
Abstract
T-cell lymphomas (TCLs) represent a heterogeneous group of lymphoid neoplasms. With the exception of ALK+ anaplastic large cell lymphoma (ALCL), early stage mycosis fungoides (MF) and T-large granular lymphocytic (LGL) leukemia, TCL respond poorly to conventional chemotherapy and have a poor prognosis. Malignant T-cells express a number of potential targets for immunotherapy, which are described in detail in this chapter. Monoclonal antibodies (mAbs) against a number of T-cell antigens have shown significant clinical activity in a variety of TCL and represent an important addition to the therapeutic toolbox, with durable remissions occurring in a subset of patients. The main toxicity of anti-T-cell mAbs is immune suppression, due to the fact that subsets of normal T-cells are also depleted during therapy. The degree and duration of T-cell lymphopenia and immune suppression produced by anti-T-cell mAbs is variable, but to date none has been shown to be completely free of these side effects. Since mAbs as single agents are not curative in TCL, they should be integrated in combination with other biological or chemotherapeutic agents, either simultaneously or sequentially. Maintenance therapy or retreatment based on the monitoring of minimal residual disease should also be considered although antigen mutation or modulation may limit repeated administration activity. Studies involving mAbs covalently bound to radioisotope or toxins to enhance their ability to destroy tumor cells show that immunoconjugates are more effective than “naked” mAbs. This multimodality strategy, together with a personalized, stage-specific approach represents the future in the management of TCL patients. Based on the spectrum of investigational mAbs now available and the activity shown, there is no doubt that mAbs therapy of TCL will continue to be an area of active interest expanding clinical trials. Long-term safety and recovery of cell-mediated immunity should be a key clinical endpoint in the ultimate risk-benefit assessment of these agents.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Overall Response Rate
- Graft Versus Host Disease
- Anaplastic Large Cell Lymphoma
- Mycosis Fungoides
- Brentuximab Vedotin
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Although the use of antibodies as tumor-targeting agents dates back to 1953 when Pressman and Korngold [1] showed that radiolabeled rabbit antisera could specifically target mouse osteogenic sarcoma cells, not until 1975, with the development of the hybridoma technology [2], adequate quantities of murine monoclonal antibodies (mAbs) became available for clinical use. These early trials demonstrated that murine mAbs had some measure of antitumor activity in hematological malignancies but also highlighted the limiting effect of the human-anti-mouse humoral immune responses on multiple mAb administrations. Despite the simplicity of the concept (antibody–antigen interaction), researchers had to confront a number of technical difficulties, including the selection of a proper and stable target, efficient delivery, and immunogenicity due to the murine components of the mAbs [3]. In 1997 the anti-CD20 antibody rituximab became the first recombinant chimeric mAb approved by the US Food and Drug Administration (FDA) for use in the treatment of cancer, demonstrating very limited immunogenicity and leading to the explosion of the field on mAb-based therapeutics. The methodology for the manufacturing of mAbs for clinical use has evolved substantially over the past 15 years, allowing the creation of “designer” antibody biopharmaceuticals that have fully human sequence and combine the desired antigen-binding specificity with unique biodistribution and pharmacodynamic properties. The nomenclature of mAb reflects this diversity. While the suffix mab defines the product as a monoclonal antibody, the letter or syllable in front of the mab reveals the antibody’s source as murine (-omab), chimeric (-ximab), humanized (-zumab), or fully human (-umab) [4]. Thus edrocolomab is a murine anti-EpCAM antibody, galiximab is a chimeric anti-CD80 antibody, alemtuzumab is a humanized anti-CD52 antibody, and ofatumumab is a fully human anti-CD20 antibody.
One of the key goals in the clinical development of mAbs for clinical use is the identification of specific surface antigens—defined according to standardized nomenclature as clusters of differentiation (CD)—whose expression is ideally restricted to the malignant cells. Unfortunately, such degree of specificity is rarely achieved and many clinically relevant antigens represent essential components of vital activation, survival, and trafficking pathways for both malignant and normal cells. Therefore, the fact that cell surface antigens expressed on lymphoma cells are virtually always present on their normal counterparts continues to represent a significant limit for the clinical application of these agents. mAbs induce tumor cell death by blocking survival pathways (direct apoptosis) along with other immunologic mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). mAbs have been used as single agent therapy or, in the attempt to enhance their ability to destroy tumor cells, in combination with chemotherapy, other antibodies or conjugated either to radioisotopes or to immunotoxins. Based on the activity in hematologic malignancies and their safety profile, mAbs are now among the most frequently used biopharmaceutical agents.
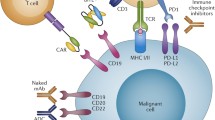
The development of mAbs for the treatment of T-cell lymphomas (TCLs) is rooted in the early functional characterization of the T-cell receptor and other surface antigens in human T-cells [3–6], and in subsequent studies aimed at the discovery of novel immune suppressive drug for use in human transplantation. The first mAb approved for clinical use was the anti-CD3 murine antibody OKT3 (muromonab), approved in 1986 for the prevention of kidney transplant rejection. Clinical experience with OKT3 showed that the drug was potently immune suppressive, achieving the desired effect against organ rejection, but also revealed a significantly higher risk of development of post-transplant lymphoproliferative disorder (PTLD), compared to conventional antirejection drugs such as antimetabolites and calcineurin inhibitors. The progressive discovery and characterization of surface markers such as CD2, CD4, CD5, CD6, CD25, CD30, and CD52 that are expressed on various functional subsets of T-cells, led to the development of corresponding murine or chimeric mAbs for clinical use. Although early investigations of these mAbs were focused on prevention of graft versus host disease (GVHD) and solid organ allograft rejection, as well as the treatment of severe autoimmune diseases, pilot studies in select subtypes of TCL we also conducted, with encouraging results. Both early and later studies of mAb-based therapy in TCL, however, have so far failed to achieve the efficacy, flexibility of use in combination, and safety profile of rituximab. Most of the anti-T-cell mAbs in clinical development or approved for TCL interfere with T-cell activation and trafficking and result in severe and prolonged T-cell depletion. Many have been associated with reactivation of latent herperviruses, such as Epstein-Barr virus (EBV) and cytomegalovirus (CMV), and a broad spectrum of bacterial and fungal opportunistic infections. Thus, in the absence of truly tumor-specific markers, the difficulty of balancing an efficient kill of malignant T-cells with an adequate preservation of normal cell-mediated immunity remains the most challenging obstacle to the development of active and safe mAbs for the treatment of TCL. Presently a number of mAbs targeting T-cell surface antigens are being evaluated in clinical trials (Table 14.1) and the purpose of this chapter is to summarize the present and potentials roles for mAbs in the treatment of TCL.
CD2 Antigen
CD2 is an approximately 50 KD transmembrane glycoprotein expressed on dendritic cells, natural killer (NK) cells, thymocytes, and mature T-cells [6–9] with much higher expression on activated T-cells than on resting T-cells [10]. Although the specificity for T-cell activation resides in the TCR/CD3 complex, the interaction of additional accessory molecules such as CD2, CD28, and lymphocyte functional antigen-1 (LFA-1) with ligands on the antigen presenting cells (APCs), is required for complete T-cell activation to occur [11]. CD2 acts as an adhesion molecule [12] through its extracellular region which has two Ig superfamily domains, the first one binds CD58 (LFA-3) [13] and the second one binds CD59 [14], helping to localize the T-cells in proximity of APCs [11, 15–17]. CD2 acts also as a signaling molecule through its large intra-cytoplasmatic domain: as a result of CD2–CD58 interaction, the CD2 intra-cytoplasmatic domain physically associates with the Wiskott-Aldrich syndrome protein [18] and the tyrosine kinase p56lck [19, 20] resulting in the initiation of the phospholipase C pathway [21, 22], in actin polymerization with further strengthening of intercellular adhesion, in the production of IL-2 and, ultimately, into T-cell activation and proliferation [23].
CD2-Targeted Therapy
The limited expression of CD2 restricted to mature T-cells, the expression of CD2 on malignant T-cells, combined with preliminary data showing that engagement of CD2 by mAbs can directly trigger apoptosis [24], provided the scientific rationale for their use in immunotherapy for TCL. A number of mAbs directed against CD2 have demonstrated the ability to inhibit T-cell activation. In particular, the rat mAb BTI-322 (MedImmune, Inc.) has been used in the prevention of allograft rejection [25] and in the therapy of GVHD [26]. Given the immunosuppressive properties of BTI-322, a humanized genetically engineered version of this molecule was developed for clinical application (Siplizumab, BioTransplant, Inc.). Siplizumab (MEDI-507) is a humanized IgG1κ monoclonal antibody initially studied in murine models of adult T-cell leukemia (ATL) [27]: severe combined immune deficiency (SCID) mice engrafted with human HTLV-1 positive T-cell leukemia cells treated with MEDI-507 survived significantly longer when compared either with mice treated with a humanized mAb direct toward CD25 or with phosphate-buffered saline (PBS) (control group). Based on this preclinical data and on the data obtained with its use in the prevention in the therapy of transplant rejection and autoimmune disease [28], phase I trials were conducted in patients with CD2+ lymphomas/leukemias [29, 30]. In the 2006 report, 16 patients diagnosed with peripheral T-cell lymphoma (PTCL) (n = 9), cutaneous T-cell lymphoma (CTCL) (n = 6), and natural killer (NK)-large granular lymphocytic (NK-LGL) leukemia (n = 1) were treated with biweekly infusion of escalating doses of intravenous (IV) siplizumab (range 0.7–4.8 mg/kg). Two responses were observed, one partial response (PR) in the NK-LGL patient and one complete response (CR) in a PTCL patient, both treated at 3.4 mg/kg. Dose-limiting toxicities (DLT) included one case of erythematous confluent dermatitis and one case of pulmonary edema, no maximum tolerated dose (MTD) was established. In the 2009 report, 29 patients with T-cell malignancies received escalating doses of IV siplizumab (range 0.2–4.8 mg/kg), either with a weekly schedule or biweekly schedule. Although the initial responses were encouraging, 4 patients (13.7%) (3 of 7 patients treated with the weekly schedule and 1 of 22 treated with biweekly schedule) developed an EBV-lymphoproliferative disease (LPD) and the trial was stopped. The weekly administration of siplizumab decreased the CD2 expressing cells to a greater extent than the biweekly schedule highlighting the importance of NK/T cells depletion in conjunction with no effect on the B cell compartment in the pathogenesis of EBV-LPD.
CD3 Antigen
The TCR heterodimer is associated with the polypeptide dimers of the CD3 complex which comprises five invariant polypeptide chains: γ, δ, ε, ζ, and η [31]. The CD3 complex stabilizes the TCR and participates in signal transduction through its extracellular (γ, δ, ε), transmembrane and intra-cytoplasmatic domain [32, 33]. Engagement of the TCR by antigen or mAbs anti-CD3 induces phosphorylation of CD3 γ, δ, ε, and ζ chains and internalization of the TCR. The outcome of these interactions, including activation and apoptosis, is influenced by the dynamic interactions between TCR and peptide-MHC complexes and, specifically, depends on slight differences in terms of affinity and dissociation rates of the antigen/TCR complex.
CD3-Targeted Therapy
The clinical utility of the first generation of mAbs specific for CD3 (such as muromonab-CD3, also called Orthoclone OKT3, Ortho Biotech, Inc.) has been limited by several drawbacks related to their composition. The first was the induction of neutralizing anti-mouse Ab response which reduces the re-treatment efficacy [34–36]. The second is a syndrome of adverse events which includes flu-like symptoms, respiratory distress, hypotension, and hypoglycemia [36]. This first generation of mAbs can induce T-cell activation through cross-linking of the CD3/TCR complex (via the F(ab′)2 arms) and Fcγ receptor (FcγR) on the surface of accessory cells (via the Fc region), with consequent release of cytokine such as tumor necrosis factor α (TNFα), interleukin-2 (IL-2), and interferon γ (IFN-γ) [37, 38]. Recently, a humanized, non-FcγR-binding, anti-CD3 mAb (visilizumab, PDL BioPharma Inc.) directed against the ε invariant chain of the CD3/TCR complex, has been evaluated for the treatment of steroid refractory acute GVHD [39]. This mAb retains the specificity for CD3 but has an engineered and modified Fc portion that fails to trigger T-cell activation and therefore is associated with significantly less cytokine release. Despite this significant structural improvement, to date, there are no published reports of the use of mAbs specific for CD3 in TCL. A phase I, multiple dose escalation trial of IV humanized anti-CD3 mAb (HuM291) in patients with CD3+ TCL has been recently completed at Stanford University (clinical trial number NCT00006009); however, the results are not yet available.
CD4 Antigen
CD4 is a 55 kD single chain glycoprotein containing four immunoglobulin-like domains that project from the T-cell surface, a hydrophobic transmembrane domain and a long cytoplasmic tail [40]. In association with the TCR, CD4 acts as a co-receptor for the major histocompatibility complex (MHC) class II molecules [41]. The main physiological importance of this surface glycoprotein is signaling which takes place through its cytoplasmic tail. The intracytoplasmic region of CD4 is constitutively and non-covalently associated with a src-like tyrosine kinase (p56lck) that initiates the transduction cascade following antigen binding [42]. CD4 is normally expressed on helper/regulatory T-cells, and, at a lower level, on monocytes, macrophages, and dendritic cells [40]. CD4 is also expressed on the malignant cells of PTCL and CTCL patients [43]. The fact that CD4 is expressed only on a subset of normal T-cells, it is brightly expressed on malignant cells from CTCL patients, it is present in all stages of disease with no down-modulation during disease progression and it is involved in cell signaling, has provided the rationale for targeting CD4 with mAbs.
CD4-Targeted Therapy
The therapeutic activity of mAbs specific for CD4, in their murine and humanized variant, has been evaluated in human inflammatory diseases such as rheumatoid arthritis and psoriasis [44–46] as well as in CTCL [43, 47, 48]. In an early study [47], seven previously treated mycosis fungoides (MF) patients were treated with a chimeric murine antibody composed of the IgG1κ human constant regions and the mouse variable regions directed against CD4 (SK3/anti-Leu3a; Becton-Dickinson, Inc.). Patients received IV doses of 10, 20, 40, or 80 mg twice a week for 3 consecutive weeks. At the 80 mg dose, the antibody was detected in skin lesions and also coating circulating CD4+ T-cells in the peripheral blood, however, no significant depletion of CD4+ cells was observed. In a second phase I study [48], 8 previously treated MF patients received a single IV dose (50, 100 or 200 mg) of a different chimeric murine anti-CD4 mAb directed to a distinct epitope on CD4 (cM-T412; Centocor, Inc.). Following the antibody infusion there was a depletion of peripheral blood CD4+ cells in 7 of the 8 patients. The overall response rate (ORR) was 88% with 5 patients achieving a PR and 2 patients achieving a minor response (MR). The time to progression (TTP) ranged from 6 to 52 weeks, with an average of 25 weeks. More recently, HuMax-CD4 (zanolimumab, Genmab, Inc.), a fully human anti-CD4 mAb, isolated from transgenic mice as a hybridoma clone and subsequently expressed in Chinese hamster ovary cells, was able to induce significant T-cell depletion in repeated dosing and was found to be safe and well tolerated in human inflammatory diseases [45, 46]. Based on its safe profile and the preliminary encouraging results obtained with murine anti-CD4 mAbs, the therapeutic activity of zanolimumab was also evaluated in two separate but otherwise identical phase 2 clinical trials in refractory CTCL [43]. In the first of the two studies, 27 patients with early-stage MF received weekly IV infusion of zanolimumab at either 280 mg (13 patients) or 560 mg (14 patients) for a total of 16 weeks. In the second study, 11 patients with advanced-stage treatment refractory MF and 9 patients with Sezary syndrome (SS) were treated with weekly IV infusion of zanolimumab at either 280 mg (7 and 4 patients, respectively) or 960 mg (4 and 5 patients, respectively). Overall, 13 of 38 patients (34.2%) with MF and 2 of 9 patients (22.2%) with SS obtained an objective response to zanolimumab (MF: 1 CR, 3 CCRs, and 9 PRs; SS: 2 PRs). 56% of the MF patients (7 of 14 at 560 mg and 3 of 4 at 980 mg) treated at the high-dose levels achieved objective response compared with only 15% at the 280 mg dose (3 of 20). In patients with SS, the response rate was 1 in 4 at the 280 mg dose and 1 in 5 at the 980 mg dose. In patients with MF, high-dose treatment resulted in an earlier time to response with 9 of 10 responses achieved within 8 weeks (median time to response 8 weeks, range 2–12 weeks) and more durable responses (median duration of response 81 weeks, range 8–91 weeks) compared with low dose treatment (three responses obtained at 4, 8, and 12 weeks; duration of response 12, 13, and 24 weeks). In patients with SS, the two responses lasted 8 weeks (280 mg) and 61 weeks (980 mg). Overall, treatment was well tolerated with infections and eczematous dermatitis being the most frequent adverse events observed. This antibody was designed to prevent the interaction between the CD4 receptor and the MHC class II molecule, thereby interfering with T-cell activation, and, recently, the mechanism through which zanolimumab may inactivate or delete T-cells has been described [49]. Ligation of CD4 by zanolimumab inhibits the necessary p56lck-mediated co-stimulation by uncoupling this kinase from the TCR and allowing this enzyme to transmit an inhibitory signal via Dok-1 and SHIP-1 inhibitory molecules that may also reduce AKT activity. Secondly, zanolimumab induces potent ADCC with no role played by CDC and, lastly, it may act by down-modulating CD4 itself. Despite its promising profile in terms of therapeutic activity and safety, future developments for zanolimumab and its clinical applications are on hold due to pharmaceutical company’s decision.
CD25 Antigen
Interleukin-2 (IL-2) is a potent immunomodulatory cytokine whose function is the activation of T- and B-lymphocytes, NK cells, and macrophages [50]. Three polypeptide chains (α, β, and γ) are non-covalently associated to form the high affinity interleukin-2 receptor (IL-2R) [51–53]. The α (CD25 or p55) and β (CD122 or p75) chains are involved in binding IL-2, while signal transduction which involves Janus kinase-1 (Jak-1), Jak-3, and STAT-5 pathways, is carried out by the γ (CD132 or p64) chain, along with the β subunit [54–56]. CD25 is a 55 kD, heavily glycosylated, integral membrane protein which, by itself, functions as a low affinity IL-2R. CD25, initially defined using the murine mAb anti-Tac [57], is a polypeptide composed of 272 aminoacids with an extracellular, transmembrane, and a very short intracellular domain. CD25 is dimly expressed on normal, resting peripheral blood T-cells, however is rapidly upregulated following antigen or cytokines stimulation [55]. Among T-cell malignancies, CD25 is strongly expressed by ATL and ALCL [55], highly variable expressed on CTCL [58]. It has been shown that CD25 expression varies depending on tissue site from CTCL patients: CD25 has been found to be highly expressed in epidermis, down-regulated in dermis, and extensively down-regulated in lymph nodes, suggesting that the microenvironment may play a role in affecting CD25 expression [58]. It has also been shown that CD25 expression correlates with advanced stage (T status) and histological grade (large cell transformation) of CTCL, suggesting a role as prognostic marker for this antigen [59]. Recently, soluble IL2-R has been identified as a reliable marker of disease activity in patients with ATL [60].
CD25-Targeted Therapy
Several studies have established the importance of CD25 as a valuable target for immunotherapy [61–63]. A number of mAbs specific for CD25 have been evaluated in several T-cell mediated disorders including prevention of rejection of transplanted organs [64], noninfectious uveitis [65], multiple sclerosis [66], and HTLV-1-associated disorders [67]. In 1997, daclizumab, a therapeutical humanized mAb specific for CD25 (zenapax, Hoffmann-La Roche) has been approved by the FDA for use in the prevention of renal allograft rejection [68]. Also among T-cell malignancies, various forms of CD25-specific mAbs have been tested in a preclinical and clinical setting. These include the murine mAb anti-Tac [57], daclizumab [69, 70], and 7G7/B6 mAb that targets CD25 at an epitope other than the IL-2 and anti-Tac binding sites [71]. These mAbs significantly prolonged the survival of MET-1 mice (murine model of ATL), however cure was not achieved [72]. For this reason, daclizumab has been combined with flavopiridol in the MET-1 murine model in which a marked synergy between these two drugs has been observed [73]. The clinical use of the murine mAbs has been limited due to their immunogenicity associated with rapid generation of neutralizing anti-mouse Ab, short in vivo survival and modest ADCC-mediated cell death due to the reduced binding to human Fc receptors [74]. However also daclizumab, as many other unmodified mAbs, has shown only modest activity as single cytotoxic agent in ATL patients. To overcome these limitations, daclizumab has been employed as a carrier of cytotoxic agents such as radionuclides or toxins. In a phase I/II trial, 18 patients with ATL were treated with 55 doses of 90Y-labeled anti-Tac mAb involving 5–15 Ci per patient [75]. Ten of the evaluable patients responded to treatment, with 8 patients achieving a PR and 2a CR. Hematologic toxicity was the limiting side effect. In an alternative approach, the antitumor activity of the anti-Tac mAb conjugated with a truncated Pseudomonas exotoxin (anti-Tac (Fv)-PE38 (LMB-2)) was evaluated in a phase I/II trial [76] in which 35 patients with a CD25+ lymphoma/leukemia were enrolled. Among these patients, 1 had CTCL/SS, 3 had PTCL unspecified and 2 ATL. Patients received escalating dose of LMB-2 on alternate days for 3 doses (1 cycle), with a starting dose of 2 μg/kg administered IV, every 3 weeks. Of the 6 patients with T-cell malignancies, 3 achieved a PR (1 with CTCL/SS, 1 with PTCL, 1 with ATL). Responding patients experienced rapid reductions of circulating malignant cells, improvement in skin lesions, and regression of lymphomatous masses and splenomegaly; however, all responses were of short duration.
CD122 Antigen
CD122 or p75 represents the β subunit shared by the heterotrimeric IL-2 and IL-15 receptors [77]. IL-15 is produced by macrophages, dendritic cells, and other non-lymphoid cells, it mainly works inducing T-cells and NK-cells maturation, enhancing the survival of CD8+ cytotoxic lymphocytes and stimulating the production of TNFα, IL-1β, and other cytokines [78]. Dysregulation of IL-15 and its receptor have been reported in CTCL, acute lymphoblastic leukemia (ALL), ATL, and T-LGL leukemia [79–81].
CD122-Targeted Therapy
In a phase I trial, 12 patients with T-LGL leukemia received escalating dose of Mik-β1 murine mAb, every 3 days, with a starting dose of 0.5 mg/kg, administered IV [81]. Greater than 95% saturation of CD122 was achieved in all patients; however, no responses in terms of decreases in T-LGL cell count were observed. A phase I safety and pharmacokinetic study of the humanized form of Mik-β1 in T-LGL leukemia is now underway.
CD26 Antigen
CD26 is a 110 kD surface glycoprotein expressed by a variety of cells including epithelial cells, T-lymphocytes, and NK cells [82]. CD26 possesses an intrinsic dipeptidyl peptidase IV (DPPIV) activity in its extracellular domain [83] and plays an important role in T-cell activation [82]. CD26 expression is tightly regulated on T-cells and it is significantly enhanced after activation [84]. CD26 is capable of providing a potent costimulatory signal which can induce several activation pathways leading to proliferation and cytokines production [82]. However the mechanism of costimulation remains unclear since the cytoplasmic domain consists of only six aminoacids and lacks a phosphorylation site, leading to the conclusion that CD26 must interact with other molecules to exert this function. CD26, through its peptidase activity, can also cleave certain chemokines involved in T-cell function and monocytes function, including RANTES and LD78β [85, 86]; it has also been shown that CD26 regulates adenosine deaminase surface expression with this complex perhaps playing a role in regulating immune function [87].
CD26-Targeted Therapy
Among TCL, CD26 is absent or dimly expressed in MF/SS [88], variably expressed in T-LGL [89], and expressed at high levels in γδ hepatosplenic and ALCL [90]. An mAb specific for CD26 (1F7) has shown to have in vitro and in vivo antitumor effect in human CD30+ ALCL cell lines by inducing G1–S cell cycle arrest and enhancement of p21Cip1 expression [91]. It has also been shown that CD26 is a marker of aggressive clinical behavior and poor prognosis for T-LGL [89] and T-cell lymphoblastic leukemia/lymphoma [92]. A phase I trial, involving a humanized anti-CD26 Mab, will be conducted in relapsed/refractory patients with CD26 positive tumors, including TCL, in the near future (Dr. NH Dang, Nevada Cancer Institute).
CD30 Antigen
CD30 is a 105–120 kD type I transmembrane glycoprotein and a member of the tumor necrosis factor (TNF) receptor superfamily [93]. The mature CD30 comprises 577 amino acids, with a 365 amino acids extracytoplasmatic domain physiologically bound by CD30 ligand (CD153), a 24 amino acids transmembrane domain and a 188 amino acids intracytoplasmatic domain which regulates signaling transduction following receptor binding through its serine/threonine phosphorylation sites [94]. In normal tissues, CD30 expression is restricted to activated B- and T-lymphocytes [95]. In the immune system, CD30 functions as a costimulatory molecule in the presence of TCR stimulation, it activates T-cells production of cytokines such as IL-2, TNF, and IFNγ and it appears to be involved in the negative selection of autoreactive lymphocytes [96]. In tumor cells, CD30 was originally identified on the surface of Reed-Sternberg cells [97]; however, it has been shown to be highly expressed also on ALCL tumor cells [98] and can be expressed at variable levels on CTCL cells [99]. A cleaved form of CD30 (sCD30) can be found in the plasma of CD30+ lymphoma patients as a result of cleavage by TNFα-converting enzyme, a zinc metalloproteinase, with serum levels of sCD30 correlating with neoplastic activity, tumor burden, response to treatment, and time to treatment failure in ALCL [100].
CD30-Targeted Therapy
Although its function has not been completely clarified, CD30-mediated signaling has been implicated in both lymphocytes death and proliferation, therefore a number of different mAbs targeting different epitopes of CD30 have been developed [101–107]. It is even more interesting that the same antibody (M67) has showed opposite effect in ALCL cell lines and Hodgkin’s disease (HD) cell lines [108]. Specifically, M67 was able to induce enhanced apoptosis in ALCL cell lines; however, it had no effects on HD cell lines. CD30 binding has been associated with NF-κB activation [109] and the difference in outcome between ALCL and HD cells following anti-CD30 treatment was attributed to the inability of ALCL cells to activate NF-κB [110], in contrast with HD cells which constitutively express NF-κB [111]. Chimeric anti-CD30 mAbs have also been tested in murine xenograft model of ALCL in which treatment with these mAbs was able to significantly prolong survival of mice bearing chemotherapy resistant human ALCL [112, 113]. In the attempt to improve its in vitro and in vivo antitumor activity, Ber-H2, an mAb specific for CD30, has been conjugated with the plant ribosome-inactivating protein saporin and showed promising initial results [114]. Based on these preclinical data, mAbs targeting CD30 have been evaluated in phase I/II clinical trials. In a phase I/II trial, MDX-060 (Medarex, Inc.), a fully human anti-CD30 mAb, was administered at doses up to 15 mg/kg to 21 patients (16 with HD, 3 with ALCL, and 2 with CD30+ TCL unspecified) [115]. Adverse events were common but primarily mild or moderate in intensity and an MTD was not defined. In phase II, an additional 51 patients were treated, 4 of which with ALCL. Of the 7 patients with ALCL, 2 (28%), with a predominantly skin disease, achieved a CR. Both CR lasted more than 1 year with both patients receiving additional cycles of therapy. In another phase I trial, SGN-30 (Seattle Genetics, Inc.), a chimeric anti-CD30 mAb, was administered at four dose levels (2, 4, 8, or 12 mg/kg) on a weekly schedule for 6 consecutive weeks, to 24 patients with refractory lymphoma (21 with HD and 3 with CD30+ NHL) [116]. Antibody treatment was well tolerated and, although antitumor activity was not an objective of this trial, of the 3 patients with CD30+ NHL, 1 patient (33%) with a history of cutaneous ALCL achieved a CR and 2 patients achieved stable disease (SD). Forero-Torres et al. [117] recently reported on 79 patients, 38 with refractory/relapsed HD and 41 with systemic ALCL (35 with ALK− ALCL), treated in a multicenter phase II trial with SGN-30 at a dose of 6 mg/kg for 6 weeks. After the first 24 patients were enrolled, the dose was escalated to 12 mg/kg per week. Overall the treatment was well tolerated, adverse events were common but primary mild or moderate. In the ALCL group, 2 patients achieved a CR and 6 patients a PR for an ORR of 21% and with a response duration ranging from 27 to 1,460+ days. Duvic et al. [118] recently reported the results from another multicenter phase II trial in which SGN-30 was administered to patients with CD30+ lymphomas. Specifically, 5 patients with primary cutaneous ALCL (pc-ALCL) and 1 patient with multiple clinical diagnosis (mixed CD30+ lesions), received 1 dose of IV SGN-30 at 4 mg/kg every 3 weeks for up to six doses. Six patients with pc-ALCL, 3 with lymphomatoid papulosis (LyP), 3 with transformed MF, and 5 with multiple clinical diagnosis (mixed CD30+ lesions), received 1 dose of IV SGN-30 at 12 mg/kg every 3 weeks for up to six doses. Eligible patients could receive two additional courses. Overall the treatment was well tolerated but adverse events were common, 15 patients (65%) experienced at least 1 adverse event with fatigue being the most common. 10 patients (43%) achieved a CR (6 with pc-ALCL, 1 with LyP, and 3 with mixed CD30+ lesions) and 6 patients (26%) achieved a PR (3 with pc-ALCL, 1 with LyP, 1 with MF, and 1 with mixed CD30+ lesions) for an ORR of 87%. However the responses were of short duration (the overall median duration of objective response, CR + PR, was 84 days, range 1–238). Overall, anti-CD30 mAbs have shown an acceptable safety profile, however, in the majority of studies, anti-CD30 mAbs as single agents showed only modest antitumor activity in most CD30+ TCL, suggesting combination strategies as an attractive alternative option. Phase I/II trials with SGN-30 either in combination with chemotherapy or conjugated with drugs/toxins are now ongoing in pretreated patients with CD30+ hematologic malignancies.
Better efficacy with CD30-targeting agents has been observed with brentuximab vedotin (SGN-35, Adcetris, Seattle Genetics, Inc), an antibody-drug conjugate which consists of the anti-CD30 monoclonal antibody cAC10 (SGN-30) conjugated with the cytotoxic agent monomethyl auristatin E (MMAE) via a protease-cleavable linker [119, 120]. Brentuximab vedotin was recently approved by the US FDA in August 2011 for the treatment of relapsed or refractory Hodgkin’s lymphoma (HL) and ALCL. In the initial phase I dose-escalation study [121], brentuximab vedotin was administered intravenously on days 1, 8, and 15, of each 28-day cycle at doses ranging from 0.4 to 1.4 mg/kg. Forty-four patients were enrolled: 38 with Hodgkin’s lymphoma, 5 with systemic ALCL, and 1 with PTCL. Doses were escalated in increments of 0.2 mg/kg until DLT was observed. Antitumor assessments were carried out every two cycles. The MTD was 1.2 mg/kg. The most common adverse events were peripheral sensory neuropathy, fatigue, nausea, diarrhea, arthralgia, and fever. The majority of events were mild to moderate in severity. Tumor regression occurred in 85% of patients and the overall objective response rate was 59% (n = 24), with 34% (n = 14) complete remissions. The median duration of response was not reached at a median follow-up of 45 weeks on study. FDA approval was based on data from two pivotal, open-label clinical trials independent review of response rate. Brentuximab vedotin was administered intravenously at a dose of 1.8 mg/kg over the course of 30 min once every 3 weeks. Results from the Hodgkin study (n = 102) showed an ORR of 73% (95% confidence interval [CI], 65–83%), including 32% with complete remission (95% CI, 23–42%). Response duration averaged 6.7 months (range, 1.3–21.9+ months). Data from the ALCL study (n = 58) revealed an 86% response rate (95% CI, 77–95%), with 57% complete remission rate (95% CI, 44–70%). Median response duration was 12.6 months (range, 0.1–15.9+ months) [122]. Based on these data, brentuximab vedotin represents a welcome addition to the therapeutic tool box for CD30+ lymphomas, including ALCL and HL, with high response rates and manageable toxicity. While the drug appears to be safe on early follow up, more recently the FDA issued a warning to health care professionals due to three cases of progressive multifocal leukoencephalopathy (PML), and a new boxed warning highlighting this risk has been added to the drug label [123]. In addition, a contraindication was added, warning against the use of brentuximab with the cancer drug bleomycin because of the increased risk for pulmonary toxicity.
CD52 Antigen
CD52 is a 21–28 kD, glycosylphosphatidylinositol (GPI)-anchored, heavily glycosilated, protein, encoded by a gene on human chromosome 1 [124]. The peptide component of CD52 is represented by a 12 aminoacids scaffold, which undergoes significant posttranslational modification [125]. Attempts to identify a ligand have not been successful and the physiologic function of CD52 remains to be clarified: it may mediate a variety of biological effects such as signal transduction including promotion of cell–cell adhesion and protection of the cell from environmental insult, however, CD52 knock-outs or congenitally CD52-deficient animals have not been described. Relatively recently, it has also been suggested that CD52 may contribute to the activation of CD4+ regulatory T-cells [126]. CD52 is highly expressed on membrane lipids raft of all B and T lymphocytes at most stages of differentiation (except plasma cells), as well as on monocytes, macrophages, eosinophils, NK cells, and dendritic cells [124, 127–130]. The antigen is also found in the male reproductive tract, where it is strongly expressed on epithelial cells lining the epididymis, vas deferens, and seminal vesicle [131]. Notably, CD52 is not expressed on hematopoietic stem cells, erythrocytes, and platelets [132]; however, it has been found on a subpopulation of CD34+CD38+ cells which are believed to represent lymphocyte-committed progenitors [133]. CD52 is also variably expressed on subsets of tumor cells, at a particular high density on T-prolymphocytic leukemia (T-PLL) cells followed by B-CLL, hairy cell leukemia (HCL), ALL, and NHL, including TCL [128, 134, 135]. However, in contrast with B-cell malignancies, there is a great variability in terms of CD52 expression in TCL. Currently the expression of CD52 is best assessed by flow cytometry [136], in fact, a reliable assay for the detection of CD52 on paraffin-embedded, formalin-fixed tissue is not available and data on CD52 expression in various subtypes of TCL are conflicting, due to inconsistent reproducibility [134, 135, 137, 138]. Alemtuzumab has shown encouraging activity in T-cell malignancies [139], however, the variable expression of CD52 among TCL and the profound lymphopenia associated with the use of this mAb, have partially limited its use in TCL.
CD52-Targeted Therapy
Campath-1H (alemtuzumab [U.S.], mabCampath [E.U.], Genzyme, and Bayer HealthCare Pharmaceuticals Inc.) is a humanized IgG1κ mAb directed against the human CD52 antigen that is FDA approved for the management of patients with pretreated [140] and untreated B-CLL [141]. The CD52 epitope recognized by alemtuzumab corresponds to the last three aminoacids of the peptide scaffold and part of the GPI anchor [139]. The mechanisms underlying the therapeutic effect of alemtuzumab in lymphoid malignancies have not been well characterized. The spectrum of biologic effects on tumor cells is heterogeneous and the dominant mechanism of antitumor activity may differ from disease to disease. In vitro and in vivo evidence show that alemtuzumab induces CDC, ADCC, and direct apoptosis in B- and T-cell malignancies [125, 142–147]. The pharmacokinetics (PK) of alemtuzumab has been studied in B-CLL, using the approved schedule consisting of a 2 h IV infusion at a starting dose of 3 mg on day 1, 10 mg on day 2 up to the target dose of 30 mg, three times per week for up to 12 weeks [148]. Considering the fact that remarkable “first-dose” reactions consisting in fever, vomiting, rigors, skin rash, dyspnea, and hypotension have been noted in the majority of patients treated with the IV route [149], Hale et al. [148] compared blood concentrations of alemtuzumab in B-CLL patients after IV and subcutaneous (SC) dosing. SC administration was not associated with “first-dose” reactions and similar peak drug concentrations were achieved; however, a higher cumulative dose of SC alemtuzumab was required to reach concentrations similar to the IV administration. PK data with alemtuzumab (IV or SC) in T-cell malignancies are not available. Multiple studies have shown single-agent alemtuzumab to be active in T-cell malignancies (T-PLL: [150]; CTCL: [151–155]; PTCL: [156]); however, clinical experience with alemtuzumab treatment in T-cell malignancies has been limited to small series and approval for this indication has not been granted yet. The first suggestion of activity in T-PLL came from a phase II trial [150] in which 39 patients with previously treated T-PLL received IV alemtuzumab (30 mg, three times a week until maximum response). The ORR was 76% (60% complete response, CR, and 16% partial response, PR). The TTP was 10 months (range 3–45) and median OS was 13 months (24 months for patients in CR). The response rate and survival were significantly longer than those reported for conventional therapies [157]. In a retrospective study of 76 patients with heavily pretreated T-PLL, alemtuzumab was administered at the standard schedule for up to 12 weeks. The ORR was 50% with 28 patients (37.5%) achieving a CR. The TTF was 4.5 months, the median OS was 7.5 months (14.8 months for patients in CR) [158]. In an early, preliminary, European experience, 50 B- and T-cell NHL patients received single-agent alemtuzumab [151]. Eight of these patients had advanced refractory MF: an ORR of 50% with 2 CRs and 2 PRs was seen. These promising results led to a multicenter phase II trial with the largest reported CTCL series [152] in which 22 patients with heavily pretreated, CD52-positive, advanced stage MF/SS, were treated with single-agent alemtuzumab. Alemtuzumab was administered IV at the standard dose of 30 mg, three times per week, for up to 12 weeks. The ORR rate was 55% (32% CR, 23% PR), with better efficacy in patients with erythroderma (69% ORR) than those with plaques/skin tumors (40% ORR). Median TTF for all responders was 12 months (range 5–32). Infectious complications were observed, including asymptomatic CMV reactivation (4 patients), generalized HSV (1 patient), 1 case of fatal aspergillosis, and 1 case of fatal Mycobacterium pneumonia. In another phase II trial [153], 8 MF/SS patients (7 of whom with refractory disease), received IV alemtuzumab according to the standard schedule; however, the ORR was only 38%, with no CR observed and a shorter TTF (4 months). In the attempt to reduce the incidence of hematologic toxicities and the risk of infectious complications associated with the standard schedule, Zinzani et al. [154] assessed the impact of a reduced-dose schedule of IV alemtuzumab (10 mg, three times per week, for 4 weeks) in 10 pretreated patients (6 PTCL and 6 MF). Despite the dose reduction, an ORR of 75% (with no CRs) among the MF patients and an ORR of 50% (with 2 CRs) among the PTCL patients were observed. More importantly, no grade 3–4 hematologic toxicities were noticed and CMV reactivation occurred in only 1 patient. More recently Bernengo et al. [155] investigated the association of a reduced-dose schedule of alemtuzumab with the SC administration route in the attempt to also eliminate the systemic reactions associated with the IV route. Eleven relapsed/refractory MF patients and 3 untreated SS patients were enrolled, 4 of them received alemtuzumab 3 mg on day 1, 10 mg on day 3, then 15 mg on alternating days, for a total of four doses. A reduced dosage (3 mg on day 1, then 10 mg on alternating days) was administered to the remaining patients. The ORR was 85.7%, with 3 CRs (21.4%). After a median follow-up of 16 months, the median TTF was 12 months. Infectious complications occurred in 4 patients. Lenihan et al. [159] described the activity of alemtuzumab, at the standard IV 12-week dosing schedule, in 5 patients with SS. Activity was modest with only 3 PR achieved, and a variety of cardiovascular adverse events were observed, including chronic heart failure, hypotension, pulmonary thromboembolism, and atrial fibrillation, even in the absence of known cardiac risk factors. However evidence of cardiac toxicity with alemtuzumab treatment has not been confirmed [160]. A few additional small studies reporting on the activity of alemtuzumab in SS have been published [161–163]. Data on PTCL patients and single-agent alemtuzumab are even scarcer than those available for T-PLL and CTCL. In the only prospective trial in PTCL patients treated with single-agent alemtuzumab published so far [156], 14 patients with advanced relapsed/refractory disease received IV alemtuzumab following the standard schedule. The ORR was 36% with 3 patients achieving a CR and 2 patients achieving a PR. The duration of response was 2, 6, and 12 months for each of the CR patients. However, in these heavily pretreated patients, alemtuzumab therapy was associated with significant toxicity, including CMV reactivation in 6 patients, pulmonary aspergillosis in 2 patients, EBV-related hemophagocytosis in 2 patients.
In the attempt to enhance the response rate and prolong the TTF, alemtuzumab has also been administered in association with chemotherapy in the treatment of TCL patients.
In the first phase II trial, 20 patients with previously untreated PTCL (10 patients with PTCL-unspecified, 3 with angioimmunoblastic lymphoma, 3 with extranodal NK/TCL, nasal type, 2 with ALK− ALCL, and 2 with subcutaneous panniculitis-like TCL) received CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) plus alemtuzumab (10 mg IV on day 1, 20 mg on day 2 of the first cycle, then 30 mg IV on day 1 in the subsequent cycles), based on 3 week interval [164]. An ORR of 80% with a 65% CR rate was observed however the trial was prematurely closed due to two treatment-related deaths and high incidence of infectious complications as well as hematologic adverse events. An Italian prospective multicenter phase II trial evaluated the activity of alemtuzumab in combination with CHOP in 24 patients with previously untreated PTCL (8 patients with PTCL-unspecified, 7 with angioimmunoblastic lymphoma, 3 with ALK− ALCL, and 1 with enteropathy-associated TCL) [165]. SC alemtuzumab was administered at 30 mg at day 1 to 8 courses of CHOP-28. An ORR of 75% with a 71% CR rate was observed. Thirteen patients were disease free with an overall median duration of response of 11 months. Neutropenia and CMV reactivation were the most common hematologic and non-hematologic complications, occurring in 59 of 176 (34%) and 15 of 176 (9%) of CHOP chemotherapy courses, respectively. Two cases of invasive aspergillosis and 1 case of PML were also noticed. Alemtuzumab was also combined with CHOP and ESHAP (ethopside, cytarabin, cisplatin, and methylpredinsolone) in 13 patients with untreated PTCL (3 patients with PTCL-unspecified, 5 with extranodal NK/TCL, nasal type, 4 with subcutaneous panniculitis-like TCL, and 1 with enteropathy-associated TCL) [166]. SC alemtuzumab was administered at 30 mg at day 1–3 of cycle 1–5 plus CHOP (day 1 of cycle 1, 3, 5) and ESHAP (day 1 of cycle 2, 4, 6) at 28-day intervals. In the 10 evaluable patients, an ORR of 90% with a 80% CR rate was observed. Infection was the major adverse event with CMV reactivation occurring in 54% of the patients.
Recently, two phase III trials (ACT1 and ACT2) combining CHOP with alemtuzumab as primary treatment for patients with PTCL, have been opened by the European Intergroup.
In a German phase II trial, alemtuzumab was combined with fludarabine, cyclophosphamide, and doxorubicin (alemtuzumab 3, 10, 30, 30 mg, days 1–4, fludarabine 25 mg/m2 days 2–4, cyclophosphamide 600 mg/m2 day 3, and doxorubicin 50 mg/m2 day 4, every 28 days) [167] to treat 19 newly diagnosed and 11 relapsed/refractory PTCL patients (13 patients with PTCL-unspecified, 9 with angioimmunoblastic lymphoma, 2 with ALK− ALCL, 2 with enteropathy-associated TCL, 2 with nasal-type NK-/TCL, 1 with an NK-cell lymphoma and 1 with a T-PLL). An ORR of 63% with a 58% CR rate and with ten ongoing remissions at the time of publication, were observed in the previously untreated patients’ group. An ORR of 45% with a 28% CR rate was observed in the relapsed/refractory group. The toxicity was significant with 65% of the patients experiencing a sustained grade III/IV neutropenia, 40% of the patients had a CMV reactivation which was symptomatic in 2.
As emerged from many clinical trials, most of the TCL patients experience variable grades of cytopenia during treatment with alemtuzumab, especially if heavily pretreated and with advanced stage of disease. Thrombocytopenia is generally most common during the first 2 weeks and neutropenia is most common during weeks 5 and 6 [152]. Importantly, grade 4 neutropenia could be an indication to postpone alemtuzumab therapy but prematurely terminating treatment should be avoided especially in SS patients because, although resolution of peripheral blood lymphocytosis occurs early, bone marrow is unlikely to be clear of disease after 4 weeks of therapy [152, 155]. Monocytes, NK cells and peripheral blood but not tissue antigen-presenting dendritic cells are also profoundly depleted but this reduction is not as pronounced as for lymphocytes. Profound and sustained T and B lymphopenia is the most significant and consistent side-effect of alemtuzumab, according to the high expression of CD52 antigen on these cells’ surface [152]. Granulocytes population usually returns to baseline levels early during the unmaintained follow-up while monocytes remain at around 50% of their baseline value for a prolonged period of time. B lymphocytes normalize relatively quickly, CD8+ T lymphocytes recover slower, and CD4+ T lymphocytes may remain subnormal for over a year [152]. The stage of the disease, the disease itself, the number of prior treatments and the myelo/immunosuppression alemtuzumab-induced, increase the susceptibility to opportunistic and other severe infections, primarily bacterial [152, 168]. The most frequent opportunistic infection is caused by CMV. The overall incidence of CMV reactivation ranges between 5 and 43% in patients treated with alemtuzumab [150–153, 155, 156] and it is usually observed shortly after the nadir in T-cell counts, between 4 and 6 weeks of therapy. Antiviral prophylaxis (acyclovir, famciclovir, valaciclovir) is a mandatory procedure in these patients but these antiviral agents do not prevent CMV reactivation, although most patients respond rapidly to IV ganciclovir. At the present, no data are available to recommend routine prophylaxis for CMV but routine weekly monitoring with CMV antigenemia is indicated. Antibacterial and anti-Pneumocystis Jiroveci prophylaxis is recommended as well. CMV monitoring and prophylaxis should be continued for 2–4 months after alemtuzumab discontinuation [168]. Recently, cases of EBV-associated B and TCL after treatment with alemtuzumab in combination with chemotherapy for TCL have been reported [169, 170]. Alemtuzumab administered with proper antimicrobial prophylaxis has shown to be active in TCL; however, hematologic toxicities and infection complications are a major concern. The infusion related events are drastically reduced by the SC administration without affecting the efficacy. Alemtuzumab as single agent does not appear to be curative, therefore combination strategies with chemotherapeutic, biological agents and emerging class of new compounds such as histone deacetylase inhibitors should be further explored.
Conclusions
TCL represent a heterogeneous group of lymphoid neoplasms, and, with the exception of ALK+ ALCL, early stage MF and T-LGL leukemia, TCL have a suboptimal response to conventional chemotherapy and a poor prognosis. Few patients achieve CR, relapse is common and usually associated with chemo-resistance. Malignant T-cells express a number of potential targets for immunotherapy. mAbs such as alemtuzumab have shown significant clinical activity in a variety of T-cell malignancies and represent a significant improvement over previous therapy considering that durable remissions have been seen in heavily pretreated patients with TCL. However, mAbs as single agents do not appear to be curative in TCL. Therefore, although active as single agents, mAbs may have a greater role in alternative strategies such as combinations with other biological or chemotherapeutic agents, either simultaneously or sequentially, with the hope to translate durable remissions into improved survival. Maintenance therapy or retreatment based on the monitoring of malignant T-cells should also be considered although antigen mutation or modulation may limit repeated administration activity. Another insight derives from studies involving mAbs covalently bound to radioisotope or toxins to enhance their ability to destroy tumor cells. This multimodality strategy together with a personalized, tailored, stage-specific approach may represent the future management of TCL patients. Based on the number of investigational mAbs now available and the activity shown by some mAbs alone or as a part of a multimodality strategy, there is no doubt that mAbs therapy of TCL will continue to be an area of active interest and expanding clinical trials.
References
Pressman D, Korngold L. The in vivo localization of anti-Wagner-osteogenic-sarcoma antibodies. Cancer. 1953;6(3):619–23.
Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–7.
Mascelli MA, Zhou H, Sweet R, et al. Molecular, biologic, and pharmacokinetic properties of monoclonal antibodies: impact of these parameters on early clinical development. J Clin Pharmacol. 2007;47(5):553–65.
Dillman RO. Monoclonal antibody therapy. In: Oldham RK, Dillman RO, editors. Principles of cancer biotherapy. 5th ed. New York: Springer; 2009. p. 303–407.
Meuer SC, Acuto O, Hercend T, et al. The human T-cell receptor. Annu Rev Immunol. 1984;2:23–50.
Kamoun M, Martin PJ, Hansen JA, et al. Identification of a human T lymphocyte surface protein associated with the E-rosette receptor. J Exp Med. 1981;153(1):207–12.
Bernard A, Gelin C, Raynal B, et al. Phenomenon of human T cells rosetting with sheep erythrocytes analyzed with monoclonal antibodies. “Modulation” of a partially hidden epitope determining the conditions of interaction between T cells and erythrocytes. J Exp Med. 1982;155(5):1317–33.
Alberola-Ila J, Places L, de la Calle O, et al. Stimulation through the TCR/CD3 complex up-regulates the CD2 surface expression on human T lymphocytes. J Immunol. 1991;146(4):1085–92.
Crawford K, Stark A, Kitchens B, et al. CD2 engagement induces dendritic cell activation: implications for immune surveillance and T-cell activation. Blood. 2003;102(5):1745–52.
Ohno H, Ushiyama C, Taniguchi M, et al. CD2 can mediate TCR/CD3-independent T cell activation. J Immunol. 1991;146(11):3742–6.
Moingeon P, Chang HC, Wallner BP. et al CD2-mediated adhesion facilitates T lymphocyte antigen recognition function. Nature. 1989;339(6222):312–4.
Peterson A, Seed B. Monoclonal antibody and ligand binding sites of the T cell erythrocyte receptor (CD2). Nature. 1987;329(6142):842–6.
Dustin ML, Sanders ME, Shaw S, et al. Purified lymphocyte function-associated antigen 3 binds to CD2 and mediates T lymphocyte adhesion. J Exp Med. 1987;165(3):677–92.
Hahn WC, Menu E, Bothwell AL, et al. Overlapping but nonidentical binding sites on CD2 for CD58 and a second ligand CD59. Science. 1992;256(5065):1805–7.
Selvaraj P, Plunkett ML, Dustin M, et al. The T lymphocyte glycoprotein CD2 binds the cell surface ligand LFA-3. Nature. 1987;326(6111):400–3.
Arulanandam AR, Kister A, McGregor MJ, et al. Interaction between human CD2 and CD58 involves the major beta sheet surface of each of their respective adhesion domains. J Exp Med. 1994;180(5):1861–71.
Zhu DM, Dustin ML, Cairo CW, et al. Mechanisms of cellular avidity regulation in CD2-CD58-mediated T cell adhesion. ACS Chem Biol. 2006;1(10):649–58.
Badour K, Zhang J, Shi F, et al. The Wiskott-Aldrich syndrome protein acts downstream of CD2 and the CD2AP and PSTPIP1 adaptors to promote formation of the immunological synapse. Immunity. 2003;18(1):141–54.
Carmo AM, Mason DW, Beyers AD. Physical association of the cytoplasmic domain of CD2 with the tyrosine kinases p56lck and p59fyn. Eur J Immunol. 1993;23(9):2196–201.
Bell GM, Fargnoli J, Bolen JB, et al. The SH3 domain of p56lck binds to proline-rich sequences in the cytoplasmic domain of CD2. J Exp Med. 1996;183(1):169–78.
Pantaleo G, Olive D, Poggi A, et al. Transmembrane signalling via the T11-dependent pathway of human T cell activation. Evidence for the involvement of 1,2-diacylglycerol and inositol phosphates. Eur J Immunol. 1987;17(1):55–60.
Hubert P, Debré P, Boumsell L, et al. Tyrosine phosphorylation and association with phospholipase C gamma-1 of the GAP-associated 62-kD protein after CD2 stimulation of Jurkat T cell. J Exp Med. 1993;178(5):1587–96.
Meuer SC, Hussey RE, Fabbi M, et al. An alternative pathway of T-cell activation: a functional role for the 50 kd T11 sheep erythrocyte receptor protein. Cell. 1984;36(4):897–906.
Dumont C, Déas O, Mollereau B, et al. Potent apoptotic signaling and subsequent unresponsiveness induced by a single CD2 mAb (BTI-322) in activated human peripheral T cells. J Immunol. 1998;160(8):3797–804.
Schad V, Greenstein JL, Giovino-Barry V, et al. An anti-CD2 monoclonal antibody that elicits alloantigen-specific hyporesponsiveness. Transplant Proc. 1996;28(4):2051–3.
Przepiorka D, Phillips GL, Ratanatharathorn V, et al. A phase II study of BTI-322, a monoclonal anti-CD2 antibody, for treatment of steroid-resistant acute graft-versus-host disease. Blood. 1998;92(11):4066–71.
Zhang Z, Zhang M, Ravetch JV, et al. Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD2 monoclonal antibody, MEDI-507. Blood. 2003;102(1):284–8.
Sorbera LA, Leeson PA, Revel L, et al. Siplizumab. Drugs Fut. 2002;27(6):558.
Casale DA, Bartlett NL, Hurd DD, et al. A phase I open label dose escalation study to evaluate MEDI-507 in patients with CD2-positive T-cell lymphoma/leukemia [ASH annual meeting abstracts]. Blood. 2006;108:2727.
O’Mahony D, Morris JC, Stetler-Stevenson M, et al. EBV-related lymphoproliferative disease complicating therapy with the anti-CD2 monoclonal antibody, siplizumab, in patients with T-cell malignancies. Clin Cancer Res. 2009;15(7):2514–22.
Manolios N, Letourneur F, Bonifacino JS, et al. Pairwise, cooperative and inhibitory interactions describe the assembly and probable structure of the T-cell antigen receptor. EMBO J. 1991;10(7):1643–51.
Chetty R, Gatter K. CD3: structure, function, and role of immunostaining in clinical practice. J Pathol. 1994;173(4):303–7.
San José E, Sahuquillo AG, Bragado R, et al. Assembly of the TCR/CD3 complex: CD3 epsilon/delta and CD3 epsilon/gamma dimers associate indistinctly with both TCR alpha and TCR beta chains. Evidence for a double TCR heterodimer model. Eur J Immunol. 1998;28(1):12–21.
Jaffers GJ, Fuller TC, Cosimi AB, et al. Monoclonal antibody therapy. Anti-idiotypic and non-anti-idiotypic antibodies to OKT3 arising despite intense immunosuppression. Transplantation. 1986;41(5):572–8.
Legendre C, Kreis H, Bach JF, et al. Prediction of successful allograft rejection retreatment with OKT3. Transplantation. 1992;53(1):87–90.
Sgro C. Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology. 1995;105(1):23–9.
Abramowicz D, Schandene L, Goldman M, et al. Release of tumor necrosis factor, interleukin-2, and gamma-interferon in serum after injection of OKT3 monoclonal antibody in kidney transplant recipients. Transplantation. 1989;47(4):606–8.
Alegre ML, Vandenabeele P, Depierreux M, et al. Cytokine release syndrome induced by the 145-2C11 anti-CD3 monoclonal antibody in mice: prevention by high doses of methylprednisolone. J Immunol. 1991;146(4):1184–91.
Carpenter PA, Appelbaum FR, Corey L, et al. A humanized non-FcR-binding anti-CD3 antibody, visilizumab, for treatment of steroid-refractory acute graft-versus-host disease. Blood. 2002;99(8):2712–9.
Springer TA. Adhesion receptors of the immune system. Nature. 1990;346(6283):425–34.
Leahy DJ. A structural view of CD4 and CD8. FASEB J. 1995;9(1):17–25.
Veillette A, Bookman MA, Horak EM, et al. The CD4 and CD8 T cell surface antigens are associated with the internal membrane tyrosine-protein kinase p56lck. Cell. 1988;55(2):301–8.
Kim YH, Duvic M, Obitz E, et al. Clinical efficacy of zanolimumab (HuMax-CD4): two phase 2 studies in refractory cutaneous T-cell lymphoma. Blood. 2007;109(11):4655–62.
Herzog C, Walker C, Müller W, et al. Anti-CD4 antibody treatment of patients with rheumatoid arthritis: I. Effect on clinical course and circulating T cells. J Autoimmun. 1989;2(5):627–42.
Choy EH, Connolly DJ, Rapson N, et al. Pharmacokinetic, pharmacodynamic and clinical effects of a humanized IgG1 anti-CD4 monoclonal antibody in the peripheral blood and synovial fluid of rheumatoid arthritis patients. Rheumatology (Oxford). 2000;39(10):1139–46.
Skov L, Kragballe K, Zachariae C, et al. HuMax-CD4: a fully human monoclonal anti-CD4 antibody for the treatment of psoriasis vulgaris. Arch Dermatol. 2003;139(11):1433–9.
Knox SJ, Levy R, Hodgkinson S, et al. Observations on the effect of chimeric anti-CD4 monoclonal antibody in patients with mycosis fungoides. Blood. 1991;77(1):20–30.
Knox S, Hoppe RT, Maloney D, et al. Treatment of cutaneous T-cell lymphoma with chimeric anti-CD4 monoclonal antibody. Blood. 1996;87(3):893–9.
Rider DA, Havenith CE, de Ridder R, et al. A human CD4 monoclonal antibody for the treatment of T-cell lymphoma combines inhibition of T-cell signaling by a dual mechanism with potent Fc-dependent effector activity. Cancer Res. 2007;67(20):9945–53.
Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–79.
Leonard WJ, Depper JM, Uchiyama T, et al. A monoclonal antibody that appears to recognize the receptor for human T-cell growth factor; partial characterization of the receptor. Nature. 1982;300(5889):267–9.
Leonard WJ, Depper JM, Crabtree GR, et al. Molecular cloning and expression of cDNAs for the human interleukin-2 receptor. Nature. 1984;311(5987):626–31.
Sharon M, Klausner RD, Cullen BR, et al. Novel interleukin-2 receptor subunit detected by cross-linking under high-affinity conditions. Science. 1986;234(4778):859–63.
Leonard WJ, Depper JM, Kanehisa M, et al. Structure of the human interleukin-2 receptor gene. Science. 1985;230(4726):633–9.
Waldmann TA. The structure, function, and expression of interleukin-2 receptors on normal and malignant lymphocytes. Science. 1986;232(4751):727–32.
Sugamura K, Asao H, Kondo M, et al. The interleukin-2 receptor gamma chain: its role in the multiple cytokine receptor complexes and T cell development in XSCID. Annu Rev Immunol. 1996;14:179–205.
Uchiyama T, Nelson DL, Fleisher TA, et al. A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. II. Expression of Tac antigen on activated cytotoxic killer T cells, suppressor cells, and on one of two types of helper T cells. J Immunol. 1981;126(4):1398–403.
Jones D, Ibrahim S, Patel K, et al. Degree of CD25 expression in T-cell lymphoma is dependent on tissue site: implications for targeted therapy. Clin Cancer Res. 2004;10(16):5587–94.
Talpur R, Jones DM, Alencar AJ, et al. CD25 expression is correlated with histological grade and response to denileukin diftitox in cutaneous T-cell lymphoma. J Invest Dermatol. 2006;126(3):575–83.
Janik JE, Morris JC, Pittaluga S, et al. Elevated serum-soluble interleukin-2 receptor levels in patients with anaplastic large cell lymphoma. Blood. 2004;104(10):3355–7.
Waldmann TA. The multi-subunit interleukin-2 receptor. Annu Rev Biochem. 1989;58:875–911.
Waldmann TA. The IL-2/IL-2 receptor system: a target for rational immune intervention. Immunol Today. 1993;14(6):264–70.
Foss FM, Waldmann TA. Interleukin-2 receptor-directed therapies for cutaneous lymphomas. Hematol Oncol Clin North Am. 2003;17(6):1449–58.
Kirkman RL, Shapiro ME, Carpenter CB, et al. A randomized prospective trial of anti-Tac monoclonal antibody in human renal transplantation. Transplant Proc. 1991;23(1 Pt 2):1066–7.
Nussenblatt RB, Fortin E, Schiffman R, et al. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase I/II clinical trial. Proc Natl Acad Sci U S A. 1999;96(13):7462–6.
Bielekova B, Richert N, Howard T, et al. Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci U S A. 2004;101(23):8705–8.
Lehky TJ, Levin MC, Kubota R, et al. Reduction in HTLV-I proviral load and spontaneous lymphoproliferation in HTLV-I-associated myelopathy/tropical spastic paraparesis patients treated with humanized anti-Tac. Ann Neurol. 1998;44(6):942–7.
Wiseman LR, Faulds D. Daclizumab: a review of its use in the prevention of acute rejection in renal transplant recipients. Drugs. 1999;58(6):1029–42.
Queen C, Schneider WP, Selick HE, et al. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci U S A. 1989;86(24):10029–33.
Junghans RP, Waldmann TA, Landolfi NF, et al. Anti-Tac-H, a humanized antibody to the interleukin 2 receptor with new features for immunotherapy in malignant and immune disorders. Cancer Res. 1990;50(5):1495–502.
Rubin LA, Kurman CC, Biddison WE, et al. A monoclonal antibody 7G7/B6, binds to an epitope on the human interleukin-2 (IL-2) receptor that is distinct from that recognized by IL-2 or anti-Tac. Hybridoma. 1985;4(2):91–102.
Phillips KE, Herring B, Wilson LA, et al. IL-2Ralpha-directed monoclonal antibodies provide effective therapy in a murine model of adult T-cell leukemia by a mechanism other than blockade of IL-2/IL-2Ralpha interaction. Cancer Res. 2000;60(24):6977–84.
Zhang M, Zhang Z, Goldman CK, et al. Combination therapy for adult T-cell leukemia-xenografted mice: flavopiridol and anti-CD25 monoclonal antibody. Blood. 2005;105(3):1231–6.
Waldmann TA, White JD, Goldman CK, et al. The interleukin-2 receptor: a target for monoclonal antibody treatment of human T-cell lymphotrophic virus I-induced adult T-cell leukemia. Blood. 1993;82(6):1701–12.
Waldmann TA, White JD, Carrasquillo JA, et al. Radioimmunotherapy of interleukin-2R alpha-expressing adult T-cell leukemia with Yttrium-90-labeled anti-Tac. Blood. 1995;86(11):4063–75.
Kreitman RJ, Wilson WH, White JD, et al. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J Clin Oncol. 2000;18(8):1622–36.
Anderson DM, Kumaki S, Ahdieh M, et al. Functional characterization of the human interleukin-15 receptor alpha chain and close linkage of IL15RA and IL2RA genes. J Biol Chem. 1995;270(50):29862–9.
Shanmugham LN, Petrarca C, Frydas S, et al. IL-15 an immunoregulatory and anti-cancer cytokine. Recent advances. J Exp Clin Cancer Res. 2006;25(4):529–36.
Döbbeling U, Dummer R, Laine E, et al. Interleukin-15 is an autocrine/paracrine viability factor for cutaneous T-cell lymphoma cells. Blood. 1998;92(1):252–8.
Cario G, Izraeli S, Teichert A, et al. High interleukin-15 expression characterizes childhood acute lymphoblastic leukemia with involvement of the CNS. J Clin Oncol. 2007;25(30):4813–20.
Morris JC, Janik JE, White JD, et al. Preclinical and phase I clinical trial of blockade of IL-15 using Mikbeta1 monoclonal antibody in T cell large granular lymphocyte leukemia. Proc Natl Acad Sci U S A. 2006;103(2):401–6.
Morimoto C, Schlossman SF. The structure and function of CD26 in the T-cell immune response. Immunol Rev. 1998;161:55–70.
von Bonin A, Hühn J, Fleischer B. Dipeptidyl-peptidase IV/CD26 on T cells: analysis of an alternative T-cell activation pathway. Immunol Rev. 1998;161:43–53.
Ishii T, Ohnuma K, Murakami A, et al. CD26-mediated signaling for T cell activation occurs in lipid rafts through its association with CD45RO. Proc Natl Acad Sci U S A. 2001;98(21):12138–43.
Oravecz T, Pall M, Roderiquez G, et al. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J Exp Med. 1997;186(11):1865–72.
Proost P, Menten P, Struyf S, et al. Cleavage by CD26/dipeptidyl peptidase IV converts the chemokine LD78beta into a most efficient monocyte attractant and CCR1 agonist. Blood. 2000;96(5):1674–80.
Kameoka J, Tanaka T, Nojima Y, et al. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science. 1993;261(5120):466–9.
Jones D, Dang NH, Duvic M, et al. Absence of CD26 expression is a useful marker for diagnosis of T-cell lymphoma in peripheral blood. Am J Clin Pathol. 2001;115(6):885–92.
Dang NH, Aytac U, Sato K, et al. T-large granular lymphocyte lymphoproliferative disorder: expression of CD26 as a marker of clinically aggressive disease and characterization of marrow inhibition. Br J Haematol. 2003;121(6):857–65.
Ruiz P, Mailhot S, Delgado P, et al. CD26 expression and dipeptidyl peptidase IV activity in an aggressive hepatosplenic T-cell lymphoma. Cytometry. 1998;34(1):30–5.
Ho L, Aytac U, Stephens LC, et al. In vitro and in vivo antitumor effect of the anti-CD26 monoclonal antibody 1F7 on human CD30+ anaplastic large cell T-cell lymphoma Karpas 299. Clin Cancer Res. 2001;7(7):2031–40.
Carbone A, Gloghini A, Zagonel V, et al. The expression of CD26 and CD40 ligand is mutually exclusive in human T-cell non-Hodgkin’s lymphomas/leukemias. Blood. 1995;86(12):4617–26.
Falini B, Pileri S, Pizzolo G, et al. CD30 (Ki-1) molecule: a new cytokine receptor of the tumor necrosis factor receptor superfamily as a tool for diagnosis and immunotherapy. Blood. 1995;85(1):1–14.
Gruss HJ, Duyster J, Herrmann F. Structural and biological features of the TNF receptor and TNF ligand superfamilies: interactive signals in the pathobiology of Hodgkin’s disease. Ann Oncol. 1996;7 Suppl 4:19–26.
Chiarle R, Podda A, Prolla G, et al. CD30 in normal and neoplastic cells. Clin Immunol. 1999;90(2):157–64.
Gilfillan MC, Noel PJ, Podack ER, et al. Expression of the costimulatory receptor CD30 is regulated by both CD28 and cytokines. J Immunol. 1998;160(5):2180–7.
Schwab U, Stein H, Gerdes J, et al. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature. 1982;299(5878):65–7.
Stein H, Mason DY, Gerdes J, et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood. 1985;66(4):848–58.
Beljaards RC, Meijer CJ, Scheffer E, et al. Prognostic significance of CD30 (Ki-1/Ber-H2) expression in primary cutaneous large-cell lymphomas of T-cell origin. A clinicopathologic and immunohistochemical study in 20 patients. Am J Pathol. 1989;135(6):1169–78.
Zinzani PL, Pileri S, Bendandi M, et al. Clinical implications of serum levels of soluble CD30 in 70 adult anaplastic large-cell lymphoma patients. J Clin Oncol. 1998;16(4):1532–7.
Hecht TT, Longo DL, Cossman J, et al. Production and characterization of a monoclonal antibody that binds Reed-Sternberg cells. J Immunol. 1985;134(6):4231–6.
Norton AJ, Isaacson PG. Detailed phenotypic analysis of B-cell lymphoma using a panel of antibodies reactive in routinely fixed wax-embedded tissue. Am J Pathol. 1987;128(2):225–40.
Bowen MA, Olsen KJ, Cheng L, et al. Functional effects of CD30 on a large granular lymphoma cell line, YT. Inhibition of cytotoxicity, regulation of CD28 and IL-2R, and induction of homotypic aggregation. J Immunol. 1993;151(11):5896–906.
Gruss HJ, Boiani N, Williams DE, et al. Pleiotropic effects of the CD30 ligand on CD30-expressing cells and lymphoma cell lines. Blood. 1994;83(8):2045–56.
Borchmann P, Treml JF, Hansen H, et al. The human anti-CD30 antibody 5F11 shows in vitro and in vivo activity against malignant lymphoma. Blood. 2003;102(10):3737–42.
Wahl AF, Klussman K, Thompson JD, et al. The anti-CD30 monoclonal antibody SGN-30 promotes growth arrest and DNA fragmentation in vitro and affects antitumor activity in models of Hodgkin’s disease. Cancer Res. 2002;62(13):3736–42.
Hu XF, Xing PX. MDX-060. Medarex. Curr Opin Investig Drugs. 2005;6(12):1266–71.
Mir SS, Richter BW, Duckett CS. Differential effects of CD30 activation in anaplastic large cell lymphoma and Hodgkin disease cells. Blood. 2000;96(13):4307–12.
Duckett CS, Gedrich RW, Gilfillan MC, et al. Induction of nuclear factor kappaB by the CD30 receptor is mediated by TRAF1 and TRAF2. Mol Cell Biol. 1997;17(3):1535–42.
Hirsch B, Hummel M, Bentink S, et al. CD30-induced signaling is absent in Hodgkin’s cells but present in anaplastic large cell lymphoma cells. Am J Pathol. 2008;172(2):510–20.
Bargou RC, Leng C, Krappmann D, et al. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood. 1996;87(10):4340–7.
Tian ZG, Longo DL, Funakoshi S, et al. In vivo antitumor effects of unconjugated CD30 monoclonal antibodies on human anaplastic large-cell lymphoma xenografts. Cancer Res. 1995;55(22):5335–41.
Pfeifer W, Levi E, Petrogiannis-Haliotis T, et al. A murine xenograft model for human CD30+ anaplastic large cell lymphoma. Successful growth inhibition with an anti-CD30 antibody (HeFi-1). Am J Pathol. 1999;155(4):1353–9.
Pasqualucci L, Wasik M, Teicher BA, et al. Antitumor activity of anti-CD30 immunotoxin (Ber-H2/saporin) in vitro and in severe combined immunodeficiency disease mice xenografted with human CD30+ anaplastic large-cell lymphoma. Blood. 1995;85(8):2139–46.
Ansell SM, Horwitz SM, Engert A, et al. Phase I/II study of an anti-CD30 monoclonal antibody (MDX-060) in Hodgkin’s lymphoma and anaplastic large-cell lymphoma. J Clin Oncol. 2007;25(19):2764–9.
Bartlett NL, Younes A, Carabasi MH, et al. A phase 1 multidose study of SGN-30 immunotherapy in patients with refractory or recurrent CD30+ hematologic malignancies. Blood. 2008;111(4):1848–54.
Forero-Torres A, Leonard JP, Younes A, et al. A phase II study of SGN-30 (anti-CD30 mAb) in Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Br J Haematol. 2009;146(2):171–9.
Duvic M, Reddy SA, Pinter-Brown L, et al. A phase II study of SGN-30 in cutaneous anaplastic large cell lymphoma and related lymphoproliferative disorders. Clin Cancer Res. 2009;15(19):6217–24.
Younes A, Yasothan U, Kirkpatrick P. Brentuximab vedotin. Nat Rev Drug Discov. 2012;11(1):19–20.
Foyil KV, Bartlett NL. Brentuximab vedotin for the treatment of CD30+ lymphomas. Immunotherapy. 2011;3(4):475–85.
Fanale MA, Forero-Torres A, Rosenblatt JD, et al. A phase I weekly dosing study of brentuximab vedotin in patients with relapsed/refractory CD30-positive hematologic malignancies. Clin Cancer Res. 2012;18(1):248–55.
Younes A, Bartlett NL, Leonard JP, et al. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med. 2010;363(19):1812–21.
Wagner-Johnston ND, Bartlett NL, Cashen A, et al. Progressive multifocal leukoencephalopathy (PML) in a patient with Hodgkin’s lymphoma treated with brentuximab vedotin. Leuk Lymphoma. 2012 [Epub ahead of print].
Hale G, Xia MQ, Tighe HP, et al. The CAMPATH-1 antigen (CDw52). Tissue Antigens. 1990;35(3):118–27.
Xia MQ, Hale G, Lifely MR, et al. Structure of the CAMPATH-1 antigen, a glycosylphosphatidylinositol-anchored glycoprotein which is an exceptionally good target for complement lysis. Biochem J. 1993;293(Pt 3):633–40.
Watanabe T, Masuyama J, Sohma Y, et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin Immunol. 2006;120(3):247–59.
Elsner J, Hochstetter R, Spiekermann K, et al. Surface and mRNA expression of the CD52 antigen by human eosinophils but not by neutrophils. Blood. 1996;88(12):4684–93.
Ginaldi L, De Martinis M, Matutes E, et al. Levels of expression of CD52 in normal and leukemic B and T cells: correlation with in vivo therapeutic responses to Campath-1H. Leuk Res. 1998;22(2):185–91.
Hale G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy. 2001;3(3):137–43.
Buggins AG, Mufti GJ, Salisbury J, et al. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood. 2002;100(5):1715–20.
Hale G, Rye PD, Warford A, et al. The glycosylphosphatidylinositol-anchored lymphocyte antigen CDw52 is associated with the epididymal maturation of human spermatozoa. J Reprod Immunol. 1993;23(2):189–205.
Gilleece MH, Dexter TM. Effect of Campath-1H antibody on human hematopoietic progenitors in vitro. Blood. 1993;82(3):807–12.
Olweus J, Lund-Johansen F, Terstappen LW. Expression of cell surface markers during differentiation of CD34+, CD38-/lo fetal and adult bone marrow cells. Immunomethods. 1994;5(3):179–88.
Rodig SJ, Abramson JS, Pinkus GS, et al. Heterogeneous CD52 expression among hematologic neoplasms: implications for the use of alemtuzumab (CAMPATH-1H). Clin Cancer Res. 2006;12(23):7174–9.
Piccaluga PP, Agostinelli C, Righi S, et al. Expression of CD52 in peripheral T-cell lymphoma. Haematologica. 2007;92(4):566–7.
Lapalombella R, Zhao X, Triantafillou G, et al. A novel Raji-Burkitt’s lymphoma model for preclinical and mechanistic evaluation of CD52-targeted immunotherapeutic agents. Clin Cancer Res. 2008;14(2):569–78.
Salisbury JR, Rapson NT, Codd JD, et al. Immunohistochemical analysis of CDw52 antigen expression in non-Hodgkin’s lymphomas. J Clin Pathol. 1994;47(4):313–7.
Jiang L, Yuan CM, Hubacheck J, et al. Variable CD52 expression in mature T cell and NK cell malignancies: implications for alemtuzumab therapy. Br J Haematol. 2009;145(2):173–9.
Dearden CE, Matutes E. Alemtuzumab in T-cell lymphoproliferative disorders. Best Pract Res Clin Haematol. 2006;19(4):795–810.
Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood. 2002;99(10):3554–61.
Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol. 2007;25(35):5616–23.
Xia MQ, Hale G, Waldmann H. Efficient complement-mediated lysis of cells containing the CAMPATH-1 (CDw52) antigen. Mol Immunol. 1993;30(12):1089–96.
Zhang Z, Zhang M, Goldman CK, et al. Effective therapy for a murine model of adult T-cell leukemia with the humanized anti-CD52 monoclonal antibody, Campath-1H. Cancer Res. 2003;63(19):6453–7.
Golay J, Manganini M, Rambaldi A, et al. Effect of alemtuzumab on neoplastic B cells. Haematologica. 2004;89(12):1476–83.
Zent CS, Chen JB, Kurten RC, et al. Alemtuzumab (CAMPATH 1H) does not kill chronic lymphocytic leukemia cells in serum free medium. Leuk Res. 2004;28(5):495–507.
Nuckel H, Frey UH, Roth A, et al. Alemtuzumab induces enhanced apoptosis in vitro in B-cells from patients with chronic lymphocytic leukemia by antibody-dependent cellular cytotoxicity. Eur J Pharmacol. 2005;514(2–3):217–24.
Lowenstein H, Shah A, Chant A, et al. Different mechanisms of Campath-1H-mediated depletion for CD4 and CD8 T cells in peripheral blood. Transpl Int. 2006;19(11):927–36.
Hale G, Rebello P, Brettman LR, et al. Blood concentrations of alemtuzumab and antiglobulin responses in patients with chronic lymphocytic leukemia following intravenous or subcutaneous routes of administration. Blood. 2004;104(4):948–55.
Wing MG, Moreau T, Greenwood J, et al. Mechanism of first-dose cytokine-release syndrome by CAMPATH 1-H: involvement of CD16 (FcgammaRIII) and CD11a/CD18 (LFA-1) on NK cells. J Clin Invest. 1996;98(12):2819–26.
Dearden CE, Matutes E, Cazin B, et al. High remission rate in T-cell prolymphocytic leukemia with CAMPATH-1H. Blood. 2001;98(6):1721–6.
Lundin J, Osterborg A, Brittinger G, et al. CAMPATH-1H monoclonal antibody in therapy for previously treated low-grade non-Hodgkin’s lymphomas: a phase II multicenter study. European Study Group of CAMPATH-1H treatment in low-grade non-Hodgkin’s lymphoma. J Clin Oncol. 1998;16(10):3257–63.
Lundin J, Hagberg H, Repp R, et al. Phase 2 study of alemtuzumab (anti-CD52 monoclonal antibody) in patients with advanced mycosis fungoides/Sezary syndrome. Blood. 2003;101(11):4267–72.
Kennedy GA, Seymour JF, Wolf M, et al. Treatment of patients with advanced mycosis fungoides and Sézary syndrome with alemtuzumab. Eur J Haematol. 2003;71(4):250–6.
Zinzani PL, Alinari L, Tani M, et al. Preliminary observations of a phase II study of reduced-dose alemtuzumab treatment in patients with pretreated T-cell lymphoma. Haematologica. 2005;90(5):702–3.
Bernengo MG, Quaglino P, Comessatti A, et al. Low-dose intermittent alemtuzumab in the treatment of Sézary syndrome: clinical and immunologic findings in 14 patients. Haematologica. 2007;92(6):784–94.
Enblad G, Hagberg H, Erlanson M, et al. A pilot study of alemtuzumab (anti-CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy-refractory peripheral T-cell lymphomas. Blood. 2004;103(8):2920–4.
Absi A, Hsi E, Kalaycio M. Prolymphocytic leukemia. Curr Treat Options Oncol. 2005;6(3):197–208.
Keating MJ, Cazin B, Coutré S, et al. Campath-1H treatment of T-cell prolymphocytic leukemia in patients for whom at least one prior chemotherapy regimen has failed. J Clin Oncol. 2002;20(1):205–13.
Lenihan DJ, Alencar AJ, Yang D, et al. Cardiac toxicity of alemtuzumab in patients with mycosis fungoides/Sézary syndrome. Blood. 2004;104(3):655–8.
Lundin J, Kennedy B, Dearden C, et al. No cardiac toxicity associated with alemtuzumab therapy for mycosis fungoides/Sézary syndrome. Blood. 2005;105(10):4148–9.
Gibbs SD, Herbert KE, McCormack C, et al. Alemtuzumab: effective monotherapy for simultaneous B-cell chronic lymphocytic leukaemia and Sézary syndrome. Eur J Haematol. 2004;73(6):447–9.
Gautschi O, Blumenthal N, Streit M, et al. Successful treatment of chemotherapy-refractory Sézary syndrome with alemtuzumab (Campath-1H). Eur J Haematol. 2004;72(1):61–3.
Capalbo S, Delia M, Dargenio M, et al. Mycosis fungoides/Sézary syndrome: a report of three cases treated with Campath-1H as salvage treatment. Med Oncol. 2003;20(4):389–96.
Kim JG, Sohn SK, Chae YS, et al. Alemtuzumab plus CHOP as front-line chemotherapy for patients with peripheral T-cell lymphomas: a phase II study. Cancer Chemother Pharmacol. 2007;60(1):129–34.
Gallamini A, Zaja F, Patti C, et al. Alemtuzumab (Campath-1H) and CHOP chemotherapy as first-line treatment of peripheral T-cell lymphoma: results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood. 2007;110(7):2316–23.
Intragumtornchai T, Bunworasate U, Nakorn TN, et al. Alemtuzumab in combination with CHOP and ESHAP as first-line treatment in peripheral T-cell lymphoma [ASH annual meeting abstracts]. Blood. 2006;108:4740.
Weidmann E, Hess G, Chow KU, et al. A phase II study of alemtuzumab, fludarabine, cyclophosphamide, and doxorubicin (Campath-FCD) in peripheral T-cell lymphomas. Leuk Lymphoma. 2010;51(3):447–55.
Osterborg A, Karlsson C, Lundin J, et al. Strategies in the management of alemtuzumab-related side effects. Semin Oncol. 2006;33(2 Suppl 5):S29–35.
Weisel KC, Weidmann E, Anagnostopoulos I, et al. Epstein-Barr virus-associated B-cell lymphoma secondary to FCD-C therapy in patients with peripheral T-cell lymphoma. Int J Hematol. 2008;88(4):434–40.
Kluin-Nelemans HC, Coenen JL, Boers JE, et al. EBV-positive immunodeficiency lymphoma after alemtuzumab-CHOP therapy for peripheral T-cell lymphoma. Blood. 2008;112(4):1039–41.
Majeau GR, Meier W, Jimmo B, et al. Mechanism of lymphocyte function-associated molecule 3-Ig fusion proteins inhibition of T cell responses. Structure/function analysis in vitro and in human CD2 transgenic mice. J Immunol. 1994;152(6):2753–67.
Branco L, Barren P, Mao SY, et al. Selective deletion of antigen-specific, activated T cells by a humanized MAB to CD2 (MEDI-507) is mediated by NK cells. Transplantation. 1999;68(10):1588–96.
Kung P, Goldstein G, Reinherz EL, et al. Monoclonal antibodies defining distinctive human T cell surface antigens. Science. 1979;206(4416):347–9.
Ravel S, Colombatti M, Casellas P. Internalization and intracellular fate of anti-CD5 monoclonal antibody and anti-CD5 ricin A-chain immunotoxin in human leukemic T cells. Blood. 1992;79(6):1511–7.
Foss FM. DAB(389)IL-2 (ONTAK): a novel fusion toxin therapy for lymphoma. Clin Lymphoma. 2000;1(2):110–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Alinari, L., Porcu, P., Coiffier, B. (2013). Monoclonal Antibodies (mAb) in the Therapy of T-Cell Lymphomas. In: Foss, F. (eds) T-Cell Lymphomas. Contemporary Hematology. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-170-7_14
Download citation
DOI: https://doi.org/10.1007/978-1-62703-170-7_14
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-169-1
Online ISBN: 978-1-62703-170-7
eBook Packages: MedicineMedicine (R0)