
Abstract
The transient receptor potential (TRP) gene superfamily, which consists of 7 subfamilies with at least 28 mammalian homologues, is known to encode a wide variety of cation channels with diverse biophysical properties, activation mechanisms, and physiological functions. Recent studies have identified multiple TRP channel subtypes, belonging to the canonical (TRPC), melastatin-related (TRPM), and vanilloid-related (TRPV) subfamilies, in pulmonary arterial smooth muscle cells (PASMCs). They operate as specific Ca2+ pathways responsive to stimuli, including Ca2+ store depletion, receptor activation, reactive oxygen species, growth factors, and mechanical stress. Increasing evidence suggests that these channels play crucial roles in agonist-induced pulmonary vasoconstriction, hypoxic pulmonary vasoconstriction, smooth muscle cell proliferation, vascular remodeling, and pulmonary arterial hypertension. This chapter highlighted and discussed these putative physiological functions of TRP channels in pulmonary vasculatures. Since Ca2+ ions regulate many cellular processes via specific Ca2+ signals, future investigations of these novel channels will likely uncover more important regulatory mechanisms of pulmonary vascular functions in health and in disease states.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
- TRP channels
- store-operated calcium channels
- receptor operated calcium channels
- hypoxia
- pulmonary hypertension
1 Introduction
In vascular smooth muscle cells (VSMCs), the Ca2+ ion serves as a multifunctional messenger responsible for numerous cellular functions, ranging from muscle contraction to gene expression. Depending on the type of agonist and physiological stimulation, [Ca2+]i can be elevated by Ca2+ influx through numerous Ca2+ pathways on the plasma membrane, including voltage-gated Ca2+ channels, receptor- and store-operated Ca2+ channels, nonselective cation channels (NSCCs), and Na+-Ca2+ exchangers, as well as by Ca2+ release from the inositol 1,4,5-trisphosphate (IP3) receptor, ryanodine receptor, and nicotinic acid adenine dinucleotide phosphate (NAADP)-gated intracellular Ca2+ stores. Voltage-dependent Ca2+ pathways in VSMCs have been well characterized, but the molecular identities and physiological properties of various voltage-independent Ca2+ pathways have long been enigmatic. Recent evidence suggests that the mammalian homologues of the Drosophila transient receptor potential (TRP) protein,1 which encode a large repertoire of cation channels, are responsible for many voltage-independent Ca2+ pathways in VSMCs.
The TRP superfamily is divided into two groups of a total of seven subfamilies. Group 1 consists of the classical/canonical (TRPC), melastatin-related (TRPM), vanilloid-related (TRPV), ankyrin-related (TRPA), and no mechanoreceptor potential C NOMPC (TRPN) subfamilies; Group 2 includes the polycystin-related (TRPP) and mucolipin-related (TRPML) subfamilies.2 All TRP proteins share the common features of having six transmembrane domains, a pore-forming loop between the fifth and sixth transmembrane segments, and the highly conserved TRP domains. But, they display remarkable diversity in physiological functions, such as cation selectivity and activation mechanisms. They play critical roles in the response to most major external stimuli, including light, sound, chemicals, temperature, and touch. To date, 28 mammalian TRP homologues have been found in a wide variety of cells and tissues, and at least 10 TRPs have been identified as functional channels in VSMCs (Table 7.1). They are implicated in many different vascular functions, such as myogenic response, agonist-induced vasoconstriction, Ca2+ and Mg2+ homeostasis, VSMC proliferation, and vascular remodeling.3
2 Expression of TRP Channels in Pulmonary Artery Smooth Muscle Cells
The classical/canonical TRPC subfamily, which is comprised of seven voltage-independent NSCCs, is the best-studied TRP family in pulmonary artery smooth muscle cells (PASMCs). Multiple TRPC messenger RNA (mRNA) transcripts and proteins have been identified. A survey of the literature showed that TRPC1 and TRPC6 are the two major TRPC channels expressed in rat, mouse, and human pulmonary arteries (PAs) and PASMCs (Table 7.2). TRPC3 and TRPC4 also were frequently detected, but their expressions varied depending on species and cell preparation (freshly isolated or cultured), whereas TRPC5 and TRPC7 were generally absent. In contrast, expression of TRPC channels is different in canine PAs. Reverse-transcription polymerase chain reaction (RT-PCR) detected TRPC4, TRPC6, and TRPC7 mRNAs in canine main PAs, with TRPC4 the major expressed isoform.4 A study showed that TRPC1 and TRPC6 levels are higher in distal than in proximal PAs of rats,5 suggesting heterogeneity of TRPC expression exists in different locations along the pulmonary vascular tree.
Compared to TRPC channels, information on other TRP subfamilies in the pulmonary vasculature is scant. We previously performed a survey on the expression of TRPM and TRPV channels in deendothelialized rat intralobar PAs and aorta.6 The mRNA of TRPM2, TRPM3, TRPM4, TRPM7, and TRPM8 of the TRPM family and TRPV1, TRPV2, TRPV3, and TRPV4 of the TRPV family were detected in both PAs and aorta. The ranks of relative expression evaluated by quantitative real-time RT-PCR were TRPV4 > TRPV2 > TRPV1 >> TRPV3, and TRPM8 > TRPM4 > TRPM7 > TRPM3 > TRPM2 > TRPM5. Expression of TRPM2, TRPM8, TRPV1, and TRPV4 proteins in PAs was also verified by Western blot. Moreover, the TRPM8 agonist menthol or the TRPV4 agonist 4α-phorbol 12,13-didecanoate (4α-PDD) evoked a Ca2+ response in PASMCs. These responses could be abolished by the removal of Ca2+ or application of Ni2+ but were unaffected by nifedipine. These results indicate that multiple TRPM and TRPV channels are expressed, and at least TRPM8 and TRPV4 channels are functional Ca2+ influx pathways in PASMCs. Expression of other TRP subfamilies (TRPA, TRPP, and TRML) has not been reported in pulmonary vasculature.
3 Physiological Functions of TRP Channels in PASMCs
3.1 Store-Operated and Receptor-Operated Ca2+ Entry
Store-operated Ca 2+ entry (SOCE) is defined as the capacitative Ca2+ entry activated by the depletion of intracellular Ca2+ stores.7 Several mechanisms, including the diffusible calcium influx factor (CIF), exocytosis, and conformational coupling, have been proposed. Studies have established that the stromal interacting molecule 1 (STIM1), a transmembrane protein with an N-terminal EF-hand Ca2+-binding domain, acts as a Ca2+ sensor of endoplasmic reticulum (ER) or sarcoplasmic reticulum (SR) Ca2+ content.8 Depletion of ER/SR Ca2+ results in rearrangement of STIM1 in the form of punctae underneath the plasma membrane and subsequent activation of Store-operated cation channel (SOCC). Moreover, the homologue STIM2 may operate as another Ca2+ sensor capable of detecting a small decrease in ER/SR [Ca2+] and triggering SOCE for feedback regulation of basal cytosolic and ER Ca2+ levels.9 In vascular smooth muscle, SOCE is mostly as nonselective cation pathways, in contrast to the highly Ca2+-selective Ca2+ release-activated current (I CRAC) recorded in T lymphocytes.10 Receptor-operated Ca2+ entry (ROCE) is usually defined loosely as voltage-independent Ca2+ entry that requires ligand binding to membrane receptors for activation. In fact, the definitions of SOCE and ROCE are not mutually exclusive, and the distinction between the two pathways sometimes is rather murky. For example, an agonist binds to a Gq protein-coupled receptor and activates phospholipase Cβ (PLC-β) to generate IP3 and diacylglycerol (DAG), leading to dual activation of SOCE and ROCE.
Since their discovery, TRPC channels have been implicated as SOCC and Receptor-operated cation channels (ROCC) because they are nonselective Ca2+-permeable channels and are activated by a PLC-dependent mechanism.11 It is now clear that TRPC1, TRPC4, and TRPC5 represent a subgroup of TRPC channels activated by Ca2+ store depletion caused by inhibition of SR Ca2+-ATPase (adenosine triphosphatase) using cyclopiazonic acid (CPA) or thapsigargin. TRPC3, TRPC6, and TRPC7 form another subgroup of channels participating in ROCE activated directly by DAG independent of protein kinase C (PKC). In a heterologous expression system, members of the TRPC1/4/5 or TRPC3/6/7 subgroup can coassemble directly to form heteromeric channels, but cross association does not occur between members of the two subgroups.2,3,12 However, some studies showed that TRPC3, TRPC4, and TRPC5 can operate as both SOCC and ROCC.13 Studies found that STIM1 not only can directly interact with TRPC1, TRPC4, and TRPC5 to activate SOCE14 but also can mediate heteromerization of TRPC3 with TRPC1 and TRPC6 with TRPC4.15 These new findings raise an important question of whether SOCE and ROCE are separate Ca2+ pathways or the same pathway due to heteromerization of various TRPC channels in native cells.
Our previous study provided evidence that SOCE and ROCE are independent Ca2+ pathways in rat intralobar PASMCs, where the putative store-operated TRPC1 and receptor-operated TRPC6 are predominantly expressed.16 Direct activation of SOCE with thapsigargin and ROCE with the DAG analogue 1-oleoyl-2-acetyl-sn-glycerol (OAG) elicited distinctive cation entries that exhibited a 1,000-fold difference in their sensitivity to La3+ (IC50 of ∼0.3 μM for SOCE and ∼300 μM for ROCE). Small interfering RNA (siRNA) knockdown of TRPC1 and TRPC6 inhibited the thapsigargin- and OAG-activated cation entry, respectively. Furthermore, TRPC1 siRNA had no effect on OAG-induced cation entry, and TRPC6 siRNA did not alter the thapsigargin-induced response. These results indicate that thapsigargin-induced SOCE and OAG-mediated ROCE are mutually independent pathways, with TRPC1 and TRPC6 the major determinants of SOCE and ROCE, respectively, in rat intralobar PASMCs.
The notion that TRPC1 is critical for SOCE is consistent with other observations that TRPC1-specific antisense oligonucleotides inhibited the TRPC1 expression and blocked SOCE in cultured human PASMCs,17 and overexpression of TRPC1 in rat PAs enhanced SOCE-induced vasoconstriction.18 Moreover, the importance of TRPC6 in ROCE is supported by the observation that OAG failed to activate ROCC in PASMCs of trpc6 -/- mice.19 However, a study in cultured rat main PASMCs showed that PDGF (platelet-derived growth factor) upregulated TRPC6 and enhanced SOCE during cell proliferation. Inhibition of TRPC6 with antisense oligonucleotides reduced the amplitude of SOCE and attenuated mitogen-mediated PASMC proliferation. Hence, TRPC6 might exhibit different properties in proliferating PASMCs, perhaps due to heteromerization with other TRPC subtypes through interactions with STIM1 or Orai1.15,20 In addition, TRPC3 and TRPC4 have been reported to mediate SOCE in cultured human PASMCs,21,22 but the TRPC subtypes responsible for ROCE have not been examined in human pulmonary myocytes.
3.2 Agonist-Induced Pulmonary Vasoconstriction
As mentioned, many vasoactive agonists and growth factors can exert their effects by binding to receptors coupled to Gq protein or receptor tyrosine kinases to activate PLC-β or PLC-γ, respectively, to generate IP3 and DAG. IP3 triggers Ca2+ release from IP3-receptor-gated Ca2+ stores, leading to Ca2+ influx through SOCC, and DAG directly activates ROCC and modulates other effectors through PKC. In addition to the PLC-IP3/DAG pathways, agonists and growth factors may activate receptor-operated TRP channels by direct tyrosine phosphorylation through Src family protein tyrosine kinases, including Src and Fyn.23,24
Vasoactive agonists, including endothelin I, angiotensin II, norepinephrine, PDGF, EGF (epidermal growth factor), and ATP (adenosine triphosphate) have been shown to activate SOCE or ROCE in VSMCs. It is generally assumed that TRPC channels, by mediating SOCE and ROCE, play significant roles in agonist-induced pulmonary vasoconstriction. However, despite evidence that SOCE is able to elicit pulmonary vasoconstriction18,25 and phenylephrine-induced contractions of PAs could be inhibited partially by the nonselectively cation channel blockers SKF-96365 and Ni2+,25,26 it is unclear which subtypes of TRPC channels are participating and what contributions TRPC channels have in a particular agonist-induced pulmonary vasoconstriction. In fact, our preliminary observations suggested that agonist-induced vasoconstriction was unabated in PAs of trpc1 -/- and trpc6 -/- mice (Ref. 27 and unpublished data), similar to a previous report on phenylephrine-induced vasoconstriction in aorta and mesenteric arteries of trpc6 -/- mice.28 It is likely that agonists activate vasoconstriction through redundant mechanisms; hence, deletion of one TRP channel subtype could be effectively compensated by other Ca2+ pathways. Moreover, 5-hydroxytryptamine or serotonin (5-HT)-induced constriction of small, pressurized rat PAs is mediated through an arachidonic acid-sensitive Ca2+ influx,29 which has properties similar to the TRPV4 channels.30 These results highlight the fact that some agonists may induce ROCE and pulmonary vasoconstriction by activating NSCCs other than TRPC channels through PLC-independent signaling pathways.
3.3 Hypoxic Pulmonary Vasoconstriction
Acute reduction in alveolar O2 tension causes reversible constriction of small resistant PAs. It serves as an adaptive mechanism for diverting blood flow from poorly ventilated to better ventilated regions of the lung to improve ventilation-perfusion matching. It has been proposed that hypoxia alters the redox state of PASMCs, leading to the inhibition of voltage-gated K+ channels, membrane depolarization, activation of voltage-gated Ca2+ channels, [Ca2+]i increase, and vasoconstriction.31 Increasing evidence suggests that voltage-independent Ca2+ entry also plays a critical role in hypoxic pulmonary vasoconstriction (HPV). It was shown first in canine PAs that hypoxia activated a nisoldipine- and ryanodine-insensitive Ca2+ influx in the presence of thapsigargin or CPA32 and later in small rat PAs that the initial phase (phase I) of HPV was carried by capacitative Ca2+ influx related to a thapsigargin-sensitive store. Phase II contraction was supported by Ca2+ influx through a separate voltage-independent pathway.33 In isolated canine and rat PASMCs, hypoxia activated a dihydropyridine-insensitive Ca2+ influx, which had a pharmacological profile similar to SOCE.34,35 Antagonists of SOCC/NSCC, including SKF-96365, Ni2+, and La3+, inhibited HPV in isolated perfused rat lungs at concentrations that inhibit vasoconstrictions induced by SOCE but not by KCl.36
A study showed that the expression levels of STIM1, TRPC1, TRPC4, and TRPC6 were higher in distal than proximal PAs of rats. Enhanced expression of these proteins was associated with a higher magnitude of CPA-induced SOCE and hypoxia-induced Ca2+ response in distal PASMCs.5 Moreover, knockdown of the SR Ca2+ sensor STIM1 with siRNA in rat PASMCs abolished SOCE and hypoxia-induced Ca2+ response.37 Collectively speaking, evidence at the organ, tissue, and cell levels suggests unequivocally that SOCCs/NSCCs participate in HPV, even though the specific TRP channel has not been determined. These observations together with other studies lead to an alternative hypothesis that HPV activates Ca2+ release from ryanodine receptor-gated Ca2+ stores, causing activation of SOCE.
Direct evidence of a critical role for TRP channels in HPV emerged from the study of trpc6 -/- mice.19 The evidence includes the following: The acute phase of HPV was completely absent in isolated perfused lungs of trpc6 -/- mice; the hypoxia-induced increase in [Ca2+]i, nonselective cation influx, and membrane currents were missing in Endothelin-1 (ET-1)-primed trpc6- /- microvascular PASMCs; and hypoxia-induced Ca2+ response was rescued by expressing TRPC6 in trpc6 -/- PASMCs.It is intriguing that the hypoxia-induced activation of TRPC6 channels required priming of PASMCs with ET-1 and was mediated by DAG accumulation. These observations are congruent with the widely accepted knowledge that priming of isolated perfused lung, PAs, or PASMCs with an agonist facilitates robust hypoxic responses, but the DAG-mediated activation of TRPC6 indicates a crucial contribution of ROCE instead of the previously suggested SOCE in HPV. It is unclear whether the discrepancy depends on animal species or the presence of priming. It will be important to evaluate the DAG/TRPC6 mechanism in agonist-primed PASMCs of rats and other species for a better understanding of ROCE involvement in HPV.
The TRPC channels may contribute to HPV by providing Ca2+ for direct activation of calmodulin/myosin light chain kinase/actin-myosin interactions and by causing membrane depolarization to activate Ca2+ influx through L-type Ca2+ channels. This is consistent with the fact that both antagonists of SOCE/NSCC and L-type Ca2+ channels attenuated independently the hypoxia-induced Ca2+ response in PASMCs and HPV in isolated perfused lungs.34,36 Moreover, hypoxia-induced activation of TRPC channels may lead to local accumulation of Na+ ions, facilitating Ca2+ influx through reverse Na+-Ca2+ exchange. However, this possibility is still controversial and requires further investigation.
3.4 PASMC Growth and Proliferation
Mitogen-induced proliferation of PASMCs requires elevation of [Ca2+]i to activate Ca2+-dependent signaling pathways and Ca2+-sensitive transcription factors. Based on a series of articles published by Yuan and associates, it is now established that PASMC proliferation is associated with augmented SOCE mediated by TRPC channel upregulation.38 Depending on species, mitogens, and physiological states, most of the TRPC subtypes expressed in PASMCs, including TRPC1, TRPC3, TRPC4, and TRPC6, have been implicated as a mediator for cell proliferation. In human PASMCs, cell proliferation induced by serum and growth factors was associated with increased resting [Ca2+]i, enhanced capacitative Ca2+ entry, and upregulation of TRPC1.17,39 Downregulation of TRPC1 using antisense oligonucleotide, removal of extracellular Ca2+, and application of Ni2+ all inhibited the enhancement of cell proliferation induced by serum and growth factors, suggesting Ca2+ influx via the upregulated TRPC1 mediates PASMC growth. In another study, incubation of human PASMCs with ATP increased phosphorylation of cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB), TRPC4 expression, SOCE, and cell proliferation. Transfection of a CREB mutant abolished ATP-induced TRPC4 upregulation, and introduction of a siRNA against TRPC4 attenuated SOCE and cell proliferation activated by ATP. These data suggested that ATP exerts its mitogenic effect via CREB-dependent upregulation of TRPC4 channels. Furthermore, PDGF stimulates Signal transducer and activator of transcription 3 (STAT3) phosphorylation, leading to the upregulation of c-Jun, which activates the transcription of TRPC6, resulting in enhanced SOCE and PASMC proliferation.40,41
It is interesting to note that the mentioned studies involved different mitogens, signaling pathways, and TRPC channels; yet they all found enhancement in SOCE and cell proliferation. The simplest explanation is that an enhanced SOCE, irrespective of the TRPC subtype involved, supports the mitogen-induced PASMC proliferation in a nondiscriminatory manner. However, Ca2+ signals carried by distinctive Ca2+ channels/pathways are known to preferentially regulate specific Ca2+-sensitive transcription factors for different physiological processes, according to the signal amplitude and frequency as well as the spatial association with their effectors.42 Indeed, PGDF-induced PASMC proliferation was unaffected by the L-type Ca2+ channel inhibitor nifedipine but was inhibited by the SOCE/NSCC inhibitor SKF-96365, suggesting a specific contribution of SOCE-dependent signaling pathways.41 Hence, it is tempting to speculate that various mitogens might regulate different TRPC subtypes to modulate specific Ca2+-dependent processes at various stages in the cell cycle.
3.5 Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH), both idiopathic (IPAH) and secondary PAH, involves numerous interacting factors that lead to massive vascular remodeling, increase in vasomotor tone, and alterations in vascular reactivity. Vascular remodeling is characterized by pronounced medial and adventitial thickening due to PASMC proliferation and migration, recruitment and differentiation of progenitor cells, and synthesis of extracellular matrix. Increase in vasomotor tone is evident by the acute reduction in pulmonary arterial pressure (Ppa) in response to vasodilators, such as prostacyclin, nitric oxide, phosphodiesterase V inhibitors, K+ channel opener, and Rho kinase inhibitors. Alterations in vascular reactivity are manifested by the enhanced responsiveness to agonists, such as endothelin 1, serotonin, and angiotensin II, and the reduced relaxation to various vasodilators.
3.5.1 Chronic Hypoxia-Induced Pulmonary Hypertension
All the salient characteristics of PAH can be either directly or indirectly related to alterations of Ca2+ homeostasis in PASMCs. It has been shown in the chronic hypoxic rat model that major changes in ionic balance occur in PASMCs, including membrane depolarization, reduction in K V currents, and increase in resting [Ca2+]i. The increase in resting [Ca2+]i in PASMCs is mainly due to Ca2+ influx through voltage-independent Ca2+ channels because removal of extracellular Ca2+ reduces resting [Ca2+]i to the level of normoxic PASMCs, but inhibition of the voltage-gated Ca2+ channel with nifedipine has little effect.16,43 In the search of alternative Ca2+ entry pathways for the disturbance of resting [Ca2+]i, we found that the expression of TRPC channels in PAs is greatly altered in rats exposed to 10% O2 for 4 weeks.16 TRPC1 and TRPC6 mRNA and protein levels were more than doubled, while TRPC3 was unchanged in hypoxic PAs. Ca2+ measurements and Mn2+-quenching experiments showed that both thapsigargin-induced SOCE and OAG-induced ROCE were enhanced proportionally in hypoxic PASMCs. More importantly, inhibition of SOCE with a low concentration of La3+ (10 μM) caused a reduction in basal [Ca2+]i similar to the removal of extracellular Ca2+, but increased La3+ concentration to inhibit ROCE failed to cause additional decline in [Ca2+]i. Similar results were obtained in parallel experiments using PA rings to evaluate basal vascular tone. Since TRPC1 is responsible for SOCE in rat PASMCs,16 these results suggested that the upregulation of TRPC1 and SOCE contribute to the elevated [Ca2+]i in PASMCs and the increase in basal vascular tone of PAs of chronic hypoxic rats.
A subsequent study further extended our findings showing that TRPC1 and TRPC6 upregulation also occurred in cultured PASMCs incubated under 4% O2 for 60 h, and in nonhypoxic PASMCs overexpressing hypoxia-inducible factor 1α (HIF-1α).44 The increase of TRPC expression was absent when mice of partial HIF-1α deficiency were exposed to hypoxia. These results clearly indicate that the upregulation of TRPC channels is a direct effect of hypoxia on PASMCs mediated by HIF-1α.
However, several important questions remain to be answered. Are TRPC1 and TRPC6 essential for the development of hypoxic PAH? Are they required specifically for vascular remodeling, increased Ppa, and enhanced vasoconstriction to agonists in PAH? It had been reported that the elevation of right ventricular systolic pressure (RVSP), right heart hypertrophy, and pulmonary vascular remodeling were unaltered in chronic hypoxic trpc6 -/- mice.19 This argued against an indispensable role for TRPC6 in chronic hypoxia-induced PAH. In contrast, our preliminary study found that hypoxia-induced PAH was significantly attenuated in trpc1 -/- mice.27 This further supports the idea that the store-operated TRPC1 channels participate in the development of hypoxic PAH.
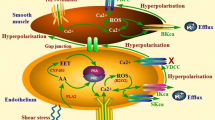
We recently have extended our investigation on the roles of TRPM and TRPV channels in chronic hypoxic PAH.45 Preliminary experiments showed that among the six subtypes of TRPV and eight subtypes of TRPM, the mechanosensitive TRPV4 was the only channel that was upregulated in PAs of rats exposed to hypoxia. Upregulation of TRPV4 was associated with enhanced Ca2+ response induced by the TRPV4 agonist 4α-PDD and hypotonicity in hypoxic PASMCs. Significant myogenic tone, sensitive to the TRPV blocker ruthenium red, was also observed in pressurized pulmonary microvessels of chronic hypoxic but not normoxic rats. Moreover, the severity of PAH was significantly attenuated in trpv4 -/- mice exposed to hypoxia. These results suggest that the mechanosensitive TRPV4 channels are upregulated in PASMCs during chronic hypoxia and play significant roles in the development of myogenic tone and PAH. A schematic is presented in Fig. 7.1 to depict our present understandings on the participation of TRPC and TRPV channels in chronic hypoxia-induced PAH.

Schematic diagram depicting the involvement of TRP channels in chronic hypoxic PAH. Hypoxia activates release of ROS (reactive oxygen species), agonists, and growth factors, leading to SOCE, ROCE, and Non-selective cation entry (NSCE) through TRPC1, TRPC6, and TRPV4 channels, respectively. Increase in intracellular [Ca2+]i enhances basal vascular tone and agonist-induced responses to cause pulmonary vasoconstriction. Increase in Ppa activates mechanosensitive TRPV4 channels to elicit myogenic response. Furthermore, HIF and other Ca2+-dependent transcriptional factors mediate gene transcription to upregulate TRPC1, TRPC6, and TRPV4 expression and promote cell proliferation and vascular remodeling
3.5.2 Idiopathic Pulmonary Arterial Hypertension
In addition to chronic hypoxic PAH, a link has also been established between TRPC and IPAH. Elevated levels of TRPC6 and TRPC3 mRNA and protein were found in cultured PASMCs isolated from patients with IPAH but not in PASMCs of patients with secondary PAH or those who were not hypertensive.46 The enhanced TRPC expression appears to be responsible for the higher activity of SOCE and cell proliferation in IPAH PASMCs21,41,46 because downregulation of TRPC6 by siRNA blocked the accelerated proliferation of these cells.46 The underlying cause for the enhanced TRPC expression in IPAH PASMCs is unclear. It is unrelated to mitogenic or autocrine factors released during PAH because elevated TRPC expression was observed in cells after multiple passages. However, it could be a specific phenotype of a population of PASMCs or myofibroblasts that are selectively expanded or recruited during the development of IPAH. It is important to note that the ET receptor antagonist bosentan suppressed TRPC6 expression and proliferation of IPAH PASMCs in the absence of agonist41; and PASMCs from normal subjects and patients with IPAH showed divergence in response to cAMP-dependent regulation on TRPC3 expression and SOCE.21 These observations suggest that aberrations in the constitutive activities of endogenous receptors and transcriptional pathways may contribute to the enhanced TRPC expression in IPAH PASMCs.
4 Conclusion
Ever since the discovery of the first TRP channel 20 years ago,1 TRP channels have continuously amazed the scientific community by their incredibly diverse complexity in structural interactions, modes of activation, molecular regulations, and physiological functions. Novel discoveries about these channels have been reported in a daily basis (>400 papers in 2008). In comparison, research on TRP channels in pulmonary vasculature is still in its infancy. The functions and regulations of most TRP channels of PASMCs, especially members of TRPV, TRPM, TRPA, TRPP, and TRPML subfamilies, have not been explored. Since Ca2+ ions regulate multiple cellular processes via specific Ca2+ signals from diverse pathways, future investigations of these unexplored TRP channels will likely uncover important regulatory pathways for pulmonary vascular functions in health and in disease states.
References
Montell C, Rubin GM (1989) Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2:1313-1323
Venkatachalam K, Montell C (2007) TRP channels. Annu Rev Biochem 76:387-417
Inoue R, Jensen LJ, Shi J et al (2006) Transient receptor potential channels in cardiovascular function and disease. Circ Res 99:119-131
Walker RL, Hume JR, Horowitz B (2001) Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am J Physiol Cell Physiol 280:C1184-C1192
Lu W, Wang J, Shimoda LA, Sylvester JT (2008) Differences in STIM1 and TRPC expression in proximal and distal pulmonary arterial smooth muscle are associated with differences in Ca2+ responses to hypoxia. Am J Physiol Lung Cell Mol Physiol 295:L104-L113
Yang XR, Lin MJ, McIntosh LS, Sham JSK (2006) Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol 290:L1267-L1276
Putney JW Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS (2001) Mechanisms of capacitative calcium entry. J Cell Sci 114:2223-2229
Roos J, DiGregorio PJ, Yeromin AV et al (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169:435-445
Brandman O, Liou J, Park WS, Meyer T (2007) STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131:1327-1339
McDonald TV, Premack BA, Gardner P (1993) Flash photolysis of caged inositol 1,4,5-trisphosphate activates plasma membrane calcium current in human T cells. J Biol Chem 268:3889-3896
Abramowitz J, Birnbaumer L (2009) Physiology and pathophysiology of canonical transient receptor potential channels. FASEB J 23:297-328
Hofmann T, Schaefer M, Schultz G, Gudermann T (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A 99:7461-7466
Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G (2000) Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem 275:17517-17526
Huang GN, Zeng W, Kim JY et al (2006) STIM1 carboxyl-terminus activates native SOC, I CRAC and TRPC1 channels. Nat Cell Biol 8:1003-1010
Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S (2007) STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat Cell Biol 9:636-645
Lin MJ, Leung GP, Zhang WM et al (2004) Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95:496-505
Sweeney M, Yu Y, Platoshyn O, Zhang S, McDaniel SS, Yuan JX-J (2002) Inhibition of endogenous TRP1 decreases capacitative Ca2+ entry and attenuates pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 283:L144-L155
Kunichika N, Yu Y, Remillard CV, Platoshyn O, Zhang S, Yuan JX-J (2004) Overexpression of TRPC1 enhances pulmonary vasoconstriction induced by capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 287:L962-L969
Weissmann N, Dietrich A, Fuchs B et al (2006) Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A 103:19093-19098
Liao Y, Erxleben C, Abramowitz J et al (2008) Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/I CRAC channels. Proc Natl Acad Sci U S A 105:2895-2900
Zhang S, Patel HH, Murray F et al (2007) Pulmonary artery smooth muscle cells from normal subjects and IPAH patients show divergent cAMP-mediated effects on TRPC expression and capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 292:L1202-L1210
Zhang S, Remillard CV, Fantozzi I, Yuan JX-J (2004) ATP-induced mitogenesis is mediated by cyclic AMP response element-binding protein-enhanced TRPC4 expression and activity in human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 287:C1192-C1201
Hisatsune C, Kuroda Y, Nakamura K et al (2004) Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279:18887-18894
Kawasaki BT, Liao Y, Birnbaumer L (2006) Role of Src in C3 transient receptor potential channel function and evidence for a heterogeneous makeup of receptor- and store-operated Ca2+ entry channels. Proc Natl Acad Sci U S A 103:335-340
McDaniel SS, Platoshyn O, Wang J et al (2001) Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol 280:L870-L880
Doi S, Damron DS, Horibe M, Murray PA (2000) Capacitative Ca2+ entry and tyrosine kinase activation in canine pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 278:L118-L130
Yang XR, Cao YN, Birnbaumer L, Sham JSK (2008) TRPC1 channels contributes to hypoxic pulmonary hypertension and right heart hypertrophy: evidence from TRPC1 knockout mice. Am J Respir Crit Care Med 177:A534
Dietrich A, Mederos YSM, Gollasch M et al (2005) Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol 25:6980-6989
Guibert C, Marthan R, Savineau JP (2004) 5-HT induces an arachidonic acid-sensitive calcium influx in rat small intrapulmonary artery. Am J Physiol Lung Cell Mol Physiol 286:L1228-L1236
Ducret T, Guibert C, Marthan R, Savineau JP (2008) Serotonin-induced activation of TRPV4-like current in rat intrapulmonary arterial smooth muscle cells. Cell Calcium 43:315-323
Archer SL, Huang J, Henry T, Peterson D, Weir EK (1993) A redox-based O2 sensor in rat pulmonary vasculature. Circ Res 73:1100-1112
Jabr RI, Toland H, Gelband CH, Wang XX, Hume JR (1997) Prominent role of intracellular Ca2+ release in hypoxic vasoconstriction of canine pulmonary artery. Br J Pharmacol 122:21-30
Robertson TP, Hague D, Aaronson PI, Ward JP (2000) Voltage-independent calcium entry in hypoxic pulmonary vasoconstriction of intrapulmonary arteries of the rat. J Physiol 525:669-680
Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT (2005) Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 288:L1059-L1069
Ng LC, Wilson SM, Hume JR (2005) Mobilization of sarcoplasmic reticulum stores by hypoxia leads to consequent activation of capacitative Ca2+ entry in isolated canine pulmonary arterial smooth muscle cells. J Physiol 563:409-419
Weigand L, Foxson J, Wang J, Shimoda LA, Sylvester JT (2005) Inhibition of hypoxic pulmonary vasoconstriction by antagonists of store-operated Ca2+ and nonselective cation channels. Am J Physiol Lung Cell Mol Physiol 289:L5-L13
Lu W, Wang J, Shimoda LA, Sylvester JT (2008) Knockdown of stromal interaction molecule 1 (STIM-1) decreases store-operated calcium entry (SOCE) and attenuates hypoxic calcium response in pulmonary artery smooth muscle cells (PASMC). FASEB J 22:1213.4
Landsberg JW, Yuan JX-J (2004) Calcium and TRP channels in pulmonary vascular smooth muscle cell proliferation. News Physiol Sci 19:44-50
Golovina VA, Platoshyn O, Bailey CL et al (2001) Upregulated TRP and enhanced capacitative Ca2+ entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol 280:H746-H755
Yu Y, Sweeney M, Zhang S et al (2003) PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am J Physiol Cell Physiol 284:C316-C330
Kunichika N, Landsberg JW, Yu Y et al (2004) Bosentan inhibits transient receptor potential channel expression in pulmonary vascular myocytes. Am J Respir Crit Care Med 170:1101-1107
Dolmetsch R (2003) Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE 2003:PE4
Shimoda LA, Sham JSK, Shimoda TH, Sylvester JT (2000) L-type Ca2+ channels, resting [Ca2+]i, and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol 279:L884-L894
Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA (2006) Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98:1528-1537
Yang XR, Hughes JM, Cao YN, Flavahan NA, Liedtke W, Sham JSK (2008) Upregulation of TRPV4 channels in pulmonary arteries (PAs) contribute to chronic hypoxia induced myogenic tone and pulmonary hypertension. FASEB J 22:1213.5 (abstract)
Yu Y, Fantozzi I, Remillard CV et al (2004) Enhanced expression of transient abstract receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci U S A 101:13861-13866
Ng LC, Gurney AM (2001) Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ Res 89:923-929
Wang J, Shimoda LA, Sylvester JT (2004) Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 286:L848-L858
McElroy SP, Gurney AM, Drummond RM (2008) Pharmacological profile of store-operated Ca2+ entry in intrapulmonary artery smooth muscle cells. Eur J Pharmacol 584:10-20
Rodat L, Savineau JP, Marthan R, Guibert C (2007) Effect of chronic hypoxia on voltage-independent calcium influx activated by 5-HT in rat intrapulmonary arteries. Pflügers Arch 454:41-51
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2010 Humana Press, a part of Springer Science+Business Media, LLC
About this paper
Cite this paper
Yang, XR., Lin, MJ., Sham, J.S.K. (2010). Physiological Functions of Transient Receptor Potential Channels in Pulmonary Arterial Smooth Muscle Cells. In: Yuan, JJ., Ward, J. (eds) Membrane Receptors, Channels and Transporters in Pulmonary Circulation. Advances in Experimental Medicine and Biology, vol 661. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60761-500-2_7
Download citation
DOI: https://doi.org/10.1007/978-1-60761-500-2_7
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-60761-499-9
Online ISBN: 978-1-60761-500-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)