
Abstract
Deep vein thrombosis (DVT) and pulmonary embolism are common mortal/morbid diseases worldwide, and improvements in prophylaxis and therapy have been elusive. The basic pathophysiology of how a thrombus forms in vivo and how it resolves are now better understood through experiments with animal models. Inflammation can both cause and incite venous thrombosis. Leukocytes, platelets, and coagulation factors coalesce locally after a thrombogenic stimulus occurs. Cell adhesion molecules, such as P-selectin, bridge the interface of thrombosis and inflammation, and are both biomarkers for, and pathogenic in DVT. Once DVT forms, natural thrombolysis occurs primarily via the urokinase-type plasminogen activator (uPA)–plasmin system. Alternative pathways also exist for venous thrombus resolution, including the matrix metalloproteinases. A new concept in DVT resolution related to sterile inflammation is clearance of procoagulant necrotic leukocytes and platelets, in part mediated by TLR9 signaling. A consequence of DVT is the proximate vein wall injury, which is dependent on the mechanism and duration of the thrombus-vein wall contact. Both vascular smooth muscle cells and endothelial cells maintain normal vein homeostasis, but adopt an injury response after thrombosis that may lead to vein fibrosis. In humans, multiple biomarkers are now available, but increased specificity for venous thrombosis is still elusive. Some of these biomarkers such as P-selectin may also play a causative role, and be attractive therapeutic targets for nonanticoagulant DVT prophylaxis and treatment.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Clinical Impact of Acute Deep Vein Thrombosis
Venous thromboembolism (VTE) is a significant health care problem in the USA, with an estimated 900,000 cases of acute deep venous thrombosis (DVT) and pulmonary embolism (PE) yearly, causing approximately 300,000 deaths each year [1]. Treatment costs for VTE are in the billions of dollars per year [2]. For the past 150 years, our view of the pathogenesis of VTE centered on Virkow’s triad of stasis, changes in the vessel wall (now recognized as injury), and thrombogenic changes in the blood. Stasis is probably permissive, and not a direct cause of VTE, while systemic infection and systemic inflammation may be more causal than previously thought [3, 4].
The late DVT sequelae, postthrombotic syndrome (PTS), affects between 400,000 and 500,000 patients with skin ulcerations and six to seven million patients with other manifestations including stasis pigmentation and stasis dermatitis. It has been reported that up to 28% of patients evaluated after having an iliofemoral DVT develop marked edema and skin changes consistent with venous stasis syndrome within 20 years [5]. Risk factors for PTS include the rate of venous recanalization, the global and anatomical level of venous reflux from dysfunction of the venous valves, and the presence of persistent venous obstruction. Finally, it must be kept in mind that mortality is increased after VTE. This is due to both overall mortality and cardiovascular mortality (especially with idiopathic thrombosis) because of the age and comorbidities of the population of patients who often develop VTE [6–12].
Overview of Thrombogenesis and Thrombolysis
Initiation of Thrombogenesis
Hemostasis is typically initiated (Fig. 3.1) by damage to the vessel wall and disruption of the endothelium, although it may be initiated in the absence of vessel wall damage, particularly in venous thrombosis [13]. Vessel wall damage simultaneously results in de-encryption (i.e., activation) of local and circulating tissue factor (TF) via protein disulfide isomerase [14]. Tissues also vary with regard to their susceptibility to thrombosis, suggesting that regional tissue-dependent mechanisms may be different. For example, hemostasis in cardiac muscle may be more dependent on the extrinsic pathway, while skeletal muscle may be more dependent on the intrinsic pathway [15], and circulating TF may be more important in venous thrombosis than in arterial thrombosis [13].

Simplified schematic of coagulation. The insult may be a local or systemic inflammatory state such as infection, which then activates the endothelium. Tissue factor is de-encrypted by protein disulfide isomerase, which then activates factor VII, then factor II (thrombin). Alternatively, direct vascular damage may activate platelets independent of thrombin, ultimately forming fibrin. PDI protein disulfide isomerase, TFe tissue factor encrypted, TFd tissue factor de-encrypted, gp glycoprotein
The adhesion of platelets to exposed subendothelial collagen is the first step in the formation of an effective hemostatic “platelet plug,” as a result of platelet activation. This platelet–vessel wall interaction is mediated by von Willebrand factor (vWF), whose platelet receptor is glycoprotein (Gp) Ib (Fig. 3.1). Similarly, fibrinogen forms bridges between platelets by binding to the GpIIb/IIIa receptor resulting in platelet aggregation [16]. Activation of platelets also leads to the release of the prothrombotic contents of platelet granules, and the expression of membrane-bound receptors for coagulation factors Va and VIIIa, as well as fibrinogen, vWF, and ADP. Platelet activation also leads to the elaboration of arachidonic acid metabolites such as thromboxane A2, further promoting platelet aggregation (as well as vasoconstriction). Platelet shape changes result in exposure of negatively charged procoagulant phospholipids normally located within caveolae of the platelet membrane [17]. Platelets also release microparticles (MP) rich in TF and other procoagulants, which accelerate and concentrate the thrombus generation.
The extrinsic pathway (Fig. 3.1) begins with de-encrypted TF forming a complex with factor VII, causing activation (VIIa). The TF–VIIa complex then activates factors IX and X to IXa and Xa in the presence of Ca2+. Feedback amplification occurs, as VIIa, IXa, and Xa are all capable of activating VII to VIIa, especially when bound to TF [18]. Factor Xa is also capable of activating factors V to Va. Factors Xa, Va, and II (prothrombin) form on the platelet phospholipid surface in the presence of Ca2+ to initiate the prothrombinase complex, which catalyzes the formation of thrombin from prothrombin. Thrombin feedback amplifies the system not only by activating factors V to Va but also by activating factors VIII (normally circulating bound to vWF) to VIIIa and XI to XIa. After activation, factor VIIIa dissociates from vWF and assembles with factors IXa and X on the platelet surface in the presence of Ca2+ to form the Xase complex, which catalyzes the activation of factor X to Xa.
Thrombin (factor II) is central to coagulation through its cleavage and release of fibrinopeptide A (FPA) from the α chain of fibrinogen and fibrinopeptide B (FPB) from the β chain of fibrinogen. This causes fibrin monomer polymerization and cross-linking, stabilizing the thrombus and the initial platelet plug. Thrombin also activates factor XIII to XIIIa, which catalyzes this cross-linking of fibrin, as well as that of other plasma proteins, such as fibronectin and α2-antitrypsin, resulting in their incorporation into the clot and increasing resistance to thrombolysis [19]. In addition, factor XIIIa activates platelets, as well as factors V and VIII, further amplifying thrombin production.
Coagulation can also be activated through the intrinsic pathway with activation of factor XI to XIa, which subsequently converts factors IX to IXa [14], promoting formation of the Xase complex and ultimately thrombin. The physiologic contribution of the intrinsic pathway is probably not as important in the venous system.
Natural Anticoagulants
Physiologic anticoagulants balance thrombin formation and limit thrombotic activity to sites of vascular injury (see Esmon for a detailed review [20]). Antithrombin (AT) is a central anticoagulant protein that binds to thrombin at the site of thrombosis and interferes with coagulation by three major mechanisms. First, inhibition of thrombin prevents the removal of FPA and FPB from fibrinogen, limiting fibrin formation. Second, thrombin becomes unavailable for factors V and VIII activation, slowing the coagulation cascade. Third, thrombin-mediated platelet activation and aggregation is inhibited. In the presence of heparin, inhibition of thrombin by AT is accelerated, resulting in systemic anticoagulation. AT has been shown to directly inhibit factors VIIa, IXa, Xa, XIa, and XIIa. Thus, patients with a genetic deficiency of AT are at much higher risk of developing VTE than the normal population.
A second natural anticoagulant mechanism is activated protein C (APC), which is produced on the surface of intact endothelium when thrombin binds to its receptor, thrombomodulin (TM) and endothelial protein C receptor (EPCR). The thrombin–TM complex inhibits the actions of thrombin, and also activates protein C to APC. APC in the presence of its cofactor, protein S, inactivates factors Va and VIIIa, therefore reducing Xase and prothrombinase activity [21].
The third innate anticoagulant is TF pathway inhibitor (TFPI). This protein binds the TF–VIIa complex, thus inhibiting the activation of factors X to Xa and formation of the prothrombinase complex. Finally, heparin cofactor II is another inhibitor of thrombin, but whose action is in the extravascular compartment. The activity of heparin cofactor II is augmented by glycosaminoglycans, including both heparin and dermatan sulfate, but its deficiency is not associated with increased VTE risk [22].
Activation and Inhibition of Thrombolysis
Thrombus formation is balanced by controlled thrombolysis in order to localize intravascular thrombosis (see Vassalli et al. for a detailed review of thrombolysis [23]). The central fibrinolytic enzyme is plasmin, a serine protease generated by the proteolytic cleavage of the proenzyme, plasminogen. Its main substrates include fibrin, fibrinogen, and other coagulation factors. Plasmin also interferes with vWF-mediated platelet adhesion by mediating proteolysis of GpIb [24].
Activation of plasminogen occurs by several mechanisms. In the presence of thrombin, vascular endothelial cells produce and release tissue plasminogen activator (tPA) as well as α2-antiplasmin, a natural inhibitor of excess fibrin-bound plasmin. As clot is formed, plasminogen, tPA, and α2-antiplasmin become incorporated into the fibrin clot. In contrast to free circulating tPA, fibrin-bound tPA is an efficient activator of plasminogen.
Another endogenous activator of plasminogen is the urokinase-type plasminogen activator (uPA), also produced by endothelial cells, but it has less affinity for fibrin. The activation of uPA in vivo is not completely understood. However, it is hypothesized that plasmin in small amounts (produced through tPA) activates uPA, leading to further plasminogen activation and amplification of fibrinolysis [25]. It is likely that uPA is more important for plasmin activation in the venous system than tPA [26].
The third mechanism of plasminogen activation involves factors of the contact activation system: activated forms of factors XII, kallikrein, and XI that can each independently convert plasminogen to plasmin. These activated factors may also catalyze the release of bradykinin from high molecular weight kininogen, which further augments tPA secretion. Finally, APC has been found to proteolytically inactivate plasminogen activator inhibitor type 1 (PAI-1), an inhibitor of plasmin activators released by endothelial cells in the presence of thrombin [20].
The degradation of fibrin polymers by plasmin ultimately results in the creation of fragment E and two molecules of fragment D, which are released as a covalently linked dimer (D-dimer) [27]. Detection of D-dimer in the circulation is a marker for ongoing thrombus metabolism and has been shown to accurately predict ongoing risk of recurrent VTE [28].
Interestingly, the activity of the fibrinolytic system within the vein wall is reduced in the area of the valvular cusps as compared with the nonvalvular area [29]. Deep veins of the lower limb have the lowest fibrinolytic activity in soleal sinuses, as well as in the popliteal and femoral vein regions, as compared with the other anatomic locations. This observation underlies a popular hypothesis as to why DVT most commonly originates in the lower limb. However, no in vivo real-time imaging studies in humans have ever shown how and where DVT actually forms.
In plasma, PAI-1 is the primary inhibitor of plasminogen activators and is likely most important in the venous system [23, 30, 31]. The primary function of PAI-1 is to inhibit plasminogen activators from converting plasminogen to plasmin, which is responsible for initiating fibrinolysis. It is secreted in an active form from liver and endothelial cells and is stabilized by binding to vitronectin (and inhibits thrombin in this form). PAI-1 is stored in the alpha-granules of quiescent platelets [32].
Mouse Models of Venous Thrombosis
Why a Mouse?
While there is no reported spontaneous venous thrombosis in animals, several experimental mouse models (Table 3.1) exist for venous thrombosis research including: photochemical [33], inferior vena cava (IVC) stasis [34–36], IVC stenosis [37], mechanical trauma [38, 39], and electrolytic models [40, 41]. Day et al. provide a review of the current options [42].
In terms of thrombotic pathways, the mouse’s physiological characteristics, which are similar to humans, make it a useful experimental tool. The mouse’s small size is convenient, making them inexpensive to house. They are relative easy to breed and are well characterized genetically, allowing for gene addition (transgenic mice) or deletion (knockout mice).
Photochemical Injury Using Rose Bengal Dye
The photochemical injury model uses Rose Bengal dye administrated at a dose of 50 mg/kg. The dye is activated by a green light laser (540 nm) placed 6 cm from the vein injury site for 15 min [33, 42, 43]. Using the jugular vein, this technique produces an occlusive thrombosis within the vein with a mean time to occlusion of 26.5 ± 9 min, or 39.7 ± 1 min if the dose of Rose Bengal is 25 mg/kg [33]. In the IVC, this technique produces a subtle endothelial injury that activates the vascular endothelium, but produces inconsistent thrombosis (author personal communication, DDM).
The IVC Ligation Model or Stasis Model
The stasis or IVC ligation mouse model of venous thrombosis involves a total occlusion or ligation of the IVC below the renal veins with a Prolene 7-0 (a nonreactive suture) to produce complete blood stasis. Back branches are cauterized and side branches are also ligated. In most species, a consolidate thrombus forms by 3 h postligation and yields quantifiable amounts of vein wall tissue and thrombus. In addition, the ligation model can be utilized to evaluate interactions between the vein wall and the occlusive thrombus and to assess the progression from acute to chronic inflammation. This model is well established and broadly used in mice [34, 35, 42, 44–46]. The major disadvantage with the stasis model is the lack of blood flow, which jeopardizes the maximal effect of administered systemic therapeutic agents on thrombi.
The IVC Stenosis Model
In the mouse stenosis model, a 4-0 silk suture is placed around the IVC and tied down on a piece of 5-0 prolene, which is then removed creating a stenosis. The reduction in blood flow, when combined with external endothelial compression with a neurosurgical vascular clip for 15 s, produces a laminar thrombus allowing the study of cellular kinetics and therapeutic agents [37, 47]. Although the technique is simple and produces a large thrombus, the degree of stenosis is inconsistent, causing large standard deviations in thrombus size that make this model less appropriate for pharmacological investigation (personal observation).
The Mechanical Injury Model
The mechanical injury model measures the kinetics of temporal thrombus growth and resolution in the mouse femoral vein. This model uses an external mechanical force to damage the endothelium. Fiber optic lights transilluminates the vein to visualize the thrombus. The main disadvantages of this model are that it only allows the study of early stages of thrombosis and the yield of thrombus, and vein wall mass is too small for most analysis. In addition, this model is expensive, since specialized optical equipment is required to visualize thrombus generation and resolution [38, 39, 48].
Electrolytic Vein Models
Mouse Femoral Vein
Venous thrombogenesis can be evaluated in a temporal fashion using a murine femoral vein electrical stimulation thrombus model [40]. Thrombosis is induced at the site of the electrode and grows in a sequential fashion. Cooley et al. [40] speculated that thrombus formation in their model is the result of either direct electrical injury (resistance heating) or a free radical-induced injury to the vein wall. When compared to IVC thrombosis models, the main limitation of this model is that the yield of thrombus and vein wall mass is too small for protein and gene expression analysis.
Mouse IVC
The electrolytic inferior vena cava model (EIM) is a newer mouse model of venous thrombosis recently published by Diaz et al. [41]. In this model, a 25G stainless-steel needle attached to a silver-coated copper wire is inserted into the exposed caudal IVC and positioned against the anterior wall (anode). Another wire is implanted subcutaneously to complete the circuit (cathode). A current of 250 μA is applied over 15 min using a Grass S48 square wave stimulator and a constant current unit. The direct current results in the formation of toxic products of electrolysis that activate the endothelial surface of the IVC, promoting a thrombogenic environment and subsequent thrombus formation. Importantly, it was demonstrated that heat does not participate in thrombus formation in the EIM model. This is the first IVC thrombosis mouse model that consistently produces thrombosis and produces a thrombus sample large enough to adequately study thrombogenesis, thrombus resolution, and pharmacologic applications in the field of venous thrombosis (Fig. 3.2).

Electrolytic injury model (EIM)-induced thrombosis of the inferior vena cava. Thrombus harvested after EIM. (a) The thrombus shape suggests thrombus formation in the presence of constant flow. (b) A hematoxylin and eosin stained histologic section of the inferior vena cava and adjacent structures demonstrates 30% patency 2 days after EIM
The Role of Inflammation in Thrombosis
An acute to chronic inflammatory response occurs in the vein wall and thrombus after the thrombus forms. This response leads to thrombus amplification, organization, and recanalization, and occurs at the expense of vein wall and vein valve damage [49]. The question that still remains is whether inflammation participates as an acute trigger of thrombosis, starting a series of thrombotic mechanisms, or if inflammation after thrombosis is a consequence.
During acute thrombosis, the primary inflammatory cells that migrate into the vein wall and thrombus are neutrophils. Cell adhesion molecules (CAMs) expressed on endothelial cells facilitate this phenomenon [50]. Neutrophil extravasation through an intact layer of endothelial cells has been described in early stages of venous thrombosis, linking acute inflammation and acute thrombosis [51–55]. Selectins (P- and E-selectin), which are CAMs that modulate leukocyte–endothelial interactions, are integrally involved in this process. Indeed, proinflammatory cytokines increase CAM expression in veins [56].
Cytokines, chemokines, and inflammatory factors such as interleukin-1 (IL-1) facilitate the inflammatory response in both the vein wall and the thrombus. Both proinflammatory and anti-inflammatory mediators are involved in the ultimate vein wall and thrombus response. Other interactions between inflammation and coagulation exist. For example, inflammation is associated with increased expression of fibrinogen, TF and membrane phospholipids, and increased platelet reactivity. Inflammation is also associated with decreased expression of thrombomodulin and vascular heparans, a decreased half-life for APC, and decreased fibrinolysis (by increasing PAI-1) [57].
CAMs and MPs in Venous Thrombosis
P-selectin is a central adhesion molecule involved in the interactions between inflammatory cells and vessels and has been linked to cardiovascular events in both the arterial and venous circulations [58]. This molecule is present in the alpha-granules of platelets and the Weibel–Palade bodies of endothelial cells. After activation by local vascular injury or platelet activation, it is translocated to the plasma membrane of these cells, mediating the initial inflammatory response [59]. Recombinant P-selectin glycoprotein ligand 1 (rPSGL-1)-Ig chimera binds and inhibits cell-associated P-selectin. Thrombin-activated platelets expressing P-selectin bind to neutrophils, and rPSGL-1-Ig effectively blocks this binding by approximately 90% [60]. Recently, a synergism between leukocytes and platelets has been identified [61], such that TF can be transferred from leukocytes to platelets in a P-selectin-mediated fashion, and even platelets have been demonstrated to express functional PSGL-1, allowing for a P-selectin-mediated mechanism for platelet rolling [53, 62].
In a mouse model of stasis DVT, P-selectin is upregulated as early as 6 h after thrombus induction, while E-selectin is upregulated at day 6 after thrombosis, with increases in gene expression preceding the protein elevations [34]. To further define the importance of the selectins to the thrombo-inflammatory response, genetically modified P- or E- selectin knockout (KO) mice were challenged with stasis DVT. Deletion of E-selectin and combined P-selectin/E-selectin deletion were associated with decreased thrombosis and significantly reduced vein wall inflammatory response [34]. A significant decrease in fibrin content is found 2 days after thrombus formation in P-selectin/E-selectin-deficient animals compared to control mice, and at 6 days after thrombus formation in E-selectin, P-selectin, and P-selectin/E-selectin-deficient animals. Mice deficient in both selectins had the smallest thrombi [63]. Together, these data suggest that selectins contribute to both thrombus generation and thrombus amplification.
When P-selectin binds to its receptor, PSGL-1, MPs are produced that can promote thrombus formation (Fig. 3.3). MPs are fragments of phospholipid cell membranes that promote coagulation and modulate a number of inflammatory cell–vessel wall interactions. For example, platelet-derived MPs are involved in the pathogenesis of heparin-induced thrombocytopenia, possibly by promoting thrombosis after the antibody binding to the platelets [65]. Platelet MPs rich in P-selectin have been found to allow flowing neutrophils to aggregate by a non-L-selectin mechanism [66]. Less is known about leukocyte-derived MPs, although they are associated with endothelial cell activation and cytokine gene induction [67]. Additionally, MPs derived from endothelial cells induce monocyte TF antigen expression and release [68].

Proposed contribution of microparticles to venous thrombosis. (1) Injury, stasis, procoagulant syndromes, or other stimuli upregulate endothelial selectin adhesion molecules that (2) bind to P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes and platelets. This results in the production of procoagulant microparticles, especially from monocytes but also from platelets and endothelial cells. (3–4) The microparticles then are concentrated in the forming thrombus where they contribute to amplification of the coagulation cascade and thrombus formation. TF tissue factor, FVII factor VII. Based on data from [64]
MPs are procoagulant and are recruited back to the developing thrombi, where they amplify coagulation [69, 70]. The PSGL-1 present on the surface of MPs can bind to P-selectin on activated platelets and endothelial cells, allowing the MPs to interact directly at the point of thrombus initiation, accumulation, and propagation. Fluorescently labeled MPs have been shown to be taken up into thrombi within 1 min of ferric chloride-induced venule thrombosis in a mouse cremaster muscle model [71]. The colocalization of fibrin, platelets, and leukocytes in the developing thrombus supports this hypothesis, as do the recent observations of the importance of P-selectin-mediated monocyte–platelet interactions to the generation of TF [72, 73].
The delta CT mouse carries transgenic expression of the extracellular (soluble) portion of P-selectin and therefore has fourfold elevated levels of soluble P-selectin (sP-selectin). A stasis model of DVT has been used in this mouse to determine the effect of sP-selectin on thrombogenesis and MPs. A 50–60% increase in thrombus size was found at days 2 and 6 after thrombosis, and this increase was associated with elevated procoagulant MPs in the circulation, most prominently of leukocyte origin. Consistently, mice deficient in P- and E-selectin had significantly smaller thrombi, and decreased levels of MPs derived from leukocytes after thrombus development [35].
E-selectin polymorphism in humans has been associated with enhanced endotoxin-triggered, TF-mediated coagulation, atherosclerosis, myocardial infarction and restenosis after angioplasty and may be associated with recurrent VTE [74–82]. Jilma et al. more recently demonstrated that homozygosity of the single nucleotide polymorphism Ser128Arg in the E-selectin gene in humans may be associated with recurrent VTE [74]. This polymorphism alters ligand affinity, enhances myeloid cellular tethering, and regulates leukocyte–endothelial cell interactions in vitro [74–77]. When 585 patients with a first idiopathic VTE were prospectively examined, 102 patients (17%) were heterozygous for the Ser128Arg mutation and 11 (2%) were homozygous. Of the total patient population, 90 patients (15% of 585 patients) demonstrated recurrent VTE. Homozygosity for this mutation appeared to increase the cumulative likelihood for early recurrent VTE and was considered an independent predictor of recurrent VTE (hazard ratio = 4.1) [74].
Endothelium and Vessel Wall Hemostasis
Most of the thrombosis–thrombolysis processes occur in juxtaposition to the endothelium, and hence the endothelium is one of the pivotal regulators of thrombotic homeostasis. Under normal conditions, endothelial cells maintain a vasodilatory and local fibrinolytic state in which coagulation, platelet adhesion, and activation are suppressed. Specifically, the nonthrombogenic endothelial surface is maintained through several mechanisms, including: (1) endothelial production of TM and subsequent activation of Protein C; (2) endothelial expression of heparan sulfate and dermatin sulfate, which accelerate AT and heparin cofactor II activity; (3) constitutive expression of TFPI, and (4) local production of tPA and uPA. In addition, the production of nitric oxide (NO) and prostacyclin by the endothelium inhibits the adhesion and activation of leukocytes and produces vasodilation [83]. TF production is also inhibited by NO [84].
During states of endothelial cell injury, a prothrombotic and proinflammatory state is supported by the endothelial surface [85]. Release of platelet activating factor (PAF) and endothelin-1 promotes vasoconstriction [85], while production of vWF, TF, PAI-1, and factor V augments thrombosis. Indeed, vWF is expressed to a greater extent on the endothelium of veins as compared with arteries, and tPA is less commonly expressed in venous endothelium [86].
In an elegant study by Zhou and colleagues, stasis thrombosis in the rat induced early endothelial damage with subendothelial collagen exposure and P-selectin expression within 6 h [87]. These experiments suggest mechanical injury results in a shift to a procoagulant milieu that drives ongoing thrombosis. Interestingly, the early thrombus matrix does facilitate the recruitment of CD34(+) cells, which may promote endothelial healing [88].
Mechanisms That Resolve Venous Thrombosis
Regardless of the location and extent of an acute DVT, the process of resolution has only recently become well understood (Fig. 3.4). The natural fibrinolytic mechanisms break down the thrombus over time and at variable rates [90, 91], with resultant vein wall damage. Venous thrombus resolution has a distinct time line and mimics wound healing, involving profibrotic growth factors, collagen deposition, matrix metalloproteinase (MMP) expression, and neovascularization (Table 3.2). Similarly, the fact that leukocytes invade the thrombus in a specific sequence suggests their importance in normal thrombus resolution [92]. The first cell type in the thrombus is the neutrophil (PMN). Although PMNs may cause vein wall injury, they appear essential for early thrombus resolution by promoting both fibrinolysis and collagenolysis [55, 93]. In a rat model of stasis DVT, neutropenia was associated with larger thrombi at 2 and 7 days, and was correlated with increased thrombus fibrosis and significantly lower thrombus levels of both uPA and MMP9 [94].

Hypothesized model of rodent venous thrombosis resolution. Early acute thrombus resolution involves release from the large clot of interleukin-1, necrotic cellular debris, platelet factors that drive neutrophil influx, and plasminogen activators, such as uPA. Concurrently, MMP9 is released, and plasmin is upregulated. At the same time, loss of endothelium integrity exposes the subendothelial matrix proteins that may further potentiate thrombosis. In the media, collagenolysis and elastinolysis occur, in addition to direct mechanical stretch. Acute clinical symptoms in the affected limb include pain and swelling. Later (usually after day 8), vein wall medial thickening occurs, resulting in decreased compliance and possibly decreased vasoreactivity. Re-endothelialization commences but is incomplete until a much later time point (>14 days). Thrombus neovascularization and organization are associated with resolution. Within the vein wall, matrix turnover occurs with increased MMP2 expression, as well as collagen-I and -III production. Interleukin-13 and TGFβ are two profibrotic growth factors that may be involved with late vein wall remodeling. Symptoms of chronic thrombosis include limb swelling, lipodermatosclerosis (LDS), dermal hyperpigmentation, and venous stasis dermal ulceration (VSU). Reprinted from [89] with permission from Elsevier
Leukocyte-derived uPA has been shown to modulate the uPA–plasmin axis in venous thrombosis [26]. In contrast, other work suggests that early (4 <day) venous thrombosis resolution may be independent of uPA. For example, in genetically deleted uPA mice, an early gamma interferon (IFNg)-dependent increase in MMP2 activity was found [95]. Mice lacking MMP2 had larger early venous thrombosis, despite no change in local plasmin levels.
The monocyte is likely the most important cell for the later stages of DVT resolution [96]. Monocyte influx into the thrombus peaks at day 8 after thrombogenesis and correlates with elevated levels of monocyte chemotactic protein-1 (MCP-1), one of the primary CC chemokines that directs monocyte chemotaxis and activation [97]. MCP-1 has also been associated with DVT resolution [98]. When mice lacking the MCP-1 receptor CC receptor-2 are challenged with stasis thrombosis, late impairment of thrombus resolution is observed, probably due to impaired IFNg-inducible vein wall proteinase activity that may be independent of monocytes [99].
Venous thrombosis is most always a sterile inflammatory nidus. Sterile inflammation and the process of resolution have only recently been investigated, but many parallels between the venous thrombosis and solid organ necrosis have been observed [100]. Experimental stasis venous thrombosis in the mouse suggests a role for TLR9 in venous thrombosis resolution, as deletion of TLR9 is associated with larger thrombus, increased thrombus leukocytes, and a decreased Th1 cytokine immune environment [101]. Interestingly, stimulation of TLR9 with an oligodeoxynucleotide (ODN) aptamer is associated with smaller thrombi in mice, suggesting that modulation of TLR9 signaling may have a possible clinical application.
Venous thrombosis resolution also involves neovascularization [52, 102]. Neovascularization occurs at later time points, usually after day 6. It is likely that local hypoxia drives hypoxia inducing factor 1alpha [HIF-1α] upregulation and production of vascular endothelial growth factor (VEGF), with generation of endothelial lined channels [103, 104]. Interestingly, circulating progenitor cells play a role in venous thrombosis resolution, but endothelial progenitor cells may not be important for neovascularization itself [105]. That is, while neovascular channels are observed in resolving experimental venous thrombosis, the progenitor cells did not specifically line these channels, suggesting another role for them in the resolution process.
Proinflammatory and Profibrotic Mediators in Vein Wall Remodeling
As the thrombus resolves, a number of proinflammatory factors are released into the local environment, including interleukin (IL)-1-α, IL-1-β, and tumor necrosis factor-alpha [TNFα] [92]. Gene expression of the anti-inflammatory cytokine IL-10 is upregulated at day 2, and remains so through day 9 after thrombosis in a stasis model of venous thrombosis, suggesting that it serves as a counterbalance to the inflammatory response. Additionally, vein wall IL-10 protein levels are elevated before mRNA upregulation, suggesting an initial increase from preformed IL-10 followed by IL-10 synthesis [34]. The cellular sources of these different mediators have not been specifically defined, but likely include leukocytes and smooth muscle-like cells within the resolving thrombus and the adjacent vein wall.
The leukocyte kinetics in the vein wall after DVT are similar to that observed in the thrombus, with an early influx of PMNs, followed by monocytes. Based on experimental studies, elastinolysis and collagenolysis occur early, as measured by an increase in vein wall stiffness, persists through 14 days, and is accompanied by elevated MMP2 and MMP9 activities [106].
An elevation of profibrotic mediators, including transforming growth factor-beta [TGFβ], interleukin (IL)-13, and monocyte chemotactic protein-1 (MCP-1), is associated with the early biomechanical injury from DVT [106, 107]. These mediators are present within the vein wall and thrombus and may drive the fibrotic response. Although exogenous MCP-1 may hasten DVT resolution, it does have profibrotic activity in vivo. The profibrotic growth factor TGFβ is also present in the thrombus and is activated with normal thrombolysis [108]. This factor may be a key to promoting vein wall fibrosis. Consistently, fibrosis with a significant increase in vein wall collagen has been observed after experimental stasis thrombogenesis [109]. An increase in collagens I and III gene expression, as well as an increase in MMP-2 and MMP-9 gene expression and activity, correlates with this increase in fibrosis [109].
Also, increased plasma levels of interleukin-6 (IL-6), a major inflammatory cytokine, have been demonstrated during venous thrombosis [110] and PTS [111]. While these studies suggest that IL-6 may be used as a marker for venous thrombosis, its role as an active cytokine in the development of PTS has been recently established [46]. IL-6 plays a critical role in the transition from acute to chronic inflammation. Antibody neutralization of IL-6 in the mouse model of venous thrombosis was associated with reduced vein wall MCP-1 gene and protein expression, as well as monocyte infiltration. Moreover, blockade of IL-6 was associated with less vein wall fibrosis at the chronic time point (14 days after thrombosis). A proposed mechanism of IL-6 induced vein wall fibrosis following venous thrombosis is shown in Fig. 3.5.

Proposed mechanism of IL-6-induced vein wall fibrosis following venous thrombosis. During venous thrombosis, endothelial cells express P-selectin and neutrophils are recruited. IL-6 is produced by activated venous endothelial cells and causes upregulation of vein wall CCL2, leading to monocyte recruitment. Recruited monocytes secrete cytokines and growth factors that stimulate fibroblasts and smooth muscle cells to produce and deposit collagen. PMN polymorphonuclear neutrophil, gp130 glycoprotein 130, CCL2 C–C motif chemokine ligand 2, IL-6 interleukin-6, sIL-6-R soluble IL-6 receptor, TGFβ transforming growth factor beta. Modified from [46] with permission from Elsevier
Further linking inflammation to fibrosis, we have shown that inhibition of the inflammatory response can decrease vein wall fibrosis. In a rat model of stasis DVT, P-selectin inhibition achieved through treatment with either low-molecular-weight heparin (LMWH) or an oral inhibitor of P-selectin 2 days after establishment of thrombosis led to significantly decreased vein wall injury (independent of thrombus size), as measured by vein wall tensiometry (stiffness), intimal thickness score, interleukin (IL)-13 levels, MCP-1 levels, and platelet-derived growth factor-β (PDGFβ) levels [107]. The mechanism accounting for this protective effect is not yet known, but probably does not involve leukocyte blockade, as no differences in vein wall monocyte influx were observed.
Loss or injury of venous endothelium also contributes to vein wall fibrosis and predisposition to recurrent thrombosis. An experimental model of DVT showed decreased expression of homeostatic endothelial genes such as NO and thrombomodulin as compared with controls, and correlated with loss of vWF-positive cell luminal staining [112]. Other investigators have found that prolonged venous stasis is associated with decreased plasminogen activators, probably related to loss of endothelium [113]. The period of time the endothelium may be impaired is not clear, although preliminary studies suggest only modest recovery by 21 days after thrombosis, as measured by endothelial markers such as CD31 and thrombomodulin.
The role of the vein wall media after thrombotic injury has not been well studied. The vascular smooth muscle cell (VSMC) found in the media contributes significantly to the neointimal process in arterial injury. As the vein wall becomes thickened after thrombosis resolution, the VSMC is likely the cell that produces collagen. Thus, early vein wall injury is associated with active matrix remodeling that seems to promote late fibrosis, not unlike many other tissue responses to inflammation. The specific mechanisms and strategies to reverse the fibrotic process are being actively investigated [114]. Preliminary studies suggest that after thrombotic injury the VSMC changes to a synthetic state from a contractile state, as measured by common antigen markers, and may contribute to the fibrotic response. The CD45+/CCR7+/Col I+ leukocyte (fibrocyte) may also contribute to both the thrombus and the vein wall response in venous thrombosis resolution. In a nonstasis model of venous thrombosis, CCR7 signaling seems important for both of these processes, although it may be less beneficial in a stasis model (Henke, unpublished data). It may be that the CCR7+ fibrocyte directs the normal matrix response for vein wall healing.
Links Between Dyslipidemia, Inflammation, and Venous Thrombosis
An interface among hyperlipidemia, vascular inflammation, and thrombosis has been suggested from a recent clinical trial showing decreased risk of VTE with use of 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibition [115]. PAI-1 levels are elevated by hyperlipidemia, and high levels of PAI-1 appear to synergize with factor V Leiden genetic abnormalities to cause thrombosis. Studies on the role of elevated levels of PAI-1 in venous thrombosis have not been consistent in suggesting increased thrombotic risk [116, 117], although it is plausible that elevated PAI-1 could suppress fibrinolysis, and thereby increase thrombosis potential. In humans, genetic polymorphisms, particularly the 4G/4G insertion/deletion in the PAI-1 promoter region, affect transcription and correlate with increased risk of VTE. Studies have found an eightfold increased VTE risk in patients with the 4G allele combined with other thrombophilic markers [118], while PE was increased 4.5-fold in 4G/4G patients with protein S deficiency [119].
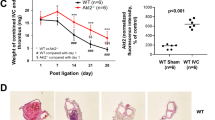
Experimentally, venous thrombi are larger in a stasis model in ApoE−/− mice, as compared with controls, suggesting a role of hyperlipidemia in venous thrombosis. Correspondingly, a significant increase in early circulating PAI-1 activity and a decrease in circulating plasmin activity was documented in ApoE−/− mice compared to controls, suggesting an impaired thrombolytic potential. In addition, ApoE−/− mice had undetectable levels of uPA in both vein wall and thrombus, compared to WT mice at both days 6 and 14 after thrombosis, suggesting impaired late thrombolysis. Loss of uPA leads to a sequence of biological events that result in impaired thrombus resolution through MMPs and MCP-1 [120]. Indeed, MMP2 and MMP9 were significantly decreased at the chronic time points (days 6 and 14) in APOE-deficient mice compared to WT mice, and MCP-1 was significantly decreased at both acute (day 2) and chronic (day 6) time points compared to WT mice. Consistently, monocyte recruitment was significantly reduced at days 6 and 14. In nonhyperlipidemic mice, loss of uPA is associated with increased MMP activity [95]. Thus, in hyperlipidemic mice with venous thrombosis, increased PAI-1 may contribute to an acute increase in thrombus size due to impaired fibrinolysis. In chronic venous thrombosis, decreased uPA is associated with impaired resolution due to decreased monocyte influx and MMP activity (Fig. 3.6). These observations may explain the clinical benefit of control of plasma lipid concentration on vascular venous inflammation.

Events that affect venous thrombosis in hyperlipidemic ApoE−/− mice. During acute venous thrombosis in hyperlipidemic ApoE−/− mice, we observed an increase in PAI-1 that contributes to increased thrombus weight due to impaired fibrinolysis. During chronic venous thrombosis in ApoE−/− mice, undetectable levels of uPA lead to decreased expression of MMP-9, MMP-2, and MCP-1, resulting in decreased monocyte recruitment and impaired thrombus resolution
Aging Alters the Risk for Venous Thrombosis
The incidence of VTE increases markedly with advancing age. In younger individuals, the incidence of venous thrombosis is less than 1 per 10,000 per year; however, this increases to approximately 1% in the elderly. Thus, aging is the most prevalent risk factor for venous thrombosis [121]. Biomarkers associated with thrombosis that increase with age are summarized in Table 3.3.
Increased risk of venous thrombosis in humans is associated with increased plasma levels of markers of intravascular coagulation such as D-dimer and prothrombin fragment [122]. Increases in fibrinogen, factors VIII and IX without a proportional increase in anticoagulant factors may also contribute to the increasing risk of DVT with age. Aging is also positively associated with IL-6 and C-reactive protein (CRP) levels, indicating that inflammation or an inflammatory state is likely an important stimulus for thrombotic events in the aging population [123].
The anatomy of the venous vessel wall is also altered with aging. The aged vein wall shows atrophy of the muscle fibers, increases in collagen, and decreased valve competency due to thickening of the values [124]. It has been hypothesized that the venous valve sinus may be a regulator of the prothrombotic state in that immediate environment. Increased levels of anticoagulant proteins such as endothelial TFPI and thrombomodulin are more active in the valvular sinus than the luminal endothelial layer, but with aging, the endothelium shifts to a lesser anticoagulant state [123]. Consistently, these factors may be decreased in the valve sinus [125].
The expression of PAI-1 is not only elevated in the aged but also induced by a multitude of pathologies associated with the process of aging, such as obesity and insulin resistance [126–128]. PAI-1 has also been shown to significantly increase with age in both plasma and murine tissues, and in aged rats [129, 130]. Recent studies performed in animal models of venous thrombosis have also shown that increased soluble P-selectin and vein wall protein levels of P-selectin in aging mice correspond to increased thrombus burden in these animals. Experimentally, soluble P-selectin and P-selectin vein wall gene expression were significantly higher 2 days post thrombosis in 18-month-old mice compared with 2- and 11-month-old mouse groups, respectively (unpublished observation). In a previous aging study, older mice (11 months) had significantly heavier thrombus weights when compared to younger animals (2 months), and decreased thrombus inflammatory cell populations. Inflammatory cells, especially monocytes, have been noted to promote thrombolysis due to their secretion of proteolytic enzymes [44, 47, 98]. Consistently, older animals had significantly higher concentrations of MPs with TF activity than those of younger animals [131].
Moving Toward the Clinic: Novel Deep Venous Thrombosis Prophylaxis and Treatment in a Primate Model
The importance of P-selectin and its receptor PSGL-1 has been demonstrated in DVT using a primate model of stasis-induced IVC thrombosis. In this model, antibody inhibition of P-selectin or rPSGL-1-Ig inhibited inflammation and thrombosis when given prophylactically [53, 132]. Further study has demonstrated a significant dose–response relationship between rPSGL-1-Ig and thrombosis and rPSGL-1-Ig and spontaneous recanalization [62]. The peri-thrombus vein wall had decreased gadolinium enhancement (marker of inflammation) in all rPSGL-1-Ig groups compared to controls, despite no significant differences in inflammatory cell extravasation. In fact, the highest dosage produced the best inhibition of thrombosis, but was associated with the greatest inflammatory cell influx, suggesting that the prevention of thrombosis does not depend on inhibiting vein wall leukocyte influx. Importantly, these effects were observed with rPSGL-1-Ig with no systemic anticoagulation, bleeding time prolongation, thrombocytopenia, or wound healing complications.
Direct selectin inhibition also effectively treats established stasis iliac DVT in a primate model. Two days after thrombus development, baboons were treated with rPSGL-1-Ig, 4 mg/kg, LMWH or saline, and treatment continued once weekly (rPSGL-1-Ig) or daily (LMWH, saline) based on drug half-life assessment [133]. The animals were examined and killed 14 or 90 days after treatment initiation. Percent spontaneous vein reopening was increased significantly in the proximal iliac vein in rPSGL-1-Ig- and LMWH-treated animals compared to controls (Fig. 3.7). There were no differences in inflammation between groups. At 90 days after thrombosis, recanalization with iliac vein valve competence was found in the rPSGL-1-Ig- and LMWH-treated animals. Thus, rPSGL-1-Ig successfully treated established DVT as well as LMWH. The most recent studies with P-selectin inhibitory aptamers for the prophylaxis and treatment of DVT are consistent with these observations (unpublished data).

Thrombus resolution in the primate following LMWH or recombinant PSGL-1 (rPSGL-1) treatment. Representative venographic examples at baseline, day 2, day 9, and day 16 from animals treated with saline (control), rPSGL-1-Ig, or low-molecular-weight heparin (LMWH). Note the improvement in recanalization in the rPSGL-1-Ig-treated animal, compared to the saline control and even the LMWH-treated animal. The unshaded rectangle indicates the evaluated area where the thrombus was created by temporary balloon occlusion. Reprinted from [133] with permission from John Wiley & Sons Inc
We have also documented the ability of an oral P-selectin inhibitor (called PSI-697) to inhibit thrombosis and inflammation. In a rodent model of stasis-induced venous thrombosis, PSI-697 preserved normal vein wall function after thrombosis compared to the LMWH [107]. In the primate iliac vein stasis occlusion model, the oral P-selectin inhibitor begun 3 days prethrombosis improved vein patency over a 6-day period better than LMWH, again with no change in coagulation function [134]. In this study, LMWH was given in a treatment dosage, although begun prethrombosis. Vein wall inflammation was decreased, as measured by MRI after gadolinium administration in response to LMWH.
Similar findings were demonstrated with another oral P-selectin inhibitor, PSI-421. Primates treated with PSI-421 had greater percent vein reopening and less vein wall inflammation than the low-molecular-weight heparin-treated and nontreated controls at day 6 (Fig. 3.8) [136]. Microparticle TF activity (MPTFA) was significantly lower in the animals receiving PSI-421 as measured immediately after thrombosis (6 h post thrombosis on the day of thrombus formation, day 0) suggesting lower potential for thrombogenesis in these animals. PSI-421 also reduced soluble P-selectin levels versus controls at 6 h post thrombosis on day 0, and on days 2 and 6. Experimental animals in all groups showed no coagulation defects, as suggested by normal bleeding time, normal prothrombin time, and normal activated partial thromboplastin time. This study was the first to demonstrate a reduction in MPTFA associated with vein reopening and reduced vein inflammation due to oral P-selectin inhibition in a baboon model of DVT.

An oral P-selectin inhibitor decreases microparticle tissue factor activity and enhances vein recanalization. Matched proximal and distal sections of thrombosed vein from a baboon model of DVT were evaluated for percent patency posttreatment with either low-molecular-weight heparin or a P-selectin inhibitor, PSI-421, by venography (left graph). Fold changes from baseline values of microparticle tissue factor activity normalized to total microparticle number. Animals receiving PSI-421 had a significant reduction in microparticle tissue factor activity compared to controls. T + 6 day 0 = 6 h after thrombogenesis; ** p < 0.05. No difference was observed at the day 2 and day 6 time points (right graph). From [135] with permission from John Wiley & Sons Inc
Improved spontaneous thrombolysis in animals treated with rPSGL-1-Ig is similar to results found in primate, porcine, and rat models of arterial and venous thrombolysis using other methods of P-selectin inhibition [45, 133, 137, 138]. This protective effect is likely due to reductions in leukocyte–platelet interactions that lead to TF release and fibrin deposition, as these are P-selectin dependent [139]. Thus, P-selectin blockade inhibits leukocyte–platelet, leukocyte–endothelial cell, leukocyte–leukocyte, and even platelet–endothelial cell interactions; all actions that potentially decrease thrombus amplification after its initiation.
In the Clinic: P-Selectin and MPs in Patients with Deep Vein Thrombosis
Endothelial- and platelet-derived MP are elevated in patients with DVT [140]. However, MPs have been found to be present in healthy individuals and may have an anticoagulant function by promoting the generation of low amounts of thrombin, which activates protein C, supporting protein C’s anticoagulant function [141]. These findings suggest that MPs have a steady-state physiologic level and that they become elevated with thrombosis.
Elevated levels of soluble P-selectin have also been associated with DVT [139, 142–144]. Most recently, a study of soluble P-selectin levels in patients with many different thromboembolic conditions found that the level of soluble P-selectin in DVT patients before beginning heparin treatment was 88.7 ± 41 ng/ml; 7 days after heparin therapy it was 54.5 ± 28.9 ng/ml; and in 30 control patients it was 22.1 ± 8.0 ng/ml [144]. Such data support the association of both MPs and soluble P-selectin with the formation of DVT.
We have also investigated the use of a combination of biomarkers to improve the ability to predict DVT. Currently, the serum marker D-dimer can reliably exclude DVT in the presence of a low clinical probability [145, 146]. However, no marker or combinations of markers improve upon the specificity of D-dimer (approximately 50%) for the diagnosis of VTE. Thus, diagnostic duplex imaging is the only practical means to diagnose DVT today and likewise spiral CT imaging to diagnose PE. Unfortunately, such imaging is not always immediately available, is costly, and is labor intensive. Recently, 62 positive and 116 negative DVT patients prospectively received a duplex scan, were evaluated for sP-selectin, D-dimer, CRP, and MP (total, leukocyte and platelet-derived and TF-MPs) concentrations, and assigned a clinical Wells score [147]. The biomarkers, or clinical scores, that differentiated DVT positives from negatives were sP-selectin, CRP, and Wells’ score. With respect to the MP analysis, only platelet-derived MP differentiated DVT positive from negative patients. Using multivariable logistic regression, a combination of sP-selectin and Wells score could establish the diagnosis of DVT (cut-point ≥90 ng/ml + Wells ≥2), with a specificity of 96% and positive predictive value (PPV) of 100%, and could exclude DVT diagnosis (cut-point ≤60 ng/ml and Wells <2) with a sensitivity of 90%, a specificity of 33%, and a negative predictive value (NPV) of 96%. This study established a biomarker and clinical profile combination that can both confirm and exclude the diagnosis of DVT.
Finally, two human studies have solidified the importance of P-selectin in venous thrombogenesis and have suggested that elevated levels of sP-selectin are a marker for VTE. In the first of these studies, a case–control study of 116 consecutive patients and 129 matched controls underwent measurement of sP-selectin. These were patients with recurrent idiopathic VTE. The mean time between the sP-selectin measurement and the first episode of VTE was 11.5 years, and between the last VTE and the selectin measurement was 2.6 years. Using a cutoff of 55.1 ng/ml, the odds ratio = 8.5 for patients vs. controls, and the odds ratio = 11 after adjusting for factor V Leiden, prothrombin 20210A gene variant, factor VIII, homocysteine, and body mass index. In those patients with a P-selectin variant that results in lower levels of sP-selectin, the mean level of sP-selectin was 31.3 ng/ml [148]. In a second study of 544 consecutive patients with VTE, 63 patients demonstrated recurrence. Those with recurrence had sP-selectin levels of 45.8 ± 16.4 mg/dl vs. 40.1 ± 13.3 mg/dl, p = 0.006. When utilizing a sP-selectin level >75th percentile, the probability of VTE recurrence was increased by 1.7-fold [149]. Both studies suggest that high circulating levels of sP-selectin are a risk factor for recurrent VTE.
Summary
Venous thrombosis is a common and significant public health issue for which the true triggering events are not well defined in humans. Recent work has shown that links between coagulation and the inflammatory system play an important role in DVT. Venous thrombosis is both triggered by inflammation and causes localized inflammation in the thrombus and vein wall. The selectin-type leukocyte adhesion molecules such as P-selectin and their ligands bridge the interface of thrombosis and inflammation by mediating leukocyte, platelet, and endothelial interactions and processes that contribute to both thrombus formation and thrombus resolution.
The role of coagulation factors, especially TF and thrombin, are well established in venous thrombus formation. Physiologic anticoagulants and thrombolytics counteract the prothrombotic process, and the endothelial cell plays a major role in modulating the local prothrombotic milieu by the production of both pro- and antithrombotic factors. In contrast to arterial thrombosis, the contribution of the platelet to venous thrombosis is not as clear. Endothelial- and monocyte-derived procoagulant MPs contribute to venous thrombus formation.
Venous thrombus resolution is a complex process that involves plasma-derived thrombolytics, but that is also partly dependent on leukocytes. Thrombus resolution is a sterile inflammatory process that involves leukocyte processing of thrombus components including coagulation proteins and necrotic cells, a process that is mediated in part by Toll-like receptor signaling. Leukocyte-derived uPA may be central to thrombus resolution, but other leukocyte factors such as certain cytokines and MMPs contribute significantly. Monocytes are particularly important to the later stages of the resolution process. In experimental models, the postthrombotic vein wall remodeling is primarily dependent on the mechanism of thrombosis instead of the absolute thrombus size. Vein remodeling and healing following DVT require contributions from both vascular smooth muscle and endothelial cells.
Recent studies have begun to translate some of these new basic science findings in DVT into the clinical setting. These studies have demonstrated that novel biomarkers for DVT such as P-selectin may be useful to increase the specificity of DVT diagnosis, and that P-selectin inhibitors may be a potential therapy for a nonanticoagulant means of DVT prophylaxis and treatment.
References
Heit JA, Cohen AT, Anderson FJ. Estimated annual number of incident and recurrent, non-fatal venous thromboembolism (vte) events in the US. Blood. 2005;106:267a.
Hull RD, Pineo GF, Raskob GE. The economic impact of treating deep vein thrombosis with low-molecular-weight heparin: outcome of therapy and health economy aspects. Haemostasis. 1998;28 Suppl 3:8–16.
Gangireddy C, Rectenwald JR, Upchurch GR, et al. Risk factors and clinical impact of postoperative symptomatic venous thromboembolism. J Vasc Surg. 2007;45:335–41. discussion 341–33.
Meissner MH, Wakefield TW, Ascher E, et al. Acute venous disease: venous thrombosis and venous trauma. J Vasc Surg. 2007;46 Suppl S:25S–53.
Heit JA, Silverstein MD, Mohr DN, et al. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–63.
Anderson Jr FA, Wheeler HB, Goldberg RJ, et al. A population-based perspective of the hospital incidence and case-fatality rates of deep vein thrombosis and pulmonary embolism. The worcester dvt study. Arch Intern Med. 1991;151:933–8.
Beyth RJ, Cohen AM, Landefeld CS. Long-term outcomes of deep-vein thrombosis. Arch Intern Med. 1995;155:1031–7.
Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrom J. Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost. 2007;5:692–9.
Savory L, Harper P, Ockelford P. Posttreatment ultrasound-detected residual venous thrombosis: a risk factor for recurrent venous thromboembolism and mortality. Curr Opin Pulm Med. 2007;13:403–8.
Young L, Ockelford P, Milne D, Rolfe-Vyson V, McKelvie S, Harper P. Post-treatment residual thrombus increases the risk of recurrent deep vein thrombosis and mortality. J Thromb Haemost. 2006;4:1919–24.
Prandoni P, Ghirarduzzi A, Prins MH, et al. Venous thromboembolism and the risk of subsequent symptomatic atherosclerosis. J Thromb Haemost. 2006;4:1891–6.
Hong C, Zhu F, Du D, Pilgram TK, Sicard GA, Bae KT. Coronary artery calcification and risk factors for atherosclerosis in patients with venous thromboembolism. Atherosclerosis. 2005;183:169–74.
Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–93.
Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–49.
Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273–81.
Savage B, Ruggeri ZM. Selective recognition of adhesive sites in surface-bound fibrinogen by glycoprotein iib-iiia on nonactivated platelets. J Biol Chem. 1991;266:11227–33.
Ferguson JJ, Waly HM, Wilson JM. Fundamentals of coagulation and glycoprotein iib/iiia receptor inhibition. Eur Heart J. 1998;19 Suppl D:D3–9.
Dahlback B. Blood coagulation. Lancet. 2000;355:1627–32.
Davie EW, Fujikawa K, Kisiel W. The coagulation cascade: initiation, maintenance, and regulation. Biochemistry. 1991;30:10363–70.
Esmon CT. The regulation of natural anticoagulant pathways. Science. 1987;235:1348–52.
Marlar RA, Kleiss AJ, Griffin JH. Mechanism of action of human activated protein c, a thrombin-dependent anticoagulant enzyme. Blood. 1982;59:1067–72.
Corral J, Aznar J, Gonzalez-Conejero R, et al. Homozygous deficiency of heparin cofactor ii: relevance of p17 glutamate residue in serpins, relationship with conformational diseases, and role in thrombosis. Circulation. 2004;110:1303–7.
Vassalli JD, Sappino AP, Belin D. The plasminogen activator/plasmin system. J Clin Invest. 1991;88:1067–72.
Adelman B, Michelson AD, Loscalzo J, Greenberg J, Handin RI. Plasmin effect on platelet glycoprotein ib-von Willebrand factor interactions. Blood. 1985;65:32–40.
Sidelmann JJ, Gram J, Jespersen J, Kluft C. Fibrin clot formation and lysis: basic mechanisms. Semin Thromb Hemost. 2000;26:605–18.
Singh I, Burnand KG, Collins M, et al. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. 2003;107:869–75.
Hassouna HI. Laboratory evaluation of hemostatic disorders. Hematol Oncol Clin North Am. 1993;7:1161–249.
Palareti G, Cosmi B, Legnani C, et al. D-dimer testing to determine the duration of anticoagulation therapy. N Engl J Med. 2006;355:1780–9.
Ljungner H, Bergqvist D. Decreased fibrinolytic activity in the bottom of human vein valve pockets. Vasa. 1983;12:333–6.
Dano K, Andreasen PA, Grondahl-Hansen J, et al. Plasminogen activators, tissue degradation, and cancer. Adv Cancer Res. 1985;44:139–266.
Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–801.
Booth NA, Simpson AJ, Croll A, et al. Plasminogen activator inhibitor (pai-1) in plasma and platelets. Br J Haematol. 1988;70:327–33.
Eitzman DT, Westrick RJ, Nabel EG, Ginsburg D. Plasminogen activator inhibitor-1 and vitronectin promote vascular thrombosis in mice. Blood. 2000;95:577–80.
Myers Jr D, Farris D, Hawley A, Wrobleski S, Chapman A, Stoolman L, Knibbs R, Strieter R, Wakefield T. Selectins influence thrombosis in a mouse model of experimental deep venous thrombosis. J Surg Res. 2002;108:212–21.
Myers DD, Hawley AE, Farris DM, et al. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–89.
Myers DD, Wrobleski SK, Henke PK, Wakefield TW. Coagulation biology. In: Souba WW, Wilmore DW, editors. Surgical research. San Diego, CA: Academic; 2001:xxxii, 1460 p
Singh I, Smith A, Vanzieleghem B, et al. Antithrombotic effects of controlled inhibition of factor viii with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. Blood. 2002;99:3235–40.
Pierangeli SS, Barker JH, Stikovac D, et al. Effect of human igg antiphospholipid antibodies on an in vivo thrombosis model in mice. Thromb Haemost. 1994;71:670–4.
Pierangeli SS, Liu XW, Barker JH, Anderson G, Harris EN. Induction of thrombosis in a mouse model by igg, igm and iga immunoglobulins from patients with the antiphospholipid syndrome. Thromb Haemost. 1995;74:1361–7.
Cooley BC, Szema L, Chen CY, Schwab JP, Schmeling G. A murine model of deep vein thrombosis: characterization and validation in transgenic mice. Thromb Haemost. 2005;94:498–503.
Diaz JA, Hawley AE, Alvarado CM, et al. Thrombogenesis with continuous blood flow in the inferior vena cava. A novel mouse model. Thromb Haemost. 2010;104:366–75.
Day SM, Reeve JL, Myers DD, Fay WP. Murine thrombosis models. Thromb Haemost. 2004;92:486–94.
Moore R, Hawley A, Sigler R, et al. Tissue inhibitor of metalloproteinase-1 is an early marker of acute endothelial dysfunction in a rodent model of venous oxidative injury. Ann Vasc Surg. 2009;23:498–505.
Henke PK, Varga A, De S, et al. Deep vein thrombosis resolution is modulated by monocyte cxcr2-mediated activity in a mouse model. Arterioscler Thromb Vasc Biol. 2004;24:1130–7.
Myers Jr DD, Rectenwald JE, Bedard PW, et al. Decreased venous thrombosis with an oral inhibitor of p selectin. J Vasc Surg. 2005;42:329–36.
Wojcik BM, Wrobleski SK, Hawley AE, Wakefield TW, Myers Jr DD, Diaz JA. Interleukin-6: a potential target for post-thrombotic syndrome. Ann Vasc Surg. 2011;25:229–39.
Burnand KG, Gaffney PJ, McGuinness CL, Humphries J, Quarmby JW, Smith A. The role of the monocyte in the generation and dissolution of arterial and venous thrombi. Cardiovasc Surg. 1998;6:119–25.
Pierangeli SS, Liu SW, Anderson G, Barker JH, Harris EN. Thrombogenic properties of murine anti-cardiolipin antibodies induced by beta 2 glycoprotein 1 and human immunoglobulin g antiphospholipid antibodies. Circulation. 1996;94:1746–51.
Henke PK, Wakefield T. Thrombus resolution and vein wall injury: dependence on chemokines and leukocytes. Thromb Res. 2009;123 Suppl 4:S72–8.
Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. 2007;101:234–47.
Roumen-Klappe EM, Janssen MC, Van Rossum J, et al. Inflammation in deep vein thrombosis and the development of post-thrombotic syndrome: a prospective study. J Thromb Haemost. 2009;7:582–7.
Wakefield TW, Linn MJ, Henke PK, et al. Neovascularization during venous thrombosis organization: a preliminary study. J Vasc Surg. 1999;30:885–92.
Wakefield TW, Strieter RM, Schaub R, et al. Venous thrombosis prophylaxis by inflammatory inhibition without anticoagulation therapy. J Vasc Surg. 2000;31:309–24.
Henke PK, Varma MR, Deatrick KB, et al. Neutrophils modulate post-thrombotic vein wall remodeling but not thrombus neovascularization. Thromb Haemost. 2006;95:272–81.
Varma MR, Varga AJ, Knipp BS, et al. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. 2003;38:1090–8.
Eriksson EE, Karlof E, Lundmark K, Rotzius P, Hedin U, Xie X. Powerful inflammatory properties of large vein endothelium in vivo. Arterioscler Thromb Vasc Biol. 2005;25:723–8.
Esmon CT. Inflammation and thrombosis. J Thromb Haemost. 2003;1:1343–8.
Ridker PM, Buring JE, Rifai N. Soluble p-selectin and the risk of future cardiovascular events. Circulation. 2001;103:491–5.
Takada M, Nadeau KC, Shaw GD, Marquette KA, Tilney NL. The cytokine-adhesion molecule cascade in ischemia/reperfusion injury of the rat kidney. Inhibition by a soluble p-selectin ligand. J Clin Invest. 1997;99:2682–90.
McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of psgl-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:485–91.
Rauch U, Bonderman D, Bohrmann B, et al. Transfer of tissue factor from leukocytes to platelets is mediated by cd15 and tissue factor. Blood. 2000;96:170–5.
Myers Jr DD, Schaub R, Wrobleski SK, et al. P-selectin antagonism causes dose-dependent venous thrombosis inhibition. Thromb Haemost. 2001;85:423–9.
Sullivan VV, Hawley AE, Farris DM, et al. Decrease in fibrin content of venous thrombi in selectin-deficient mice. J Surg Res. 2003;109:1–7.
Myers DD, Wakefiend TW. Inflammation dependent thrombosis. Front Biosci. 2005;10:2750–7.
Walenga JM, Jeske WP, Messmore HL. Mechanisms of venous and arterial thrombosis in heparin-induced thrombocytopenia. J Thromb Thrombolysis. 2000;10 Suppl 1:13–20.
Kumar A, Villani MP, Patel UK, Keith Jr JC, Schaub RG. Recombinant soluble form of psgl-1 accelerates thrombolysis and prevents reocclusion in a porcine model. Circulation. 1999;99:1363–9.
Mesri M, Altieri DC. Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a jnk1 signaling pathway. J Biol Chem. 1999;274:23111–8.
Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood. 2002;99:3962–70.
Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000;95:1317–23.
Kirchhofer D, Tschopp TB, Steiner B, Baumgartner HR. Role of collagen-adherent platelets in mediating fibrin formation in flowing whole blood. Blood. 1995;86:3815–22.
Breimo ES, Osterud B. Generation of tissue factor-rich microparticles in an ex vivo whole blood model. Blood Coagul Fibrinolysis. 2005;16:399–405.
Hrachovinova I, Cambien B, Hafezi-Moghadam A, et al. Interaction of p-selectin and psgl-1 generates microparticles that correct hemostasis in a mouse model of hemophilia a. Nat Med. 2003;9:1020–5.
Vandendries ER, Furie BC, Furie B. Role of p-selectin and psgl-1 in coagulation and thrombosis. Thromb Haemost. 2004;92:459–66.
Jilma B, Kovar FM, Hron G, et al. Homozygosity in the single nucleotide polymorphism ser128arg in the e-selectin gene associated with recurrent venous thromboembolism. Arch Intern Med. 2006;166:1655–9.
Ruvelle BM, Scott D, Beck PJ. Single amino acid residues in the e- and p-selectin epidermal growth factor domains can determine carbohydrate binding specificity. J Biol Chem. 1996;271:16160–70.
Rao RM, Clarke JL, Ortlepp S, Robinson MK, Landis RC, Haskard DO. The s128r polymorphism of e-selectin mediates neuraminidase-resistant tethering of myeloid cells under shear flow. Eur J Immunol. 2002;32:251–60.
Yoshida M, Takano Y, Sasaoka T, Izumi T, Kimura A. E-selectin polymorphism associated with myocardial infarction causes enhanced leukocyte-endothelial interactions under flow conditions. Arterioscler Thromb Vasc Biol. 2003;23:783–8.
Wenzel K, Blackburn A, Ernst M, et al. Relationship of polymorphisms in the renin-angiotensin system and in e-selectin of patients with early severe coronary heart disease. J Mol Med. 1997;75:57–61.
Ghilardi G, Biondi ML, Turri O, Guagnellini E, Scorza R. Ser128arg gene polymorphism for e-selectin and severity of atherosclerotic arterial disease. J Cardiovasc Surg (Torino). 2004;45:143–7.
Ellsworth DL, Bielak LF, Turner ST, Sheedy 2nd PF, Boerwinkle E, Peyser PA. Gender- and age-dependent relationships between the e-selectin s128r polymorphism and coronary artery calcification. J Mol Med. 2001;79:390–8.
Mlekusch W, Exner M, Schillinger M, et al. E-selectin and restenosis after femoropopliteal angioplasty: prognostic impact of the ser128arg genotype and plasma levels. Thromb Haemost. 2004;91:171–9.
Jilma B, Marsik C, Kovar F, Wagner OF, Jilma-Stohlawetz P, Endler G. The single nucleotide polymorphism ser128arg in the e-selectin gene is associated with enhanced coagulation during human endotoxemia. Blood. 2005;105:2380–3.
Becker MD, O’Rourke LM, Blackman WS, Planck SR, Rosenbaum JT. Reduced leukocyte migration, but normal rolling and arrest, in interleukin-8 receptor homologue knockout mice. Invest Ophthalmol Vis Sci. 2000;41:1812–7.
Yang Y, Loscalzo J. Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation. 2000;101:2144–8.
Gross PL, Aird WC. The endothelium and thrombosis. Semin Thromb Hemost. 2000;26:463–78.
Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–73.
Zhou J, May L, Liao P, Gross PL, Weitz JI. Inferior vena cava ligation rapidly induces tissue factor expression and venous thrombosis in rats. Arterioscler Thromb Vasc Biol. 2009;29:863–9.
de Boer HC, Verseyden C, Ulfman LH, et al. Fibrin and activated platelets cooperatively guide stem cells to a vascular injury and promote differentiation towards an endothelial cell phenotype. Arterioscler Thromb Vasc Biol. 2006;26:1653–9.
Henke PK, Comerota AJ. An update on etiology, prevention, and therapy of postthrombotic syndrome. J Vasc Surg. 2011;53:500–9.
Meissner MH, Manzo RA, Bergelin RO, Markel A, Strandness Jr DE. Deep venous insufficiency: the relationship between lysis and subsequent reflux. J Vasc Surg. 1993;18:596–605. discussion 606–598.
Killewich LA, Macko RF, Cox K, et al. Regression of proximal deep venous thrombosis is associated with fibrinolytic enhancement. J Vasc Surg. 1997;26:861–8.
Wakefield TW, Strieter RM, Wilke CA, et al. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:258–68.
Stewart GJ. Neutrophils and deep venous thrombosis. Haemostasis. 1993;23 Suppl 1:127–40.
Varma MR, Moaveni DM, Dewyer NA, et al. Deep vein thrombosis resolution is not accelerated with increased neovascularization. J Vasc Surg. 2004;40:536–42.
Sood V, Luke CE, Deatrick KB, et al. Urokinase plasminogen activator independent early experimental thrombus resolution: Mmp2 as an alternative mechanism. Thromb Haemost. 2010;104:1174–83.
Ali T, Humphries J, Burnand K, et al. Monocyte recruitment in venous thrombus resolution. J Vasc Surg. 2006;43:601–8.
Hogaboam CM, Steinhauser ML, Chensue SW, Kunkel SL. Novel roles for chemokines and fibroblasts in interstitial fibrosis. Kidney Int. 1998;54:2152–9.
Humphries J, McGuinness CL, Smith A, Waltham M, Poston R, Burnand KG. Monocyte chemotactic protein-1 (mcp-1) accelerates the organization and resolution of venous thrombi. J Vasc Surg. 1999;30:894–9.
Henke PK, Pearce CG, Moaveni DM, et al. Targeted deletion of ccr2 impairs deep vein thombosis resolution in a mouse model. J Immunol. 2006;177:3388–97.
Barton GM. A calculated response: control of inflammation by the innate immune system. J Clin Invest. 2008;118:413–20.
Henke PK, Mitsuya M, Luke CE, et al. Toll-like receptor 9 signaling is critical for early experimental deep vein thrombosis resolution. Arterioscler Thromb Vasc Biol. 2011;31:43–9.
Modarai B, Burnand KG, Humphries J, Waltham M, Smith A. The role of neovascularisation in the resolution of venous thrombus. Thromb Haemost. 2005;93:801–9.
Waltham M, Burnand KG, Collins M, McGuinness CL, Singh I, Smith A. Vascular endothelial growth factor enhances venous thrombus recanalisation and organisation. Thromb Haemost. 2003;89:169–76.
Evans CE, Humphries J, Mattock K, et al. Hypoxia and upregulation of hypoxia-inducible factor 1{alpha} stimulate venous thrombus recanalization. Arterioscler Thromb Vasc Biol. 2010;30:2443–51.
Modarai B, Burnand KG, Sawyer B, Smith A. Endothelial progenitor cells are recruited into resolving venous thrombi. Circulation. 2005;111:2645–53.
Henke PK, Varma MR, Moaveni DK, et al. Fibrotic injury after experimental deep vein thrombosis is determined by the mechanism of thrombogenesis. Thromb Haemost. 2007;98:1045–55.
Myers Jr DD, Henke PK, Bedard PW, et al. Treatment with an oral small molecule inhibitor of p selectin (psi-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. 2006;44:625–32.
Grainger DJ, Wakefield L, Bethell HW, Farndale RW, Metcalfe JC. Release and activation of platelet latent tgfb in blood clots during dissolution with plasmin. Nature Med. 1995;1:932–7.
Deatrick KB, Eliason JL, Lynch EM, et al. Vein wall remodeling after deep vein thrombosis involves matrix metalloproteinases and late fibrosis in a mouse model. J Vasc Surg. 2005;42:140–8.
Roumen-Klappe EM, den Heijer M, van Uum SH, et al. Inflammatory response in the acute phase of deep vein thrombosis. J Vasc Surg. 2002;35:701–6.
Shbaklo H, Holcroft CA, Kahn SR. Levels of inflammatory markers and the development of the post-thrombotic syndrome. Thromb Haemost. 2009;101:505–12.
Moaveni DK, Lynch EM, Luke C, et al. Vein wall re-endothelialization after deep vein thrombosis is improved with low-molecular-weight heparin. J Vasc Surg. 2008;47:616–24.
Stenberg B, Bylock A, Risberg B. Effect of venous stasis on vessel wall fibrinolysis. Thromb Haemost. 1984;51:240–2.
Deroo S, Deatrick KB, Henke PK. The vessel wall: a forgotten player in post thrombotic syndrome. Thromb Haemost. 2010;104:681–92.
Glynn RJ, Danielson E, Fonseca FA, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–61.
Schulman S, Wiman B. The significance of hypofibrinolysis for the risk of recurrence of venous thromboembolism. Duration of anticoagulation (durac) trial study group. Thromb Haemost. 1996;75:607–11.
Crowther MA, Roberts J, Roberts R, et al. Fibrinolytic variables in patients with recurrent venous thrombosis: a prospective cohort study. Thromb Haemost. 2001;85:390–4.
Segui R, Estelles A, Mira Y, et al. Pai-1 promoter 4g/5g genotype as an additional risk factor for venous thrombosis in subjects with genetic thrombophilic defects. Br J Haematol. 2000;111:122–8.
Zoller B, Garcia de Frutos P, Dahlback B. A common 4g allele in the promoter of the plasminogen activator inhibitor-1 (pai-1) gene as a risk factor for pulmonary embolism and arterial thrombosis in hereditary protein s deficiency. Thromb Haemost. 1998;79:802–7.
Gossage JA, Humphries J, Modarai B, Burnand KG, Smith A. Adenoviral urokinase-type plasminogen activator (upa) gene transfer enhances venous thrombus resolution. J Vasc Surg. 2006;44:1085–90.
Engbers MJ, van Hylckama Vlieg A, Rosendaal FR. Venous thrombosis in the elderly: incidence, risk factors and risk groups. J Thromb Haemost. 2010;8:2105–12.
Esmon CT. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009;23:225–9.
Wilkerson WR, Sane DC. Aging and thrombosis. Semin Thromb Hemost. 2002;28:555–68.
Chopard RP, Miranda Neto MH, Biazotto W, Molinari SL. Age-related changes in the human renal veins and their valves. Ital J Anat Embryol. 1994;99:91–101.
Brooks EG, Trotman W, Wadsworth MP, et al. Valves of the deep venous system: an overlooked risk factor. Blood. 2009;114:1276–9.
Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (pai-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276–85.
Yamamoto K, Takeshita K, Shimokawa T, et al. Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci U S A. 2002;99:890–5.
Mari D, Coppola R, Provenzano R. Hemostasis factors and aging. Exp Gerontol. 2008;43:66–73.
Takeshita K, Yamamoto K, Ito M, et al. Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “klotho” mouse. Semin Thromb Hemost. 2002;28:545–54.
Hashimoto Y, Kobayashi A, Yamazaki N, Sugawara Y, Takada Y, Takada A. Relationship between age and plasma t-PA, PA-inhibitor, and PA activity. Thromb Res. 1987;46:625–33.
McDonald AP, Meier TR, Hawley AE, et al. Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. 2010;125:72–8.
Downing LJ, Wakefield TW, Strieter RM, et al. Anti-p-selectin antibody decreases inflammation and thrombus formation in venous thrombosis. J Vasc Surg. 1997;25:816–27. discussion 828.
Myers D, Wrobleski S, Londy F, et al. New and effective treatment of experimentally induced venous thrombosis with anti-inflammatory rpsgl-ig. Thromb Haemost. 2002;87:374–82.
Myers Jr DD, Wrobleski SK, Longo C, et al. Resolution of venous thrombosis using a novel oral small-molecule inhibitor of p-selectin (psi-697) without anticoagulation. Thromb Haemost. 2007;97:400–7.
Meier TR, Myers Jr DD, Wrobleski SK, et al. Prophylactic p-selectin inhibition with psi-421 promotes resolution of venous thrombosis without anticoagulation. Thromb Haemost. 2008;99:343–51.
Meier T, Myers Jr DD, Wrobleski SK, Zajkowski PJ, Hawley AE. Prophylactic p-selectin inhibition with psi-421 promotes resolution of venous thrombosis without anticoagulation. Thromb Haemost. 2008;99:343–51.
Falati S, Liu Q, Gross P, et al. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle p-selectin glycoprotein ligand 1 and platelet p-selectin. J Exp Med. 2003;197:1585–98.
Toombs CF, DeGraaf GL, Martin JP, Geng JG, Anderson DC, Shebuski RJ. Pretreatment with a blocking monoclonal antibody to p-selectin accelerates pharmacological thrombolysis in a primate model of arterial thrombosis. J Pharmacol Exp Ther. 1995;275:941–9.
Palabrica T, Lobb R, Furie BC, et al. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by p-selectin on adherent platelets. Nature. 1992;359:848–51.
Biro E, Sturk-Maquelin KN, Vogel GM, et al. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J Thromb Haemost. 2003;1:2561–8.
Blann AD, Noteboom WM, Rosendaal FR. Increased soluble p-selectin levels following deep venous thrombosis: cause or effect? Br J Haematol. 2000;108:191–3.
Yang LC, Wang CJ, Lee TH, et al. Early diagnosis of deep vein thrombosis in female patients who undergo total knee arthroplasty with measurement of p-selectin activation. J Vasc Surg. 2002;35:707–12.
Bucek RA, Reiter M, Quehenberger P, Minar E, Baghestanian M. The role of soluble cell adhesion molecules in patients with suspected deep vein thrombosis. Blood Coagul Fibrinolysis. 2003;14:653–7.
Papalambros E, Sigala F, Travlou A, Bastounis E, Mirilas P. P-selectin and antibodies against heparin-platelet factor 4 in patients with venous or arterial diseases after a 7-day heparin treatment. J Am Coll Surg. 2004;199:69–77.
Motykie GD, Zebala LP, Caprini JA, et al. A guide to venous thromboembolism risk factor assessment. J Thromb Thrombolysis. 2000;9:253–62.
Wells PS, Anderson DR, Rodger M, et al. Evaluation of d-dimer in the diagnosis of suspected deep-vein thrombosis. N Engl J Med. 2003;349:1227–35.
Ramacciotti E, Clark M, Sadeghi N, et al. Contaminants in heparin: review of the literature, molecular profiling, and clinical implications. Clin Appl Thromb Hemost. 2011;17:425–31.
Cihan AY, Jungbauer LV, Sailer T, Tengler T, Koder S, Kaider A. High levels of soluble p-selectin are associated with the risk of venous thromboembolism and the p-selectin. Blood. 2006;108:555–68.
Kyrle PA, Hron G, Eichinger S, Wagner O. Circulating p-selectin and the risk of recurrent venous thromboembolism. Thromb Haemost. 2007;97:880–3.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media New York
About this chapter
Cite this chapter
Henke, P.K., Diaz, J.A., Myers, D.D., Wakefield, T.W. (2012). Recent Insights into the Molecular and Cellular Contributions to Venous Thrombosis. In: Homeister, J., Willis, M. (eds) Molecular and Translational Vascular Medicine. Molecular and Translational Medicine. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-61779-906-8_3
Download citation
DOI: https://doi.org/10.1007/978-1-61779-906-8_3
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-61779-905-1
Online ISBN: 978-1-61779-906-8
eBook Packages: MedicineMedicine (R0)