
Abstract
Considerable progress with respect to donor source has been achieved in allogeneic stem cell transplant for patients with hemoglobin disorders, with matched sibling donors in the 1980s, matched unrelated donors and cord blood sources in the 1990s, and haploidentical donors in the 2000s. Many studies have solidified hematopoietic progenitors from matched sibling marrow, cord blood, or mobilized peripheral blood as the best source—with the lowest graft rejection and graft versus host disease (GvHD), and highest disease-free survival rates. For patients without HLA-matched sibling donors, but who are otherwise eligible for transplant, fully allelic matched unrelated donor (8/8 HLA-A, B, C, DRB1) appears to be the next best option, though an ongoing study in patients with sickle cell disease will provide data that are currently lacking. There are high GvHD rates and low engraftment rates in some of the unrelated cord transplant studies. Haploidentical donors have emerged in the last decade to have less GvHD; however, improvements are needed to increase the engraftment rate. Thus the decision to use unrelated cord blood units or haploidentical donors may depend on the institutional expertise; there is no clear preferred choice over the other. Active research is ongoing in expanding cord blood progenitor cells to overcome the limitation of cell dose, including the options of small molecule inhibitor compounds added to ex vivo culture or co-culture with supportive cell lines. There are inconsistent data from using 7/8 or lower matched unrelated donors. Before routine use of these less matched donor sources, work is needed to improve patient selection, conditioning regimen, GvHD prophylaxis, and/or other strategies.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The first reported bone marrow transplant for sickle cell disease (SCD) was published in 1984 by the group at St. Jude Children’s Research Hospital [1]. An 8 year-old girl with SCD developed acute myeloid leukemia (AML) and underwent sibling donor myeloablative transplantation as part of her AML therapy. She was ultimately cured of both and remains alive almost three decades later. This proof-of-principle report paved the way for a series of small pilot studies also demonstrating that transplantation from HLA-matched siblings could cure SCD [2,3,4,5]. Later, the multicenter trial published by Walters and colleagues in 1996 was instrumental in solidifying transplant as a bonafide treatment [6]. In this landmark trial, 22 children with very symptomatic SCD underwent sibling marrow transplantation; overall and event-free survival estimates at 4 years were 91 and 73%, respectively. Since then, the procedure has become even safer and more effective. This success is evidenced by the 95% event-free survival (EFS) in 44 patients undergoing refined transplant procedures after January 2000, as described in the French experience [7]. Such excellent results have prompted physicians to reconsider the methods and timing of treatment.
The concept of transplantation earlier in the disease course was already being considered in the 1980s, as reported by investigators in Belgium [8]. In this study, 50 transplanted patients belonged to one of two groups: permanent residents of a European country who had already developed severe sickle cell phenotype before transplant, or visiting patients who were transplanted much earlier in the disease course due to a desire to return to their country of origin. The combined rate of non-engraftment, mixed chimerism, and death was significantly higher in the more diseased group (25% vs. 7%, p < 0.001). While transplanting patients earlier in their disease course was controversial in the 1980s, current understanding of the devastating nature of SCD has led providers to accept early transplant in less severe patients.
Traditionally, myeloablative conditioning was used in SCD transplants to maximize engraftment. This often excluded patients older than 16 years of age who had significant sequelae of SCD, including considerable organ dysfunction. Today, less intense preparative regimens have made curative approaches available. Increased rates of stable mixed donor chimerism are accepted as part of these less intense regimens because the red cell compartment becomes predominantly donor cells and symptoms of SCD resolve with time. The largest, most successful report to date of such non-myeloablative transplantation was described by Hsieh et al. where 30 adults with severe sickle cell disease safely underwent matched sibling transplant with an 87% success rate using a lower-intensity protocol (manuscript under review).
While the field continues to make matched sibling transplant safer and available to more patients with SCD regardless of disease severity and age, there remains the inherent problem of donor availability. The likelihood of two siblings being HLA identical is only 25%, so the odds of finding such a donor are already limited. Notably, a fraction of siblings themselves will have SCD, further limiting the chance of having a suitable match. Some families of patients who do not have siblings have pursued fertility treatments including in vitro fertilization with pre-implantation genetic testing to select embryos that are HLA-matched and disease-free. There are significant ethical and financial considerations to this approach, and it is unlikely that it will become an option for the majority of patients. Therefore, the field has moved to investigate alternative donor-graft sources. Improved outcomes have already been described with matched unrelated donor (MUD) , umbilical cord blood (CB), and haploidentical transplantation techniques for malignancy and immunodeficiency in the last two decades [9,10,11,12]. This success is confirmed by the 2012 Center for International Blood and Marrow Transplant Research (CIBMTR) data on allogeneic transplants performed in the United States from 2007 to 2011, which shows that approximately 60% of all transplants are from alternative donor sources [13].
Similarly, the first bone marrow transplant to correct β-thalassemia major was reported by E.D. Thomas and colleagues in 1982 [14]. A 16 month-old boy received minimal red blood cell transfusion prior to transplant and underwent dimethyl busulfan and cyclophosphamide conditioning followed by infusion of matched sibling bone marrow. Two months later, his hepatosplenomegaly had completely resolved; at 6 months post-transplant, the patient had a normal hemoglobin level. The authors concluded that transplant could cure not only thalassemia but also other genetic diseases of the marrow such as SCD. In the 1980s, a series of reports from the group in Pesaro, Italy, described varying preparative regimens used in matched related donor bone marrow transplantation for β-thalassemia [15,16,17]. In 1990 they reported a large experience of 222 matched related donor transplants for homozygous β-thalassemia in patients under the age of 16 years [18]. EFS was an impressive 94% for the patients with least disease severity (Pesaro Class 1), but this was in stark contrast to the only 53% EFS in the most diseased patients (Pesaro Class 3). This same group of investigators subsequently spent several years working on improving outcomes in the highest-risk patients. Ultimately, they published a regimen, referred to as protocol 26 (hydroxyurea, azathioprine, fludarabine, busulfan, cyclophosphamide), that resulted in an improved EFS of 85% for these high-risk Class 3 patients [19]. A major driver behind pursuing curative transplant in thalassemia is the resulting improvement in quality of life post-transplant, provided there are no long-lasting complications such as severe chronic graft-versus-host disease (GvHD) [20]. Also, life expectancy in developing countries where chelation is not optimal results in a significantly shorter lifespan, so curative transplant becomes a more attractive treatment modality in these areas. However, relying on a matched sibling donor again leaves the majority of patients without a donor, leading to the search for an alternative graft source.
The transplant field is continuing to make improvements in the approach for hemoglobin disorders with better outcomes on the horizon for unrelated donor and cord transplants, but the major limitation remains donor availability. In one report from 2003 that evaluated searches done by the National Marrow Donor Program (NMDP) , the chance of finding a potential 6/6 HLA-matched MUD for a patient with thalassemia or SCD was approximately 60% in each case [21]. In the same report, the chances of finding a 5/6 or 6/6 HLA-matched cord donor were approximately 62 and 30%, respectively. However, this matching was done at the serological level for HLA-A and -B and at the potential allele level for -DRB1. A recent look at this scenario that incorporated more detailed matching demonstrated that the chances of a sickle cell patient finding a potential allele 8/8 HLA-A,-B,-C and -DRB1 MUD was only 19% [22]. Combining 5/6 and 6/6 HLA-matched cords (HLA-A,-B antigen; DRB1 potential allele) with higher cell doses as needed for hemoglobin disorder transplants (total nucleated cell count of at least 5 × 106/kg) could increase the alternative donor option. When this was examined, the probability of a sickle cell patient finding a potential 8/8 MUD, 5/6 or 6/6 unrelated cord improved but remained relatively low at 45% [22]. This leaves a significant number of patients without a potential donor if only using HLA matched siblings, 8/8 MUDs, 5/6 and 6/6 cords with good cell doses. In both of these reports describing donor availability, over 95% of patients would have a donor option if double cord transplants or 7/8 MUD donors were used.
Over the last 2 decades, there has been significant progress in transplantation using umbilical CB units, haploidentical donors, and unrelated donors, to make this curative procedure more accessible to those who are eligible.
Umbilical Cord Blood Transplantation for Patients with Hemoglobin Disorders
Since the first report of a successful umbilical cord blood (CB) transplant in 1989 in a pediatric patient with Fanconi Anemia [23], this option has increasingly been used to treat patients with malignant and nonmalignant hematologic diseases . While CB grafts are smaller in cellularity and volume as compared to bone marrow, the proliferative capacity and numbers of progenitor cells contained within the graft are often greater [24, 25]. Further, due to immunological differences such as lower numbers of CD3+, CD4+, and CD8+ T-cells with a higher CD4/CD8 ratio and a higher percentage of naïve CD45RA+ T-cells [26], CB transplants have been associated with a lower risk of GvHD as compared to bone marrow transplants. Besides the lower risk of GvHD, other advantages associated with CB grafts include easy availability, reduced time to complete the pre-transplant process, little or no donor morbidity, a decreased risk of transmitting latent viral infections, possibility of directed sibling banking, transplant at earlier disease stages, potential for greater HLA-mismatching, an expandable donor pool, and a greater frequency of rare HLA haplotypes in the donor pool as compared to bone marrow registries [27,28,29,30,31,32].
For Patients with Sickle Cell Disease (SCD)
Table 5.1 describes the results to date for patients with SCD who have undergone related and unrelated CB transplantation (CBT).
Related Umbilical Cord Blood Transplantation
The first report of CBT in a patient with SCD occurred in 1996 [33]. The patient received a myeloablative conditioning regimen consisting of busulfan, cyclophosphamide, and anti-thymocyte globulin (ATG) with cyclosporine for prophylaxis against GvHD. The patient engrafted without evidence of GvHD and had hemoglobin electrophoresis results consistent with sickle trait donor by 9 months post-transplant. The largest study was recently reported by the Eurocord and European Blood and Marrow Transplantation group [34]. A total of 325 patients with β-thalassemia major (TM) and 160 patients with SCD underwent HLA-identical CBT or bone marrow transplantation (BMT); among patients with SCD, 30 underwent CBT and 130 received BMT. The 6-year disease-free survival (DFS) was 90 ± 5% after CBT and 92 ± 2% after BMT. Of all patients transplanted, those who received a CBT had significantly longer time to neutrophil and platelet engraftment as compared to those who underwent BMT (p < 0.005). None who received CBT developed grade IV acute GvHD as compared to 8 (2%) who received BM cells. Twenty-nine percent of the BMT patients experienced chronic GvHD as compared to none of the CBT recipients.
A subgroup analysis that included all of the CBT recipients revealed that MTX affects outcome [34] as noted in their previous report [35]. Patients who did not versus did receive MTX had disease-free survivals of 90 ± 4% versus 60 ± 11%, respectively (p < 0.001). The time in which transplant was performed also influenced outcome, with CBT performed after 1999 faring significantly better as compared to those transplanted earlier (hazard ratio 0.033, confidence interval 0.12–0.89, p = 0.02). The use of thiotepa and patients belonging to Pesaro Class 1 also did better, though those variables were not significant in multivariate analysis. The total nucleated cell dose infused did not influence outcome in patients that received CBT, though median TNC was sufficient at 3.9 × 107 cells/kg.
To date, 44 children or young adult patients with SCD have received related CBT as reported in the literature (Table 5.1, top). The grafts have primarily been 6/6 HLA-matched. The conditioning regimens have been mixed, but most contained busulfan and cyclophosphamide with or without ATG. Related CBT have been successful in patients with SCD, with an overall survival of 91% and DFS of 86%. Eleven percent of patients have developed grade 2–4 acute GvHD, but none of the patients has experienced chronic extensive GvHD. Therefore, while a standard conditioning regimen has not been developed, the DFS results of related CBT for patients with SCD are at least as good as HLA-matched sibling BMT and with a lower incidence of chronic GvHD .
Unrelated Umbilical Cord Blood Transplantation
In contrast, unrelated CBT have been much less successful. The largest study was reported in 2011, and includes 35 patients with TM and 16 patients with SCD [36]. Nine of the SCD patients received myeloablative regimens while seven underwent reduced intensity conditioning. While the overall survival was 94%, DFS was only 50%. The authors did not distinguish the incidence of GvHD based on hemoglobinopathy type, but 22% of all patients experienced grade 2–4 acute GvHD and 3% extensive chronic GvHD.
A total of 41 patients with SCD have been reported to undergo unrelated CBT (Table 5.1, bottom). The conditioning regimens varied in each study and ranged from myeloablative to reduced-intensity. Compared to the related setting, the majority of patients received 1 or 2 HLA-mismatched grafts. Overall survival was 85%, DFS about 50%, but was reported as low as 37.5% in one study despite only selecting 0 or 1 HLA-mismatched grafts [37]. About one third of patients has developed grade 2–4 acute GvHD, and 7% extensive chronic GvHD. Therefore, the results of unrelated CBT for patients with SCD are suboptimal. As these data reported the composite results of 6/6, 5/6, and 4/6 matches, there is insufficient information to decipher if 6/6 HLA matches would render better outcomes. More effective conditioning, better GvHD prophylaxis, and/or other graft sources should be sought for patients who do not have an HLA -matched sibling donor.
For Patients with Thalassemia Major (TM)
Table 5.2 shows the results for patients with TM who have undergone related and unrelated CBT.
Related Umbilical Cord Blood Transplantation
There are many more reports of related than unrelated CBT for patients with TM. The first patient with TM who underwent CBT was reported in 1995 [38]. The cord was 6/6 HLA-matched from a sibling, and the patient received busulfan, cyclophosphamide, and ATG. The transplant was successful, and the patient did not experience GvHD.
In the study mentioned previously [34], 66 patients with TM underwent CBT, and 259 received BMT. A greater percentage of patients categorized as Pesaro Class 2 and 3 underwent BMT (44%) as compared to those who received CBT (39%, p = 0.01). In contrast to patients with SCD (92 ± 2%), the 6-year DFS in patients with TM was 84 ± 2% (p = 0.04). Upon multivariate analysis, a diagnosis of SCD was the only variable which favorably influenced the DFS probability for all patients transplanted in the study (hazard ratio 0.52, 95% confidence interval 0.28–0.97, p = 0.04). The 6-year DFS was 86 ± 2% in patients with TM who received BMT as compared to 80 ± 5% in patients who underwent CBT. This largest study to date does not report a significant difference in DFS in patients who receive CBT as compared to BMT. On the other hand, the incidence of grade 2–4 acute GvHD was similar at 15%, and the rate of chronic extensive GvHD was close to non-existent. Therefore, the incidence of GvHD is significantly lower in patients with TM who undergo CBT as compared to BMT.
Currently, 124 patients with TM have undergone related CBT as reported in the literature (Table 5.2, top). Similar to patients with SCD, the majority of conditioning regimens have included busulfan and cyclophosphamide with or without ATG. Patients have had all stages of disease with Pesaro Class ranging from 1 to 3. The majority of the studies have employed 6/6 HLA-matched grafts. The overall survival is excellent at 94%. However, as compared to an 86% DFS found in patients with SCD, DFS in patients with TM appears to be lower at 76%. Notably, while hypertransfusion and cytoreductive therapy pre-transplant has been associated with improved DFS in subjects with Pesaro Class 3 disease [19], these additional therapies were only employed in one study [39]. Patients with TM may require more intensive therapy pre- and post-transplant to suppress the enhanced erythropoietic drive which may out-compete the immature immune and progenitor cells located within the CB graft. The contribution of hypertransfusion and the remaining portion of the ‘pre-transplant conditioning regimen’ to differences in DFS should be further evaluated .
Unrelated Umbilical Cord Blood Transplantation
Two relatively large studies involving patients with TM who have undergone unrelated CBT have been reported. The first study described above included 35 patients with TM [36]. Thirty patients received myeloablative conditioning, and five reduced-intensity conditioning. Pesaro classification was not available in the majority of the subjects. Most received 1 or 2 HLA-mismatched grafts, and 1 patient received a 3 HLA-mismatched graft. Thirty-four percent of the patients died: 7 patients died from transplant-related complications and 5 died as a result of graft failure. The transplant was successful in only 8 patients (21%). As described above, 22% of the total patients transplanted experienced grade 2–4 acute GvHD while only 3% developed extensive chronic GvHD.
Conversely, another group reported their results for 35 patients with TM who underwent unrelated CBT [40]. They received busulfan, cyclophosphamide, ATG, and cyclosporine and methylprednisolone for prophylaxis against GvHD. Pesaro classification was not reported, and again the majority received 1 or 2 HLA-mismatched cords with one patient receiving a 3 HLA-mismatched cord. Overall survival was 89%. By the end of the study, about 80% of patients were free of their disease. However, 6 of the patients required a second transplant: 5 received second CBT and 1 patient underwent peripheral blood stem cell transplantation. All of the patients experienced acute GvHD: 18% grade 1, 35% grade 2, 44% grade 3, and 3% grade 4. However, only 3% developed extensive chronic GvHD.
A total of 75 patients with TM have been reported to have received unrelated CBT (Table 5.2, bottom). The regimens are varied, but most patients are administered busulfan, cyclophosphamide, ATG, and/or fludarabine. Most of the patients received mismatched grafts. The overall survival is 79% with a DFS of 53%. About half of the patients developed grade 2–4 acute GvHD, and only 2.5% extensive chronic GvHD. Therefore, mortality (21%) associated with unrelated CBT is high in patients with TM, and should not be routinely applied to patients with Pesaro Class 1 who have <10% mortality with transfusions and iron chelation [41,42,43]. Since many patients died from graft failure, survival may be improved with second transplants, even using a second CB graft. The incidence of grade 2 and 3 acute GvHD is high, though the rates of grade 4 acute and extensive chronic GvHD are low. Again these studies reported composite results—6/6 HLA matches with 5/6 and 4/6 matches—thus there are no sufficient data to convincingly decipher 6/6 matches would lead to better outcomes. Conditioning regimens to induce tolerance, more intensive supportive care post-transplant, and other donor sources are necessary for patients with TM who do not have an HLA -matched sibling donor .
Summary and Future Directions for Umbilical Cord Blood Transplantation
The most encouraging results are from related donor CBT ; this option is most favorable when the cord is 6/6 HLA-matched and the TNC is greater than 4 × 107/kg [28]. It is disappointing that the transplant results from using unrelated CB grafts have been suboptimal to date. The major obstacles with unrelated CBT remain graft failure in SCD, and low DFS and excessively high mortality in TM. Thus CB grafts have not emerged as a clear alternative cell source for patients without matched sibling donors. While cord blood banking should be discussed among patients, parents, and providers, the enthusiasm for routine cord blood banking, including directed to siblings with hemoglobin disorders, has been dampened by favorable results obtained only when using fully matched related units. Donation of cord blood units for research purposes, however, should be encouraged.
Methotrexate should not be used as GvHD prophylaxis due to the associated inferior results [30]. The incidence of graft failure may be decreased by infusing bone marrow cells from the same sibling donor [39, 44, 45]. These results have led us to conclude that improvements to CBT outcome can come from several areas currently in active research.
Optimizing Supportive Care and Conditioning Regimen
One study reported that CBT performed more recently had a higher success rate [34], suggesting that greater experience, better supportive care, and improved antibiotic prophylaxis has led to better results. Indeed, improved success over the past few decades in patients with hematologic malignancies has been at least partially attributed to better selection of CB units, more experienced supportive care, and better selection of eligible transplant recipients [29, 46], and those principles will need to be optimized in the non-malignant hematologic disease setting as well. Additional factors that may contribute to graft failure and should be evaluated are the effects of red cell alloimmunization and donor-specific HLA antibodies.
Cord blood transplants for hemoglobin disorders have been plagued by graft failure as demonstrated in the BMT CTN 0601 SCURT trial (NCT00745420), where the premature closure of the cord donor arm was related to an unacceptably high graft failure rate [37]. This study suggests that even one antigen-mismatched grafts and optimal cell dose may not be sufficient [37]. Thus adding more chemotherapy agents was a reasonable next step to overcome the engraftment barrier. Preliminary data suggest that improvement in engraftment has been achieved by adding hydroxyurea and thiotepa to the preparative regimen of alemtuzumab, fludarabine, and melphalan used in the SCURT study [47]. While 5 out of 8 patients showed graft rejection in the SCURT trial, only 1 of 12 rejected with the new regimen that included thiotepa and hydroxyurea. Further follow-up of these early findings is warranted. The use of thiotepa has also shown some preliminary success in other studies [34, 48]. However, increasingly intensive conditioning was not sufficient to significantly improve the graft failure rate in nine patients with TM who received related CBT [49].
Regimens that instead focus on tolerance induction, such as promoting regulatory T-cells (sirolimus) or eliminating alloreactive T-cells following graft infusion (post-transplant cyclophosphamide), may decrease the risk of graft failure. Mixed donor chimerism has been found to be associated with a decreased incidence of GvHD in patients with SCD or TM who receive BMT or peripheral blood stem cell transplantation (PBSCT) [50, 51]. Further, mixed donor chimerism (10–95%) was not found to be associated with an increased risk of graft failure in 27 patients with TM given CBT from a related donor [48]. All 27 patients were alive, transfusion-independent, off immunosuppression, and did not have GvHD. Therefore, a low intensity regimen which leads to stable mixed chimerism and tolerance induction may be optimal. This approach in the unrelated donor setting would need to be balanced with the higher risks of graft failure and GvHD .
Allelic HLA Matching
There has been an international effort to expand allelic level typing for all matched unrelated donors, and recently this is increasingly performed for cord blood units. In a recent report from CIBMTR, where they performed such allelic level HLA-typing in single unit CBT for hematologic malignancies, neutrophil recovery was not more delayed in one and two antigen mismatched units. However, non-relapse mortality was higher, evidenced by higher hazard ratios, in any mismatched setting (even one antigen mismatched) [52]. Due to these apparently conflicting results and the novelty of the data, further investigation is warranted. A marrow donor recruitment group from Washington, DC, performed DNA sequencing and assigned HLA haplotypes, and showed that there were novel HLA-B, -C,-DRB1, and -DQB1 associations in African-American mothers and their cord blood units, again highlighting HLA diversity in this population [53]. Thus higher resolution HLA-typing could provide detailed information for the possibility of better HLA matching.
Double Cord Blood Transplant
Double cord transplant , while mostly described in the malignant transplant realm, remains the subject of research for hemoglobin disorder transplants. The currently open trial NCT00920972, which evaluates alemtuzumab, fludarabine, and melphalan conditioning in non-malignant transplants, has strata dedicated to single and double cord transplants for hemoglobin disorders [54].
Ex vivo Expansion
Another significant limiting factor has been cord size/cell dose. A study published on behalf of the Eurocord Registry, the CIBMTR, and the New York Blood Center describing outcomes of unrelated cord transplants for hemoglobin disorders recommended that a total nucleated cell count of at least 5 × 106/kg be targeted for single cord transplants to maximize engraftment [36]. Such high cell doses can significantly restrict cord options for patients, especially as adulthood is approached and cord units become too small. In the last few years, the transplant field has seen a bevy of approaches used to expand hematopoietic stem cells in the basic science lab now being translated to clinical practice. The most impressive report to date describes 31 adults with hematologic malignancy who received unrelated double umbilical cord transplants [55]. In this trial, one of the cords was co-cultured ex vivo with mesenchymal stromal cells prior to infusion, resulting in a 12-fold increase in total nucleated cell doses and a 30-fold increase in CD34+ cell content. The rate of neutrophil engraftment by day 42 was significantly improved in patients who received the expanded cord as part of their transplant (96%) when compared to two separate control cohorts who received unmanipulated double cords (83 and 78%). While the expanded cord transplant contributed to hematopoiesis early on, at 1 year the predominant donor-derived chimerism was from the unmanipulated unit in the vast majority of the patients. The finding that the unmanipulated cord is the one ultimately responsible for long-term engraftment brings into question whether expanded units, despite having increased numerical cell doses, behave functionally similar to unmanipulated grafts of similar size and are capable of life-long engraftment. Promising preliminary data in transplants for malignancy have supported the NiCord® trial for SCD (NCT01590628), which is evaluating the use of double cord transplantation after myeloablative conditioning where one of the cord units is expanded ex vivo in culture with cytokines and nicotinamide [56]. The study is estimated to complete in the spring of 2014.
Haploidentical (and Mismatch Related) Transplantation for Patients with Hemoglobin Disorders
Haploidentical transplantation has increasingly been explored as a viable treatment option for patients with hemoglobin disorders. Many benefits exist including a readily available donor pool, since parents, children, and half-matched siblings can serve as donors , relatively short time to collect the graft as compared to unrelated donors, and the option for repeat collections as opposed to a CB graft. However, due to the higher immunologic barrier, there has traditionally been a high risk for graft rejection, GvHD, and transplant-related mortality.
For Patients with Sickle Cell Disease (Table 5.3)
The first report of a haploidentical BMT in a patient with SCD occurred in 2004 [57]. The 14 year-old patient was conditioned with fludarabine and 200 cGy TBI, and received CSA and MMF for prophylaxis against GvHD. The patient survived free of SCD and did not develop GvHD. In 2012, the Johns Hopkins group reported their experience [58]. Their regimen was based on the success seen in their patients with hematologic malignancies who underwent haploidentical BMT and included post-transplant infusion of cyclophosphamide to decrease the incidence of GvHD [59, 60]. Fourteen patients, predominantly adults, were treated. While the rate of graft rejection was expectedly high due to the non-myeloablative regimen, 7 of the 14 patients are completely free of SCD. One additional patient displays a mixed donor and recipient erythroid phenotype with severe anemia and sickle hemoglobin elevated more than would be expected in a subject with sickle cell trait. Importantly, none of the patients developed GvHD and all of the patients are living. Further, six of the patients with complete donor chimerism have been weaned off of immunosuppressive therapy.
Recently, Dallas and colleagues described eight pediatric patients who received haploidentical PBSCT [61]. The patients underwent reduced-intensity conditioning consisting of fludarabine or pre-transplant cyclophosphamide, busulfan, thiotepa, and muromonab-CD3 with or without ATG. They were treated with MMF for GvHD prophylaxis. The patients received CD34+-selected PBSC on day 0 and CD3+-depleted PBSC on day +1. These patients were compared to 14 pediatric patients who received myeloablative conditioning: 13 patients underwent matched sibling BMT and 1 patient received matched sibling CBT. The overall survival and DFS were 93% for patients who underwent matched related donor transplantation. Conversely, for patients who received haploidentical transplants, overall survival was 75% and DFS 38%. Two patients (25%) developed grade 2 acute and two patients chronic extensive GvHD; both patients died from GvHD.
Therefore, haploidentical transplantation is associated with a high risk of graft rejection in patients with SCD, with a total DFS rate of 43% (Table 5.3). While the most recent report was associated with a prohibitive rate of transplant-related mortality, both as a result of GvHD, the Hopkins group transplanted 14 patients, including 12 adults, and none of the patients died or developed GvHD. The improved survival and absence of GvHD may be due to the bone marrow stem cell source and/or the use of post-transplant cyclophosphamide. Additional studies to attempt to decrease the rate of graft rejection and GvHD should be performed in the context of a clinical trial.
For Patients with Thalassemia Major (Table 5.4)
In two earlier reports, up to three antigen mismatch family donors were used and the majority of the patients belonged to Pesaro Class 2 or 3. In the report by Gaziev et al. [62], reduced doses of busulfan down to 8 mg/kg were used to reduce the potential liver and other transplant-related toxicity, which was one explanation of the surprisingly high graft rejection rate (55%). There was also an equally high rate of acute and chronic GvHD, 47 and 38% respectively. Separately, Sullivan and colleagues also reported a high rejection rate at 28%, but rates of GvHD were not reported [63]. Both reports contained mortality rates close to 30%, which dampened the enthusiasm of relying on this donor source for transplantation. However, in the most recent report of 16 patients, the results were much improved with only one death and GvHD rates of 15–20% [64].
In 2004, a patient with Pesaro Class three disease was reported to have undergone haploidentical PBSCT after being conditioned with busulfan, fludarabine, anti-lymphocyte globulin (ALG), and 500 cGy TBI [65]. The patient survived free of disease but developed grade 2 acute GvHD and extensive chronic GvHD. The largest study to date was reported in 2011 [66]. Thirty-one patients with TM received myeloablative conditioning with fludarabine or cyclophosphamide, busulfan, thiotepa, ATG, and CSA for prophylaxis against GvHD. Donor grafts consisted of T-cell depleted BM and PBSCs to achieve mega doses of enriched CD34+ cells. Overall survival was 94% with a DFS of 70%. None of the patients developed acute or chronic GvHD.
To date, a total of 159 patients with TM have been reported to undergo haploidentical transplantation (Table 5.3). Conditioning regimens are mixed, but all except one contain fludarabine. Total overall survival is 93% and DFS is 74%. Despite most donor grafts including PBSCs, GvHD rate was low: 13% grade 2–4 acute and 4% extensive chronic GvHD.
Interestingly, unlike in CBT where DFS is higher in patients with SCD as compared to TM, DFS appears to be higher in patients with TM as compared to SCD when undergoing haploidentical transplantation. The mega doses of CD34+-cells in more than half of the patients with TM may have helped to overcome the engraftment barrier. Therefore, future studies should consider mega doses of CD34+ cells (>10 × 106 cells/kg) to help decrease the rate of graft rejection along with ex vivo or in vivo T-cell depletion and/or post-transplant cyclophosphamide to help decrease the risk of GvHD and again should be performed as part of a clinical trial.
Matched Unrelated Donor Transplantation for Patients with Hemoglobin Disorders (Table 5.5)
Due to the lack of donor availability, matched unrelated donor (MUD) marrow transplants have not been performed in sufficient number of patients with SCD. There is one report from Germany that included two children who received matched unrelated marrow transplant successfully [67]. The Sickle Cell Unrelated Transplant (SCURT) trial (NCT00745420), which has been open since 2008, has transplanted close to 20 patients from MUD (European Bone Marrow Transplant meeting 2012). When published, the study results will provide useful information about this donor option for patients with SCD.
With respect to MUD marrow transplants in TM, there have been slightly over 130 patients transplanted. La Nasa et al. reported MUD marrow transplants from 10/10 donors, but included more Pesaro Class 3 (more severe) patients [68, 69]. Rates of acute GvHD, chronic GvHD, and mortality were 35, 20, and 24%, respectively. A more recent cohort was described in mostly Pesaro Class 2 patients where they allowed up to one allele HLA-mismatch donors [70]. There was only 8% acute GvHD (grade 2 not reported), no extensive chronic GvHD (limited GvHD not reported), and 8% mortality. Having more Class 2 patients, using reduced doses of cyclophosphamide, and employing more current supportive care certainly contributed to the better results reported in this recent cohort. There is preliminary report from an international collaborative collection of 23 children with thalassemia, 16 of them received transplant from unrelated donors. The DFS was 78%, with acute and chronic GvHD rates of 30 and 9% respectively (Shenoy et al., American Society of Hematology Annual Meeting, 2013 ).
Summary and Recommendations
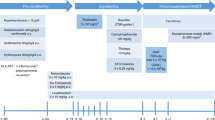
With the passing of each decade, substantial progress in allogeneic transplantation from the different donor options has been made for hemoglobin disorders: matched sibling/related donors in the 1980s, matched unrelated and cord blood in the 1990s, and haploidentical donors in the 2000s. These alternative options have greatly expanded transplantation to those who were otherwise eligible for a conventional matched sibling transplant. From our review of the literature, improvements in supportive care and high resolution HLA-typing have further made transplantation safer, and modifications to the conditioning regimen have allowed those with end-organ damage or higher Pesaro classification to receive this curative procedure (Fig. 5.1).

(a) Summary of transplants performed in sickle cell disease. The average rates of event-free survival (Alive, no disease), acute GvHD, chronic GvHD, and mortality are plotted with respect to the different donor sources. *There are insufficient matched unrelated donor (MUD) data to date, but the sickle cell unrelated transplant (SCURT) trial results will be available soon. (b) Summary of transplants performed in thalassemia. The average rates of event-free survival (Alive, no disease), acute GvHD, chronic GvHD, and mortality are plotted with respect to the different donor sources
The conditioning regimens have gradually been adapted from the most common backbone of busulfan (14–16 mg/kg), cyclophosphamide (200 mg/kg), anti-thymocyte globulin, and cyclosporine for GvHD prophylaxis. Busulfan has mostly been switched to intravenous for consistent pharmacokinetic measurements and less risk of sinusoidal obstructive syndrome (veno-occlusive disorder). Cyclophosphamide dose can be reduced to as low as 120 mg/kg to prevent significant liver injury peri-transplant. For haploidentical or allelic mismatched donors, thiotepa and/or fludarabine have been added to enhance engraftment, and post-transplant cyclophosphamide and other immunosuppressants (e.g. sirolimus) to reduce GvHD.
The results of these transplant studies that we surveyed can be summarized into the following recommendations, which can be applied with children or adults, and in β-thalassemia or SCD (Fig. 5.2).
-
1.
Marrow, peripheral blood derived, or cord blood progenitor cells from matched sibling/related donors offer the best results of transplantation, thus this donor source should be sought whenever possible.
-
2.
Fully matched unrelated marrow (8/8 or 10/10 allelic match) would be the next best option (Table 5.5), as the evidence for this donor choice in patients with thalassemia has been reported in two reasonably sized studies in different populations [68, 70]. There are insufficient data in SCD thus far, but the results from SCURT trial (BMT CTN 0601, NCT00745420) should be available soon. This donor source may be more difficult to find for patients with SCD, compared to patients with thalassemia.
Table 5.5 Mismatch related marrow or matched unrelated donor transplantation for patients with thalassemia major -
3.
While fully matched unrelated cord blood units (6/6 match) would appear to be the next reasonable option, the reports only included limited number of patients and the results were combined 6/6 match with lower HLA matches. There are favorable results in some reports but high rates of GvHD with low rates of engraftment in others (Tables 5.1 and 5.2). Thus at this time, this source (whether 6/6 match or lower) is not clearly more preferable than haploidentical donors. The institutional expertise would likely dictate which source is better suited. Additionally there are other factors that make this source less desirable than match unrelated marrow.
-
a.
Since total nucleated cell count can vary, this source is better suited for children.
-
b.
Fully matched unrelated cord blood units are rare for patients with SCD.
-
c.
Delayed platelet engraftment that is typical with CBT and may lead to excessive morbidity should be considered: hypersplenism in patients with thalassemia with risk of platelet transfusion refractoriness, or CNS vasculopathy in patients with SCD with risk of intracranial hemorrhage.
-
a.
-
4.
Haploidentical (or mismatch related) donors have emerged as a reasonable and safe alternative from the studies published in the last decade (Tables 5.3 and 5.4), but the cumulative experience is less than that of cord blood transplants. The typical conditioning regimen backbone of busulfan, cyclophosphamide, and ATG has been adapted to include fludarabine and/or low dose radiation in thalassemia. Although at a glance, the outcomes from this source appear to be suboptimal, the data from the most recent series have better engraftment and lower GvHD rates [64]. The conditioning regimens in SCD are significantly different from busulfan, cyclophosphamide, and ATG. The addition of post-transplant cyclophosphamide has been shown to decrease the incidence of GvHD, but more optimization is needed to improve this donor option.
-
5.
Although data from other nonmalignant diseases (immune deficiencies or aplastic anemia) are encouraging, the results from using mismatched unrelated marrow (7/8 or less) in hemoglobin disorders are overall inferior to the other options based on studies to date. Therefore, this donor source should be studied in the context of clinical trials, testing newer conditioning regimens, other immunosuppressive combinations, and/or other strategies.

Suggested priority of alternative donors . Hematopoietic progenitors from matched sibling donors remain the best option. From the studies reviewed in this work, fully matched unrelated donor is the next best option for patients with thalassemia. The corresponding data in sickle cell disease will be available soon. Since the results from unrelated cord blood and haploidentical/mismatched related transplant are variable, there is no one preferred source at this time. Enrollment in clinical trials is strongly encouraged. Mismatched unrelated marrow source is not recommended until after further optimization.
References
Johnson F, Look A, Gockerman J, Ruggiero M, Dalla-Pozza L, Billings Fr. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med. 1984;311(12):780-3.
Vermylen C, Fernandez Robles E, Ninane J, Cornu G. Bone marrow transplantation in five children with sickle cell anaemia. Lancet. 1988;1(8600):1427-8.
Vermylen C, Cornu G. Bone marrow transplantation for sickle cell disease. The European experience. Am J Pediatr Hematol Oncol. 1994;16(1):18-21.
Ferster A, De Valck C, Azzi N, Fondu P, Toppet M, Sariban E. Bone marrow transplantation for severe sickle cell anaemia. Br J Haematol. 1992;80(1):102-5.
Johnson FL, Mentzer WC, Kalinyak KA, Sullivan KM, Abboud MR. Bone marrow transplantation for sickle cell disease. The United States experience. Am J Pediatr Hematol Oncol. 1994;16(1):22-6.
Walters M, Patience M, Leisenring W, Eckman J, Scott J, Mentzer W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369-76.
Bernaudin F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749-56.
Vermylen C, Cornu G, Ferster A, Brichard B, Ninane J, Ferrant A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998;22(1):1-6.
Hahn T, McCarthy PL, Jr., Hassebroek A, Bredeson C, Gajewski JL, Hale GA, et al. Significant improvement in survival after allogeneic hematopoietic cell transplantation during a period of significantly increased use, older recipient age, and use of unrelated donors. J Clin Oncol. 2013;31(19):2437-49.
Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013;122(4):491-8.
Gooley TA, Chien JW, Pergam SA, Hingorani S, Sorror ML, Boeckh M, et al. Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med. 2010;363(22):2091-101.
Cavazzana-Calvo M, Andre-Schmutz I, Fischer A. Haematopoietic stem cell transplantation for SCID patients: where do we stand? Br J Haematol. 2013;160(2):146-52.
Pasquini MC WZ. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2012.
Thomas ED, Buckner CD, Sanders JE, Papayannopoulou T, Borgna-Pignatti C, De Stefano P, et al. Marrow transplantation for thalassaemia. Lancet. 1982;2(8292):227-9.
Lucarelli G, Polchi P, Izzi T, Manna M, Agostinelli F, Delfini C, et al. Allogeneic marrow transplantation for thalassemia. Exp Hematol. 1984;12(8):676-81.
Lucarelli G, Polchi P, Galimberti M, Izzi T, Delfini C, Manna M, et al. Marrow transplantation for thalassaemia following busulphan and cyclophosphamide. Lancet. 1985;1(8442):1355-7.
Lucarelli G, Galimberti M, Polchi P, Giardini C, Politi P, Baronciani D, et al. Marrow transplantation in patients with advanced thalassemia. N Engl J Med. 1987;316(17):1050-5.
Lucarelli G, Galimberti M, Polchi P, Angelucci E, Baronciani D, Giardini C, et al. Bone marrow transplantation in patients with thalassemia. N Engl J Med. 1990;322(7):417-21.
Sodani P, Gaziev D, Polchi P, Erer B, Giardini C, Angelucci E, et al. New approach for bone marrow transplantation in patients with class 3 thalassemia aged younger than 17 years. Blood. 2004;104(4):1201-3.
La Nasa G, Caocci G, Efficace F, Dessi C, Vacca A, Piras E, et al. Long-term health-related quality of life evaluated more than 20 years after hematopoietic stem cell transplantation for thalassemia. Blood. 2013;122(13):2262-70.
Krishnamurti L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for hematopoietic stem cell transplantation for hemoglobinopathies. Bone Marrow Transplant. 2003;31(7):547-50.
Justus D, Perez E, Dioguardi J, Abraham A. Allogeneic Donor availability for Hematopoietic Stem Cell Transplantation in Patients with Sickle Cell Disease. World Cord Blood Congress IV and Innovative Therapies for Sickle Cell Disease. Monaco; October 24-27, 2013.
Gluckman E, Broxmeyer HA, Auerbach AD, Friedman HS, Douglas GW, Devergie A, et al. Hematopoietic reconstitution in a patient with Fanconi's anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med. 1989;321(17):1174-8.
Mayani H, Lansdorp PM. Biology of human umbilical cord blood-derived hematopoietic stem/progenitor cells. Stem Cells. 1998;16(3):153-65.
Broxmeyer HE, Hangoc G, Cooper S, Ribeiro RC, Graves V, Yoder M, et al. Growth characteristics and expansion of human umbilical cord blood and estimation of its potential for transplantation in adults. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(9):4109-13.
Bradley MB, Cairo MS. Cord blood immunology and stem cell transplantation. Human Immunology. 2005;66(5):431-46.
Agarwal MB. Umbilical cord blood transplantation: newer trends. The Journal of the Association of Physicians of India. 2006;54:143-7.
Pinto FO, Roberts I. Cord blood stem cell transplantation for haemoglobinopathies. British Journal of Haematology. 2008;141(3):309-24.
Kanathezhath B, Walters MC. Umbilical cord blood transplantation for thalassemia major. Hematology/Oncology Clinics of North America. 2010;24(6):1165-77.
Boncimino A, Bertaina A, Locatelli F. Cord blood transplantation in patients with hemoglobinopathies. Transfusion and apheresis science: official journal of the World Apheresis Association: official journal of the European Society for Haemapheresis. 2010;42(3):277-81.
Thompson LM, Ceja ME, Yang SP. Stem cell transplantation for treatment of sickle cell disease: bone marrow versus cord blood transplants. American journal of health-system pharmacy: AJHP: official journal of the American Society of Health-System Pharmacists. 2012;69(15):1295-302.
Shenoy S. Umbilical cord blood: an evolving stem cell source for sickle cell disease transplants. Stem cells translational medicine. 2013;2(5):337-40.
Brichard B, Vermylen C, Ninane J, Cornu G. Persistence of fetal hemoglobin production after successful transplantation of cord blood stem cells in a patient with sickle cell anemia. The Journal of pediatrics. 1996;128(2):241-3.
Locatelli F, Kabbara N, Ruggeri A, Ghavamzadeh A, Roberts I, Li CK, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122(6):1072-8.
Locatelli F, Rocha V, Reed W, Bernaudin F, Ertem M, Grafakos S, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101(6):2137-43.
Ruggeri A, Eapen M, Scaravadou A, Cairo MS, Bhatia M, Kurtzberg J, et al. Umbilical cord blood transplantation for children with thalassemia and sickle cell disease. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2011;17(9):1375-82.
Kamani NR, Walters MC, Carter S, Aquino V, Brochstein JA, Chaudhury S, et al. Unrelated donor cord blood transplantation for children with severe sickle cell disease: results of one cohort from the phase II study from the Blood and Marrow Transplant Clinical Trials Network (BMT CTN). Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2012;18(8):1265-72.
Issaragrisil S, Visuthisakchai S, Suvatte V, Tanphaichitr VS, Chandanayingyong D, Schreiner T, et al. Brief report: transplantation of cord-blood stem cells into a patient with severe thalassemia. N Engl J Med. 1995;332(6):367-9.
Sun X, Hao WG, Liu S, Xia T, Li Y, Liao C. [Combined transplantation of umbilical cord blood and bone marrow from same sibling donor in children with beta-thalassemia major]. Zhongguo shi yan xue ye xue za zhi/Zhongguo bing li sheng li xue hui = Journal of experimental hematology/Chinese Association of Pathophysiology. 2007;15(4):801-4.
Jaing TH, Hung IJ, Yang CP, Chen SH, Chung HT, Tsay PK, et al. Unrelated cord blood transplantation for thalassaemia: a single-institution experience of 35 patients. Bone Marrow Transplantation. 2012;47(1):33-9.
Olivieri NF, Nathan DG, MacMillan JH, Wayne AS, Liu PP, McGee A, et al. Survival in medically treated patients with homozygous beta-thalassemia. The New England journal of medicine. 1994;331(9):574-8.
Modell B, Khan M, Darlison M, Westwood MA, Ingram D, Pennell DJ. Improved survival of thalassaemia major in the UK and relation to T2* cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance. 2008;10.
Roudbari M. The survival analysis of beta thalassemia major patients in the South East of Iran Reply. Saudi Medical Journal. 2008;29(11):1680-1.
Walters MC, Quirolo L, Trachtenberg ET, Edwards S, Hale L, Lee J, et al. Sibling donor cord blood transplantation for thalassemia major: Experience of the Sibling Donor Cord Blood Program. Annals of the New York Academy of Sciences. 2005;1054:206-13.
Goussetis E, Petrakou E, Theodosaki M, Kitra V, Peristeri I, Vessalas G, et al. Directed sibling donor cord blood banking for children with beta-thalassemia major in Greece: usage rate and outcome of transplantation for HLA-matched units. Blood cells, molecules & diseases. 2010;44(2):107-10.
Shahrokhi S, Menaa F, Alimoghaddam K, McGuckin C, Ebtekar M. Insights and hopes in umbilical cord blood stem cell transplantations. Journal of biomedicine & biotechnology. 2012;2012:572821.
Shenoy S, Murray L, Abraham A, Kamani N. Unrelated Donor Umbilical Cord Blood Transplantation (UCBT) for Hemoglobinopathy using Reduced Intensity Conditioning (RIC). World Cord Blood Congress IV and Innovative Therapies for Sickle Cell Disease, October 24-27, 2013. Monaco.
Lisini D, Zecca M, Giorgiani G, Montagna D, Cristantielli R, Labirio M, et al. Donor/recipient mixed chimerism does not predict graft failure in children with beta-thalassemia given an allogeneic cord blood transplant from an HLA-identical sibling. Haematologica. 2008;93(12):1859-67.
Fang J, Huang S, Chen C, Zhou D, Li CK, Li Y, et al. Umbilical cord blood transplantation in Chinese children with beta-thalassemia. Journal of Pediatric Hematology/Oncology. 2004;26(3):185-9.
Hsieh MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361(24):2309-17.
Alimoghaddam K, Ghaffari H, Foroughi F, Chardouli B, Sanaat Z, Bahar B, et al. Effects of chimerism on graft-versus-host disease, disease recurrence, and survival after HLA-identical marrow transplantation in Iran. Archives of Iranian medicine. 2006;9(2):99-103.
Eapen M, Klein JP, Ruggeri A, Spellman S, Lee SJ, Anasetti C, et al. Impact of allele-level HLA matching on outcomes after myeloablative single unit umbilical cord blood transplantation for hematologic malignancy. Blood. 2013.
Tu B, Leahy N, Yang R, Cha N, Kariyawasam K, Hou L, et al. Extensive haplotype diversity in African American mothers and their cord blood units. Tissue antigens. 2013;81(1):28-34.
Campath/Fludarabine/Melphalan Transplant Conditioning for Non-Malignant Diseases. 2013.
de Lima M, McNiece I, Robinson SN, Munsell M, Eapen M, Horowitz M, et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N Engl J Med. 2012;367(24):2305-15.
Pilot Study Evaluating Safety & Efficacy of a DCBT: NiCord® & UNM CBU to SCD Patients After Myeloablative Therapy. 2013.
Raj A, Bertolone S, Cheerva A. Successful treatment of refractory autoimmune hemolytic anemia with monthly rituximab following nonmyeloablative stem cell transplantation for sickle cell disease. Journal of Pediatric Hematology/Oncology. 2004;26(5):312-4.
Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285-91.
Munchel AT, Kasamon YL, Fuchs EJ. Treatment of hematological malignancies with nonmyeloablative, HLA-haploidentical bone marrow transplantation and high dose, post-transplantation cyclophosphamide. Best practice & research. Clinical haematology. 2011;24(3):359-68.
Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2008;14(6):641-50.
Dallas MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, et al. Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2013;19(5):820-30.
Gaziev D, Galimberti M, Lucarelli G, Polchi P, Giardini C, Angelucci E, et al. Bone marrow transplantation from alternative donors for thalassemia: HLA-phenotypically identical relative and HLA-nonidentical sibling or parent transplants. Bone Marrow Transplant. 2000;25(8):815-21.
Sullivan KM, Anasetti C, Horowitz M, Rowlings PA, Petersdorf EW, Martin PJ, et al. Unrelated and HLA-nonidentical related donor marrow transplantation for thalassemia and leukemia. A combined report from the Seattle Marrow Transplant Team and the International Bone Marrow Transplant Registry. Ann N Y Acad Sci. 1998;850:312-24.
Gaziev J, Marziali M, Isgro A, Sodani P, Paciaroni K, Gallucci C, et al. Bone marrow transplantation for thalassemia from alternative related donors: improved outcomes with a new approach. Blood. 2013;122(15):2751-6.
Hongeng S, Pakakasama S, Chaisiripoomkere W, Ungkanont A, Jootar S. Nonmyeloablative stem cell transplantation with a haploidentical donor in a class 3 lucarelli severe thalassemia patient. Bone Marrow Transplantation. 2004;34(3):271-2.
Sodani P, Isgro A, Gaziev J, Paciaroni K, Marziali M, Simone MD, et al. T cell-depleted hla-haploidentical stem cell transplantation in thalassemia young patients. Pediatric reports. 2011;3 Suppl 2:e13.
Mynarek M, Bettoni da Cunha Riehm C, Brinkmann F, Weissenborn K, Tell-Luersen M, Heuft HG, et al. Normalized transcranial Doppler velocities, stroke prevention and improved pulmonary function after stem cell transplantation in children with sickle cell anemia. Klin Padiatr. 2013;225(3):127-32.
La Nasa G, Argiolu F, Giardini C, Pession A, Fagioli F, Caocci G, et al. Unrelated bone marrow transplantation for beta-thalassemia patients: The experience of the Italian Bone Marrow Transplant Group. Ann N Y Acad Sci. 2005;1054:186-95.
La Nasa G, Giardini C, Argiolu F, Locatelli F, Arras M, De Stefano P, et al. Unrelated donor bone marrow transplantation for thalassemia: the effect of extended haplotypes. Blood. 2002;99(12):4350-6.
Li C, Wu X, Feng X, He Y, Liu H, Pei F, et al. A novel conditioning regimen improves outcomes in beta-thalassemia major patients using unrelated donor peripheral blood stem cell transplantation. Blood. 2012;120(19):3875-81.
Miniero R, Rocha V, Saracco P, Locatelli F, Brichard B, Nagler A, et al. Cord blood transplantation (CBT) in hemoglobinopathies. Eurocord. Bone Marrow Transplantation. 1998;22 Suppl 1:S78-9.
Gore L, Lane PA, Quinones RR, Giller RH. Successful cord blood transplantation for sickle cell anemia from a sibling who is human leukocyte antigen-identical: implications for comprehensive care. Journal of Pediatric Hematology/Oncology. 2000;22(5):437-40.
Matthes-Martin S, Lawitschka A, Fritsch G, Lion T, Grimm B, Breuer S, et al. Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. European Journal of Haematology. 2013;90(4):308-12.
Mazur M, Kurtzberg J, Halperin E, Ciocci G, Szabolcs P. Transplantation of a child with sickle cell anemia with an unrelated cord blood unit after reduced intensity conditioning. Journal of Pediatric Hematology/Oncology. 2006;28(12):840-4.
Adamkiewicz TV, Szabolcs P, Haight A, Baker KS, Staba S, Kedar A, et al. Unrelated cord blood transplantation in children with sickle cell disease: review of four-center experience. Pediatric Transplantation. 2007;11(6):641-4.
Sauter C, Rausen AR, Barker JN. Successful unrelated donor cord blood transplantation for adult sickle cell disease and Hodgkin lymphoma. Bone Marrow Transplantation. 2010;45(7):1252.
Radhakrishnan K, Bhatia M, Geyer MB, Del Toro G, Jin Z, Baker C, et al. Busulfan, fludarabine, and alemtuzumab conditioning and unrelated cord blood transplantation in children with sickle cell disease. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2013;19(4):676-7.
Chik KW, Shing MM, Li CK, Yuen PM, Tsang KS, Li K. Autologuous marrow recovery in a multitransfused beta-thalassemia major patient after umbilical cord blood transplantation. Blood. 1996;88(2):755.
Lau YL, Ma ES, Ha SY, Chan GC, Chiu D, Tang M, et al. Sibling HLA-matched cord blood transplant for beta-thalassemia: report of two cases, expression of fetal hemoglobin, and review of the literature. Journal of Pediatric Hematology/Oncology. 1998;20(5):477-81.
Chan LL, Lin HP. Cure of beta-thalassaemia major by umbilical cord blood transplantation--a case report of Malaysia's first cord blood transplantation. Journal of Tropical Pediatrics. 1999;45(4):243-5.
Hongeng S, Pakakasama S, Hathirat P, Ajjimakorn S, Jaovisidha A, Tardtong P, et al. Mismatched related cord blood transplantation in a severe thalassemia patient. Bone Marrow Transplantation. 2000;25(12):1322-3.
Zhou X, Ha SY, Chan GC, Luk CW, Chan V, Hawkins B, et al. Successful mismatched sibling cord blood transplant in Hb Bart's disease. Bone Marrow Transplantation. 2001;28(1):105-7.
Vanichsetakul P, Wacharaprechanont T, R OC, Seksarn P, Kupatawintu P. Umbilical cord blood transplantation in children with beta-thalassemia diseases. Journal of the Medical Association of Thailand = Chotmaihet thangphaet. 2004;87 Suppl 2:S62-7.
Sun X, Liu S, Hao WG, Chen ZX, Guo NL. [Umbilical cord blood transplantation for patients with beta-thalassemia major]. Zhonghua er ke za zhi. Chinese journal of pediatrics. 2005;43(3):178-82.
Fang J, Huang S, Chen C, Zhou D. Unrelated umbilical cord blood transplant for beta-thalassemia major. Journal of Tropical Pediatrics. 2003;49(2):71-3.
Tan PL, Shek PC, Lim LC, How GF, Tan P, Yeoh AE, et al. Umibilical cord blood stem cell from unrelated donors is a feasible alternate stem cell source for transplant in patients with genetic diseases. Annals of the Academy of Medicine, Singapore. 2004;33(5 Suppl):S82-3.
Hall JG, Martin PL, Wood S, Kurtzberg J. Unrelated umbilical cord blood transplantation for an infant with beta-thalassemia major. Journal of Pediatric Hematology/Oncology. 2004;26(6):382-5.
Bradley MB, Satwani P, Baldinger L, Morris E, van de Ven C, Del Toro G, et al. Reduced intensity allogeneic umbilical cord blood transplantation in children and adolescent recipients with malignant and non-malignant diseases. Bone Marrow Transplantation. 2007;40(7):621-31.
Gumuscu B, Thompson EI, Grovas AC, Zach TL, Warkentin PI, Coccia PF. Successful Unrelated Cord Blood Transplantation For Homozygous alpha-Thalassemia. Journal of Pediatric Hematology/Oncology. 2013;35(7):570-2.
Jaing TH, Sun CF, Lee WI, Wen YC, Yang CP, Hung IJ. Successful unmanipulated peripheral blood progenitor cell transplantation from an HLA haploidentical 2-locus-mismatched mother in a thalassemic patient with primary graft failure after transplantation of bone marrow and cord blood from unrelated donors. Pediatric Transplantation. 2008;12(2):232-4.
Hao WG, Sun X, Liu S, Zhao Z, Chen ZX. [Haploidentical hematopoietic stem cell transplantation for beta-thalassemia major in children]. Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatrics. 2009;11(7):546-8.
Wang SB, Hu DM, Li L, Yang YH, Pan XH, Liu L, et al. [HLA haploidentical peripheral blood stem cells transplantation for beta thalassemia major]. Zhonghua xue ye xue za zhi = Zhonghua xueyexue zazhi. 2011;32(12):844-7.
Feng Z, Sun E, Lan H, Zhang C, Li Q, Zhu W. Unrelated donor bone marrow transplantation for beta-thalassemia major: an experience from China. Bone Marrow Transplant. 2006;37(2):171-4.
Resnick IB, Aker M, Tsirigotis P, Shapira MY, Abdul-Hai A, Bitan M, et al. Allogeneic stem cell transplantation from matched related and unrelated donors in thalassemia major patients using a reduced toxicity fludarabine-based regimen. Bone Marrow Transplant. 2007;40(10):957-64.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this chapter

Cite this chapter
Fitzhugh, C.D., Abraham, A., Hsieh, M.M. (2017). Alternative Donor/Unrelated Donor Transplants for the β-Thalassemia and Sickle Cell Disease. In: Malik, P., Tisdale, J. (eds) Gene and Cell Therapies for Beta-Globinopathies. Advances in Experimental Medicine and Biology(), vol 1013. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7299-9_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7299-9_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7297-5
Online ISBN: 978-1-4939-7299-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)