
Abstract
The monogenic hypocholesterolaemic lipid disorders are classified depending on the lipid biochemical phenotype, gene involved, and mode of inheritance of the condition, together with the severity of the mutation or mutations present. These disorders may or may not be associated with clinical manifestations such as fat malabsorption, growth failure, fat-soluble vitamin deficiency, fatty liver disease, and neuro-ophthalmological dysfunction. We have reviewed the molecular basis, pathogenesis, and clinical aspects of these disorders of apolipoprotein (apo) B production and catabolism, focusing on abetalipoproteinaemia, familial hypobetalipoproteinaemia, and chylomicron retention disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Abetalipoproteinaemia
- Apolipoprotein B
- Cholesterol
- Chylomicron retention disease
- Familial hypobetalipoproteinaemia
- Genetics
- Hypobetalipoproteinaemia
- Hypocholesterolaemia
- Low-density lipoprotein
- Microsomal triglyceride transfer protein
- Molecular mechanisms
- Regulation
- Triglyceride
- Very low-density lipoprotein
Introduction
Inherited diseases of lipoprotein metabolism may give rise to marked hypocholesterolaemia with low or absent levels of betalipoproteins, depending on the gene involved and mode of inheritance of the condition, together with the severity of the mutation or mutations present [1, 2].
The most extreme form of these disorders is abetalipoproteinaemia (Online Mendelian Inheritance in Man (OMIM) 200100), a very rare recessive disorder characterised by the absence of apolipoprotein (apo) B-containing lipoproteins in plasma, leading to a variable clinical phenotype that presents in early childhood with fat malabsorption, steatorrhoea, and failure to thrive, and may include progressive neurological and ophthalmological abnormalities as the patient ages [3]. The molecular basis of this disorder is the inheritance of two mutations in the microsomal triglyceride transfer protein gene (MTTP), a chaperone protein critical for the assembly and secretion of apoB in the formation of very low-density lipoprotein (VLDL) and chylomicrons.
Low plasma concentrations of low-density lipoprotein (LDL) -cholesterol and apoB are observed in familial hypobetalipoproteinaemia (OMIM 107730), a codominant disorder of lipoprotein metabolism caused by the inheritance of a mutation in APOB, usually giving rise to a truncated apoB protein [3]. Patients are generally asymptomatic, but may be at increased risk of fatty liver disease. Inheritance of two such mutations in APOB is known as homozygous familial hypobetalipoproteinaemia and is clinically indistinguishable from abetalipoproteinaemia.
Chylomicron retention disease (OMIM 246700) is characterised by the selective absence of apoB-48 containing particles. Instead of being incorporated into chylomicrons, lipid droplets accumulate within the enterocytes [5]. Clinical findings include fat malabsorption, diarrhoea, abdominal distension, vomiting, and failure to thrive. Patients with chylomicron retention disease inherit two defective copies of secretion associated, Ras related GTPase 1B (SAR1B), the product of which is critical for the intracellular trafficking of chylomicron particles.
In recent years, additional causes of inherited hypobetalipoproteinaemia have been found in the proprotein convertase subtilisin kexin 9 (PCSK9) and the angiopoietin-like 3 (ANGPTL3) genes, although these have not been associated with any clinical symptoms. Loss-of-function mutations in PCSK9 result in reduced concentrations of LDL-cholesterol in a gene dose-dependent manner, leading to a lifetime low risk of cardiovascular disease [6]. Mutations in ANGPTL3 are associated with recessive familial combined hypolipidaemia, characterised by a reduction in all plasma lipids, including high-density lipoprotein (HDL)-cholesterol [7, 8].
This chapter reviews the molecular basis, pathogenesis, and clinical aspects of these disorders of apoB production and catabolism, focusing on abetalipoproteinaemia, familial hypobetalipoproteinaemia, and chylomicron retention disease.
History
Abetalipoproteinaemia was originally named Bassen–Kornzweig syndrome, after the two physicians who in 1950 described the clinical association of peripheral blood acanthocytosis, atypical retinitis pigmentosa, and ataxia [9]. An autosomal recessive mode of inheritance was suggested, but it was not until 1958 that the observation of low levels of serum cholesterol was made, and in 1960 the absence of beta-migrating lipoproteins by electrophoresis (betalipoproteins) described, leading to the name of the disease being changed to abetalipoproteinaemia [10]. The distinction was then made between abetalipoproteinaemia and homozygous familial hypobetalipoproteinaemia , whereby carriers of familial hypobetalipoproteinaemia have marked hypocholesterolaemia while carriers of abetalipoproteinaemia do not [11].
In 1987, a truncated apoB was found in the plasma of a family with familial hypobetalipoproteinaemia [12], leading to a four-nucleotide deletion being identified in the APOB gene [13]. The finding of absent MTTP in hepatic and intestinal microsomes in 1992 suggested that defects in this protein were the cause of abetalipoproteinaemia [14], with mutations subsequently identified in the MTTP gene the following year [15, 16].
Chylomicron retention disease was originally named Anderson’s disease, after the physician who in 1961 described an infant with fat malabsorption and fat-laden enterocytes on histology, in whom chylomicrons were absent from plasma after meals, and plasma lipids and fat-soluble vitamin levels were low [17]. It was subsequently shown that in Anderson’s disease, the enterocytes reacted intensely to monoclonal antibodies to apoB-48, but not to those selectively reactive with apoB-100 [18]. In 1991, the APOB gene was excluded as a cause of Anderson’s disease [19]. In 2003, mutations in the SAR1B gene were identified in patients with Anderson’s disease and chylomicron retention disease, revealing that they were, in fact, the same disease [20].
Epidemiology
Data from the Framingham Offspring Study identified persistent hypobetalipoproteinaemia in 1.9 % of over 3800 individuals, and a truncated apoB species causing familial hypobetalipoproteinaemia in only one of these subjects [21]. This gives an estimated prevalence of the condition at ~ 1 in 3000, taking into account that the immunoblot testing method used does not detect circulating plasma apoB of a size less than 30 % of the full-length protein. The prevalence of abetalipoproteinaemia and chylomicron retention disease is unknown, but these conditions appear to be extremely rare ( < 1 in 1 million).
Heterozygous PCSK9 nonsense mutations can be found in ~ 2 % of Africans and African-Americans [22, 23], which predicts homozygosity in ~ 1 in 10,000 in these populations.
Etiology and Pathogenesis
Abetalipoproteinaemia
Patients with abetalipoproteinaemia carry two defective copies of the MTTP gene on chromosome 4q22–24. The role of MTTP in this disorder was first implicated in 1992, when its activity was not detected in intestinal biopsies of patients with abetalipoproteinaemia [14]; mutations in the MTTP gene were subsequently described [16]. MTTP encodes an 894 amino acid protein, which forms a heterodimer with the ubiquitous endoplasmic reticulum enzyme protein disulfide isomerise (PDI) [24]. The function of the MTTP heterodimer is to facilitate the transfer of lipids to nascent apoB by a shuttle mechanism [24]; lack of MTTP activity results in insufficient lipidation of nascent apoB and targets the apoB to a degradation pathway [25], preventing the secretion of triglyceride-rich lipoproteins. Mutations in MTTP associated with abetalipoproteinaemia may disrupt production of the normal MTTP protein, disrupt its binding with the PDI subunit, or affect MTTP’s lipid transfer activity [26–28].
Familial Hypobetalipoproteinaemia
In familial hypobetalipoproteinaemia , mutations in the APOB gene on chromosome 2p23–24 either abolish the expression of apoB or interfere with the translation of full-length apoB leading to formation of prematurely truncated apoB forms [1, 30–32]. These apoB truncations have traditionally been named according to the centile system that also gave the name to apoB-48. The majority of mutations are nucleotide substitutions and deletions in exon 26, which at over 7500 nucleotides is one of the largest exons in the human genome. Several missense mutations in the N-terminal βα1 domain of apoB causing familial hypobetalipoproteinaemia have also been described [33–35]. The mutation R463W was shown to cause impaired secretion of VLDL by impaired endoplasmic reticulum exit and enhanced binding of the mutant protein to MTTP [33]. It is worth noting that missense mutations in the LDL-receptor-binding domain in the carboxyl-terminus of apoB cause familial ligand-defective apoB-100, a form of familial hypercholesterolaemia [36].
In patients with familial hypobetalipoproteinaemia , truncated forms of apoB are produced at lower rates (about 25 %) than apoB-100 [37]. The secretion rate was shown to be reduced by about 1.4 % for each 1 % of apoB truncated [38]. However, for every 10 % decrease in apoB length, there is a 13 % reduction in the lipoprotein core volume, indicating that the lipid content of secreted apoB-containing lipoproteins is decreased as apoB is shortened [39]. Truncated apoB species shorter than apoB-30 are not detectable in plasma; this appearing to be the minimum length of apoB that is required for MTTP-dependent lipoprotein assembly. Shorter apoB species are unable to acquire sufficient lipid, leading to intracellular degradation rather than secretion [40]. In addition, clearance of the truncated apoB species is faster than the clearance of apoB-100, particularly for the longer truncations such as apoB-89 that contain the LDL-receptor-binding domain, resulting in enhanced LDL-receptor-binding [41].
Linkage analysis in families with familial hypobetalipoproteinaemia without mutations in APOB has identified chromosomes 3p21.1–22 and 10q25.1–10q26.11 as susceptibility loci, but the genes responsible remain to be identified [42, 43].
Chylomicron Retention Disease
Mutations in SAR1B, a member of the Sar1-adenosine diphosphate (ADP)-ribosylation factor family of small GTPases that control the intracellular trafficking of proteins, are the cause of chylomicron retention disease [20]. SAR1B is needed for the fusion of the intestine specific pre-chylomicron transport vesicle to the Golgi apparatus, allowing transport of chylomicrons through the cellular secretory pathways [44]. Mutations in SAR1B result in the inability to secrete chylomicrons, resulting in the accumulation of lipid droplets within the enterocytes.
Other Molecular Causes of Hypobetalipoproteinaemia
PCSK9 is an important regulator of plasma LDL-cholesterol concentrations [45]. It is a protease that binds to the LDL-receptor and targets it for lysosomal degradation within hepatocytes. Gain-of-function missense mutations in PCSK9 can cause a severe autosomal dominant form of hypercholesterolaemia [46]. In contrast, nonsense and loss-of-function PCSK9 missense mutations increase the number of LDL-receptors on the cell surface and, therefore, the number of LDL particles able to be internalised by the cell, reducing both circulating LDL-cholesterol concentrations and coronary heart disease risk (Fig. 14.1).

Distribution of plasma LDL cholesterol levels and incidence of coronary heart disease, according to the presence or absence of a PCSK9 142X or PCSK9 679X allele. a the distribution of plasma LDL-cholesterol levels at baseline among 3278 black subjects who did not carry a PCSK9 nonsense mutation (top) compared to the 85 subjects who carried either PCSK9 142X or PCSK9 679X. b the percentage of participants from these two groups who had no evidence of coronary heart disease (CHD) at baseline and in whom CHD developed during 15 years of follow-up. LDL low-density lipoprotein, PCSK9 proprotein convertase subtilisin kexin 9. (Request permission, Cohen 2007) [5]
ANGPTL3 gene mutations are associated with a combined hypolipidaemia. The function of ANGPTL3 appears to be the reversible inhibition of lipase activity, involving endothelial lipase [47], lipoprotein lipase [48], or hepatic lipase [49]. The disruption of ANGPTL3 production would therefore increase lipolysis, enhancing clearance of lipoproteins and decreasing circulating lipid concentrations.
Classification
Lipoprotein disorders causing primary hypocholesterolaemia—abetalipoproteinaemia and hypobetalipoproteinaemia—are classified depending on the lipid biochemical phenotype, gene involved, and mode of inheritance of the condition, together with the severity of the mutation or mutations present (Table 14.1).
Clinical and Physical Findings
Abetalipoproteinaemia
Abetalipoproteinaemia is associated with multisystem manifestations. Patients with abetalipoproteinaemia typically present in childhood with failure to thrive, growth failure, malabsorption of fat, acanthocytosis, and low plasma cholesterol and vitamin E concentrations [50]. Later in life, retinitis pigmentosa, spinocerebellar ataxia, and myopathy have complicated most of the cases.
Gastrointestinal manifestations of abetalipoproteinaemia include steatorrhoea and fat-soluble vitamin deficiency. Fat malabsorption is a central feature of abetalipoproteinaemia and is usually observed in the neonatal period with diarrhoea, vomiting, and failure to thrive. The severity of the intestinal symptoms relates to the fat content of the diet, and usually decreases with age, in part, due to the avoidance of dietary fat in these patients [51]. A yellow colour of the duodenal mucosa has been observed on endoscopy as a result of intestinal lipid accumulation [52]. A characteristic intestinal histology shows normal villi with enterocytes that are distended with lipid droplets (Fig. 14.2).

Haematoxylin and eosin-stained light micrograph of the duodenal biopsy from a patient with homozygous familial hypobetalipoproteinaemia, showing marked cytoplasmic microvacuolization of enterocytes (magnification, × 400). (Reprinted with permission, Vongsuvanh [81])
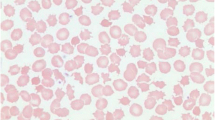
Haematological manifestations of abetalipoproteinaemia include acanthocytosis (Fig. 14.3) with acanthocytes comprising 50 % or more of circulating erythrocytes [51]. Of interest, normal erythrocytes become acanthocytic after transfusion into abetalipoproteinaemia patients and circulate in plasma [50]. Acanthocytosis in abetalipoproteinaemia could be a result of either vitamin E deficiency or an altered membrane lipid composition. Other abnormalities include low erythrocyte sedimentation rates, decreased red cell survival [53], anaemia, hyperbilirubinaemia and haemolysis [54], and increased international normalised ratio due to vitamin K deficiency [55].

Acanthocytes in abetalipoproteinaemia and familial hypobetalipoproteinaemia. a ‘Normal’ subject. b homozygous familial hypobetalipoproteinaemia. c and d abetalipoproteinaemia. e and f two different subjects with heterozygous familial hypobetalipoproteinaemia carrying apoB-6.9 mutation
Hepatic manifestations of abetalipoproteinaemia include abnormal liver transaminases with hepatomegaly. Liver biopsies have shown marked hepatic steatosis that may or may not be associated with increased liver transaminase concentrations [50]. Steatosis can progress to steatohepatitis and fibrosis [56], and, importantly, cirrhosis has been reported in abetalipoproteinaemia [57].
Neuromuscular manifestations of abetalipoproteinaemia typically begin in the first or second decade of life, affecting both the central and peripheral nervous system, with either upper or lower motor neuron abnormalities or both. Neurological signs related to the deficiency of vitamin E include the progressive loss of deep tendon reflexes, vibratory sense and proprioception, muscle weakness and, eventually, a Friedrich’s-like ataxia [58].
Ophthalmological manifestations of abetalipoproteinaemia are variable with the most prominent being an atypical pigmentation of the retina characterised by small, irregularly distributed white spots on fundoscopy [59]. Although visual acuity can be affected during the first decade, many patients are asymptomatic until adulthood, with loss of night vision and/or colour vision occurring early in the course of disease. Patients develop annular scotomas with macular sparing that slowly enlarge with progression of the retinopathy. Without treatment, complete visual loss can occur.
Familial Hypobetalipoproteinaemia
Patients with heterozygous APOB-linked familial hypobetalipoproteinaemia are often asymptomatic; however, most develop fatty liver (Figs. 14.4 and 14.5), and mild acanthocytosis and fat malabsorption can occur [60, 61]. The clinical and biochemical features of familial hypobetalipoproteinaemia in homozygous and compound heterozygous form are, in general, indistinguishable from those of abetalipoproteinaemia [62]. However, the clinical features appear to depend on the apoB truncation length, with longer truncations (i.e., greater than apoB-48), as well as those with rare missense mutations who may be asymptomatic, having a milder phenotype.

Liver biopsy showing severe macrovesicular steatosis from a patient with heterozygous familial hypobetalipoproteinaemia (H&E, 100 ×). PT indicates portal tract

Liver proton nuclear magnetic resonance spectroscopy (1H MRS) in a normal and familial hypobetalipoproteinaemia subject. Liver 1H MRS is shown for a normal subject (upper panel), and a heterozygous familial hypobetalipoproteinaemia subject with 28 % liver fat (lower panel). The area for water and fat peaks are labelled as ‘I:’. Percentage liver fat was calculated by 100*fat/(water + fat)
Familial hypobetalipoproteinaemia might represent a longevity syndrome and be associated with cardiovascular protection due to resistance to atherosclerosis due to a lower lifetime exposure to atherogenic apoB-containing lipoproteins [63]. However, the cardioprotective effects of familial hypobetalipoproteinaemia in humans have, to date, only been shown using the surrogate markers carotid intima-media thickness and distal common carotid arterial wall stiffness [64].
Chylomicron Retention Disease
Chylomicron retention disease presents shortly after birth with diarrhoea, fat malabsorption, and failure to thrive, with vomiting and abdominal distension often present [5, 65]. Acanthocytosis is rare and may be transient. Hepatomegaly and hepatic steatosis may develop in some patients, but do not correlate with liver transaminase levels, along with fat-soluble vitamin deficiencies and their corresponding manifestations. In contrast to abetalipoproteinaemia and familial hypobetalipoproteinaemia , cirrhosis has not been reported in chylomicron retention disease. A white colour of the duodenal mucosa has been observed on endoscopy with histology like in abetalipoproteinaemia showing vacuolisation of enterocytes in intestinal villi of normal structure (Figs. 14.6 and 14.7).

Endoscopy of a chylomicron retention disease patient. Upper endoscopy reveals a white duodenal mucosa in chylomicron retention disease (a) compared with normal subject (b). (Reproduced with permission from Peretti et al. [5])

Histology of a jejunal biopsy from a chylomicron retention disease patient. Left panel: photomicrograph of haematoxylin–eosin staining showing vacuolization of enterocytes and well-preserved villous structure. The distribution of vacuolization, which corresponds to lipid droplets, is heterogeneous: Fat filled enterocytes (black arrow) in the upper part of the villus are associated with normal enterocytes in the crypts (white arrow) (× 20). Inset: Higher magnification (× 100) of the same patient’s biopsy. Right panel: Electronic microscopy. The pictures show the apical pole of the enterocytes exhibiting well-preserved microvilli (black arrow), numerous chylomicrons (CM) and fat droplets of homogenous size gorging the cytoplasm (Cy). The intercellular membranes demonstrate a complete juxtaposition of intercellular membranes where lipid particles are absent (white arrow) (× 4000). (Reproduced with permission from Peretti et al. [5])
Approach to the Patient
The usual biochemical trigger for the investigation of genetic abetalipoproteinaemia and hypobetalipoproteinaemia is the finding of marked hypocholesterolaemia with plasma LDL-cholesterol and apoB concentrations below the fifth percentile for age and sex [1]. A personal and family history should be taken and a physical examination conducted. Secondary causes of hypobetalipoproteinaemia should be excluded (see differential diagnosis below) and clinical manifestations such as fat malabsorption, growth failure, fat-soluble vitamin deficiency, fatty liver disease, and neuro-ophthalmological dysfunction sought. A suggested algorithm for the investigation of marked hypocholesterolaemia is shown in Fig. 14.8.

The diagnosis of genetic abetalipoproteinaemia and hypobetalipoproteinaemia. LDL-C low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, apoB apolipoprotein B, TG triglyceride
Laboratory Tests
A full fasting lipid profile including total cholesterol, HDL-cholesterol, triglyceride, LDL-cholesterol, and apoB should be performed where genetic abetalipoproteinaemia or hypobetalipoproteinaemia is suspected.
Patients with abetalipoproteinaemia or homozygous familial hypobetalipoproteinaemia will have very low plasma total cholesterol and generally low-triglyceride concentrations. LDL-cholesterol, when measured by direct methods, and apoB, will be absent or very low. Vitamin E levels will also be very low, and acanthocytosis may be observed on peripheral blood smear (Fig. 14.3).
Patients with heterozygous familial hypobetalipoproteinaemia typically have plasma LDL-cholesterol and apoB concentrations that are one quarter to one third of normal. The reasons for these lower-than-expected levels may include decreased hepatic secretion of the apoB-containing lipoproteins, or the up-regulation of the LDL-receptor, resulting in an enhanced clearance rate for VLDL and LDL particles produced by the normal allele [37].
Subjects who carry a single MTTP mutation (i.e. abetalipoproteinaemia ‘carriers’) may have normal plasma lipids or may have LDL-cholesterol and apoB concentrations similar to those seen in heterozygous familial hypobetalipoproteinaemia [66].
In chylomicron retention disease, total cholesterol, LDL-cholesterol, and HDL-cholesterol are low, but triglyceride levels are generally normal. The low plasma LDL- and HDL-cholesterol are a consequence of low rates of apoB-100 and apoA-I production [67]. High creatine kinase (4–5x normal) may be seen from infancy, along with deficiencies in fat-soluble vitamins [5].
Molecular Tests
Molecular testing of MTTP, APOB, SAR1B, PCSK9, and ANGPTL3 genes is available in specialist laboratories [68]. Target exonic regions in genomic DNA should be amplified by polymerase chain reaction, ensuring at least 20 base pairs of flanking intronic sequence is included in order to capture any potential splice site mutations. Sequencing should be bidirectional and, if possible, testing of the patient’s parents is recommended to confirm that where two mutations are identified, that they originate from two different chromosomes.
In patients with abetalipoproteinaemia where the inheritance pattern is unclear or MTTP mutations unable to be identified, then the APOB gene should also be sequenced given the clinical and biochemical similarities with homozygous familial hypobetalipoproteinaemia . Likewise, in patients with homozygous familial hypobetalipoproteinaemia, the MTTP gene could be sequenced in the event where APOB mutations are unable to be found. Alternatively, high-throughput sequencing technology is emerging as a means for screening multiple genes for mutations; massively parallel sequencing could potentially analyse a panel of the genes associated with low cholesterol [69]. This may prove more cost-effective than traditional Sanger sequencing, particularly where multiple genes may need to be sequenced and when the APOB gene is involved, which needs over 40 primer sets to cover the whole coding region.
Western blotting can be used as a screening method for mutations in APOB, to detect truncated apoB species that are > 30 % of full-length protein size. DNA sequencing of the region where the mutation is estimated to occur can then be performed. However, truncated apoB species shorter than apoB-30 are not detectable in plasma, so if Western blotting fails to detect an apoB truncation, then sequencing of the first 30 % of the APOB gene (exons 1–25) should be performed.
Differential Diagnosis
Illnesses and diseases associated with secondary causes of hypobetalipoproteinaemia include severe chronic liver disease, chronic pancreatitis, cystic fibrosis, end-stage renal disease, hyperthyroidism, cachexia, and malabsorption [32, 51]. A vegan diet is associated with ~ 50 % of the general population levels of plasma LDL-cholesterol and triglyceride [70].
Prognosis, Clinical Course, and Complications
Abetalipoproteinaemia and Homozygous Familial Hypobetalipoproteinaemia
The impact on prognosis of age at diagnosis, commencement of a low-fat diet and vitamin replacement therapy, and genotype is variable in abetalipoproteinaemia and homozygous familial hypobetalipoproteinaemia [3, 4, 71]. Early treatment with high-oral doses of vitamin E and A, which are thought to bypass the intestinal chylomicron pathway via the portal circulation, can reduce the potential severity of neuropathy and retinopathy [71–75]. Patients need to be followed regularly for evaluation of symptoms, complications, and to monitor compliance with therapy. A relative paucity of data makes it difficult to predict clinical outcomes based on MTTP or APOB genotype.
Heterozygous Familial Hypobetalipoproteinaemia
Although the majority of familial hypobetalipoproteinaemia heterozygotes are asymptomatic, most have increased liver transaminases and hepatic steatosis, the long-term consequences of which are unknown [60, 61, 76–78]. Familial hypobetalipoproteinaemia heterozygotes have three- to fivefold greater liver fat content compared to control subjects with no difference in adiposity or insulin resistance [61, 76, 78, 79]. It would seem prudent to monitor biochemically and by imaging techniques the livers of these individuals given a potential increased risk of progression to cirrhosis, particularly in the presence of known risk factors, such as alcohol, caloric excess, and liver injury [32, 77, 80, 81].
Chylomicron Retention Disease
Patients with chylomicron retention disease get better within a few days or weeks with a low-fat diet [5]. No relationship has been found between liver transaminases, hepatomegaly, and hepatic steatosis . Neurological manifestations include hyporeflexia and loss of proprioception in teenagers through to ataxia, myopathy, and sensory neuropathy in adults.
Treatment
Abetalipoproteinaemia
The cornerstone of treatment for abetalipoproteinaemia is dietary modification and the replacement of fat-soluble vitamins [1, 50, 51, 71]. A low-fat diet eliminates steatorrhoea and allows absorption of other nutrients essential for growth and development. Oral vitamin E supplementation (100–300 mg/kg/day orally) in abetalipoproteinaemia is recommended to halt the progression of the neurological disease; however, despite this high dose, serum levels do not fully normalise [50, 51]. Most adult patients with abetalipoproteinaemia who have not received supplements exhibit neuro-ophthalmological complications [50]. Supplementation with a combination of vitamins E and A has been shown to be effective in reducing retinal degeneration [72]. Patients treated with very large oral doses of vitamin E from the age of 16 months do not develop neurological or retinal features, while progression is halted or sometimes even reversed in older patients who already show symptoms of neurological dysfunction [83]. Although serum vitamin E is usually undetectable in untreated abetalipoproteinaemia, supplementation results in trace concentrations, with normal levels in adipose tissue [84]. Erythrocyte and platelet vitamin E have also been used to assess tissue vitamin E status [85]. Oral supplementation of two to four times the recommended daily allowance of vitamin A normalises serum levels. Vitamin D deficiency is not a consistent finding; however, vitamin D replacement should be considered in abetalipoproteinaemia patients, along with other supplementary nutrients such as iron and folate.
There is a need for novel therapeutic approaches to abetalipoproteinaemia as vitamin therapy alone fails to completely control or cure this disease.
Familial Hypobetalipoproteinaemia
In familial hypobetalipoproteinaemia homozygotes, dietary fats should be restricted to prevent steatorrhoea. Long-term high-dose vitamin E and A supplementation should prevent or slow progression of the neuromuscular and retinal degenerative disease [51, 72]. Moderate-dose vitamin E supplementation in familial hypobetalipoproteinaemia heterozygotes with low serum vitamin E concentrations has been recommended to prevent neurological disease [30]. However, this recommendation has been called into question [85].
Chylomicron Retention Disease
There are no specific recommendations for follow-up or treatment of chylomicron retention disease , with therapeutic regimens based on those recommended for abetalipoproteinaemia . Vomiting, diarrhoea, and abdominal distension improve on a low-fat diet of polyunsaturated fatty acids and supplementation with fat-soluble vitamins, particularly vitamin E, can prevent neurological complications [5].
Conclusion
The monogenic hypocholesterolaemic lipid disorders are classified depending on the lipid biochemical phenotype, gene involved, and mode of inheritance of the condition, together with the severity of the mutation or mutations present. These disorders may or may not be associated with clinical manifestations such as fat malabsorption, growth failure, fat-soluble vitamin deficiency, fatty liver disease, and neuro-ophthalmological dysfunction. We have reviewed the molecular basis, pathogenesis, and clinical aspects of these disorders of apoB production and catabolism, focusing on abetalipoproteinaemia, familial hypobetalipoproteinaemia, and chylomicron retention disease.
References
Hooper AJ, van Bockxmeer FM, Burnett JR. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit Rev Clin Lab Sci. 2005;42(5):515–45.
Hooper AJ, Burnett JR. Update on primary hypobetalipoproteinaemia.Curr Atheroscler Rep2014;16(7):423.
Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: abetalipoproteinaemia - Update 2014. Eur J Hum Genet. 2014 Oct 22. doi:10.1038/ ejhg.2014.224.
Burnett JR, Bell DA, Hooper AJ, Hegele RA. Clinical utility gene card for: familial hypobetalipoproteinaemia (APOB). Eur J Hum Genet. 2012;20(8). doi:10.1038/ejhg.2012.85.
Peretti N, Sassolas A, Roy CC, Deslandres C, Charcosset M, Castagnetti J, et al. Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J Rare Dis. 2010;5:24.
Cohen JC, Boerwinkle E, Mosley TH Jr., Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72.
Hooper AJ, Burnett JR. Recent developments in the genetics of LDL deficiency. Curr Opin Lipidol. 2013;24(2):111–5.
Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363(23):2220–7.
Bassen FA, Kornzweig AL. Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood. 1950;5(4):381–7.
Sturman RM. The Bassen-Kornzweig syndrome: 18 years in evolution. J Mt Sinai Hosp N Y. 1968;35(5):489–517.
Wolff JA, Bauman WA. Studies concerning acanthocytosis: a new genetic syndrome with absent beta lipoprotein. Am J Dis Child. 1961;102:478.
Young SG, Bertics SJ, Curtiss LK, Witztum JL. Characterization of an abnormal species of apolipoprotein B, apolipoprotein B-37, associated with familial hypobetalipoproteinemia. J Clin Invest. 1987;79(6):1831–41.
Young SG, Northey ST, McCarthy BJ. Low plasma cholesterol levels caused by a short deletion in the apolipoprotein B gene. Science. 1988;241(4865):591–3.
Wetterau JR, Aggerbeck LP, Bouma ME, Eisenberg C, Munck A, Hermier M, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258(5084):999–1001.
Sharp D, Blinderman L, Combs KA, Kienzle B, Ricci B, Wager-Smith K, et al. Cloning and gene defects in microsomal triglyceride transfer protein associated with abetalipoproteinaemia. Nature. 1993;365(6441):65–9.
Shoulders CC, Brett DJ, Bayliss JD, Narcisi TM, Jarmuz A, Grantham TT, et al. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum Mol Genet. 1993;2(12):2109–16.
Anderson CM, Townley RR, Freemanm, Johansen P. Unusual causes of steatorrhoea in infancy and childhood. Med J Aust. 1961;48(2):617–22.
Bouma ME, Beucler I, Aggerbeck LP, Infante R, Schmitz J. Hypobetalipoproteinemia with accumulation of an apoprotein B-like protein in intestinal cells. Immunoenzymatic and biochemical characterization of seven cases of Anderson’s disease. J Clin Invest. 1986;78(2):398–410.
Pessah M, Benlian P, Beucler I, Loux N, Schmitz J, Junien C, et al. Anderson’s disease: genetic exclusion of the apolipoprotein-B gene in two families. J Clin Invest. 1991;87(1):367–70.
Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34(1):29–31.
Welty FK, Lahoz C, Tucker KL, Ordovas JM, Wilson PW, Schaefer EJ. Frequency of apoB and apoE gene mutations as causes of hypobetalipoproteinemia in the Framingham offspring population. Arterioscler Thromb Vasc Biol. 1998;18(11):1745–51.
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37(2):161–5.
Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis. 2007;193(2):445–8.
Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44(1):22–32.
Pariyarath R, Wang H, Aitchison JD, Ginsberg HN, Welch WJ, Johnson AE, et al. Co-translational interactions of apoprotein B with the ribosome and translocon during lipoprotein assembly or targeting to the proteasome. J Biol Chem. 2001;276(1):541–50.
Ohashi K, Ishibashi S, Osuga J, Tozawa R, Harada K, Yahagi N, et al. Novel mutations in the microsomal triglyceride transfer protein gene causing abetalipoproteinemia. J Lipid Res. 2000;41(8):1199–204.
Rehberg EF, Samson-Bouma ME, Kienzle B, Blinderman L, Jamil H, Wetterau JR, et al. A novel abetalipoproteinemia genotype. Identification of a missense mutation in the 97-kDa subunit of the microsomal triglyceride transfer protein that prevents complex formation with protein disulfide isomerase. J Biol Chem. 1996;271(47):29945–52.
Wang J, Hegele RA. Microsomal triglyceride transfer protein (MTP) gene mutations in Canadian subjects with abetalipoproteinemia. Hum Mutat. 2000;15(3):294–5.
Miller SA, Burnett JR, Leonis MA, McKnight CJ, van Bockxmeer FM, Hooper AJ. Novel missense MTTP gene mutations causing abetalipoproteinaemia. Biochim Biophys Acta 2014;1842(10):1548–54.
Linton MF, Farese RV, Young SG. Familial hypobetalipoproteinemia. J Lipid Res. 1993;34(4):521–41.
Schonfeld G, Lin X, Yue P. Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci. 2005;62(12):1372–8.
Tarugi P, Averna M. Hypobetalipoproteinemia: genetics, biochemistry, and clinical spectrum. Adv Clin Chem. 2011;54:81–107.
Burnett JR, Shan J, Miskie BA, Whitfield AJ, Yuan J, Tran K, et al. A novel nontruncating APOB gene mutation, R463W, causes familial hypobetalipoproteinemia. J Biol Chem. 2003;278(15):13442–52.
Burnett JR, Zhong S, Jiang ZG, Hooper AJ, Fisher EA, McLeod RS, et al. Missense mutations in APOB within the betaalpha1 domain of human APOB-100 result in impaired secretion of apoB and apoB-containing lipoproteins in familial hypobetalipoproteinemia. J Biol Chem. 2007;282(33):24270–83.
Zhong S, Magnolo AL, Sundaram M, Zhou H, Yao EF, Leo ED, et al. Nonsynonymous mutations within APOB in human familial hypobetalipoproteinemia—evidence for feedback inhibition of lipogenesis and post-endoplasmic reticulum degradation of apolipoprotein B. J Biol Chem. 2010;285(9):6453–64.
Boren J, Ekstrom U, Agren B, Nilsson-Ehle P, Innerarity TL. The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J Biol Chem. 2001;276(12):9214–8.
Elias N, Patterson BW, Schonfeld G. Decreased production rates of VLDL triglycerides and apoB-100 in subjects heterozygous for familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 1999;19(11):2714–21.
Parhofer KG, Barrett PHR, Aguilar-Salinas CA, Schonfeld G. Positive linear correlation between the length of truncated apolipoprotein B and its secretion rate: in vivo studies in human apoB-89, apoB-75, apoB-54.8, and apoB-31 heterozygotes. J Lipid Res. 1996;37(4):844–52.
Yao Z, McLeod RS. Synthesis and secretion of hepatic apolipoprotein B-containing lipoproteins. Biochim Biophys Acta. 1994;1212(2):152–66.
Yao ZM, Blackhart BD, Linton MF, Taylor SM, Young SG, McCarthy BJ. Expression of carboxyl-terminally truncated forms of human apolipoprotein B in rat hepatoma cells. Evidence that the length of apolipoprotein B has a major effect on the buoyant density of the secreted lipoproteins. J Biol Chem. 1991;266(5):3300–8.
Parhofer KG, Barrett PHR, Bier DM, Schonfeld G. Lipoproteins containing the truncated apolipoprotein, Apo B-89, are cleared from human plasma more rapidly than Apo B-100-containing lipoproteins in vivo. J Clin Invest. 1992;89(6):1931–7.
Sherva R, Yue P, Schonfeld G, Neuman RJ. Evidence for a quantitative trait locus affecting low levels of apolipoprotein B and low density lipoprotein on chromosome 10 in Caucasian families. J Lipid Res. 2007;48(12):2632–9.
Yuan B, Neuman R, Duan SH, Weber JL, Kwok PY, Saccone NL, et al. Linkage of a gene for familial hypobetalipoproteinemia to chromosome 3p21.1–22. Am J Hum Genet. 2000;66(5):1699–704.
Siddiqi SA, Gorelick FS, Mahan JT, Mansbach CM 2nd. COPII proteins are required for Golgi fusion but not for endoplasmic reticulum budding of the pre-chylomicron transport vesicle. J Cell Sci. 2003;116(Pt 2):415–27.
Lambert G, Sjouke B, Choque B, Kastelein JJ, Hovingh GK. The PCSK9 decade. J Lipid Res. 2012;53(12):2515–24.
Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6.
Shimamura M, Matsuda M, Yasumo H, Okazaki M, Fujimoto K, Kono K, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27(2):366–72.
Shan L, Yu XC, Liu Z, Hu Y, Sturgis LT, Miranda ML, et al. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 inhibit lipoprotein lipase activity through distinct mechanisms. J Biol Chem. 2009;284(3):1419–24.
Nakajima K, Kobayashi J, Mabuchi H, Nakano T, Tokita Y, Nagamine T, et al. Association of angiopoietin-like protein 3 with hepatic triglyceride lipase and lipoprotein lipase activities in human plasma. Ann Clin Biochem. 2010;47(Pt 5):423–31.
Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma M, Wetterau JR. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr. 2000;20:663–97.
Kane JP, Havel RJ. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler K, et al., editors. The metabolic and molecular bases of inherited disease. 8th edn. New York: McGraw-Hill; 2001. pp. 2717–52.
Delpre G, Kadish U, Glantz I, Avidor I. Endoscopic assessment in abetalipoproteinemia (Bassen-Kornzweig-syndrome). Endoscopy. 1978;10(1):59–62.
Kayden HJ, Bessis M. Morphology of normal erythrocyte and acanthocyte using Nomarski optics and the scanning electron microscope. Blood. 1970;35(4):427–36.
Mier M, Schwartz SO, Boshes B. Acanthrocytosis, pigmentary degeneration of the retina and ataxic neuropathy: a genetically determined syndrome and associated metabolic disorder. Blood. 1960;16:1586–608.
Caballero FM, Buchanan GR. Abetalipoproteinemia presenting as severe vitamin K deficiency. Pediatrics. 1980;65(1):161–3.
Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolaemia. J Hepatol 2014;61(4):891–902.
Partin JS, Partin JC, Schubert WK, McAdams AJ. Liver ultrastructure in abetalipoproteinemia: evolution of micronodular cirrhosis. Gastroenterology. 1974;67(1):107–18.
Tanyel MC, Mancano LD. Neurologic findings in vitamin E deficiency. Am Fam Physician. 1997;55(1):197–201.
Cogan DG, Rodrigues M, Chu FC, Schaefer EJ. Ocular abnormalities in abetalipoproteinemia. A clinicopathologic correlation. Ophthalmology. 1984;91(8):991–8.
Ogata H, Akagi K, Baba M, Nagamatsu A, Suzuki N, Nomiyama K, et al. Fatty liver in a case with heterozygous familial hypobetalipoproteinemia. Am J Gastroenterol. 1997;92(2):339–42.
Schonfeld G, Patterson BW, Yablonskiy DA, Tanoli TS, Averna M, Elias N, et al. Fatty liver in familial hypobetalipoproteinemia: triglyceride assembly into VLDL particles is affected by the extent of hepatic steatosis. J Lipid Res. 2003;44(3):470–8.
Schonfeld G. Familial hypobetalipoproteinemia: a review. J Lipid Res. 2003;44(5):878–83.
Glueck CJ, Gartside P, Fallat RW, Sielski J, Steiner PM. Longevity syndromes: familial hypobeta and familial hyperalpha lipoproteinemia. J Lab Clin Med. 1976;88(6):941–57.
Sankatsing RR, Fouchier SW, de Haan S, Hutten BA, de Groot E, Kastelein JJ, et al. Hepatic and cardiovascular consequences of familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2005;25:1979–84.
Peretti N, Roy CC, Sassolas A, Deslandres C, Drouin E, Rasquin A, et al. Chylomicron retention disease: a long term study of two cohorts. Mol Genet Metab. 2009;97(2):136–42.
Narcisi TM, Shoulders CC, Chester SA, Read J, Brett DJ, Harrison GB, et al. Mutations of the microsomal triglyceride-transfer-protein gene in abetalipoproteinemia. Am J Hum Genet. 1995;57(6):1298–310.
Ouguerram K, Zair Y, Kasbi-Chadli F, Nazih H, Bligny D, Schmitz J, et al. Low rate of production of apolipoproteins B100 and AI in 2 patients with Anderson disease (chylomicron retention disease). Arterioscler Thromb Vasc Biol. 2012;32(6):1520–5.
Tarugi P, Averna M, Di Leo E, Cefalu AB, Noto D, Magnolo L, et al. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007;195(2):e19–27.
Johansen CT, Dubé JB, Loyzer MN, MacDonald A, Carter DE, McIntyre AD, et al. LipidSeq: a nextgeneration clinical resequencing panel for monogenic dyslipidemias. J Lipid Res. 2014;55(4):765–72.
De Biase SG, Fernandes SF, Gianini RJ, Duarte JL. Vegetarian diet and cholesterol and triglycerides levels. Arq Bras Cardiol. 2007;88(1):35–9.
Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis. 2008;3:19.
Chowers I, Banin E, Merin S, Cooper M, Granot E. Long-term assessment of combined vitamin A and E treatment for the prevention of retinal degeneration in abetalipoproteinaemia and hypobetalipoproteinaemia patients. Eye. 2001;15(Pt 4):525–30.
Muller DP, Lloyd JK. Effect of large oral doses of vitamin E on the neurological sequelae of patients with abetalipoproteinemia. Ann N Y Acad Sci. 1982;393:133–44.
Muller DP, Lloyd JK, Wolff OH. The role of vitamin E in the treatment of the neurological features of abetalipoproteinaemia and other disorders of fat absorption. J Inherit Metab Dis. 1985;8(Suppl 1):88–92.
Runge P, Muller DP, McAllister J, Calver D, Lloyd JK, Taylor D. Oral vitamin E supplements can prevent the retinopathy of abetalipoproteinaemia. Brit J Ophthalmol. 1986;70(3):166–73.
Tanoli T, Yue P, Yablonskiy D, Schonfeld G. Fatty liver in familial hypobetalipoproteinemia: roles of the APOB defects, intra-abdominal adipose tissue, and insulin sensitivity. J Lipid Res. 2004;45(5):941–7.
Tarugi P, Lonardo A, Ballarini G, Erspamer L, Tondelli E, Bertolini S, et al. A study of fatty liver disease and plasma lipoproteins in a kindred with familial hypobetalipoproteinemia due to a novel truncated form of apolipoprotein B (APO B-54.5). J Hepatol. 2000;33(3):361–70.
Whitfield AJ, Barrett PHR, Robertson K, Havlat MF, van Bockxmeer FM, Burnett JR. Liver dysfunction and steatosis in familial hypobetalipoproteinemia. Clin Chem. 2005;51(1):266–9.
Visser ME, Lammers NM, Nederveen AJ, van der Graaf M, Heerschap A, Ackermans MT, et al. Hepatic steatosis does not cause insulin resistance in people with familial hypobetalipoproteinaemia. Diabetologia. 2011;54(8):2113–21.
Hooper AJ, Adams LA, Burnett JR. Genetic determinants of hepatic steatosis in man. J Lipid Res. 2011;52(4):593–617.
Lonardo A, Tarugi P, Ballarini G, Bagni A. Familial heterozygous hypobetalipoproteinemia, extrahepatic primary malignancy, and hepatocellular carcinoma. Dig Dis Sci. 1998;43(11):2489–92.
Welty FK. Hypobetalipoproteinaemia and abetalipoproteinaemia.Curr Opin Lipidol 2014;25(3):161–8.
Muller DP. Vitamin E and neurological function. Mol Nutr Food Res. 2010;54(5):710–8.
Kayden HJ, Hatam LJ, Traber MG. The measurement of nanograms of tocopherol from needle aspiration biopsies of adipose tissue: normal and abetalipoproteinemic subjects. J Lipid Res. 1983;24(5):652–6.
Clarke MW, Hooper AJ, Headlam HA, Wu JH, Croft KD, Burnett JR. Assessment of tocopherol metabolism and oxidative stress in familial hypobetalipoproteinemia. Clin Chem. 2006;52(7):1339–45.
Vongsuvanh R, Hooper AJ, Coakley JC, Macdessi JS, O’Loughlin EV, Burnett JR, et al. Novel mutations in abetalipoproteinaemia and homozygous familial hypobetalipoproteinaemia. J Inherit Metab Dis. 2007;30(6):990.
Acknowledgements
This work was supported by the National Health and Medical Research Council Project Grant 1010133 (to AJH and JRB) and a Practitioner Fellowship from the Royal Perth Hospital Medical Research Foundation (to JRB).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Humana Press
About this chapter
Cite this chapter
Hooper, A., Burnett, J. (2015). Genetic Abetalipoproteinaemia and Hypobetalipoproteinaemia. In: Garg, A. (eds) Dyslipidemias. Contemporary Endocrinology. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60761-424-1_14
Download citation
DOI: https://doi.org/10.1007/978-1-60761-424-1_14
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-60761-423-4
Online ISBN: 978-1-60761-424-1
eBook Packages: MedicineMedicine (R0)