
Abstract
Beta-thalassemia and sickle cell anemia are two of the most common diseases related to the hemoglobin protein. In these diseases, the beta-globin gene is mutated, causing severe anemia and ineffective erythropoiesis. Patients can additionally present with a number of life-threatening co-morbidities, such as stroke or spontaneous fractures. Current treatment involves transfusion and iron chelation; allogeneic bone marrow transplant is the only curative option, but is limited by the availability of matching donors and graft-versus-host disease. As these two diseases are monogenic diseases, they make an attractive setting for gene therapy. Gene therapy aims to correct the mutated beta-globin gene or add back a functional copy of beta- or gamma-globin. Initial gene therapy work was done with oncoretroviral vectors, but has since shifted to lentiviral vectors. Currently, there are a few clinical trials underway to test the curative potential of some of these lentiviral vectors. This review will highlight the work done thus far, and present the challenges still facing gene therapy, such as genome toxicity concerns and achieving sufficient transgene expression to cure those with the most severe forms of thalassemia.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Beta-thalassemia
- Sickle cell anemia
- Hemoglobinopathies
- Hemoglobin disorders
- Gene therapy
- Oncoretrovirus
- Lentivirus
- Hematopoietic stem cells
- Cell-based therapy
- Mixed chimerism
Introduction
Sickle cell anemia (SCA) and beta-thalassemia are the most frequently inherited blood disorders worldwide. Altogether, roughly 100,000 Americans are affected by these disorders. Both disorders are characterized by mutations in the beta-globin gene, a subunit of hemoglobin (Hb). SCA is an inherited disorder characterized by only a single mutation in the beta-globin gene, leading to the formation of hemoglobin S (HbS) [1]. HbS exhibits a marked decrease in solubility, and an increase in viscosity and polymer formation. Ischemic stroke, caused by large vessel arterial obstruction with superimposed thrombosis is one of SCA’s most devastating complications. Blood transfusions are administered to prevent thrombosis. Unfortunately, periodic blood transfusions are associated with significant risks of iron overload and other complications, and must be accompanied by iron chelation [2, 3]. Beta-thalassemia on the other hand is characterized by one or more of over 300 various mutations in the beta-globin gene . Based on the combinations of these mutations, patients might be affected by a milder form, indicated as beta-thalassemia intermedia or non-transfusion dependent thalassemia (NTDT), or the most severe form, beta-thalassemia major [4, 5]. Beta-thalassemia major requires regular transfusions to sustain life. However, due to the negative progression of this disease, very often NTDT patients become transfusion dependent as well [6, 7]. Major problems are progressive splenomegaly from extra medullary hematopoiesis and iron build-up in the heart and other organs, often resulting in fatal outcomes for some patients in their teens or early 20s [8, 9]. Current palliative therapeutic options to treat these two disorders are red blood cell transfusion and iron chelation [2, 10].
In addition to life-threatening anemia, patients may present with inherent and treatment-related complications that exacerbate the pathology. For patients with SCA, common complications include painful episodes, acute chest syndrome, and stroke [1]; in patients with thalassemia, hepato-splenomegaly, recurrent infections, and spontaneous fractures [11,12,13]. In both cases, transfusion-associated infections and organ damage are side effects of long-term treatment and unsatisfactory iron chelation. Iron overload is observed also in NTDT patients because of ineffective erythropoiesis [6, 14]. Ineffective erythropoiesis triggers a cascade of compensatory mechanisms resulting in erythroid marrow expansion, extramedullary hematopoiesis, splenomegaly, and increased gastrointestinal iron absorption [15]. Ineffective erythropoiesis triggers increased iron absorption by reducing the expression of hepcidin, the hormone that controls dietary iron absorption [14, 16,17,18].
Although both transfusion and iron chelation treatments have remarkably improved over the years and, thus, improving the quality of life, they do not provide a definitive cure, as they do not address the inherent genetic cause. To this end, hematopoietic stem cell (HSC) transplantation is the only presently available cure. Allogeneic bone marrow transplant (BMT) can be curative, but only a small proportion of patients have suitable donors. Furthermore, myeloablative HSC transplantation carries a 5–10% mortality rate. Graft-vs-host disease and adverse immune reactions can limit the success of allogenic BMT as well [9]. Given these limitations, gene therapy using a patient’s own HSCs represents an alternative and potential cure because it aims at the direct recovery of the hemoglobin protein function via the addition of a functional copy of the beta- or gamma-globin gene . The development of gene therapy tools for SCA and beta-thalassemia has been the object of research of the last few decades and has been proved successful in mouse model studies, in vitro human cell studies, and thus far in one clinical trial. This review will highlight key findings from these gene-addition studies.
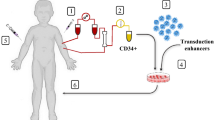
The conditions for a clinical-grade gene therapy vector can be summarized as follows: (1) controlled transgene expression: erythroid-specific, stage-restricted, elevated, position-independent, and sustained over time; (2) effective targeting of HSCs; (3) highly efficient and stable transduction; (4) absent or low genomic toxicity; and (5) correction of the phenotype in preclinical models. Figure 6.1 provides a schematic of gene therapy and key issues found in each stage.

Schematic of gene therapy for beta-thalassemia and sickle cell anemia
Oncoretroviral Vectors
The first studies of gene addition were done with oncoretroviral vectors and helped paved the way for current lentiviral vectors. Oncoretroviruses , like lentiviruses, belong to the Retroviridae family and are RNA-based viruses [19]. Multiple studies showed oncoretroviruses are capable of transferring genetic material without transferring any viral material, and are able to achieve expression of human beta-globin in murine cells. However, the expression of beta-globin was extremely low and nowhere near therapeutic [20,21,22].
Studies then moved on to determining what other elements needed to be incorporated to achieve higher beta-globin expression. Discovery of the locus control region, or LCR, brought significant advancements to vector design. The LCR is a regulatory region upstream of the beta-globin locus and is critical for high-level, sustained, erythroid-specific, and position-independent globin expression [23, 24]. The LCR is made up of four DNaseI hypersensitive sites (HS) that contain many motifs for transcription factors and chromatin remodeling factors. It is thought to regulate globin gene expression through a looping mechanism, bringing various transcriptional modifiers to the globin promoter [25]. Incorporation and modification of these HS sites was done in a number of works. Work by Plavec et al. in 1993 [26] showed HS2, HS3, and HS4 elements increased beta-globin expression by 10-fold in mouse erythroleukemia (MEL) cells, but still remained relatively low for mice transplanted with oncoretrovirus -transduced cells: 0.04–3.2% of endogenous mouse beta-globin RNA. Furthermore, there were problems with stability and high viral titer production. Leboulch and coworkers [27] undertook modification of the LCR in order to overcome these problems. They saw instability in all combinations of LCR sequences, with HS2 alone conferring a single common rearrangement and other combinations showing multiple rearrangements. Beta-globin gene mutagenesis and elimination of a 372 base pair intronic sequence and multiple reverse polyadenylation and splice sites resulted in higher titer viruses and more stable proviral transmission. Sadelain saw additional success in 1995 [28] with producing a high-titer retroviral beta-globin vector, but unfortunately the vector did not give high position-independent expression and large clonal variation was seen.
Other methods were also tested for their ability to achieve therapeutic levels of expression : addition of a chromatin insulator [29]; use of the ankyrin promoter driving the gamma-globin gene [30]; use of a mutant gamma-globin enhancer characterized from patients with hereditary persistence of fetal hemoglobin (HPFH) driving gamma globin [31]; addition of the HS40 regulatory region from the human alpha-globin gene locus [32]; and use of an anti-sickling beta-globin [33]. Many important insights were gained by these experiments, however, as with other oncoretroviral studies, their success was limited. Eventually oncoretroviral studies gave way to the lentiviral studies discussed below .
Lentiviral Vectors
In the mid-1990s, lentiviral vectors based on the human immunodeficiency virus (HIV-1 ) arose as an option for gene transfer. Engineered to be devoid of any pathogenic or replication competency, these viruses are efficiently able to encompass large therapeutic transgene cassettes. Like all retroviruses, lentiviruses exhibit receptor-mediated entry, capsid uncoating, reverse transcription, and integration into the host genome [19]. However, lentiviruses have a more intricate genome, notably the Rev response element, or RRE. The RRE helps stabilize the proviral RNA by interacting with the viral protein Rev. This allows for stronger unspliced RNA export from the nucleus [19, 34, 35]. Another important element discovered in lentiviruses is the central polypurine tract/central termination sequence element. The cPPT/CTS element is a short noncoding part of the pol gene sequence that increases lentiviral transduction efficiency.
Importantly, for hematological gene therapy purposes, lentiviruses are able to infect dividing and non-diving cells [36]. Gene therapy for hematological disorders typically infect HSCs , so that the genetic modification is passed on short term to differentiating cells, and long-term to more stem cells through self-renewal. HSCs, however, are difficult to culture and transduce ex vivo due to a delicate balance between dividing/proliferation and engraftment potential. Normally, proliferation and engraftment potential are negatively correlated: increasing the proliferation is detrimental because the cells subsequently do not engraft [37]. Thus, ability to infect non-dividing cells that retain engraftment potential is an extremely beneficial property of lentiviruses. Lentiviruses containing regulatory elements, promoters, enhancers, and beta-globin or gamma-globin have been successful with correction of mouse models of thalassemia and SCA, and with in vitro correction of human CD34+ peripheral blood (PB) cells. A list of beta-globin vectors can be found in Table 6.1.
In 2000, May [38] and colleagues used the TNS9 vector to correct a mouse model of thalassemia intermedia. Later in 2003, Rivella [39] and colleagues showed that this same vector could be used to rescue lethality in a new model of Cooley’s anemi a (thalassemia major). TNS9 exhibits position-effect variation though, and in the Cooley’s anemia model, was unable to be therapeutic in all mice, with average human beta-globin expression between 3.6 and 9.4 g/dL. Two mice models of SCA were corrected in 2001 by Pawliuk [40] and colleagues: the S-Antilles-D Punjab model (SAD ) and the Berkeley (BERK) model. The SAD mouse model expresses human alpha and a human “super S” beta-globin that has two point mutations [41]. The BERK model expresses human alpha and human sickle beta-globin, but additionally does not express any endogenous mouse alpha or mouse beta globin [42]. As a result, the BERK model has a more severe phenotype, in part because of suboptimal expression of the human beta-globin gene as compared to the endogenous mouse gene. Pawliuk used a transgenic “βT87Q” form of beta-globin, an anti-sickling mutant form which has an amino acid substitution at the 87th position. With this vector, Pawliuk saw transgenic Hb could make up to 12% and 52% total Hb for the SAD and BERK models, respectively. In 2013, another anti-sickling mutant form was tested with three point mutations: T87Q for blocking the lateral contact with HbS, E22A to disrupt axial contacts with HbS, and G16D, which confers a competitive advantage over HbS for interaction with alpha-globin [43]. Named “CCL-βAS3-FB”, this vector could reduce the relative amount of sickled red blood cells differentiated in vitro; and, using vector copy numbers of 0.5–2, could make up 15–25% of total Hb.
In order to increase the safety of lentiviral vectors and improve expression, insulators were tested by Puthenveetil in 2004 [44]. An insulator is a genetic element which usually has two properties: (1) enhancer-blocking activity, when placed between an enhancing element and a promoter and (2) preventing the spread of heterochromatin into the integrated transgenic cassette from a nearby heterochromanized region [45,46,47]. By adding an insulator, one can prevent the beta-globin LCR from acting on nearby oncogenes it might have integrated near. It can also help reduce vector silencing , to ensure sustained and high transgene expression. Puthenveetil et al.’s vector, named BG-1, added a 1.2 kb cHS4 insulator, taken from the chicken beta-globin hypersensitive site 4. As reviewed by Nienhuis and Persons [48], the cHS4 insulator has a “core” that contains five footprints. The footprints are involved in: recruiting CTCF, an enhancer blocking protein; binding USF proteins in order to recruit histone-modification enzymes that make transcription-activation marks; and binding VEZF1, which prevents DNA methylation in the transcribed region. Human beta-thalassemic cells treated with BG-1 and differentiated in vitro showed similar amounts of hemoglobin as non-thalassemic controls . Upon transplantation into immunodeficient mice, treated cells underwent effective erythropoiesis and expressed normal amounts of beta-globin. In 2007, Arumugam [49] and colleagues compared vectors with the cHS4 insulator to those without and consistently saw approximately double the beta-globin expression with the insulator in vitro with MEL cells and in vivo with transplanted and transduced murine HSCs . While beneficial to expression, the 1.2 kb cHS4 insulator causes low viral titers. Thus, in 2009, Arumugam [50] identified a 400 bp extended core region of the cHS4, that still exhibits full insulator activity but does not have a severe an impact on titer. They found the previously identified core only reduced clonal variegation.
Further studies have also been done on the LCR . In 2007, Lisowski et al. [51] showed addition of HS1 from the LCR to HS2-4 significantly increased globin expression. Miccio in 2008 [52] used a “GLOBE” vector containing the HS2 and HS3 regions without the HS4 region. They used it to rescue Cooley’s anemia lethality, but high copy numbers were required for correction [34]. Interestingly, upon transplantation, transduced cells expressing a high level of beta-globin were preferentially selected in vivo. Roselli [53] produced preclinical data using the “GLOBE” vector in 2010 on a diverse set of CD34+ patient samples to restore adult Hb (HbA) synthesis. Integration analysis revealed integration preference in transcriptionally active regions but no preference for cancer-related regions. “GLOBE” was later modified into “G-GLOBE” by adding the HS2 enhancer of the GATA-1 gene [54]; it too could achieve high expression of beta-globin. The GATA-1 HS2 bound GATA1 and CBP acetyltransferase, leading to the establishment of an open chromatin region.
Lentiviral transduction of gamma-globin has additionally been shown to be therapeutic. Table 6.2 lists gamma-globin vectors that have been successfully used. In 2003, Persons et al. [32] developed the “d432βAγ” vector, which expressed gamma-globin under beta-globin LCR elements. Although they saw expression of fetal hemoglobin, they still saw high variation due to position and vector copy number. Hanawa in 2004 [55] used a longer LCR to achieve more consistent expression across animals. In 2006, Samakoglu [56] and colleagues showed that a combination vector could be made that expressed gamma-globin and concurrently knocked-down sickle beta-globin via small hairpin RNA. They tested this vector on HeLa and MEL cells stably expressing sickle beta globin. The discovered that the placement of the shRNA was critical, as it affected interferon response, siRNA production, and the amount of gamma-globin expression. Pestina [57] tested a gamma-globin vector in vivo on the BERK SCA model. The gamma-globin was modified to contain a 3′UTR from beta-globin, since proteins are believed to bind the beta-globin 3′UTR and increase mRNA stability. This was further modified by Wilber in 2011 [58] with the addition of the 400bp core of the cHS4 insulator, and tested on human CD34+ PB cells from three beta-thalassemia patients. They saw fetal Hb (HbF) production ranged between 45 and 60% of total Hb, and up to a threefold increase in total Hb content. Papanikolaou in 2012 [59] published a report of a gamma-globin virus without the LCR, but instead containing a gamma-globin promoter with a -117 point mutation associated with HPFH, the HS40 enhancer from the alpha-globin locus, the HPFH-2 enhancer, and the cHS4 insulator. They saw mild improvement of HbF synthesis compared to mock-transduced controls.
Most recently, our laboratory has done work to find new insulators. The aforementioned cHS4 insulator is subject to rearrangements and loss (see Clinical Trials, below). To this end, we generated a new lentiviral vector, which we have named AnkT9W. AnkT9W contains the erythroid-specific ankyrin 5′ hyper-sensitive barrier insulator [60, 61]. This insulator does not exhibit enhancer-blocking activity, but does prevent the spread of heterochromatin, and significantly increases expression of beta-globin as compared to vectors without this insulator. This vector was able to maintain high, yet stable levels of Hb synthesis in MEL cells and human CD34+ PBMCs. Analysis indicated that in MEL cells, AnkT9W expressed the transgenic mRNA and hemoglobin at higher levels than the parental T9W (a modified TNS9). Interestingly, AnkT9W was additionally able to correct the phenotype of SCA cells by modifying the proportion of sickling vs functional Hb, without changing the overall Hb content. This could be clinically relevant since there is a concern that adding transgenic beta-globin into SCD HSC s increases the total amount of beta-chains, both sick and transgenic. This new total amount might exceed the amount of α-chains, leading to an alpha-thalassemia like phenotype.
Addition of Non-globin Genetic Elements
On top of the numerous studies to add back beta and gamma globin genetic sequences, there are have been additional studies based on adding other genetic elements which can modify beta or gamma-globin gene expression. The gamma-globin repressor BCL11A has been identified as target to increase gamma-globin gene expression. Xu and colleagues were able to demonstrate that affecting BCL11A alone was able to increase endogenous gamma-globin expression and ameliorate the sickle cell phenotype in mice [62]. Since Bcl11A knockout is postnatal lethal, they used a floxed Bcl11A mice crossed with the EpoR-GFP Cre mice, which express Cre recombinase under the erythropoietin receptor promoter. As in the full Bcl11A knockout, the switch from HbF to HbA did not occur. These mice were then bred with SCD mice. In the combination SCD/Bcl11A fl/fl mice, sickle cells were absent and blood parameters were markedly improved, thus showing that Bcl11A deletion alone was sufficient to ameliorate SCD. Along the same lines, Wilber and colleagues [58] tested a lentiviral construct encoding a BCL11A shRNA on CD34+ human PB cells and saw a 3-fold increase in gamma-globin expression. Most recently, Bauer and colleagues [63] have done a genome-wide association study concerning Bcl11A. They found a sequence in intron-2 that causes developmentally restricted, erythroid-specific lacZ reporter expression in mice. Disruption of this sequence with transcription activator-like effector nucleases (TALENs) in MEL cells lead to reduced expression of Bcl11A. As such, this sequence might be a new target to lower Bcl11A expression and increase HbF production.
Oct-1 is another gene that negatively regulates gamma-globin gene expression. Oct-1 is a transcription factor that recognizes the octamer ATGCAAAT. The gamma-globin promoter contains three Oct-1 consensus sequences. The -175 consensus sequence has been shown to be associated with HPFH and mutagenesis of the -280 consensus sequence leads to increased gamma-globin expression. Xu and colleagues [64] tested the ability of a “decoy oligonucleotide” to compete for Oct-1 binding, therefore reducing Oct-1 binding at the endogenous gamma-globin locus. In K562 cells, they saw an increase in gamma-globin gene expression after addition of the decoy oligonucleotide.
Genetic elements can also be engineered to affect beta and gamma-globin gene expression. Advances in zinc-finger (ZF) development have allowed the creation of domains able to recognize any 18 base pair DNA sequence . ZF domains can be paired with transcriptional activation domains to create “artificial transcription factors”. In 2010, Wilber and colleagues [65] extensively examined one such engineered ZF transcription factor, termed GG1-VP64. GG1-VP64 recognizes the -117 position of the gamma-globin promoter. The -117 position is the site of a naturally occurring mutation which causes HPFH, and is thus a known region important for modulating gamma-globin gene expression [66]. They discovered in wild-type CD34 PB cells that up to 20% HbF could be produced, as compared to 2% in untransduced controls. Later in 2011 [58], they tested beta-thalassemic samples and found a therapeutic 20-fold increase in gamma-globin could be achieved. In 2012, Deng et al. published a paper concerning an artificial ZF linked to the protein Ldb1 [25]. Ldb1 is a critical part of GATA1-mediated chromatin looping of the LCR to the beta-globin promoter. Deng and colleagues created Ldb1-ZFs that recognized the beta-major promoter (P-ZF) or the HS2 site of the LCR (L-ZF). They showed in murine GATA-1 null cells that beta-major expression could be induced by P-ZF alone or P-ZF and L-ZF , but not by L-ZF alone. Furthermore, they showed that the self-association domain of Ldb1 was sufficient for this activity as well [25].
Beta-thalassemia is characterized by over 300 mutations. A subset of these mutations creates new cryptic splice sites and, even though the original splice sites are intact, leads to incorrect splicing. The most common splice mutations involve the creation of a splice site in intron 1 or intron 2 of the beta-globin gene, and are termed IVS1 or IVS2 for intravenous sequence as such. Specific mutations are followed with a number denoting the site of the mutation, such as IVS1-110 or IVS2-654. Since the correct splice sites are still intact, approaches have been made to create splice switching oligonucleotides, which cover the aberrant splice site and restore splicing to the original sites . A splice switching oligonucleotide has to achieve a number of goals: (a) it must bind to the aberrant splice site and prevent it from being recognized by the splicing machinery and (b) the duplex it creates must not be recognized by RNaseH, as to prevent degradation of the RNA. To this end, Svasti and colleagues [67] have developed a morpholino oligomer conjugated to the cell-penetrating peptide P005. The conjugation leads to efficient uptake of the oligomer into the cell. The oligomer targeted the aberrant splice site in the IVS2-654 mutation, and upon in vivo delivery, improved hemoglobin synthesis in an IVS2-654 mouse. Laccera et al. [68] investigated this in vitro on human CD34+ cells as well. They found dose-dependent and sequence-specific correction for one IVS2–654 and two IVS2–745 thalassemic patient samples. The IVS2–745 pre-mRNA splicing was corrected more efficiently than that of IVS2–654 pre-mRNA. The authors say this coincides with the clinical phenotype of the two diseases, in that IVS2-654 is more severe .
Clinical Trials
The first successful gene therapy trial for beta-thalassemia was done in Paris and reported by Leboulch in 2010 [69]. It was a small trial involving only two patients. The first patient failed to engraft due to technical issues unrelated to the vector. The second patient, however, has been transfusion independent now for several years. The patient is a compound heterozygote (βE/β0), in which one allele (β0) is nonfunctioning and the other (βE) is an HbE mutant allele whose mRNA may either be spliced correctly (producing a mutated βE-globin) or incorrectly (producing no beta-globin).
The βT87Q LentiGlobin vector was used for this trial (see Table 6.1). As with the βT87Q vector, this vector expresses a mutated beta-globin distinguishable from transfused beta-globin due to an anti-sickling mutation at the 87th amino acid. It also contains two core copies of the cHS4 insulator. Analysis of the patient’s transduced cells revealed an intact coding sequence for the vector, however, with the loss of one copy of the cHS4. Of the twenty-four chromosomal integration sites (IS) found, one of the sites, high mobility group AT-hook 2 (HMGA2), caused transcriptional activation of HMGA2 and became the dominant clone. Cells from the clinical trial patient with a HGMA2 IS showed loss of the 3′UTR of HGMA2, preventing the binding of let-7 miRNAs to complementary sequences. Erythroid cells from the HMGA2 clone exhibited a dominant, myeloid-biased cell clone. HMGA2 mRNA was undetectable in granulocyte-monocytes, thus the expression was reported to be erythroblast-specific. However, the clonal dominance of HMGA2 was represented in all populations in similar proportions (erythroblasts, granulocyte-monocyte and LTC-IC cells). The authors hypothesize that this dominance is due to a transient expression of HMGA2 in a myeloid-restricted LT-HSC during β-LCR priming, before the β-LCR becomes restricted to the erythroid lineage.
Overexpression of HMGA2 is found in a number of benign and malignant tumors and can lead to a clonal growth advantage [70]. Overexpression is often associated with mutations affecting the 3′ untranslated region (UTR), which contains binding sites for the regulatory miRNA let-7 [70]. Let-7 miRNA binding to the 3′UTR of HMGA2 negatively regulates HMGA2 mRNA and thus the level of protein expression [71]. Transgenic mice carrying a HMGA2 with a shortened 3′UTR expressed increased levels of HMGA2 protein in multiple tissues including hematopoietic cells. These mice showed splenomegaly, erythropoietin-independent erythroid colony formation, and an increased number of peripheral blood cells in all lineages. Furthermore, BM cells derived from these animals had a growth advantage over wild-type cells. Thus, overexpression of HMGA2 is associated with clonal expansion at the stem cell and progenitor levels [70].
At the time of reporting, the patient had been transfusion-independent for 2 years, and showed stable Hb levels from 9 to 10 g/dL−1. The patient has undergone frequent phlebotomies to increase iron clearance. Therapeutic Hb-βT87Q LentiGlobin however only accounted for 1/3 of the total Hb, with endogenous HbE and HbF making up the rest. Without the additive effect of these endogenous Hb’s, this first trial might not have been a success. This suggests that we not only need a predictive in vitro model with which to evaluate potential trial patients, but better vectors that can achieve higher therapeutic Hb expression.
Currently the first United States phase I clinical trial has received FDA approval and is enrolling patients. The strategy was briefly described by Sadelain and colleagues in 2010 with the main goals of assessing insertional oncogenesis and replication-competent lentivirus safety, and determining levels of engraftment and vector expression [72]. The study plans to use CD34+ cells mobilized by granulocyte colony-stimulating factor (G-CSF ). Using G-CSF, Sadelain et al. have already achieved successful mobilization of CD34+ cells in three beta-thalassemia patients in amounts sufficient for transduction. In 2002 Li et al. [73] studied G-CSF peripheral blood stem cell mobilization in beta-thalassemia patients and found up to a 21.5 fold increase in CD34+ cells could be collected. In 2012, Yannaki and colleagues studied different mobilization methods in 23 patients with beta-thalassemia [74]. They studied patients with or without splenectomy, and found that non-splenectomized patients tolerated G-CSF, but splenectomized patients could not tolerate it without a 1-month pretreatment with hydroxyurea. They additionally examined Plerixafor, which reversibly inhibits the CXCR4-SDF1 interaction with the BM microenvironment, to mobilize HSC s. Plerixafor proved successful for both splenectomized and non-splenectomized patients .
For the Sadelain trial, the previously described TNS9 vector [38, 39] will be used to induce transgenic expression of beta-globin. Small unpublished modifications have been made to this vector to increase titer, but the gene, promoter, enhancers, and LCR remain intact. Two more trials are also in the works. (1) A trial St. Jude Children’s Research Hospital is planned using gamma-globin coding sequences under control of the beta-globin promoter. (2) The company Bluebird Bio, a company specializing in genetic and orphan diseases, is planning a trial in the San Francisco area using a LentiGlobin BB305 T87Q virus. The identifiers for the TNS9, gamma-globin, and LentiGlobin trials are NCT01639690, NCT00669305, and NCT01745120, respectively; and at the time of writing were all recruiting participants.
Mixed Chimerism and In Vivo Selection of Transduced Cells
When undergoing autologous stem cell transplant, patients first need to undergo a myeloablative conditioning regimen. The success of conditioning regimen intensity depends on a balance between toxicity and the amount of mixed transgenic-chimerism, i.e. the amount of BM made up of transplanted cells that carry the vector (Fig. 6.2). Full myeloablation can, theoretically, result in a complete transgenic-chimerism, where the BM is made entirely of cultured cells, with a greater amount of therapeutic vector-transduced cells than in a partial myeloablation setting. However, full myeloablation is more toxic and puts the patient at greater risk, especially in the case of graft failure. A reduced-intensity conditioning regimen can be less toxic; however it can also lead to a lower composition of therapeutic vector-transduced cells due to lower transgenic-chimerism. When a reduced-intensity conditioning regimen is used, beta- globin expression from the vector must be high enough to give transduced cells a survival advantage compared to untransduced cells. In studies done of traditional allogeneic BMT, patients with as low as 20% donor contribution were still able to achieve transfusion independence and normal hemoglobin levels. However, a high initial engraftment (>90% at 60 days post-transplant) is necessary for good chances of stable mixed chimerism [9].

Mixed chimerism . White: starting bone marrow of the patient before procedure; Textured Grey: Bone marrow that has been cultured but not transduced; Black: Bone marrow that has been cultured and successfully transduced with therapeutic vector. Ex-vivo expansion would increase the total amount of bone marrow that been cultured. More toxic myeloablation would decrease the amount of residual bone marrow. Selection with MGMT or MDR1 would increase the amount of vector transduced cells
Lucarelli and colleagues identified three Pesaro risk classes for beta-thalassemia patients based on previous iron chelation , hepato- and splenomegaly, and liver fibrosis [75]. Patients with irregular iron chelation and more liver damage fall into class 3 while those patients with less iron overload and liver damage fall into class 1 and 2. A study of 886 beta-thalassemia patients who received transplants from HLA-matched siblings or parents showed a 91% and 84% probability of Thalassemia-free survival with a normal conditioning regimen for class 1 and class 2 patients, respectively [76]. There has been a recent trend to lower-intensity, non-myeloablative conditioning regimens though based on the following data: (1) lower morbidity and mortality is associated with these regimens, (2) patients not eligible for the traditional full myeloablative regimen have been safely transplanted with these regimens, and (3) mixed chimerism can be sustained and still lead to amelioration of disease in patients with allografts [77]. Multiple groups have had success with reduced-intensity regimens with lower doses of busulfan, or by using alternatives such as thiotepa, treosulfan, fludarabine, busulfex, or antithmocyte globulin (as reviewed in [77, 78]). All of these myeloablation reports, however, relate to beta-thalassemia transplants with HLA-matched donors and not to autologous transplants done with vector-transduced cells. The TNS9 trial with vector-transduced cells will use a reduced-intensity regimen based off of data from successful allogeneic transplants [72] and data from autologous transplants with vector-transduced cells in immunodeficiency disorders [79, 80].
If after transplant a patient were to have a non-therapeutic level mixed chimerism, or suboptimal transgene expression, it would be helpful to have an in vivo strategy to increase the chimerism. Conferring a cytoprotective drug resistance to lentiviral-transduced cells is one of these strategies. The human multidrug resistance 1 (MDR1) gene encodes a P-glycoprotein drug efflux pump that confers resistance to several chemotherapy drugs, including paclitaxel and doxorubicin, both of which under normal conditions are hematopoietically toxic. Researchers discovered a modest positive selection of peripheral blood progenitor cells that had been transduced with MDR1 could be achieved by giving paclitaxel [81, 82]. In a second set of studies, researchers used a mutated MGMT gene. MGMT encodes the enzyme O6-methylguanine-DNA methyltransferase, and confers resistance to nitrosoureas and O6-benzylguanine drugs. A dual vector was created encoding both MGMT under a constitutive promoter and gamma globin under erythroid control elements (V5 from Table 6.1). Murine wild-type [83] and murine beta-thalassemic bone marrow [84] were transduced with this vector. This dual vector was able to increase the number of fetal hemoglobin expressing cells in vivo after treatment with drug. Furthermore, for thalassemic bone marrow, researchers could achieve amelioration of the anemia. Researchers also showed that ex vivo selection of lentiviral-transduced and transplantable cells was possible. They pretreated the cells with drug prior to transplantation and saw that a greater number a mice achieved therapeutic fetal hemoglobin levels as compared to untreated controls [84].
MGMT has been studied in both dogs [85] and non-human primates . One study with non-human primates showed mostly mild and transient enrichment of MGMT-transduced cells [86], while another group showed more stable enrichment [87]. The dog [85] and one non-human primate study [87] showed no significant enrichment for vector integration sites near proto-oncogenes after drug treatment. In the dog study, two dogs had to be euthanized due to health complications, but these complications seemed to be unrelated to the MGMT-transduced cells. The other non-human primate study [86] did not extensively study genome toxicity, but also saw no evidence of clonal dominance or leukemic transformation. MGMT and MDR1 have additionally been combined, with bicistronic vectors encoding both genes . The stoichiometry between the two genes has even been examined. With an MDR1-IRES-MGMT vector, Maier and colleagues [88] saw a similar cytoprotective effect for monotherapy with paclitaxel or O6-BG/temozolomide, and a greater cytoprotective effect with the combination therapy. Later studies showed that a F2A provided the best stoichiometry between the two drug resistant genes for the best cytoprotective effect [89].
Concerns and Genome Toxicity
Although lentiviral vectors offer a number of benefits, there are still many unmet concerns. Sustained, high expression is still difficult to achieve, as transgene silencing by chromatin modifications is still a problem. Insulators have helped this situation, but it has not been solved entirely. Additionally, there is a mild concern with replication-competent lentivirus; although as generations of lentiviruses progress, they resemble the original HIV-1 genome less [90]. The SIN, or self-inactivating design for a lentivirus removes a 400bp region from the 3′ long terminal repeat. This deletion abolishes the enhancer/promoter activity of the virus, therefore reducing transcriptional interference. It is less likely to recombine with cells that have been infected with HIV-1 or make replication-competent lentivirus as it has less similarity [91].
Several studies have been done on non-viral methods to achieve gene transfer [92]. However, these methods have not been as efficient and still have difficulty achieving sustained and stable expression. The Sleeping Beauty transposase (SB) system is a non-viral method; it is a synthetic transposon system, reverse engineered from defective copies in fish [93]. Sjeklocha and coworkers used SB to transduce human CD34+ cord blood cells. They saw integration and expression of the beta-globin gene, and in studies with K562 cells, saw sustained transgene expression [94].
One of the greatest concerns with lentiviral gene therapy is random integration. Random transgene integration can potentially disrupt a tumor suppressor or cause activation of an oncogene. In trials for X-linked severe combined immunodeficiency, leukemia developed as a result of aberrant gene activation from random integration [95]. One method of preventing malignancy with lentiviruses is to analyze the insertion sites before transplantation and select those “safe harbor” sites which are least likely to cause endogenous gene perturbation. A safe harbor is an integration site that is more that 50–100 kb away from known coding, miRNA, and ultraconserved regions. In 2011, Papapetrou published a paper concerning genomic “safe harbors” and induced pluripotent stem cells (iPSCs). They found that about 10% of integrations occurred in safe harbors and permitted beta-globin gene expression [96, 97].
Another method is to have a failsafe way of getting rid of vector-transduced cells should they become malignant. In addition to the therapeutic gene, a suicide gene can also be transduced at the same time. If malignancy occurs, this suicide gene can be induced with drugs to cause apoptosis and ablate vector-transduced cells in the body. Two such suicide genes studied in the context of gene therapy are the herpes simplex virus type 1 thymidine kinase (HSVtk) and inducible caspase 9 (iCasp9 ) [98]. HSVtk-transduced cells can be eliminated with the phosphorylation of acyclovir or ganciclovir by HSVtk. iCasp9 is expressed as a monomer, but upon addition of AP1903, dimerizes and causes apoptosis. In a study with T-cells, iCasp9 effected immediate death, but HSVtk needed 3 days of treatment [99].
Last, site-specific integration—which does not disrupt other genes—is a new area being explored. Site-specific correction of the beta-globin gene has been done with iPSCs. Making iPSCs from thalassemic cells usually requires the addition of four factors: Oct4, Sox2, Klf4, and c-Myc, although a number of other gene combinations have been successfully tried [100, 101]. This field alone is a large area of research, and reprogramming can be done in a variety of ways: lentiviruses, episomes, nonintegrating viruses, synthetic RNA, or proteins. In 2009, Ye and colleagues showed iPSCs could be successfully generated from thalassemic patients and upon differentiation, could be stained for HbF [102]. Zou [103] and Sebastiano [104] showed in two separate papers that thalassemic iPSCs could undergo site-specific correction of beta-globin using zinc finger nucleases and homologous recombination . This provided the scientific basis for potentially non-integrating in situ correction. While the iPSCs generated in the above papers were done with random integration, it is possible to combine a non-integrating method of iPSC generation and site-specific correction, thus avoiding integration-associated genome toxicity. Most recently, Ma in 2013 used non-integrating episomal technology to create iPSCs and non-integrating TALEN to perform in situ correction [105]. All of these studies have been met with very limited success though, as iPSCs express extremely low and nowhere near therapeutic levels of beta-globin upon differentiation. Site-specific insertion into the adeno-associated virus preferred integration site (AAVS1) has also been done with limited success. In 2008, Howden and colleagues used bacterial artificial chromosomes and components from adeno-associated virus to preferentially insert beta-globin into the AAVS1 in K562 cells [106]. Of the 36 insertion sites analyzed, only 6 of them (17%) occurred in AAVS1, and 5 out of those 6 were intact and functional.
Conclusion
Beta-globin gene addition strategies have come a long way in the past 25 years. Many different vectors with a wide-range of genetic elements have proven successful in preclinical tests and some will be tested in clinical trials. However, work still needs to be done to improve the safety and efficacy. Genomic toxicity and malignancy are some of the largest hurdles to overcome in order to move gene therapy to widespread clinical application. Consistently therapeutic transgene expression for those with thalassemia major is an additional problem. For those vectors that do prove safe and effective, research into increasing the number of engraftable lentiviral transduced cells would help with cases of insufficient mixed chimerism.
References
Rees DC, Williams TN, Gladwin MT: Sickle-cell disease. Lancet, 376:2018-2031.
Cappellini MD, Bejaoui M, Agaoglu L, Canatan D, Capra M, Cohen A, Drelichman G, Economou M, Fattoum S, Kattamis A, et al: Iron chelation with deferasirox in adult and pediatric patients with thalassemia major: efficacy and safety during 5 years' follow-up. Blood 2011, 118:884-893.
Gardenghi S, Grady RW, Rivella S: Anemia, Ineffective Erythropoiesis, and Hepcidin: Interacting Factors in Abnormal Iron Metabolism Leading to Iron Overload in beta-Thalassemia. Hematol Oncol Clin North Am 2010, 24:1089-1107.
Musallam KM, Cappellini MD, Wood JC, Taher AT: Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Rev 2012, 26 Suppl 1:S16-19.
Taher AT, Porter JB, Viprakasit V, Kattamis A, Chuncharunee S, Sutcharitchan P, Siritanaratkul N, Galanello R, Karakas Z, Lawniczek T, et al: Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol 2013, 92:1485-1493.
Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA: Non-transfusion-dependent thalassemias. Haematologica 2013, 98:833-844.
Rivella S: The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev 2012, 26 Suppl 1:S12-15.
Dong A, Rivella S, Breda L: Gene therapy for hemoglobinopathies: progress and challenges. Transl Res 2013, 161:293-306.
Angelucci E: Hematopoietic stem cell transplantation in thalassemia. Hematology Am Soc Hematol Educ Program 2010, 2010:456-462.
Taher AT, Temraz S, Cappellini MD: Deferasirox for the treatment of iron overload in non-transfusion-dependent thalassemia. Expert Rev Hematol 2013, 6:495-509.
Porter JB: Pathophysiology of transfusional iron overload: contrasting patterns in thalassemia major and sickle cell disease. Hemoglobin 2009, 33 Suppl 1:S37-45.
Vogiatzi MG, Tsay J, Verdelis K, Rivella S, Grady RW, Doty S, Giardina PJ, Boskey AL: Changes in Bone Microarchitecture and Biomechanical Properties in the th3 Thalassemia Mouse are Associated with Decreased Bone Turnover and Occur During the Period of Bone Accrual. Calcif Tissue Int 2010.
Haidar R, Musallam KM, Taher AT: Bone disease and skeletal complications in patients with beta thalassemia major. Bone 2011, 48:425-432.
Ginzburg Y, Rivella S: beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118:4321-4330.
Rivella S: Ineffective erythropoiesis and thalassemias. Curr Opin Hematol 2009, 16:187-194.
Guo S, Casu C, Gardenghi S, Booten S, Aghajan M, Peralta R, Watt A, Freier S, Monia BP, Rivella S: Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest 2013, 123:1531-1541.
Parrow NL, Gardenghi S, Ramos P, Casu C, Grady RW, Anderson ER, Shah YM, Li H, Ginzburg YZ, Fleming RE, Rivella S: Decreased hepcidin expression in murine beta-thalassemia is associated with suppression of Bmp/Smad signaling. Blood 2012, 119:3187-3189.
Gardenghi S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, Liu Y, Amariglio N, Rechavi G, Rachmilewitz EA, et al: Ineffective erythropoiesis in beta-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007, 109:5027-5035.
John M Coffin SHH, and Harold E Varmus: Retroviruses. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997.
Dzierzak EA, Papayannopoulou T, Mulligan RC: Lineage-specific expression of a human beta-globin gene in murine bone marrow transplant recipients reconstituted with retrovirus-transduced stem cells. Nature 1988, 331:35-41.
Karlsson S, Papayannopoulou T, Schweiger SG, Stamatoyannopoulos G, Nienhuis AW: Retroviral-mediated transfer of genomic globin genes leads to regulated production of RNA and protein. Proc Natl Acad Sci U S A 1987, 84:2411-2415.
Karlsson S, Papayannopoulou T, Stamatoyannopoulos G, Nienhuis AW: Regulated expression of human globin genes following transfer with retroviral vectors. Prog Clin Biol Res 1987, 251:595-603.
Grosveld F, van Assendelft GB, Greaves DR, Kollias G: Position-independent, high-level expression of the human beta-globin gene in transgenic mice. Cell 1987, 51:975-985.
Li Q, Peterson KR, Fang X, Stamatoyannopoulos G: Locus control regions. Blood 2002, 100:3077-3086.
Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA: Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 2012, 149:1233-1244.
Plavec I, Papayannopoulou T, Maury C, Meyer F: A human beta-globin gene fused to the human beta-globin locus control region is expressed at high levels in erythroid cells of mice engrafted with retrovirus-transduced hematopoietic stem cells. Blood 1993, 81:1384-1392.
Leboulch P, Huang GM, Humphries RK, Oh YH, Eaves CJ, Tuan DY, London IM: Mutagenesis of retroviral vectors transducing human beta-globin gene and beta-globin locus control region derivatives results in stable transmission of an active transcriptional structure. Embo J 1994, 13:3065-3076.
Sadelain M, Wang CH, Antoniou M, Grosveld F, Mulligan RC: Generation of a high-titer retroviral vector capable of expressing high levels of the human beta-globin gene. Proc Natl Acad Sci U S A 1995, 92:6728-6732.
Emery DW, Yannaki E, Tubb J, Stamatoyannopoulos G: A chromatin insulator protects retrovirus vectors from chromosomal position effects. Proc Natl Acad Sci U S A 2000, 97:9150-9155.
Sabatino DE, Seidel NE, Cline AP, Anderson SM, Gallagher PG, Bodine DM: Development of a stable retrovirus vector capable of long-term expression of gamma-globin mRNA in mouse erythrocytes. Ann N Y Acad Sci 2001, 938:246-261.
Fragkos M, Anagnou NP, Tubb J, Emery DW: Use of the hereditary persistence of fetal hemoglobin 2 enhancer to increase the expression of oncoretrovirus vectors for human gamma-globin. Gene Ther 2005, 12:1591-1600.
Persons DA, Hargrove PW, Allay ER, Hanawa H, Nienhuis AW: The degree of phenotypic correction of murine beta -thalassemia intermedia following lentiviral-mediated transfer of a human gamma-globin gene is influenced by chromosomal position effects and vector copy number. Blood 2003, 101:2175-2183.
Oh IH, Fabry ME, Humphries RK, Pawliuk R, Leboulch P, Hoffman R, Nagel RL, Eaves C: Expression of an anti-sickling beta-globin in human erythroblasts derived from retrovirally transduced primitive normal and sickle cell disease hematopoietic cells. Exp Hematol 2004, 32:461-469.
Arumugam P, Malik P: Genetic therapy for beta-thalassemia: from the bench to the bedside. Hematology Am Soc Hematol Educ Program 2010, 2010:445-450.
Breda L, Gambari R, Rivella S: Gene therapy in thalassemia and hemoglobinopathies Mediterranean Journal of Hematology and Infectious Diseases 2009, Vol 1, No 1.
Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D: In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector [see comments]. Science 1996, 272:263-267.
Millington M, Arndt A, Boyd M, Applegate T, Shen S: Towards a clinically relevant lentiviral transduction protocol for primary human CD34 hematopoietic stem/progenitor cells. PLoS One 2009, 4:e6461.
May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, Sadelain M: Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature 2000, 406:82-86.
Rivella S, May C, Chadburn A, Riviere I, Sadelain M: A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood 2003, 101:2932-2939.
Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, et al: Correction of sickle cell disease in transgenic mouse models by gene therapy. Science 2001, 294:2368-2371.
Trudel M, Saadane N, Garel MC, Bardakdjian-Michau J, Blouquit Y, Guerquin-Kern JL, Rouyer-Fessard P, Vidaud D, Pachnis A, Romeo PH, et al.: Towards a transgenic mouse model of sickle cell disease: hemoglobin SAD. Embo J 1991, 10:3157-3165.
Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM: Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 1997, 278:876-878.
Romero Z, Urbinati F, Geiger S, Cooper AR, Wherley J, Kaufman ML, Hollis RP, de Assin RR, Senadheera S, Sahagian A, et al: beta-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest 2013.
Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L, Li S, Yu Y, Hiti AL, Yee JK, Malik P: Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood 2004, 104:3445-3453.
Rivella S, Callegari JA, May C, Tan CW, Sadelain M: The cHS4 insulator increases the probability of retroviral expression at random chromosomal integration sites. J Virol 2000, 74:4679-4687.
Emery DW: The use of chromatin insulators to improve the expression and safety of integrating gene transfer vectors. Hum Gene Ther 2011, 22:761-774.
Rivella S, Sadelain M: Genetic treatment of severe hemoglobinopathies: the combat against transgene variegation and transgene silencing. Semin Hematol 1998, 35:112-125.
Nienhuis AW, Persons DA: Development of gene therapy for thalassemia. Cold Spring Harb Perspect Med 2012, 2.
Arumugam PI, Scholes J, Perelman N, Xia P, Yee JK, Malik P: Improved human beta-globin expression from self-inactivating lentiviral vectors carrying the chicken hypersensitive site-4 (cHS4) insulator element. Mol Ther 2007, 15:1863-1871.
Arumugam PI, Urbinati F, Velu CS, Higashimoto T, Grimes HL, Malik P: The 3' region of the chicken hypersensitive site-4 insulator has properties similar to its core and is required for full insulator activity. PLoS One 2009, 4:e6995.
Lisowski L, Sadelain M: Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood 2007, 110:4175-4178.
Miccio A, Cesari R, Lotti F, Rossi C, Sanvito F, Ponzoni M, Routledge SJ, Chow CM, Antoniou MN, Ferrari G: In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc Natl Acad Sci U S A 2008, 105:10547-10552.
Roselli EA, Mezzadra R, Frittoli MC, Maruggi G, Biral E, Mavilio F, Mastropietro F, Amato A, Tonon G, Refaldi C, et al: Correction of beta-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol Med 2010, 2:315-328.
Miccio A, Poletti V, Tiboni F, Rossi C, Antonelli A, Mavilio F, Ferrari G: The GATA1-HS2 enhancer allows persistent and position-independent expression of a beta-globin transgene. PLoS One 2011, 6:e27955.
Hanawa H, Hargrove PW, Kepes S, Srivastava DK, Nienhuis AW, Persons DA: Extended beta-globin locus control region elements promote consistent therapeutic expression of a gamma-globin lentiviral vector in murine beta-thalassemia. Blood 2004, 104:2281-2290.
Samakoglu S, Lisowski L, Budak-Alpdogan T, Usachenko Y, Acuto S, Di Marzo R, Maggio A, Zhu P, Tisdale JF, Riviere I, Sadelain M: A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat Biotechnol 2006, 24:89-94.
Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA: Correction of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther 2009, 17:245-252.
Wilber A, Hargrove PW, Kim YS, Riberdy JM, Sankaran VG, Papanikolaou E, Georgomanoli M, Anagnou NP, Orkin SH, Nienhuis AW, Persons DA: Therapeutic levels of fetal hemoglobin in erythroid progeny of beta-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood 2011, 117:2817-2826.
Papanikolaou E, Georgomanoli M, Stamateris E, Panetsos F, Karagiorga M, Tsaftaridis P, Graphakos S, Anagnou NP: The new self-inactivating lentiviral vector for thalassemia gene therapy combining two HPFH activating elements corrects human thalassemic hematopoietic stem cells. Hum Gene Ther 2012, 23:15-31.
Gallagher PG, Steiner LA, Liem RI, Owen AN, Cline AP, Seidel NE, Garrett LJ, Bodine DM: Mutation of a barrier insulator in the human ankyrin-1 gene is associated with hereditary spherocytosis. J Clin Invest 2010, 120:4453-4465.
Yocum AO, Steiner LA, Seidel NE, Cline AP, Rout ED, Lin JY, Wong C, Garrett LJ, Gallagher PG, Bodine DM: A tissue-specific chromatin loop activates the erythroid ankyrin-1 promoter. Blood 2012, 120:3586-3593.
Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, Ippolito GC, Fujiwara Y, Ebert BL, Tucker PW, Orkin SH: Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011, 334:993-996.
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, et al: An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342:253-257.
Xu XS, Hong X, Wang G: Induction of endogenous gamma-globin gene expression with decoy oligonucleotide targeting Oct-1 transcription factor consensus sequence. J Hematol Oncol 2009, 2:15.
Wilber A, Tschulena U, Hargrove PW, Kim YS, Persons DA, Barbas CF, 3rd, Nienhuis AW: A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary human adult erythroblasts. Blood 2010, 115:3033-3041.
Graslund T, Li X, Magnenat L, Popkov M, Barbas CF, 3rd: Exploring strategies for the design of artificial transcription factors: targeting sites proximal to known regulatory regions for the induction of gamma-globin expression and the treatment of sickle cell disease. J Biol Chem 2005, 280:3707-3714.
Suwanmanee T, Sierakowska H, Lacerra G, Svasti S, Kirby S, Walsh CE, Fucharoen S, Kole R: Restoration of human beta-globin gene expression in murine and human IVS2-654 thalassemic erythroid cells by free uptake of antisense oligonucleotides. Mol Pharmacol 2002, 62:545-553.
Lacerra G, Sierakowska H, Carestia C, Fucharoen S, Summerton J, Weller D, Kole R: Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients. Proc Natl Acad Sci U S A 2000, 97:9591-9596.
Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, et al: Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467:318-322.
Ikeda K, Mason PJ, Bessler M: 3'UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood 2011, 117:5860-5869.
Young AR, Narita M: Oncogenic HMGA2: short or small? Genes Dev 2007, 21:1005-1009.
Sadelain M, Riviere I, Wang X, Boulad F, Prockop S, Giardina P, Maggio A, Galanello R, Locatelli F, Yannaki E: Strategy for a multicenter phase I clinical trial to evaluate globin gene transfer in beta-thalassemia. Ann N Y Acad Sci 2010, 1202:52-58.
Li CK, Luk CW, Ling SC, Chik KW, Yuen HL, Li CK, Shing MM, Chang KO, Yuen PM: Morbidity and mortality patterns of thalassaemia major patients in Hong Kong: retrospective study. Hong Kong Med J 2002, 8:255-260.
Yannaki E, Papayannopoulou T, Jonlin E, Zervou F, Karponi G, Xagorari A, Becker P, Psatha N, Batsis I, Kaloyannidis P, et al: Hematopoietic stem cell mobilization for gene therapy of adult patients with severe beta-thalassemia: results of clinical trials using G-CSF or plerixafor in splenectomized and nonsplenectomized subjects. Mol Ther 2012, 20:230-238.
Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, Politi P, Durazzi SM, Muretto P, Albertini F: Bone marrow transplantation in patients with thalassemia. N Engl J Med 1990, 322:417-421.
Lucarelli G, Andreani M, Angelucci E: The cure of thalassemia by bone marrow transplantation. Blood Rev 2002, 16:81-85.
Bertaina A, Bernardo ME, Mastronuzzi A, La Nasa G, Locatelli F: The role of reduced intensity preparative regimens in patients with thalassemia given hematopoietic transplantation. Ann N Y Acad Sci, 1202:141-148.
Bernardo ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, Pagliara D, Contoli B, Pinto RM, Caocci G, et al: Allogeneic hematopoietic stem cell transplantation in thalassemia major: results of a reduced-toxicity conditioning regimen based on the use of treosulfan. Blood 2012, 120:473-476.
Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Diez IA, Dewey RA, Bohm M, Nowrouzi A, Ball CR, Glimm H, et al: Stem-cell gene therapy for the Wiskott-Aldrich syndrome. N Engl J Med 2010, 363:1918-1927.
Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, Glimm H, Kuhlcke K, Schilz A, Kunkel H, et al: Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med 2006, 12:401-409.
Abonour R, Williams DA, Einhorn L, Hall KM, Chen J, Coffman J, Traycoff CM, Bank A, Kato I, Ward M, et al: Efficient retrovirus-mediated transfer of the multidrug resistance 1 gene into autologous human long-term repopulating hematopoietic stem cells. Nat Med 2000, 6:652-658.
Moscow JA, Huang H, Carter C, Hines K, Zujewski J, Cusack G, Chow C, Venzon D, Sorrentino B, Chiang Y, et al: Engraftment of MDR1 and NeoR gene-transduced hematopoietic cells after breast cancer chemotherapy. Blood 1999, 94:52-61.
Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, Nienhuis AW, Sorrentino BP: Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood 2003, 102:506-513.
Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW, Persons DA: Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood 2009, 113:5747-5756.
Beard BC, Sud R, Keyser KA, Ironside C, Neff T, Gerull S, Trobridge GD, Kiem HP: Long-term polyclonal and multilineage engraftment of methylguanine methyltransferase P140K gene-modified dog hematopoietic cells in primary and secondary recipients. Blood 2009, 113:5094-5103.
Larochelle A, Choi U, Shou Y, Naumann N, Loktionova NA, Clevenger JR, Krouse A, Metzger M, Donahue RE, Kang E, et al: In vivo selection of hematopoietic progenitor cells and temozolomide dose intensification in rhesus macaques through lentiviral transduction with a drug resistance gene. J Clin Invest 2009, 119:1952-1963.
Beard BC, Trobridge GD, Ironside C, McCune JS, Adair JE, Kiem HP: Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J Clin Invest 2010, 120:2345-2354.
Maier P, Spier I, Laufs S, Veldwijk MR, Fruehauf S, Wenz F, Zeller WJ: Chemoprotection of human hematopoietic stem cells by simultaneous lentiviral overexpression of multidrug resistance 1 and O(6)-methylguanine-DNA methyltransferase(P140K). Gene Ther 2010, 17:389-399.
Maier P, Heckmann D, Spier I, Laufs S, Zucknick M, Allgayer H, Fruehauf S, Zeller WJ, Wenz F: F2A sequence linking MGMT(P140K) and MDR1 in a bicistronic lentiviral vector enables efficient chemoprotection of haematopoietic stem cells. Cancer Gene Ther 2012, 19:802-810.
Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L: A third-generation lentivirus vector with a conditional packaging system. J Virol 1998, 72:8463-8471.
Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D: Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol 1998, 72:9873-9880.
Papapetrou EP, Zoumbos NC, Athanassiadou A: Genetic modification of hematopoietic stem cells with nonviral systems: past progress and future prospects. Gene Ther 2005, 12 Suppl 1:S118-130.
Ivics Z, Hackett PB, Plasterk RH, Izsvak Z: Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91:501-510.
Sjeklocha LM, Park CW, Wong PY, Roney MJ, Belcher JD, Kaufman DS, Vercellotti GM, Hebbel RP, Steer CJ: Erythroid-specific expression of beta-globin from Sleeping Beauty-transduced human hematopoietic progenitor cells. PLoS One 2011, 6:e29110.
Wang GP, Berry CC, Malani N, Leboulch P, Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M, Bushman FD: Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial. Blood 2010, 115:4356-4366.
Papapetrou EP, Sadelain M: Generation of transgene-free human induced pluripotent stem cells with an excisable single polycistronic vector. Nat Protoc 2011, 6:1251-1273.
Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, Kadota K, Roth SL, Giardina P, Viale A, et al: Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol 2011, 29:73-78.
Sadelain M: Eliminating cells gone astray. N Engl J Med 2011, 365:1735-1737.
Marin V, Cribioli E, Philip B, Tettamanti S, Pizzitola I, Biondi A, Biagi E, Pule M: Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum Gene Ther Methods 2012, 23:376-386.
Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126:663-676.
Yamanaka S: Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 2012, 10:678-684.
Ye L, Chang JC, Lin C, Sun X, Yu J, Kan YW: Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc Natl Acad Sci U S A 2009, 106:9826-9830.
Zou J, Mali P, Huang X, Dowey SN, Cheng L: Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118:4599-4608.
Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LF, et al: In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29:1717-1726.
Ma N, Liao B, Zhang H, Wang L, Shan Y, Xue Y, Huang K, Chen S, Zhou X, Chen Y, et al: TALEN-mediated gene correction in integration-free beta-thalassemia iPSCs. J Biol Chem 2013.
Howden SE, Voullaire L, Wardan H, Williamson R, Vadolas J: Site-specific, Rep-mediated integration of the intact beta-globin locus in the human erythroleukaemic cell line K562. Gene Ther 2008, 15:1372-1383.
Acknowledgements
This work is supported by the Children’s Cancer and Blood Foundation and NIH grant NHLBI-R01HL102449-03 (to S. Rivella)
Competing Interests
S. Rivella is a consultant for Novartis, Biomarin and Isis Pharmaceuticals. In addition, he is a co-inventor for the patents US8058061 B2 C12N 20111115 and US7541179 B2C12N 20090602. The consulting work and intellectual property of S. Rivella did not affect in any way the design, conduct, or reporting of this research.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this chapter

Cite this chapter
Dong, A.C., Rivella, S. (2017). Gene Addition Strategies for β-Thalassemia and Sickle Cell Anemia. In: Malik, P., Tisdale, J. (eds) Gene and Cell Therapies for Beta-Globinopathies. Advances in Experimental Medicine and Biology(), vol 1013. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7299-9_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7299-9_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7297-5
Online ISBN: 978-1-4939-7299-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)