacheux L, Salles G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754–8.
Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116(6):926–34.
Mossner E, Brunker P, Moser S, et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115(22):4393–402.
Alduaij W, Ivanov A, Honeychurch J, et al. Novel type II anti-CD20 monoclonal antibody (GA101) evokes homotypic adhesion and actin-dependent, lysosome-mediated cell death in B-cell malignancies. Blood. 2011;117(17):4519–29.
Bologna L, Gotti E, Manganini M, et al. Mechanism of action of type II, glycoengineered, anti-CD20 monoclonal antibody GA101 in B-chronic lymphocytic leukemia whole blood assays in comparison with rituximab and alemtuzumab. J Immunol. 2011;186(6):3762–9.
Dalle S, Reslan L, Besseyre de Horts T, et al. Preclinical studies on the mechanism of action and the anti-lymphoma activity of the novel anti-CD20 antibody GA101. Mol Cancer Ther. 2011;10(1):178–85.
Klein C, Lammens A, Schafer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22–33.
Niederfellner G, Lammens A, Mundigl O, et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118(2):358–67.
Morschhauser FA, Cartron G, Thieblemont C, et al. Obinutuzumab (GA101) monotherapy in relapsed/refractory diffuse large b-cell lymphoma or mantle-cell lymphoma: results from the phase II GAUGUIN study. J Clin Oncol. 2013;31(23):2912–9.
Coiffier B, Haioun C, Ketterer N, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood. 1998;92(6):1927–32.
Vitolo U, Trneny M, Belada D, et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J Clin Oncol. 2017;35(31):3529–37.
de Romeuf C, Dutertre CA, Le Garff-Tavernier M, et al. Chronic lymphocytic leukaemia cells are efficiently killed by an anti-CD20 monoclonal antibody selected for improved engagement of FcgammaRIIIA/CD16. Br J Haematol. 2008;140(6):635–43.
Teeling JL, French RR, Cragg MS, et al. Characterization of new human CD20 monoclonal antibodies with potent cytolytic activity against non-Hodgkin lymphomas. Blood. 2004;104(6):1793–800.
Teeling JL, Mackus WJ, Wiegman LJ, et al. The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. J Immunol. 2006;177(1):362–71.
Bleeker WK, Munk ME, Mackus WJ, et al. Estimation of dose requirements for sustained in vivo activity of a therapeutic human anti-CD20 antibody. Br J Haematol. 2008;140(3):303–12.
Du J, Yang H, Guo Y, Ding J. Structure of the Fab fragment of therapeutic antibody Ofatumumab provides insights into the recognition mechanism with CD20. Mol Immunol. 2009;46(11–12):2419–23.
van Imhoff GW, McMillan A, Matasar MJ, et al. Ofatumumab versus rituximab salvage chemoimmunotherapy in relapsed or refractory diffuse large B-cell lymphoma: the ORCHARRD study. J Clin Oncol. 2017;35(5):544–51.
Viardot A, Goebeler ME, Hess G, et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood. 2016;127(11):1410–6.
Ansell SM, Hurvitz SA, Koenig PA, et al. Phase I study of ipilimumab, an anti-CTLA-4 monoclonal antibody, in patients with relapsed and refractory B-cell non-Hodgkin lymphoma. Clin Cancer Res. 2009;15(20):6446–53.
Menter T, Bodmer-Haecki A, Dirnhofer S, Tzankov A. Evaluation of the diagnostic and prognostic value of PDL1 expression in Hodgkin and B-cell lymphomas. Hum Pathol. 2016;54:17–24.
Armand P, Nagler A, Weller EA, et al. Disabling immune tolerance by programmed death-1 blockade with pidilizumab after autologous hematopoietic stem-cell transplantation for diffuse large B-cell lymphoma: results of an international phase II trial. J Clin Oncol. 2013;31(33):4199–206.
Chao MP, Alizadeh AA, Tang C, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142(5):699–713.
Takeda K, Kojima Y, Uno T, et al. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J Immunol. 2010;184(10):5493–501.
Kohrt HE, Houot R, Goldstein MJ, et al. CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117(8):2423–32.
Gopal AK, Bartlett NL, Levy R, et al. A phase I study of PF-05082566 (anti-4-1BB) + rituximab in patients with CD20+ NHL. J Clin Oncol. 2015;33(15_suppl):3004.
Tutt AL, O'Brien L, Hussain A, Crowther GR, French RR, Glennie MJ. T cell immunity to lymphoma following treatment with anti-CD40 monoclonal antibody. J Immunol. 2002;168(6):2720–8.
Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27(6):466–74.
Poirot L, Philip B, Schiffer-Mannioui C, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75(18):3853–64.
Wang K, Wei G, Liu D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp Hematol Oncol. 2012;1(1):36.
Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–71.
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509.
Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–5.
Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111–7.
Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum Interleukin-15 levels. J Clin Oncol. 2017;35(16):1803–13.
Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119(12):2709–20.
Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–54.
Abramson JS, Lia Palomba P, Gordon LI, et al. High durable CR rates in relapsed/refractory (R/R) aggressive B-NHL treated with the CD19-directed CAR T cell product JCAR017 (TRANSCEND NHL 001): defined composition allows for dose-finding and definition of pivotal cohort 59th annual meeting & exposition of the American Society of Hematology. Atlanta. 2017.
Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498–508.
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–5.
Morsut L, Roybal KT, Xiong X, et al. Engineering customized cell sensing and response behaviors using synthetic Notch receptors. Cell. 2016;164(4):780–91.
John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology. 2013;2(10):e26286.
Fraietta JA, Beckwith KA, Patel PR, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127(9):1117–27.
Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–83.
Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350(6258):aab4077.
Rodgers DT, Mazagova M, Hampton EN, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(4):E459–68.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cartron, G., Frontzek, F., Schmitz, N. (2019). Immunological Approaches. In: Lenz, G., Salles, G. (eds) Aggressive Lymphomas. Hematologic Malignancies. Springer, Cham. https://doi.org/10.1007/978-3-030-00362-3_16
Download citation
DOI: https://doi.org/10.1007/978-3-030-00362-3_16
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-00361-6
Online ISBN: 978-3-030-00362-3
eBook Packages: MedicineMedicine (R0)