
Abstract
Monogenic hypercholesterolemias (MHs) are a heterogeneous group of diseases characterized by a high concentration of low-density lipoprotein (LDL) cholesterol and a high risk of premature cardiovascular disease (CVD). There are autosomal dominant and recessive MHs, although the latter are very rare. The most common and best studied MH is familial hypercholesterolemia (FH), an autosomal codominant disease caused by mutations in the LDL receptor gene (LDLR). FH is genetically heterogeneous because mutations in APOB and proprotein convertase subtilisin/kexin type 9 (PCSK9) are accompanied by an indistinguishable phenotype. FH is underdiagnosed and undertreated today, so it is a medical priority to improve the detection and treatment of affected subjects. Appropriate lipid-lowering treatment significantly improves the prognosis of these patients. In this chapter, MH classification, main clinical forms, etiology, clinical presentation, and management are reviewed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Familial hypercholesterolemia
- Autosomal dominant hypercholesterolemia
- LDL receptor
- Familial defective apo B-100
- PCSK9
- LDLRAP1
- APOE
Definition and Classification
Monogenic hypercholesterolemias (MHs) are a heterogeneous group of single-gene defects with Mendelian transmission in the family characterized by elevated plasma low-density lipoprotein (LDL) cholesterol levels and very high risk for premature atherosclerotic disease, especially coronary heart disease (CHD) [1] (Table 10.1).
Approximately, one in 200–500 people is affected by MH in most populations explored so far, so this group of diseases is among the most frequent genetic metabolic defects [2]. The study of MH has provided decisive evidence of the linkage between high LDL cholesterol concentration and atherosclerosis development in humans. Furthermore, the metabolic and genetic characterization of MH in the past decades has supplied crucial information about the cholesterol homeostasis, metabolism, and regulatory pathways. The scientific information generated around the MH has contributed decisively to the development of many drugs in common use today, such as hydroxy-methyl-glutaryl coenzyme A reductase (HMG-CoAR) inhibitors or statins, which have contributed to change the evolution of arteriosclerotic disease. The information generated around the MH remains very active today, and the discovery of new genes responsible for high LDL cholesterol is promoting the development of very promising new drugs, such as inhibitors of proprotein convertase subtilisin/kexin type 9 (PCSK9) , and others.
MH traditionally included two common diseases of autosomal dominant inheritance: familial hypercholesterolemia (FH), due to mutations in the LDL receptor gene (LDLR, Online Mendelian Inheritance in Man (OMIM) 143890) causing isolated high LDL cholesterol (type IIa hyperlipoproteinemia) and familial combined hyperlipidemia (FCHL) of unknown etiology (OMIM 144250) usually associated with mixed hyperlipidemia secondary to high concentrations of very LDLs (VLDL) and LDL particles (type IIb hyperlipoproteinemia); as well as several rare recessive diseases such as sitosterolemia (OMIM 210250) and autosomal recessive hypercholesterolemia (ARH;OMIM 603813) [3]. However, FH is heterogeneous from the genetic standpoint, and mutations in the LDLR are found only in 60–80 % of patients with a clinical diagnosis of FH [4]. Functional mutations in other genes produce indistinguishable clinical phenotypes of FH, including a missense mutation p.(Arg3527Gln) located in the LDLR-binding domain of apolipoprotein (apo) B-100 that produces familial defective apo B-100 (FDB, OMIM 144010) [5]; gain-of-function mutations in PCSK9, a protein that binds to the LDL receptor inducing its degradation along with the LDL particle, and are termed FH3 (OMIM 603776) [6, 7]; and, a deletion in a codon of APOE (p.Leu167del) has been recently associated with autosomal dominant hypercholesterolemia in two different studies [8, 9]. Very high levels of lipoprotein(a) ((Lp(a)), named as hyperLp(a), is a single-gene condition also causing MH [10]. Except for the presence of apo(a) in the LDL particle surface, Lp(a) is essentially indistinguishable from LDL, so Lp(a) carries in some cases substantial amounts of cholesterol. Lp(a) varies from 0.1 to 300 mg/dL among individuals due to the LPA gene locus which codes for the apo(a) [11]. LPA kringle IV-2 sequence is present in a variable number of identical repeated copies (from 3 to > 60) and the number of kringle IV-2 repeats is inversely correlated with the Lp(a) concentration [12]. Lp(a) is discussed in extension in another chapter of this book.
The genetic heterogeneity of isolated high LDL cholesterol in MH, the clinical similarities among them, which make their clinical diagnosis in most cases indistinguishable, their common high cardiovascular risk, and their uniform response to the different lipid lowering treatments, mean that all of them are referred to collectively as FH, regardless of the presence of mutations in the LDLR [13, 14]. Hence, FH should be defined as a group of monogenic genetic defects resulting in severely elevated serum LDL cholesterol concentrations with autosomal codominant transmission pattern of inheritance.
In contrast, most cases of FCHL, the most common genetic form of hyperlipidemia identified in survivors of myocardial infarction [15, 16], do not correspond to a monogenic disease, rather they are complex genetic diseases resulting from the interaction of multiple genetic and environmental factors mainly overweight, obesity , saturated fat- and sugar-enriched diets, and physical inactivity [17]. Many families with FCHL combine adipose tissue dysfunction [18, 19], insulin resistance [20], hepatic overproduction of VLDL particles [21], and peripheral slow clearance of triglycerides-rich lipoproteins [22]. Different association and linkage studies have shown more than 40 different genes associated with FCHL that have been recently reviewed by Brouwers et al. [23] although with great differences among studies. Therefore, FCHL is currently considered to be a complex phenotype consequence of multiple genetic defects, each one mostly with minor effects, which differ among families, and among populations. In over 50 % of the families with the clinical diagnosis of FCHL, all effected members are overweight or obese, and with high frequency they develop diabetes mellitus with time [24]. This predisposition within certain families, to develop mixed hyperlipidemia only in the presence of increased body fat deposits, we have proposed to be named as “adiposity-related familial hyperlipidemia” [25]. However, the actual definition of FCHL, familial transmission of high apo B levels with high plasma total cholesterol and/or triglycerides, also includes some forms in which the lipid phenotype is largely determined by a single gene [23]. These less common forms of autosomal dominant FCHL demonstrate that, in some cases, the FCHL phenotype is largely determined by a single genetic defect, and behaves as an MH [8, 26] (Table 10.1).
Familial Hypercholesterolemia
As defined above, FHs are a group of monogenic genetic defects resulting in severely elevated serum LDL cholesterol concentrations with autosomal codominant transmission pattern of inheritance. Hence, patients with two defective alleles (FH homozygotes or compound heterozygotes) have much higher LDL cholesterol than those with one mutant allele (FH heterozygotes). Lifelong elevated plasma concentrations of LDL cholesterol are responsible for the major clinical manifestation of FH: premature CHD and extravascular cholesterol deposits as tendon xanthomas or corneal arcus [27]. The frequency of FH heterozygotes, (1 in 500 individuals) is much higher than FH homozygotes ( < 1 in 1 million). However, some populations such as French Canadians [28], Afrikaners in South Africa [29], Lebanese, and Finns [30] have a much higher prevalence due to a founder effect.
Etiology and Pathogenesis
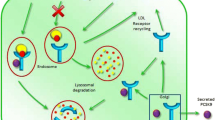
FH caused by mutations in LDLR was the first disease of lipid metabolism to be genetically defined, the best known at present time, and the most frequent MH in most countries around the world. Different mutations in the LDLR affect the LDL receptor protein functionality [27]. LDL receptor in cell membranes binds LDL and the complexes enter the cell by endocytosis [31]. The LDL receptor is synthesized as a 120-kDa precursor protein, which is converted to a mature form of apparent molecular mass of 160 kDa. The increase in molecular mass is correlated with extensive N- and O-glycosylation in the Golgi apparatus during transfer to the cell surface [32]. In addition to the glycosylation, in the endoplasmic reticulum (ER), the 21-amino-acid signal peptide of the LDL receptor is cleaved to give rise to a mature receptor. The transmembrane LDL receptor (glycoprotein of 839 amino acids) is present at the surface of most cell types and mediates the transport of lipoproteins containing apo B or apo E into cells, through receptor-mediated endocytosis. The mature LDL receptor reaches the cell surface and is directed towards clathrin-coated pits where it binds to apo B- and apo E-enriched lipoproteins via its extracellular domain [33]. The lipoprotein–LDL receptor complex is endocytosed and migrates to endosomes. At the acidic pH of the lysosomes, LDL is released, allowing LDL receptor to return to the membrane and entering into a new cycle [34] (Fig. 10.1). Although the LDL receptor was initially thought to play the single role of helping to achieve cholesterol homeostasis, its expression in neurons suggests it may also play other functional roles [35, 36].

Schematic representation of the itinerary of the low-density lipoprotein (LDL) receptor in human cells. The LDL receptor is synthesized in the endoplasmic reticulum (ER) as a precursor of apparent molecular weight of 120 kd and transported at the Golgi complex where the N-linked carbohydrates are processed. Once transferred to the surface of the cell, the receptor recognizes the apolipoprotein B-100 component of the LDL. Binding leads to cellular uptake and lysosomal degradation of the LDL by receptor-mediated endocytosis. This uptake process satisfies the cholesterol needs of the cells, and hence keeps cholesterol synthesis suppressed
Cholesterol homeostasis is among the most regulated processes in biology. Cellular cholesterol balance is achieved by both synthesis and uptake through LDL receptor. When cellular cholesterol levels rise, LDLR transcription is reduced and de novo synthesis is inhibited. When cellular cholesterol storage is depleted, LDLR transcription is activated and de novo synthesis activated. Two major transcriptional LDLR regulation pathways have evolved in mammals to coordinate responses to both elevated and reduced cellular cholesterol content: the sterol regulatory element-binding proteins (SREBPs) and the liver X receptors (LXRs). SREBP-2 promotes the expression of the LDLR, thereby increasing LDL uptake and cholesterol delivery to cells [37]. The SREBP-2 precursor protein resides in the ER and is transported to the Golgi apparatus under low intracellular cholesterol content, where it undergoes proteolytic processing. The mature SREBP protein translocates to the nucleus and switches on the transcription of LDLR, as well as other genes involved in cholesterol biosynthesis, including HMGCoAR and HMGCoA synthase (HMGCoAS) [38]. An additional modulator of LDL receptor-dependent cholesterol uptake independent of the SREBP pathway is the LXR [39]. LXR induces expression of E3 ubiquitin ligase inducible degrader of the LDL receptor (Idol), which in turn catalyzes the ubiquitination of the LDL receptor and targets it for degradation [40].
Characterization of Idol-deficient cells has also provided insights into to the functional relationship between PCSK9 and Idol pathways. PCSK9 and Idol share the same protein substrates, but PCSK9 is still able to induce LDL receptor degradation in Idol−/− cells, suggesting that Idol and PCSK9 may be complementary but independent pathways [41]. PCSK9 is secreted into plasma and binds to the first domain (EGF-A) of epidermal growth factor (EGF) homology repeats of LDL receptor [42–44]. Although the C-terminal domain of PCSK9 is not required for LDL receptor binding, it is required for LDL receptor degradation [33]. The complete mechanism by which PCSK9 binding to the LDL receptor targets the receptor for degradation is not understood. Although PCSK9 is a protease, it does not cleave LDL receptor, nor is the proteolysis of LDL receptor required to downregulate LDLR. The LDL receptor–PCSK9 complex is internalized via clathrin-mediated endocytosis and then routed to lysosomes via a mechanism that does not require ubiquitination and is distinct from the autophagy and proteosomal degradation pathways [45].
Posttranscriptional regulation of LDLR expression is also a major determinant of lipoprotein metabolism. LDLR adaptor protein 1 (LDLRAP1) is a protein required for the efficient activity of LDL receptor. It has been demonstrated that LDLRAP1 is essential for the efficient internalization of the LDL–LDL receptor complex and cells from patients with ARH fail to internalize the LDL receptor because they carry two defective alleles of LDLRAP1, a gene that encodes a specific clathrin adaptor protein [46]. LDLRAP1 is an endocytic sorting adaptor that actively participates in the internalization of the LDL–LDL receptor complex, possibly enhancing the efficiency of its packaging into the endocytic vesicles [47]. LDLRAP1 is required not only for internalization of the LDL-LDL receptor complex but also for efficient binding of LDL to the receptor. LDLRAP1 stabilizes the associations of the receptor with LDL and with the invagination portion of the budding pit, thereby increasing the efficiency of LDL internalization [48].
The LDLR Gene
The LDLR is mapped to chromosome 19p13.1-13.3 and spans 45 kb and contains 18 exons and 17 introns encoding the six functional domains of the mature protein: signal peptide, ligand-binding domain, EGF-like, O-linked sugar, transmembrane, and cytoplasmic domain [49, 50] (Fig. 10.2). The human LDLR complementary DNA (cDNA) and gene were cloned and characterized in 1984 and 1985, respectively [51, 52]. The gene sequencing of the LDLR suggested that the LDL receptor is a mosaic protein built up of exons shared with different proteins, and it therefore belongs to several supergene families [52].

Schematic representation of the five domains in the structure of the human low-density lipoprotein (LDL)-receptor protein and their corresponding exons in the LDLR gene. Red dots represent the six cysteine residues that form three disulfide bonds in each tandem structurally homologous repeats or class A repeats in the ligand-binding domain of the LDL receptor protein
Exon 1 encodes a hydrophobic sequence of 21 amino acids that correspond to the signal peptide, which is cleaved from the protein into the ER during the translocation process. Around 4.5 % of the total LDLR mutations described including frameshift, missense, and nonsense sequence variants have been located in this exon (http://www.ucl.ac.uk/fh; http://www.umd.necker.fr).
Exons 2–6 encode the ligand-binding domain, a cysteine-rich sequence of seven tandem structurally homologous repeats of 40 amino acids each, which is responsible for binding lipoproteins. The structure of the ligand-binding domain has been partially elucidated. Each repeat contains a cluster of negatively charged amino acids, Asp-X-Ser-Asp-Glu and six cysteine residues that form three disulfide bonds [53–55]. Binding of lipoproteins to the LDL receptor appears to be mediated by an interaction between acidic residues in the LDL receptor binding domain and basic residues of apo E and apo B-100 [53, 56]. Repeats R3–R7 are necessary for LDL binding (apo B-100-mediated), whereas remnant lipoproteins binding (apo E-mediated) is impaired only when R5 is deleted. Repeats R4 and R5 are sufficient to bind to apo E-phospholipids vesicles [54]. We have proposed a new mechanism for the release of LDL particles in the endosome; it is based on the instability of repeat 5 at endosome low pH and low Ca2+ [57]. Under these conditions, repeat 5 is unable to bind Ca2+ and appears in an unfolded conformation not expected to bind LDL particles. In the ligand-binding domain, 40 % of the total allelic variants associated with FH have been found to date (http://www.ucl.ac.uk/fh; http://www.umd.necker.fr).
Exons 7–14 encode a region that shares 33 % sequence identity to the human EGF gene. This domain consists of a 411-amino-acid sequence, encoded by exons 7–14. Like the ligand-binding domain, this region also contains three repeats of 40–50 amino acids with cysteine-rich sequences. The first two repeats, designated A and B and encoded by exons 7 and 8, are contiguous and separated from the third repeat, C encoded by exon 14, by a 280-amino-acid sequence that contains five copies of the conserved motif Tyr-Trp-Thr-Asp, encoded by exons 9–13. PCSK9 binds to EGF-A repeat, decreasing receptor recycling and increasing degradation [43]. The EGF-like domain is required for the acid-dependent dissociation of the LDL particles from the LDL receptor and clathrin-coated pits during receptor recycling. When the EGF-like domain is deleted from the LDL receptor, the receptor can no longer bind LDL particles but it still binds lipoproteins that contain apo E [58]. The majority of FH mutations described (55 % of total) have been associated with the EGF homology region.
Exon 15 encodes an LDL receptor domain of 58 amino acids rich in Thr and Ser residues. The function of this domain is unknown, but it has been observed that this region serves as an attachment site for O-linked carbohydrate chains and it is thought that it plays a role in the stabilization of the receptor. This domain shows minimal sequence conservation among six species analyzed, and Davis et al. reported that deletion of clustered O-linked carbohydrates does not impair function and turnover of human LDL receptor in Chinese hamster ovary (CHO) cells transfected with the human LDLR gene [59]. However, from the analysis of LDL receptors in CHO mutant cells with defective uridine diphosphate (UDP)-galactose and UDP-N-acetylgalactosamine 4-epimerase, Kingsley et al. proposed that O-linked carbohydrate chains may be crucial for receptor stability [60]. A total of 41 allelic variants within exon 15 are registered in LDLR databases.
Exon 16 and the 5′-end of exon 17 encode a domain of 22 hydrophobic amino acids that is essential for anchoring the LDL receptor to the cell membrane.
The cytoplasmic domain of the LDL receptor, that compromises 50-amino-acid residues, is encoded by the remainder 3′ region of the exon 17 and the 5′ end of the exon 18 [27]. This domain contains two sequence signals for targeting the LDL receptor to the surface and for localizing the receptor in coated pits [61]. This domain is the most conserved region of the LDL receptor, which is more than 86 % identical among six species [27]. Only a few allelic variants, 6 % of the total, have been identified within these domains.
The DNA motifs essential for the transcriptional regulation of the LDLR are located within 177 bp of the proximal promoter. The LDL receptor production is tightly regulated by a sophisticated feedback mechanism that controls the transcription of the LDLR in response to variations in the intracellular sterol concentration and the cellular demand for cholesterol [62]. The promoter region contains all the cis-acting elements for basal expression and sterol regulation and includes three imperfect direct repeats of 16 bp each, repeats 1–3. Repeats 1 and 3 contain binding sites for Sp1 transcription factor, and contribute to the basal expression of the gene, requiring the contribution of the repeat 2 for a strong expression. Repeat 2 contains a regulatory element, sterol regulatory element (SRE)-1, that enhances transcription when the intracellular sterol concentration is low through interaction with SREBP [63]. Several naturally occurring mutations have been mapped to the transcriptional regulatory elements of the LDLR.
Nowadays, over 1500 naturally occurring LDLR mutations have been described in FH patients (http://www.ucl.ac.uk/fh; http://www.umd.necker.fr). The LDLR mutations can produce defects in transcription, posttranscription processes, translation, and posttranslation processes. FH mutations have been classified into five classes depending on phenotypic behavior of mutant protein [64]. Class 1 mutations are known as “null alleles,” which fail to produce immune-precipitable LDL receptor protein. Most of them are due to LDLR promoter deletion, rearrangements, frameshift, nonsense, or splicing mutations in a way that messenger RNA (mRNA) is not produced [64]. Class 2 mutations are transport-defective alleles which encode for proteins that cannot adopt an adequate tridimensional structure after being synthesized and keep them blocked, completely or partially (2A and 2B, respectively) in transport process between ER and Golgi apparatus. This defect is caused, usually, by missense mutations or small deletions in LDLR avoiding partially or completely the folding of the protein. These mutations are the most common at the LDLR locus [64]. Class 3 mutations are binding-defective alleles which encode for LDL receptor that are synthesized and transported to cell surface but fail to bind LDL particles. This is a heterogeneous group, because LDL binding activity goes from 2 to 30 % of normal. This defect is due to rearrangements in repeat cysteine residues in binding ligand domain or repeat deletions in EGF-like domain [65]. Class 4 mutations are known as internalization-defective alleles. These alleles produce proteins that are unable to cluster into clathrin-coated pits, therefore LDL receptor is not internalized [66]. Finally, class 5 mutations result in receptors that are able to bind and internalize LDL, but they fail to release LDL in the sorting endosomes and fail to recycle. Instead, they are rerouted to the lysosomes for degradation [67, 68].
Several studies have shown that different mutations are associated with differences in lipid levels, and it is likely that these will be associated with clinically different effects [29, 69]. In addition, the phenotypic effect of the mutation is modulated by other genetic or environmental factors [29, 70]. Even the LDL lowering effect of statins in FH patients may depend on the nature of the LDLR mutation [71, 72]. Our group observed that FH patients with a molecular diagnosis show different advanced carotid and femoral atherosclerosis in relation to LDLR mutational class, thus FH patients with null allele mutations of LDLR show a more severe clinical phenotype and worse advanced carotid atherosclerosis than those with receptor-defective mutations, independently of age, gender, lipid, and nonlipid risk factors [73, 74].
APOB Gene
APOB was the second locus identified to be responsible for MH. A group of individuals with a clinical phonotype similar to FH and also reduced LDL catabolism were found to have normal LDL receptor activity . The disease was secondary to a defective apo B that displays low affinity for the LDLR, and was named FDB. FDB is as common as FH in some European populations [75, 76] but much higher in Old Order Amish living in the USA [77]. The interaction between LDL and the LDL receptor is essential for the regulation of plasma cholesterol in humans. Apo B-100, the major protein component of LDL, is also a ligand for the LDL receptor; therefore, apo B-100 mediates the binding of LDL particle to the LDL receptor [78]. Studies using immunoelectron microscopy have shown that the N-terminal 89 % of apo B-100 enwraps the LDL particle like a belt and that the –COOH terminal 11 % constitutes a bow that crosses over the belt, bringing residues 4154–4189 and 4507–4513 close to amino acid 3527 [79].
Vega and Grundy observed that a group of patients with hypercholesterolemia have reduced clearance of LDL because of a defect in the structure or composition of LDL that reduces its affinity for receptors [80]. Innerarity et al. found that this type of hypercholesterolemia could be attributed to a defective receptor binding of a genetically altered apo B-100 to the LDL receptor [81].
The first mutation found in APOB as FDB cause was demonstrated by Soria et al. They observed a mutation in the codon for amino acid 3527 that results in the substitution of Gln for Arg (p.R3527Q) [5]. So far, ten true mutations at the APOB locus have been identified that alter the binding properties of apo B-100 indicating that FDB is more heterogeneous than previously assumed. Two mutations causing FDB were described in 1995: a change of Gln for a Trp in the amino acid 3527 (p.R3527W) and a substitution of Arg for Cys in 3558 codon (p.R3558C) [82, 83].
The binding affinities of p.R3527Q and p.R3558C to the LDL receptor are reduced to 30 and 70 %, respectively. Nevertheless, the deleterious effect of the p.R3558C variation was reconsidered not sufficient to cause hypercholesterolemia suggesting that it is more a susceptibility variation than a causative mutation [84].
The p.E3432Q mutation binds to LDL receptor at the same rate as normal LDL, but LDL particles containing this mutant protein are taken up and degraded at significantly reduced rates [85]. The APOB mutation p.R3507W has been associated with FDB because of its position near Trp 4396 that was shown to interact with Arg 3527 and facilitate the protein conformation required for normal receptor binding of LDL [86].
Another APOB mutation was found in codon 3543, located between known FDB mutations at codons 3527 and 3558, this mutation, p.N3543K, introduces a positively charged amino acid Lys, while other FDB mutations remove a positively charged residue Arg. The p.N3543K mutation influences conformation of LDL apo B and its interaction with the LDL receptor [82].
Other four APOB mutations, p.H3570Y, p.R3527L, p.R4385H, and p.V4394L were detected in hypercholesterolemic patients [87, 88].
However, the causative effect of these four mutations has not been yet demonstrated.
Recent data reveal that compared with FH patients with LDLR mutations, FDB patients have lower LDL cholesterol levels by 20–25 %, respond better to statins, and have lower risk of CHD [88, 89]. This difference could be due to normal clearance of VLDL remnants through apo E-mediated uptake in FDB [90].
Proprotein Convertase Subtilisin/Kexin Type 9 Gene
In 1999, Varret et al. identified by linkage analysis a third major autosomal dominant locus (HCHOLA3) at 1q34.1-p32 chromosome and showed that HCHOLA3 was in fact PCSK9 [6, 91]. The PSCK9 gene comprises 12 exons transcribed into a cDNA that spans 3617 bp. PCSK9 was first identified as a member of proprotein convertase family with hepatic, intestine, and kidney expression. PCSK9 is a 692-amino-acid glycoprotein that contains a 22-residue signal sequence followed by a pro-domain and a catalytic domain that shares structural homology with the proteinase K family of subtilisin-like serine proteases [7]. PCSK9 is a secreted protein that promotes degradation of the LDL receptor, and variants in PCSK9 gene that cause hypercholesterolemia in humans are gain-of-function mutations [3, 92].
Initially, it was thought that PCSK9 has a role in LDL receptor degradation at the cell surface [92]. As we have described above, there is enough evidence to think that PCSK9 participates in LDL receptor lysosomal degradation via a mechanism that does not require ubiquitination and is distinct from the autophagy and proteosomal degradation pathways [45] (Fig. 10.3).

Proprotein convertase subtilisin/kexin type 9 (PCSK9) is involved in the metabolism of low-density lipoprotein (LDL) receptor. PCSK9 is synthesized in hepatocytes and released into blood. On the hepatocyte surface PCSK9 binds to the LDL receptor. The binding of LDL particles with the complex PCSK9/LDL receptor produces its internalization by endocytosis and lysosomal degradation of the LDL receptor. In the absence of PCSK9, LDL receptor degradation does not occur and the LDL receptor is recycled back to the cell surface
PCSK9 mutations have been also classified into five classes, including “null alleles,” mutations that affect autocatalytic scission, avoiding the protein transport through ER or from the ER to cell surface, alleles that affect PCSK9 stability and, finally, mutations that produce gain of function because of gene overexpression [93–96] (Fig. 10.4). By contrast, mutations in PCSK9 that produce loss of function (Y142X, C679X, and R46 L) are associated with low LDL cholesterol [97, 98].

Effect of proprotein convertase subtilisin/kexin type 9 (PCSK9) mutations on plasma low-density lipoprotein (LDL) cholesterol concentration (Modified from reference [96])
Other FH Loci
The proportion of individuals with autosomal dominant hypercholesterolemia without a mutation in LDLR, APOB, or PCSK9 ranges from 12 to 60% (ADH-) [88, 99–102]. This variability is due mainly to the clinical–biological criteria used to select FH subjects as well as due to ethnicity [103]. Our group has demonstrated that hyperLp(a) is responsible for FH phenotype in approximately 6 % of nonLDLR/nonAPOB subjects [10].
More recently, two groups independently have demonstrated that a rare mutation in APOE , c.500_502delTCC/p.Leu167del causes a lipid phenotype indistinguishable from classical FH in Spain and France [8, 9]. In the Spanish study, the mutation was found in probands with the clinical diagnosis of FCHL, but the family studies demonstrated that the most common phenotype in mutation carriers’ family members was isolated high LDL cholesterol rather than combined hyperlipidemia [8]. The mechanism of high LDL cholesterol associated with this mutation is unknown, but is predicted to interrupt an alpha-helix in the binding domain of apo E and reduce the catabolism of particles containing apo E, including LDL [9].
Several genome-wide linkage scan have suggested susceptibility FH loci on chromosomes 3q25–26, 8q24.22, 16q22.1, and 21q22. However, no gene nor disease-causing mutation was identified in these loci so far [104–106].
Clinical Findings
Atherosclerotic Vascular Disease in FH
Approximately 80 % of FH heterozygotes and almost 100 % of homozygotes will suffer and die of atherosclerosis vascular disease if they are not treated to lower their LDL cholesterol during long periods of time [107]. Symptomatic atherosclerosis disease presents as CHD before age 55 and 60, in over 50 % of FH heterozygotes, men and women, respectively, while homozygotes with much higher LDL cholesterol typically suffer CHD very early in life and usually die before age 20 without treatment. In homozygotes, atherosclerosis begins in the aortic root, causing CHD and supravalvular aortic stenosis [27]. The mean age of onset of a cardiovascular event in men with heterozygous FH is in the early 40s and in women with FH in the early 50s. Approximately, 85 % of males will suffer a coronary event before 65 years if they are not treated. Atherosclerotic vascular disease in FH is mostly CHD. FH represents 1–2 % of all premature ( < 55 years of age in men and < 65 years of age in women) myocardial infarctions in most countries [13, 14], and up to 9 % of total premature CHD in Eastern Finland [108] and Germany [109] are caused by FH. The mechanism of this excess of coronary lesions in FH with respect to other vascular beds such as lower limbs or carotid arteries is not known, but is probably related to the type of LDL particle which accumulates in plasma.
Risk factors associated with CHD in heterozygous FH are the traditional risk factors for the general population, but however, the effect of each risk factor is greater in FH [2, 13]. Besides, FH specific clinical or molecular features as tendon xanthomas or receptor-negative or null mutations in the LDLR gene have also been reported to increase risk of CHD in FH heterozygotes. Major risk factors for CVD in heterozygous FH are presented in Table 10.2 [2, 13]. The presence or absence of these factors modifies the LDL cholesterol treatment goals in FH [13].
Extravascular Cholesterol Deposits
The presence of tendon xanthomas is common in people over age 40 in FH heterozygotes and almost constant in the first decade of life in FH homozygotes. The most characteristic location of tendon xanthomas is the Achilles tendon, but they are also common in elbows and fingers (Fig. 10.5). The presence of tendon xanthomas is associated with an increased risk of premature CVD, especially in women [110], and increased risk of tendinitis. In recent years, due to the availability of effective treatments for hypercholesterolemia from youth, the prevalence of tendon xanthomas has dropped sharply. Achilles tendon sonography improves the detection of xanthomas, and facilitates the clinical diagnosis [111] (Fig. 10.6).

Xanthomas in familial hypercholesteromia (FH). a Xanthomas on the extensor tendons of the hand in FH heterozygote. b and c tendon and tuberous xanthomas on the elbows and knees in FH homozygote. d Achilles tendon xanthomas in FH heterozygote. (Courtesy Prof. Francisco Carapeto)

Achilles tendon sonographic longitudinal images. a normal. b presence of tendon xanthoma. Calipers are located in the proximal and distal borders of the Achilles tendons
The corneal arcus in the first decades of life is another surface lipid deposition characteristic of FH (Fig. 10.7), which is sometimes used as a criterion in some diagnostic algorithms.

Corneal arcus in familial hypercholesterolemia (FH)
Coronary Diease-Genotype Correlations in FH
Because of the large number of allelic variants (more than 1500) differently affecting LDL receptor clearance function, LDLR allele-specific differences may be predicted for the FH phenotype. LDLR mutations may be classified into different functional types: (1) receptor-negative or null alleles, which include disruptions of the promoter sequence, large rearrangements, nonsense, frameshift, or mutations resulting in a deletion of the translation initiation signal and early stop codons, which result in no protein synthesis; (2) receptor-defective alleles, that is, transcription and missense defects that do not completely suppress the function of the protein, which has residual receptor activity; and (3) undetermined receptor activity alleles, which are splicing defects with an unknown effect on protein function [27].
Different studies have analyzed [29, 112–120] whether LDLR mutational class affects the prevalence of CHD risk in heterozygous FH by comparing receptor-negative versus receptor-defective alleles (Table 10.3). In population with large genetic heterogeneity, most of the studies have usually found higher prevalence of xanthomas and CHD in patients with receptor-negative alleles than in those with receptor-defective alleles. However, this association was partially due to higher total and LDL cholesterol in receptor-negative subjects [119].
Low-density Lipoprotein Concentration in FH
FH is defined by severely elevated serum LDL cholesterol concentrations from birth onwards. LDL cholesterol usually ranges from 200 to 400 mg/dL in heterozygous adults, and over 500 mg/dL in homozygous subjects from childhood. LDLR mutations usually present higher LDL cholesterol concentration than subjects with APOB or PCSK9 mutations; and receptor-negative alleles higher LDL cholesterol than defective alleles. Those subjects with clinical diagnosis of FH, but without mutation in LDLR, APOB, or PCSK9 tend to have lower LDL cholesterol than genetically well-defined FH, 237 ± 49 mg/dL versus 302 ± 69 mg/dL, respectively [4].
FH Diagnosis
An early diagnosis of ADH is extremely important since lipid-lowering drugs are highly effective, safe, and cost-effective in FH. The diagnosis of FH has been traditionally performed based on blood lipid values within a family, deposition of cholesterol in extravascular tissues such as tendon xanthomas or corneal arcus, and personal and family history of premature CHD.
Homozygous FH
The diagnosis of homozygous is typically based on the presence of very high LDL cholesterol, in absence of secondary causes of hypercholesterolemia , and high LDL cholesterol in both parents. Appearance of cutaneous xanthomas, especially interdigital planar xanthomas, or tendon xanthomas prior to age 10 years is almost with high LDL cholesterol is almost pathognomonic of homozygous FH. The genetic confirmation of two mutated LDLR, APOB, or PCSK9 alleles and the genetic diagnosis of true homozygosity, compound heterozygosity, or double heterozygosity for FH genes is highly recommended in these subjects. The most common diagnostic criteria are presented in Table 10.4 [121].
Heterozygous FH
Three important diagnostic criteria have been extensively used for the clinical diagnosis of FH: The Simon Broome Register Group in the UK [122], the US Make Early Diagnosis to Prevent Early Deaths (MedPed) Program [123], and the Dutch Lipid Clinic Network [124] (Table 10.5). The accuracy of these three diagnostic methods has not been evaluated in large, independent cohorts. Tendon xanthomas are pathognomonic of FH; however, their identification is not always easy and they are considered insensitive diagnostic markers. A high variability of xanthoma presence has been reported in FH patients [125]. Besides, tendon xanthomas may appear in patients with cerebrotendinous xanthomatosis, sitosterolemia, or dysbetalipoproteinemia. Variability in the frequency of xanthomas observed in different studies depends in part on the clinical criteria used for FH (some of them included the presence of xanthomas), as well as the methods used for the identification of xanthomas.
There are not absolutely predictive clinical criteria for the diagnosis of FH, and arbitrary criteria must be used. The criteria established by the Simon Broome Register Group from the UK for FH were based on elevated total plasma cholesterol concentration greater than 7.5 mmol/L (300 mg/dL) in the proband, together with either tendon xanthomas in the proband or in a first-degree relative or the presence of premature CHD or hypercholesterolemia in a first-degree relative [122]. The US MedPed program focuses the diagnostic criteria, principally, on high LDL cholesterol levels in the individual, and on the family history of hypercholesterolemia with evidence for a dominant transmission [123]. The presence of children with hypercholesterolemia increases the diagnostic probability. The US National Lipid Association Expert Panel on FH advises that LDL cholesterol levels > 250 mg/dL in a patient aged 30 or more > 220 mg/dL for patients aged 20–29; and > 190 mg/dL in patients under age 20, should prompt the clinician to strongly consider a diagnosis of FH and obtain further family information [13]. With those LDL cholesterol criteria, the sensitivity is 70 % while specificity is 82 % for genetically defined FH [4]. The Dutch MedPed Group described a clinical scoring system for the diagnosis of heterozygous FH patients. These criteria include personal and familial LDL cholesterol levels, history of CVD (coronary, carotid, and peripheral arteries), the presence of corneal arcus before the age of 45, and tendon xanthomas. By weighing the occurrence of these clinical signs, alone or in combination with others, a diagnostic scoring table has been constructed in the Netherlands (Table 10.5). These criteria seem to be easy to use in clinical practice and include all the clinical and laboratory features for the diagnosis of FH; and they have recently been proposed as the preferred diagnostic tool by the European Atherosclerosis Society (EAS) [14].
Molecular biology techniques have dramatically improved in recent years and more and more have become highly specific tools to improve the diagnosis of many medical conditions, including FH. Furthermore, the genetic diagnosis is the preferable diagnostic method in FH in most situations because it provides an unequivocal diagnosis. The National Institute for Health and Clinical Excellence (NICE) guidelines on the identification and management of FH recommend cascade screening using a combination of genetic testing and LDL cholesterol concentration measurement [126]. This approach has also been recommended by the European Atherosclerosis Society (EAS). [14].
Several methods are currently used to identify sequence changes in LDLR, APOB, and PCSK9 genes including direct sequencing and high-throughput FH resequencing arrays [127]. A microarray for the detection of common point mutations and small deletions in the LDLR and APOB genes has been developed by our group [117]. By providing either a positive (presence of LDLR, PCSK9, or APOB mutations) or negative (absence of defects in these genes) diagnosis, this platform has allowed the genetic characterization of > 8000 Spanish patients [128]. Even though the diagnosis of FH based on the detection of a functional mutation on a causative gene is the recommended procedure in highly suspicious cases, it cannot be recommended for all cases of hypercholesterolemia. The genetic testing is still complex, expensive, and should be used as complementary to the clinical diagnosis. A group of criteria have been proposed to maximize the likelihood of genetic confirmation in subjects with clinical suspicion of FH based on age, tendon xanthomas presence, and LDL cholesterol levels (Table 10.6) [4].
Lipid-Lowering Therapy for Heterozygous FH
Excess CHD in FH is attributable to high LDL cholesterol in this population; consequently, LDL cholesterol reduction to normal levels is a priority in the management of FH. Long-term therapy is the only way, at this time, to substantially reduce or remove the excess lifetime risk of CHD due to their genetic disorder. A healthy lifestyle is also important in the FH treatment. Lifestyle comprises a healthy diet, ideal body weight, no smoking, and adequate physical activity [4]. A healthy lifestyle provides many benefits beyond LDL cholesterol lowering, and can increase the LDL cholesterol lowering effect of drugs. Although LDL cholesterol is the fundamental CHD risk factor in FH, these subjects are very responsive to other risk factors such as smoking, which should be carefully explored and treated.
Different medical societies and expert panels have published guidelines for the management of FH, and without exception, they highly recommend aggressive LDL cholesterol lowering in all adults and less intensive treatment in children > 10 years of age (Table 10.7).
International Panel on Management of Familial Hypercholesterolemia (2004) [2]. Promoted by the Spanish Society of Arteriosclerosis, a panel of international experts proposed the first global recommendations for the diagnosis and treatment of FH: early diagnosis of the disease, a screening strategy based on finding family affected members, CHD risk stratification in heterozygous subjects according to the presence of other risk factors, early detection of atherosclerosis in preclinical phase, the establishment of three LDL cholesterol treatment goals based on baseline risk, and a therapeutic strategy based on lifestyle and pharmacological treatment with potent statins as first choice. LDL apheresis was recommended after drug treatment when LDL cholesterol is above 200 mg/dL in the presence of coronary artery disease , or above 300 mg/dL without CHD.
NICE in the UK (2008) [126] recommends a clinical diagnosis based on the criteria of Simon Broome British Register. Interestingly, a specific target LDL cholesterol target is not recommended, instead an advice to reduce LDL cholesterol by more than 50 %. Baseline risk stratification before beginning the treatment was not considered. Affected children should start drug treatment after 10 years of age.
Belgian consensus for the FH treatment in children and young adults (2011) [129] focused on the diagnosis and treatment of children. They recommend the diagnosis and diet treatment of children from 2 years of age and consider drug treatment after 10 years of age when the LDL is > 190 mg/dL or > 160 mg/dL in the presence of premature CHD in the family or cardiovascular risk factors. LDL cholesterol goal of treatment is to obtain > 30 % LDL cholesterol reduction between 10 and 14 years, and < 130 mg/dL onwards.
Lipid National Association Expert Panel on Familial Hypercholesterolemia (2011) [130, 131] from the USA published a special issue of the Journal of Clinical Lipidology in 2011 dedicated to FH. The documents recommend preferably clinical diagnosis based on LDL cholesterol concentrations adjusted for age, and a general population screening in subjects with LDL cholesterol levels > 190 mg/dL in adulthood, or > 160 mg/dL in children. Drug treatment is recommended when LDL cholesterol is > 190 mg/dL, with different therapeutic targets depending on individual risk factors.
Consensus Statement of the European Atherosclerosis Society (2013) [14]. This document emphasizes that FH is underdiagnosed and undertreated in the general population, defines all FH heterozygotes as high-risk patients, promotes the FH screening in subjects with plasma total cholesterol ≥ 310 mg/dL in adults or ≥ 230 mg/dL in children, premature CHD in the subject or family members, presence of tendon xanthomas in the subject or family member(s), or sudden premature cardiac death in a family member. The LDL cholesterol goals for lipid-lowering treatment are < 135 mg/dL for children, < 100 mg/dL for adults, and < 70 mg/dL for adults with known CHD or diabetes. Therefore, high doses of potent statins, combined drug regimens usually with ezetimibe or bile acid sequestrans, and, in some cases, LDL-apheresis are required to reach such exigent targets.
Despite current maximal treatment, many heterozygous FH subjects remain with undesired high LDL cholesterol concentration. PCSK9 inhibition with two different monoclonal antibodies against PCSK9 (evolucumab and alirocumab) has been studied in two double-blind, randomized, placebo-controlled trials in heterozygous FH insufficiently controlled with standard treatment [132, 133]. In both studies, the inhibition of PCSK yielded rapid and over 50 % reductions in LDL cholesterol with good tolerability, so this therapeutic approach seems very promising in the future treatment of heterozygous FH.
Homozygous FH Treatment
Statins, bile acid sequestrants, and ezetimibe have lesser lipid-lowering effect in homozygous FH than in heterozygous subjects, because these drugs need some LDL receptor functionality to be fully effective. However, they are remarkably safe in homozygous FH and should be tested because in some cases LDL cholesterol reductions between 20 and 40 s% can be obtained especially in LDL receptor-defective patients by increasing residual LDL receptor activity, and by inhibition of cholesterol synthesis [134].
The current treatment of choice for homozygous FH is LDL-apheresis at weekly or biweekly intervals, usually in children over the age of 7. Most homozygous FH obtain substantial reductions in LDL cholesterol, usually > 50 %, with periodical LDL-apheresis and is the only treatment that substantially lowers Lp(a) in these patients [121].
New drugs have been recently tested for the treatment of homozygous FH: mipomersen, lomitapide, and PCSK9 inhibitors. Mipomersen is an antisense apo B-100 mRNA recently approved for the treatment of homozygous FH. It is administered subcutaneously as a weekly injection. In a randomized, double-blind, placebo-controlled study with 34 patients, the mean LDL cholesterol reduction with this inhibitor of the apo B synthesis was 25 %, although the lipid-lowering response was highly variable among individuals [135].
Lomitapide is an inhibitor of the microsomal triglyceride transfer protein (MTTP), a key protein in the assembly of apo B-containing lipoproteins in the liver and intestine. It is a highly potent lipid-lowering drug recently approved for the homozygous FH treatment in the USA and Europe. In a single-arm, open-label study with homozygous FH, aged 18 years or older with a median dose of 40 mg a day, lomitapide reduced LDL cholesterol by 50 % after 26 weeks of treatment. Lomitapide’s adverse effects included accumulation of hepatic fat. Mean hepatic fat was 1.0 % at baseline and increased to 8.6 and 8.3 % at week 56 and at week 78, respectively. The long-term consequence of this accumulation is unknown which seems to stabilize, or even decrease, with time [136].
PCSK9 inhibition has also been evaluated in homozygous FH. Alirocumab (AMG 145) a monoclonal antibody to PCSK9 demonstrates reductions of 19.2 % in LDL cholesterol in patients with defective LDL receptor activity, but no efficacy in those who were receptor negative [137].
The prognosis of intensive treated FH subjects has drastically improved in recent years reducing the CHD [138]. The key challenge in the coming years is to expand the diagnosis and treatment to this group of patients in whom cardiovascular prevention is paradigmatic and cost-effective [139].
Autosomal Recessive Hypercholesterolemia
In 1973, a Lebanese family with an autosomal recessive form of severe hypercholesterolemia, clinically indistinguishable from FH, was described by Khachadurian et al. [140]. Afterwards, subjects with similar phenotype were identified in Sardinia [141], in subjects of Turkish and Asian-Indian origin [142], and in Japan [143], and this entity was named ARH. The causative gene, LDLRAP1 , on chromosome 1, which encodes LDLRAP1, was identified by linkage analysis in 2001 by Garcia et al. [46]. In this gene, both homozygous and compound heterozygous mutations can be found. Most of the ARH-causing mutations are due to premature stop codons, producing no mRNA or truncated proteins [46]. The mutations identified in LDLRAP1 gene causing ARH can be found in http://www.ucl.ac.uk/ldlr/.
LDLRAP1 is a 32-kDa and 308-amino-acid endocytic adaptor protein required for the function of LDLR in hepatocytes. LDLRAP1 protein serves as an adaptor for LDL receptor endocytosis in the liver and a deficiency in this protein results in a decrease in the LDL cholesterol catabolism [144]. The N-terminal domain of LDLRAP1 contains a phosphotyrosine-binding (PTB) domain, which binds to the internalization sequence (FDNPVY) in the cytoplasmic tail of the LDL receptor. This domain can also simultaneously interact with cell membrane phosphoinositides. Specific sequences within the C-terminal region of LDLRAP1 bind clathrin and its adaptor AP2 [145]. All these interactions together enable LDLRAP1 to function as an endocytic adaptor for the clathrin-mediated endocytosis of LDL receptor in the liver. Accordingly, LDLRAP1 has been classified as a clathrin-associated sorting protein (CLASP) [146], a group of proteins that serve as a molecular bridge between receptors and the clathrin machinery for their endocytic internalization.
ARH subjects have severely elevated plasma LDL cholesterol, tuberous and tendon xanthomata, corneal arcus, and premature atherosclerosis , with severe CHD, that make it clinically indistinguishable from FH [147]. Plasma cholesterol levels and clinical symptoms that present subjects with ARH are intermediate between those of FH heterozygotes and FH homozygotes. The age of onset of symptomatic coronary artery disease in these patients is later and tendon xanthomas tend to be large and bulky [148]. In a phenotypic comparison study between 42 ARH subjects and 42 homozygous FH subjects, Pisciotta et al. [149] reported that in ARH subjects, plasma LDL cholesterol (550 ± 88.6 mg/dL) was lower than in receptor-negative homozygous FH (827 ± 138 mg/dL) but similar to that found in receptor-defective homozygous FH (601 ± 92.5 mg/dL). The risk of coronary artery disease was ninefold lower in ARH patients [150].
However, LDL receptor activity and LDL binding ability in cultured fibroblasts are normal. All LDLRAP1 mutations characterized to date preclude the synthesis of full-length LDLRAP1, and this LDLRAP1 is required for normal LDLR function in lymphocytes and hepatocytes, but not in fibroblasts. Residual LDL receptor function in cells that do not require LDLRAP1 could explain the reason why ARH subjects have lower plasma LDL cholesterol levels than homozygous FH patients, who have no functional LDL receptor [150].
LDL turnover studies have demonstrated that the rate of clearance of LDL from plasma is similar in subjects with ARH and in subjects with homozygous FH, and markedly reduced compared with normolipidemic controls [143], suggesting that LDLRAP1 is essential for LDL receptor-mediated uptake of LDL. Despite comparable reductions in the fractional catabolic rate of LDL, the metabolic and clinical phenotype of ARH is less severe than that of homozygous FH, as stated above [151]. ARH patients also respond to lipid-lowering drugs, as statins, with a greater reduction in plasma levels of LDL cholesterol than that observed in homozygous FH patients [152]. As the clearance rates of LDL are similarly decreased in ARH and FH, the less severe clinical phenotype in ARH points to LDL production. LDL is produced as a metabolic product of VLDL, and it has been proposed that the molecular basis for this milder phenotype is the increased removal of VLDL remnants from the circulation [153]. The increased clearance of remnant lipoproteins could contribute to the great responsiveness to statins of ARH patients [154]. Also, studies demonstrate that clearance of postprandial remnant lipoproteins is preserved in ARH in contrast to FH. This preservation of postprandial remnant particles catabolism could also contribute to the mild phenotype of ARH compared with FH [155].
Lysosomal Acid Lipase Deficiency
Deficiency of the enzyme lysosomal acid lipase results in two distinct diseases in humans: Wolman disease and cholesteryl ester storage disease (CESD;OMIM 278000). Wolman disease is a severe lipid infiltration of the liver, spleen, and other organs early in life, causing early death in infants. Wolman disease is very rare, with an incidence of less than one in 100,000 live births. CESD is also a rare disease, around one case in 40,000 people, possibly underdiagnosed, and characterized by the accumulation of cholesterol esters in different organs of the body, especially liver. Clinically, CESD presents as a mixed hyperlipidemia in a young patient with no family history of hyperlipidemia, with hepatosplenomegaly and elevated liver enzymes. Ultrasound typically shows steatosis, but the diagnosis is usually suspected by finding a microvesicular steatosis on liver biopsy. A definitive diagnosis is made by detecting a low lysosomal acid lipase activity, or in the presence of functional mutations in the gene encoding this enzyme, LIPA [156].
The accumulation of cholesterol esters and free cholesterol reduction in the liver of these subjects leads to increased endogenous cholesterol production, and this is the believed mechanism. Evolution of these patients is to chronic liver disease, liver cirrhosis, and increased incidence of atheromatous disease [157]. Recombinant lysosomal acid lipase replacement was shown to be effective in animal models, and recently, a phase I/II clinical trial demonstrated its safety and indicated its potential metabolic efficacy [158].
Cholesterol-7-Alpha-Hydroxylase Deficiency (OMIM 118455)
Pullinger et al. described in 2002 that mutations in the CYP7A1 gene, encoding the enzyme cholesterol-7-alpha-hydroxylase produced hypercholesterolemia associated with heterozygosity and homozygosity, so it is considered an autosomal codominant hypercholesterolemia OMIM 118455 [159]. The enzyme cholesterol-7-alpha-hydroxylase controls the rate of conversion of cholesterol into bile acids . CYP7A1 deficiency would cause a decrease in the production of bile acids and cholesterol accumulation in the liver, causing a decrease in the expression of LDL receptors, and therefore, an increase in plasma concentration of LDL. In these patients, as liver cholesterol content is increased, statins have the desired effect of inducing the expression of LDL receptors [159].
Summary
MHs are the most frequent inherited metabolic diseases and common causes of premature cardiovascular death and disability in most countries. They are genetically heterogeneous in which similar phenotypes may be caused by mutations in different genes, commonly LDLR, APOB, and PCSK9. However, there are some MH families in whom the responsible gene/s are unknown. Very high LDL cholesterol, familial presentation, and high prevalence of premature coronary disease are the clinical features to suspect MH. Early diagnosis of MH is very important so that therapy can be initiated as soon as possible. Combination of clinical and genetic test is the preferable diagnostic method. Familial cascade screening, once an index patient is diagnosed, is mandatory. MHs are frequently underdiagnosed and undertreated, so there is a need for a much better diagnostic screening worldwide. High doses of potent statins, combination therapy of statins with ezetimibe, or bile acid sequestrants, and, in some cases, LDL-apheresis are required to reach LDL cholesterol goals. PCSK9, apo B, and MTTP inhibitors are novel and very promising drugs that can substantially improve the treatment in the highest risk patients.
References
Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–803.
Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68.
Varret M, Abifadel M, Rabe’s J-P, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet. 2008;73:1–13.
Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol. 2008;102:1187–93.
Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci U S A. 1989;86:587–91.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003:34:154–6.
Seidah NG, Benjannet S, Wickham L, Marcinkiewicz J, Jasmin SB, Stifani S, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928–33.
Solanas-Barca M, de Castro-Orós I, Mateo-Gallego R, Cofán M, Plana N, Puzo J, et al. Apolipoprotein E gene mutations in subjects with mixed hyperlipidemia and a clinical diagnosis of familial combined hyperlipidemia. Atherosclerosis. 2012;222:449–55.
Marduel M, Ouguerram K, Serre V, Bonnefont-Rousselot D, Marques-Pinheiro A, Erik Berge K, et al. Description of a large family with autosomal dominant hypercholesterolemia associated with the APOE p.Leu167del mutation. Hum Mutat. 2013;34:83–7.
Meriño-Ibarra E, Puzo J, Jarauta E, Cenarro A, Recalde D, García-Otín AL, et al. Hyperlipoproteinaemia(a) is a common cause of autosomal dominant hypercholesterolaemia. J Inherit Metab Dis. 2007;30:970–7.
Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a) gene accounts for greater than 90 % of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90:52–60.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9.
Hopkins PN, Toth PP, Ballantyne CM, Rader DJ. National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3 Suppl):S9–17.
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–90.
Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–68.
Genest JJ Jr, Martin-Munley SS, McNamara JR, Ordovas JM, Jenner J, Myers RH, et al. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation. 1992;85:2025–33.
Paramsothy P, Knopp R, Bertoni AG, Tsai MY, Rue T, Heckbert SR. Combined hyperlipidemia in relation to race/ethnicity, obesity, and insulin resistance in the Multi-Ethnic Study of Atherosclerosis. Metabolism. 2009;58:212–9.
Arner P, Bernard S, Salehpour M, Possnert G, Liebl J, Steier P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478:110–3.
Cabré A, Lázaro I, Cofán M, Jarauta E, Plana N, Garcia-Otín AL, et al. FABP4 plasma levels are increased in familial combined hyperlipidemia. J Lipid Res. 2010;51:1173–8.
Martinez-Hervas S, Fandos M, Real JT, Espinosa O, Chaves FJ, Saez GT, et al. Insulin resistance and oxidative stress in familial combined hyperlipidemia. Atherosclerosis. 2008;199:384–9.
Venkatesan S, Cullen P, Pacy P, Halliday D, Scott J. Stable isotopes show a direct relation between VLDL apoB overproduction and serum triglyceride levels and indicate a metabolically and biochemically coherent basis for familial combined hyperlipidemia. Arterioscler Thromb. 1993;13:1110–8.
Verseyden C, Meijssen S, Castro Cabezas M. Postprandial changes of apoB-100 and apoB-48 in TG rich lipoproteins in familial combined hyperlipidemia. J Lipid Res. 2002;43:274–80.
Brouwers MC, van Greevenbroek MM, Stehouwer CD, de Graaf J, Stalenhoef AF. The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol. 2012;8:352–62.
Brouwers MC, van der Kallen CJ, Schaper NC, van Greevenbroek MM, Stehouwer CD. Five-year incidence of type 2 diabetes mellitus in patients with familial combined hyperlipidaemia. Neth J Med. 2010;68:163–7.
Civeira F, Cenarro A. DNA oxidation is a potential cardiovascular risk factor in combined familiar hyperlipemia. Med Clin (Barc). 2008;131(1):14–5. (Spanish).
Marcil M, Vu H, Cui W, Dastani Z, Engert JC, Gaudet D, et al. Identification of a novel C5L2 variant (S323I) in a French Canadian family with familial combined hyperlipemia. Arterioscler Thromb Vasc Biol. 2006;26:1619–25.
Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, AB, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 2863–913.
Moorjani S, Roy M, Gagné C, Davignon J, Brun D, Toussaint M, et al. Homozygous familial hypercholesterolemia among French Canadians in Québec Province. Arteriosclerosis. 1989;9:211–6.
Kotze MJ, De Villiers WJ, Steyn K, Kriek JA, Marais AD, Langenhoven E, et al. Phenotypic variation among familial hypercholesterolemics heterozygous for either one of two Afrikaner founder LDL receptor mutations. Arterioscler Thromb. 1993;13:1460–8.
Gylling H, Aalto-Setälä K, Kontula K, Miettinen TA. Serum low density lipoprotein cholesterol level and cholesterol absorption efficiency are influenced by apolipoprotein B and E polymorphism and by the FH-Helsinki mutation of the low density lipoprotein receptor gene in familial hypercholesterolemia. Arterioscler Thromb. 1991;11:1368–75.
Anderson RG, Goldstein JL, Brown MS. Localization of low density lipoprotein receptors on plasma membrane of normal human fibroblasts and their absence in cells from a familial hypercholesterolemia homozygote. Proc Natl Acad Sci U S A. 1976;73:2434–8.
Cummings RD, Kornfeld S, Schneider WJ, Hobgood KK, Tolleshaug H, Brown MS, et al. Biosynthesis of N- and O-linked oligosaccharides of the low density lipoprotein receptor. J Biol Chem. 1983;258:15261–73.
Zhang DW, Garuti R, Tang WJ, Cohen JC, Hobbs HH. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc Natl Acad Sci U S A. 2008;105:13045–50.
Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232:34–47.
Willnow TE, Nykjaer A, Herz J. Lipoprotein receptors: new roles for ancient proteins. Nat Cell Biol. 1999;1:E157–62.
Beffert U, Stolt PC, Herz J. Functions of lipoprotein receptors in neurons. J Lipid Res. 2004;45:403–9.
Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30.
Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40.
Zelcer N, Tontonoz P. Liver X receptors as integrators of metabolic and inflammatory signaling. J Clin Invest. 2006;116:607–14.
Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–4.
Scotti E, Hong C, Yoshinaga Y, Tu Y, Hu Y, Zelcer N, et al. Targeted disruption of the idol gene alters cellular regulation of the low-density lipoprotein receptor by sterols and liver x receptor agonists Mol Cell Biol. 2011;31:1885–93.
Sawamura T. New idol for cholesterol reduction? Clin Chem. 2009;55:2082–4.
Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, et al. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–12.
Shan L, Pang L, Zhang R, Murgolo NJ, Lan H, Hedrick JA. PCSK9 binds to multiple receptors and can be functionally inhibited by an EGF-A peptide. Biochem Biophys Res Commun. 2008;375:69–73.
Wang Y, Huang Y, Hobbs HH, Cohen JC. Molecular characterization of proprotein convertase subtilisin/kexin type 9-mediated degradation of the LDLR. J Lipid Res. 2012;53:1932–43.
Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, et al. Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science. 2001;292:1394–8.
Sirinian MI, Belleudi F, Campagna F, Ceridono M, Garofalo T, Quagliarini F, et al. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein (LDL) binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem. 2005;280:38416–2.
Michaely P, Li WP, Anderson RG, Cohen JC, Hobbs HH. The modular adaptor protein ARH is required for low density lipoprotein (LDL) binding and internalization but not for LDL receptor clustering in coated pits. J Biol Chem. 2004;279:34023–31.
Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem. 1974;249:5153–62.
Sudhof TC, Van der Westhuyzen DR, Goldstein JL, Brown MS, Russell DW. Three direct repeats and a TATA-like sequence are required for regulated expression of the human low density lipoprotein receptor gene. J Biol Chem. 1987;262:10773–9.
Yamamoto T, Davis CG, Brown MS, Schneider WJ, Casey ML, Goldstein JL, et al. The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell. 1984;39:27–38.
Südhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228:815–22.
Russell DW, Brown MS, Goldstein JL. Different combinations of cysteine-rich repeats mediate binding of low density lipoprotein receptor to two different proteins. J Biol Chem. 1989;264:21682–8.
Fisher C, Abdul-Aziz D, Blacklow SC. A two-module region of the low-density lipoprotein receptor sufficient for formation of complexes with apolipoprotein E ligands. Biochemistry. 2004;43:1037–44.
Abdul-Aziz D, Fisher C, Beglova N, Blacklow SC. Folding and binding integrity of variants of a prototype ligand-binding module from the LDL receptor possessing multiple alanine substitutions. Biochemistry. 2005;44:5075–85.
Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J Biol Chem. 1988;263:13282–90.
Arias-Moreno X, Velázquez-Campoy A, Rodríguez JC, Pocoví M, Sancho J. Mechanism of low density lipoprotein (LDL) release in the Endosome. J Biol Chem. 2008;283:22670–9.
Brown MS, Herz J, Goldstein JL. Calcium cages, acid baths and recycling receptors. Nature. 1997;388:629–30.
Davis CG, Elhammer A, Russell DW, Schneider WJ, Kornfeld S, Brown MS, et al. Deletion of clustered O-linked carbohydrates does not impair function of low density lipoprotein receptor in transfected fibroblasts. J Biol Chem. 1986;261:2828–38.
Kingsley DM, Sege RD, Kozarsky KF, Krieger M. DNA-mediated transfer of a human gene required for low-density lipoprotein receptor expression and for multiple Golgi processing pathways. Mol Cell Biol. 1986;6:2734–7.
Yokode M, Pathak RK, Hammer RE, Brown MS, Goldstein JL, Anderson RG. Cytoplasmic sequence required for basolateral targeting of LDL receptor in livers of transgenic mice. J Cell Biol. 1992;117:39–46.
Dawson PA, Hofmann SL, van der Westhuyzen DR, Sudhof TC, Brown MS, Goldstein JL. Sterol-dependent repression of low density lipoprotein receptor promoter mediated by 16-base pair sequence adjacent to binding site for transcription factor Sp1. J Biol Chem. 1988;263:3372–9.
Smith JR, Osborne TF, Goldstein JL, Brown MS. Identification of nucleotides responsible for enhancer activity of sterol regulatory element in low density lipoprotein receptor gene. J Biol Chem. 1990;265:2306–10.
Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–66.
Lehrman MA, Goldstein JL, Brown MS, Russell DW, Schneider WJ. Internalization-defective LDL receptors produced by genes with nonsense and frameshift mutations that truncate the cytoplasmic domain. Cell. 1985;41:735–43.
Lehrman MA, Schnedier WJ, Südhof TC, Brown MS, Goldstein JL, Russell DW. Mutation in LDL receptor: Alu-Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science. 1985;227:140–6.
Brown MS, Anderson RG, Goldstein JL. Recycling receptors: the round-trip itinerary of migrant membrane proteins. Cell. 1983;32:663–7.
Strøm TB, Tveten K, Holla ØL, Cameron J, Berge KE, Leren TP. The cytoplasmic domain is not involved in directing Class 5 mutant LDL receptors to lysosomal degradation. Biochem Biophys Res Commun. 2011;408:642–6.
Gudnason V, Day IN, Humphries SE. Effect on plasma lipid levels of different classes of mutations in the low-density lipoprotein receptor gene in patients with familial hypercholesterolemia. Arterioscler Thromb. 1994;14:1717–22.
Hobbs HH, Leitersdorf E, Leffert CC, Cryer DR, Brown MS, Goldstein JL. Evidence for a dominant gene that suppresses hypercholesterolaemia in a family with defective low density lipoprotein receptors. J Clin Invest. 1989;84:656–64.
Jeenah M, September W, Graadt van Roggen F, de Villiers W, Seftel H, Marais D. Influence of specific mutations at the LDL-receptor gene locus on the response to simvastatin therapy in Afrikaner patients with heterozygous familial hypercholesterolaemia. Atherosclerosis. 1993;98:51–8.
Leitersdorf E, Eisenberg S, Eliav O, Friedlander Y, Berkman N, Dann EJ, et al. Genetic determinants of responsiveness to the HMG-CoA reductase inhibitor fluvastatin in patients with molecularly defined heterozygous familial hypercholesterolemia. Circulation. 1993;87(4 Suppl):III35–44.
Junyent M, Gilabert R, Zambón D, Pocoví M, Mallén M, Cofán M, Núñez I, Civeira F, Tejedor D, Ros E. Femoral atherosclerosis in heterozygous familial hypercholesterolemia: influence of the genetic defect. Arterioscler Thromb Vasc Biol. 2008;28:580–6.
Junyent M, Gilabert R, Jarauta E, Núñez I, Cofán M, Civeira F, et al. Impact of low-density lipoprotein receptor mutational class on carotid atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis. 2010;208:437–41.
Myant NB. Familial defective apolipoprotein B-100: a review, including some comparisons with familial hypercholesterolaemia. Atherosclerosis. 1993;104:1–18.
Innerarity TL, Mahley RW, Weisgraber KH, Bersot TP, Krauss RM, Vega GL, et al. Familial defective apolipoprotein B-100: a mutation of apolipoprotein B that causes hypercholesterolemia. J Lipid Res. 1990;31:1337–49.
Shen H, Damcott CM, Rampersaud E, Pollin TI, Horenstein RB, McArdle PF, et al. Familial defective apolipoprotein B-100 and increased low-density lipoprotein cholesterol and coronary artery calcification in the old order amish. Arch Intern Med. 2010;170;1850–5.
Kane J, Havel R. Disorders of the biogenesis and secretion of lipoproteins containing the B apolipoproteins. In: Scriver C, Beaudet A, Sly W, editors. The metabolic and molecular bases of inherited disease. New York: Mc Graw-Hill; 1995, pp. 1853–86.
Chatterton JE, Phillips ML, Curtiss LK, Milne R, Fruchart JC, Schumaker VN. Immuno-electron microscopy of low density lipoproteins yields a ribbon and bow model for the conformation of apolipoprotein B on the lipoprotein surface. J Lipid Res. 1995;36:2027–37.
Vega GL, Grundy SM. In vivo evidence for reduced binding of low density lipoproteins to receptors as a cause of primary moderate hypercholesterolemia. J Clin Invest. 1986;78:1410–4.
Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A. 1987;84:6919–23.
Gaffney D, Pullinger CR, O’Reilly DS, Hoffs MS, Cameron I, Vass JK, et al. Influence of an asparagine to lysine mutation at amino acid 3516 of apolipoprotein B on low-density lipoprotein receptor binding. Clin Chim Acta. 2002;321:113–21.
Pullinger CR, Hennessy LK, Chatterton JE, Liu W, Love JA, Mendel CM, et al. Familial ligand-defective apolipoprotein B.Identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest. 1995;95:1225–34.
Rabès JP, Varret M, Devillers M, Aegerter P, Villéger L, Krempf M, et al. R3531C mutation in the apolipoprotein B gene is not sufficient to cause hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:E76–82.
Fisher E, Scharnagl H, Hoffmann MM, Kusterer K, Wittmann D, Wieland H, et al. Mutations in the apolipoprotein (apo) B-100 receptor-binding region: detection of apo B-100 (Arg3500–.Trp) associated with two new haplotypes and evidence that apo B-100 (Glu3405–Gln) diminishes receptor-mediated uptake of LDL. Clin Chem. 1999;45:1026–38.
Boren J, Ekstrom U, Agren B, Nilsson-Ehle P, Innerarity TL. The molecular mechanism for the genetic disorder familial defective apolipoprotein B100. J Biol Chem. 2001;276:9214–8.
Soufi M, Sattler AM, Maerz W, Starke A, Herzum M, Maisch B, et al. A new but frequent mutation of apoB-100 apoB His3543Tyr. Atherosclerosis. 2004;174:11–6.
Fouchier SW, Kastelein JJ, Defesche JC. Update of the molecular basis of familial hypercholesterolemia in the Netherlands. Hum Mutat. 2005:26:550–6.
Fouchier SW, Kastelein JJ, Sijbrands EJ. Familial defective apolipoprotein B versus familial hypercholesterolemia: an assessment of risk. Semin Vasc Med. 2004;4:259–64.
Schaefer JR, Scharnagl H, Baumstark MW, Schweer H, Zech LA, Seyberth H, et al. Homozygous familial defective apolipoprotein B-100. Enhanced removal of apolipoprotein E-containing VLDLs and decreased production of LDLs. Arterioscler Thromb Vasc Biol. 1997;17:348–3.
Varret M, Rabès JP, Saint-Jore B, Cenarro A, Marinoni JC, Civeira F, et al. A third major locus for autosomal dominant hypercholesterolemia maps to 1p34.1-p32. Am J Hum Genet. 1999;64:1378–87.
Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069–74.
Cameron J, Holla ØL, Ranheim T, Kulseth MA, Berge KE, Leren TP. Effect of mutations in the PCSK9 gene on the cell surface LDLreceptors. Hum Mol Genet. 2006;15:1551–8.
Cunningham D, Danley DE, Geoghegan KF, Griffor MC, Hawkins JL, Subashi TA, et al. Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat Struct Mol Biol. 2007;14:413–9.
Pandit S, Wisniewski D, Santoro JC, Ha S, Ramakrishnan V, Cubbon RM, et al. Functional analysis of sites within PCSK9 responsible for hypercholesterolemia. J Lipid Res. 2008;49:1333–43.
Poirier S, Mayer G. The biology of PCSK9 from the endoplasmic reticulum to lysosomes: new and emerging therapeutics to control low-density lipoprotein cholesterol. Drug Des Devel Ther. 2013;7:1135–48.
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia C, Hobbs H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–5.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Damgaard D, Jensen JM, Larsen ML, Soerensen VR, Jensen HK, Gregersen N, et al. No genetic linkage or molecular evidence for involvement of the PCSK9, ARH or CYP7A1 genes in the Familial Hypercholesterolemia phenotype in a sample of Danish families without pathogenic mutations in the LDL receptor and apoB genes. Atherosclerosis. 2004:177:415–22.
Wang J, Ban MR, Hegele RA. Multiplex ligationdependent probe amplification of LDLR enhances molecular diagnosis of familial hypercholesterolemia. J Lipid Res. 2005;46:366–72.
Tosi I, Toledo-Leiva P, Neuwirth C, Naoumova RP, Soutar AK, et al. Genetic de fects causing familial hypercholesterolaemia: identification of deletions and duplications in the LDL-receptor gene and summary of all mutations found in patients attending the Hammersmith Hospital Lipid Clinic. Atherosclerosis. 2007;194:102–11.
Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet. 2007;43:943–9.
Ahmad Z, Adams-Huet B, Chen C, Garg A. Low prevalence of mutations in known loci for autosomal dominant hypercholesterolemia in a multiethnic patient cohort. Cir Cardiovasc Genet. 2012;5:666–75.
Wang X, Li X, Zhang YB, Zhang F, Sun L, Lin J, et al. Genome-wide linkage scan of a pedigree with familial hypercholesterolemia suggests susceptibility loci on chromosomes 3q25–26 and 21q22. PLoS One. 2011;6:e24838.
Cenarro A, García-Otín AL, Tejedor MT, Solanas M, Jarauta E, Junquera C, et al. A presumptive new locus for autosomal dominant hypercholesterolemia mapping to 8q24.22. Clin Genet. 2011;79:475–81.
Marques-Pinheiro A, Marduel M, Rabès JP, Devillers M, Villéger L, Allard D, et al. A fourth locus for autosomal dominant hypercholesterolemia maps at 16q22.1. Eur J Hum Genet. 2010;18:1236–42.
Hirobe K, Matsuzawa Y, Ishikawa K, Tarui S, Yamamoto A, Nambu S, et al. Coronary artery disease in heterozygous familial hypercholesterolemia. Atherosclerosis. 1982;44:201–10.
Koivisto UM, Hämäläinen L, Taskinen MR, Kettunen K, Kontula K. Prevalence of amilial hypercholesterolemia among young north Karelian patients with coronary eart disease: a study based on diagnosis by polymerase chain reaction. J Lipid Res. 1993;34:269–77.
Schuster H. High risk/high priority: familial hypercholesterolemia-a paradigm for molecular medicine. Atheroscler Suppl. 2002;2:27–30.
Civeira F, Castillo S, Alonso R, Meriño E, Cenarro A, Artieda M, et al. Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the LDL receptor gene mutation. Arterioscler Thromb Vasc Biol. 2005;25:1960–5.
Junyent M, Gilabert R, Zambón D, Núñez I, Vela M, Civeira F, et al. The use of Achilles tendon sonography to distinguish familial hypercholesterolemia from other genetic dyslipidemias. Arterioscler Thromb Vasc Biol. 2005;25:2203–8.
Vohl MC, Gaudet D, Moorjani S, Tremblay G, Perron P, Gagné C, et al. Comparison of the effect of two low-density lipoprotein receptor class mutations on coronary heart disease among French-Canadian patients heterozygous for familial hypercholesterolaemia. Eur J Clin Invest. 1997;27:366–73.
Bertolini S, Cantafora A, Averna M, Cortese C, Motti C, Martini S, et al. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol. 2000;20:E41–52.
Dedoussis GV, Skoumas J, Pitsavos C, Choumerianou DM, Genschel J, Schmidt H, et al. FH clinical phenotype in Greek patients with LDL-R defective vs. negative mutations. Eur J Clin Invest. 2004;34:402–9.
Real JT, Chaves FJ, Ejarque I, García-García AB, Valldecabres C, Ascaso JF, et al. Influence of LDL receptor gene mutations and the R3500Q mutation of the apoB gene on lipoprotein phenotype of familial hypercholesterolemic patients from a South European population. Eur J Hum Genet. 2003;11:959–65.
Koeijvoets KC, Wiegman A, Rodenburg J, Defesche JC, Kastelein JJ, Sijbrands EJ. Effect of low-density lipoprotein receptor mutation on lipoproteins and cardiovascular disease risk: a parent-offspring study. Atherosclerosis. 2005;180:93–9.
Tejedor D, Castillo S, Mozas P, Jiménez E, López M, Tejedor MT, et al. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem. 2005;51:1137–44.
Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muñiz O, et al. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis. 2008;200:315–21.
Bertolini S, Pisciotta L, Rabacchi C, Cefalù AB, Noto D, Fasano T, et al. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis. 2013;227:342–8.
Souverein OW, Defesche JC, Zwinderman AH, Kastelein JJ, Tanck MW. Influence of LDL-receptor mutation type on age at first cardiovascular event in patients with familial hypercholesterolaemia. Eur Heart J. 2007;28:299–304.
Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment Atherosclerosis. 2012;223:262–8.
No Authors. Scientific Steering Committee on behalf of the Simon Broome Register Group Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ. 1991;303:893–6.
Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–6.
Defesche J. Familial hypercholesterolemia. In: Betteridge J, editor. Lipids and vascular disease, vol. 6. London: Martin Dunitz; 2000, pp. 65–76.
Descamps OS, Leysen X, Van Leuven F, Heller FR. The use of Achilles tendon ultrasonography for the diagnosis of familial hypercholesterolemia. Atherosclerosis. 2001;157:514–8.
Wierzbicki AS, Humphries SE, Minhas R. Guideline development group. Familial hypercholesterolaemia: summary of NICE guidance. BMJ. 2008;337:a1095.
Chiou KR, Charng MJ, Chang HM. Array-based resequencing for mutations causing familial hypercholesterolemia. Atherosclerosis. 2011;216:383–9.
Palacios L, Grandoso L, Cuevas N, Olano-Martín E, Martinez A, Tejedor D, et al. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis. 2012;221:137–42.
Descamps OS, Tenoutasse S, Stephenne X, Gies I, Beauloye V, Lebrethon MC, et al. Management of familial hypercholesterolemia in children and young adults: consensus paper developed by a panel of lipidologists, cardiologists, paediatricians, nutritionists, gastroenterologists, general practitioners and a patient organization. Atherosclerosis. 2011;218:272–80.
Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, et al. National Lipid Association Expert Panel on Familial Hypercholesterolemia. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3Suppl):S1–8.
Robinson JG, Goldberg AC, National Lipid Association Expert Panel on Familial Hypercholesterolemia. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5(3Suppl):S18–29.
Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–17.
Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36.
Gagne C, Gaudet D, Bruckert E, Ezetimibe Study Group. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105:2469–75.
Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Cuchel M, Meagher EA, du Toit Theron H, Blom DJ, Marais AD, Hegele RA, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the PCSK9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20.
Elis A, Zhou R, Stein EA. Effect of lipid-lowering treatment on natural history of heterozygous familial hypercholesterolemia in past three decades. Am J Cardiol. 2011;108:223–6.
Sibley C, Stone NJ. Familial hypercholesterolemia: a challenge of diagnosis and therapy. Cleve Clin J Med. 2006;73:57–64.
Khachadurian AK, Uthman SM. Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients. Nutr Metab. 1973;15:132–40.
Zuliani G, Vigna GB, Corsini A, Maioli M, Romagnoni F, Fellin R. Severe hypercholesterolaemia: unusual inheritance in an Italian pedigree. Eur J Clin Invest. 1995;25:322–31.
Norman D, Sun X-M, Bourbon M, Knight BL, Naoumova RP, Soutar AK. Characterization of a novel cellular defect in patients with phenotypic homozygous familial hypercholesterolemia. J Clin Invest. 1999;104:619–28.
Harada-Shiba M, Tajima S, Yokoyama S, Miyake Y, Kojima S, Tsushima M, et al. Siblings with normal LDL receptor activity and severe hypercholesterolemia. Arterioscler Thromb. 1992;12:1071–8.
Garuti R, Jones C, Li WP, Michaely P, Herz J, Gerard RD, et al. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem. 2005;280:40996–1004.
Dvir H, Shah M, Girardi E, Guo L, Farquhar MG, Zajonc DM. Atomic structure of the autosomal recessive hypercholesterolemia phosphotyrosine-binding domain in complex with the LDL-receptor tail. Proc Natl Acad Sci U S A. 2012;109:6916–21.
Mishra SK, Keyel PA, Edeling MA, Dupin AL, Owen DJ, Traub LM. Functional dissection of an AP-2 beta2 appendage-binding sequence within the autosomal recessive hypercholesterolemia protein. J Biol Chem. 2005;280:19270–80.
Soutar AK, Naoumova RP, Traub LM. Genetics, clinical phenotype, and molecular cell biology of autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2003;23:1963–70.
Arca M, Zuliani G, Wilund K, Campagna F, Fellin R, Bertolini S, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet. 2002;359:841–7.
Pisciotta L, Priore Oliva C, Pes GM, Di Scala L, Bellocchio A, et al. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): a phenotypic comparison. Atherosclerosis. 2006;188:398–405.
Wilund KR, Yi M, Campagna F, Arca M, Zuliani G, Fellin R, et al. Molecular mechanisms of autosomal recessive hypercholesterolemia. Hum Mol Genet. 2002;11:3019–30.
Zuliani G, Arca M, Signore A, Bader G, Fazio S, Chianelli M, et al. Characterization of a new form of inherited hypercholesterolemia: familial recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1999;19:802–9.
Soutar AK, Naoumova RP. Autosomal recessive hypercholesterolemia. Semin Vasc Med. 2004;4:241–8.
Jones C, Garuti R, Michaely P, Li WP, Maeda N, Cohen JC, et al. Disruption of LDL but not VLDL clearance in autosomal recessive hypercholesterolemia. J Clin Invest. 2007;117:165–74.
Tada H, Kawashiri MA, Ikewaki K, Terao Y, Noguchi T, Nakanishi C, et al. Altered metabolism of low-density lipoprotein and very-low-density lipoprotein remnant in autosomal recessive hypercholesterolemia: results from stable isotope kinetic study in vivo. Circ Cardiovasc Genet. 2012;5:35–41.
Tada H, Kawashiri MA, Tanaka A, Nakano T, Nakajima K, Inoue T, et al. Post-prandial remnant lipoprotein metabolism in autosomal recessive hypercholesterolaemia. Eur J Clin Invest. 2012;42:1094–9.
Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol. 2013;24:332–8.
Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol. 2013;58:1230–43.
Balwani M, Breen C, Enns GM, Deegan PB, Honzík T, Jones S, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013;58:950–7.
Pullinger CR, Eng C, Salen G, Shefer S, Batta AK, Erickson SK, et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest. 2002;110:109–17.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Humana Press
About this chapter
Cite this chapter
Civeira, F., Pocovi, M. (2015). Monogenic Hypercholesterolemias. In: Garg, A. (eds) Dyslipidemias. Contemporary Endocrinology. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-60761-424-1_10
Download citation
DOI: https://doi.org/10.1007/978-1-60761-424-1_10
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-60761-423-4
Online ISBN: 978-1-60761-424-1
eBook Packages: MedicineMedicine (R0)