
Abstract
Cytochrome P450 epoxygenases metabolize a spectrum of ω-6 and ω-3 polyunsaturated fatty acids such as arachidonic acid, linoleic acid, eicosapentaenoic acid, and docosahexenoic acid to generate bioactive lipid epoxide mediators. The epoxides thus generated demonstrate potent antihypertensive, angiogenic and anti-inflammatory properties. Endogenous epoxide levels are largely regulated by the soluble epoxide hydrolase (sEH), and the inhibition or genetic deletion of this enzyme increases epoxide concentrations and potentiates their biological actions. This review summarizes the mechanisms regulating epoxygenase and sEH, as well as the signaling event known to be regulated by the fatty acid epoxides and diols that can account for their vascular actions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 The Cytochrome P450 (CYP)/Soluble Epoxide Hydrolase Axis
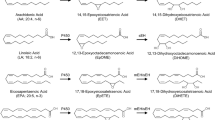
Cytochrome P450 (CYP) enzymes are membrane-bound, heme-containing terminal oxidases in a multi-enzyme complex that also includes an flavin adenine dinucleotide/flavin mononucleotide (FAD/FMN)-containing nicotinamide adenine dinucleotide phosphate (NADPH)-CYP reductase and cytochrome b5. CYP enzymes oxidize, peroxidize and/or reduce cholesterol, vitamins, steroids, xenobiotics, and numerous pharmacological substances in an oxygen- and NADPH-dependent manner. Some isoforms are fairly specific in their choice of substrates but many catalyze a large number of chemical reactions and can use an almost unlimited number of biologically occurring and synthetic compounds. Hepatic CYP enzymes are responsible for the metabolism of xenobiotica and many pharmaceuticals, but they also utilize endogenous compounds as substrates, such as cholesterol and fatty acids. Even though many CYP isozymes can oxidize a spectrum of ω-6 and ω-3 polyunsaturated fatty acids (PUFAs) such as retinoic acid, linoleic acid, eicosapentaenoic acid (EPA), and docosahexenoic acid (DHA; Fig. 9.1), they are often referred to as the third pathway of arachidonic acid metabolism, mainly because more is known about the biological actions of these products [1].

Cytochrome P450 (CYP)-dependent metabolism of n-6 (arachidonic and linoleic acid) and n-3 (eicosapentaenoic and docosahexaenoic acid), and metabolism of the epoxides generated to the corresponding diols by the soluble epoxide hydrolase (sEH). DHDP dihydroxydocosapentaenoic acid, DHEQ dihydroxyeicosatetraenoic acid, DHET dihydroxyeicosatrienoic acid, DiHOME dihydroxyoctadecenoic acid, EDP epoxydocosapentaenoic acid, EEQ epoxyeicosatetraenoic acid, EET epoxyeicosatrienoic acid, EpOME epoxyoctadecenoic acid. Reproduced from Fleming [9], with permission
Interest in the vascular actions of CYP enzymes followed reports that the epoxides of arachidonic acid (the epoxyeicosatrienoic acids [EETs]) were endothelium-derived hyperpolarizing factors (EDHFs) [2, 3], while the 20-hydroxyeicosatetraenoic acid (HETE) generated by ω-hydroxylases belonging to the CYP4A family were potent vasoconstrictors (for review see Harder et al. and Imig et al. [4, 5]). Initial reports focused on the effects of arachidonic acid metabolites on membrane potential, but it is now generally appreciated that these compounds mediate a number of membrane potential-independent effects and regulate angiogenesis [6, 7]. The arachidonic acid-metabolizing CYP enzymes with prominent roles in vascular regulation are the epoxygenases of the CYP2 gene family (e.g. CYP2B, 2C8, 2C9, 2C10, and 2J2 in humans; 2C34 in pigs; 2C11, 2C23, and 2J4 in rats) and the arachidonic acid ω-hydroxylases belonging to the CYP4A family which form subterminal and ω-terminal HETEs [8, 9].
Epoxide generation is thought to be determined by both the level of epoxygenase expression and the availability of the PUFA substrate, which in the case of arachidonic acid is determined by the activity of phospholipases such as phospholipase A2. Intracellular levels of the epoxides are tightly regulated and metabolism occurs relatively rapidly by hydrolysis, β-oxidation, and chain elongation [10]. The soluble epoxide hydrolase (sEH) is the most important epoxide-metabolizing enzyme that generates dihydroxy fatty acids (or diols). For a long time, the latter were considered to be less active that the parent epoxides but recent evidence has challenged this assumption (see section 9.6). There are, of course, exceptions to every rule and some epoxides are not great sEH substrates—the best-studied exception is probably 5,6-EET which is more rapidly metabolized by cyclooxygenases (COXs) [11, 12].
2 Regulation of CYP Expression and Activity
EET production may change as a consequence of altered CYP expression (by induction or repression) or altered activity. Little is known about the regulation of vascular CYP expression, and although CYP2C protein has been convincingly demonstrated in native endothelial cells, messenger RNA (mRNA) and protein levels rapidly decrease after cell isolation, so that in passaged cultured endothelial cells, mRNA can only be detected using reverse transcriptase-polymerase chain reaction (RT-PCR) [3, 13]. Such findings indicate that CYP2C proteins are relatively unstable and that transcriptional processes play an important role in determining CYP expression levels, and at the same time highlight the importance of physiological stimuli in the control of CYP levels. Indeed, the exposure of cultured endothelial cells to either cyclic stretch or fluid shear stress can restore CYP2C protein expression as well as endothelial EET production [14].
The expression of several CYP enzymes is modulated by changes in oxygen tension; for example, hypoxia downregulates CYP2J2 [15] but upregulates CYP2C8/9 expression in cultured human endothelial cells [16], while transient cerebral ischemia induces CYP2C11 in rats [17]. The promoter regions of several CYP2C genes contain hypoxia-responsive elements, a finding which may explain the observation that the myogenic contraction, as well as the constrictor response to phenylephrine, is attenuated in mesenteric arteries from rats exposed to hypoxia for 48 h [18]. This phenomenon can be attributed to the hypoxia-induced induction of CYP expression as CYP2C protein was elevated above control in arteries from rats exposed to hypoxia, and both the vascular smooth muscle hyperpolarization and the hypoxia-induced decrease in the myogenic response were normalized by the CYP2C inhibitor sulfaphenazole [19].
Given the size of the CYP family of proteins, it is not surprising that there is considerable inter-isoform variation in the regulation of gene expression and mRNA stability, as well as post-translational modification of the CYP protein. Regulation of the CYP2 family involves nuclear receptors related to the steroid hormone receptor superfamily, such as the constitutive androstane receptor and the retinoic acid receptor. Retinoic acid is a CYP2C (CYP2C8) substrate, thus the regulation of CYP2C8 expression by a receptor that is activated by an endogenous substrate such as retinoic acid is not surprising. CYP2C9, which is highly homologous to CYP2C8, is inducible in primary human hepatocytes by xenobiotics, including dexamethasone and phenobarbital [20]. The CYP2C8 and 2C9 promoters contain a glucocorticoid-responsive element that is recognized and transactivated by human glucocorticoid receptor [21–23]. Identification of this functional element provides a rational mechanistic basis for the induction of CYP2C protein, and an increase in EDHF-mediated responses in porcine coronary arteries by cortisol [24]. Retinoic acid is not the only CYP substrate that affects CYP protein levels, and the expression of many of the CYP enzymes can be induced by a substrate excess. Indeed, a number of cardiovascular drugs currently in clinical use are metabolized, at least in part, by CYP2C family members. While this process mainly occurs in the liver and is associated with the induction of the metabolizing enzyme, the expression of CYP2C enzymes in endothelial cells can also be affected. For example, the HMG-CoA reductase inhibitor fluvastatin, which is metabolized by CYP2C9 in the liver [25, 26], also increases CYP2C expression in cultured and native porcine coronary artery endothelial cells [13]. The L-type Ca2+ channel blocker nifedipine also elicits a very marked increase in the expression of CYP2C in porcine coronary arteries, and enhances EDHF-mediated responses [13, 27].
Once the protein is expressed, CYP activity is thought to be determined mainly by the availability of its substrates [28]. Since phospholipase A2 inhibitors attenuate CYP-dependent EDHF responses, the activation cascade is thought to involve a stimulus-induced increase in intracellular Ca2+, followed by the activation of phospholipase A2, which then liberates the PUFA substrate (i.e. arachidonic acid) from membrane phospholipids. The increase in substrate immediately results in the activation of CYP enzymes (when expressed) and the generation of vasoactive products. While this sequence of events is certainly plausible, it is highly likely that additional mechanisms, such as phosphorylation, play a role in regulating CYP activity. Indeed, some CYP enzymes (CYP2B1, 2B2, and 2E1) are reported to be phosphorylated by protein kinase A (PKA), and the consequences of CYP phosphorylation range from the regulation of activity [29] and subcellular localization [30, 31] to proteasome degradation [30, 32].
An additional mechanism thought to modulate CYP activity is nitrosation by nitric oxide (NO), which can interact with CYP enzymes in two ways. NO reversibly binds to the heme moiety of CYP enzymes, forming iron-nitrosyl complexes, and it can irreversibly react with cysteine residues [33]. Both NO–CYP adducts are enzymatically inactive in vitro. As endothelial CYP enzymes of the 2C family were found to be inhibited by NO, the role of EETs in the regulation of vascular tone in the healthy vasculature which constantly generates NO was suggested to be of minor importance compared with that in circumstances of an endothelial dysfunction in which the bioavailability of NO is impaired [34]. However, there are clear physiological consequences of EET activation in endothelial cells (e.g. on Akt, PKA, and transient receptor potential [TRP] channels) that can be demonstrated, even in the presence of a fully functional endothelial NO synthase. Thus, whether or not physiologically relevant (low nmol/L) levels of NO really affect CYP epoxygenase activity in vivo, remains to be determined.
3 The Soluble Epoxide Hydrolase
The sEH protein is a homodimer composed of two 60 kDa monomers joined by a proline-rich bridge [35], with each monomer consisting of an N-terminal domain that displays lipid phosphatase activity and a larger C-terminal that processes classical α/β-hydrolase activity [36, 37]. Surprisingly little is known about the mechanisms that regulate sEH activity. There have been a number of studies linking changes in sEH expression with inflammatory or hormonal stimuli [38, 39]. Two tyrosine residues (Tyr383 and Tyr466) in the active site of the hydrolase are reportedly essential for enzyme activity [40], and these were recently shown to be nitrated by peroxynitrite in vitro and in vivo in mouse models of type 1 and type 2 diabetes , leading to a decrease in sEH activity [41]. It is currently only possible to speculate about the involvement of sEH tyrosine nitration in the amplification of inflammation associated with diabetes, but at least one sEH polymorphism, which results in decreased enzymatic activity, has previously been associated with human insulin resistance [42]. The sEH was also recently reported to be nitrosated in leptin-stimulated wild-type but not endothelial NO synthase knockout mice, suggesting that the effects of NO on PUFA metabolism may be partly related to the modulation of sEH activity [43].
Inhibition or deletion of the sEH increases tissue and circulating levels of the PUFA epoxides at the same time as decreasing diol production, and has pronounced effects on blood pressure [44, 45], inflammation [46], progenitor cell proliferation, angiogenesis and vascular repair [47]. The particular effectiveness of sEH inhibitors against hypertension associated with activation of the renin-angiotensin system is most likely related to the fact that angiotensin II markedly increases sEH expression in vivo [39]. Interestingly, hypoxia does the opposite and markedly downregulates sEH promoter activity and thus protein expression in the lung [48]. There are other examples of hypertension being associated with elevated sEH expression and/or activity, such as the spontaneously hypertensive rat. In these animals, elevated sEH expression is linked to an increase in the renal metabolism of EETs to dihydroxyeicosatrienoic acids (DHETs), and sEH inhibitors blunt the development of hypertension [44]. Initial reports also documented that sEH−/− mice have lower blood pressure and elevated EET levels than their wild-type littermates [49]. However, the blood pressure phenotype now seems to be controversial as the loss of the hydrolase can be compensated by elevated concentrations of the pressor and vasoconstrictor eicosanoid, 20-HETE, as well as increased lipoxygenase-derived hydroxylation and prostanoid production [50]. Despite the lack of alteration in blood pressure, hearts from these sEH−/− animals show improved recovery of left ventricular contractility and less infarction than hearts from wild-type mice after ischemia [51], and have a survival advantage following acute systemic inflammation [50]. Several of the metabolites generated by the sEH, such as the DHETs generated from the EETs, are also biologically active but generally less so than the parent epoxides. However, the DHETs are not as readily incorporated into membrane lipids as the EETs, and the latter are thought to be the form in which the majority of endothelium-derived EETs leave the cell [52].
The exact physiological role of the lipid phosphatase activity associated with the N-terminal domain of the sEH is currently unclear as there are currently no selective inhibitors of this domain (sEH inhibitors act on the hydrolase domain and do not affect the phosphatase activity [36]). However, the lipid phosphatase has been associated with cholesterol-related disorders, peroxisome proliferator-activated receptor (PPAR) activity, and the isoprenoid/cholesterol biosynthesis pathway [53]. Indeed, in addition to demonstrating enhanced circulating EET levels [49], male sEH−/− mice exhibit decreased plasma cholesterol and testosterone levels [54]. Moreover, it seems that isoprenoid pyro- and monophosphates are substrates for the N-terminal domain of the enzyme [55, 56], and these lipid phosphates are metabolic precursors of cholesterol biosynthesis and are also utilized for isoprenylation of small G-proteins involved in multiple cell signaling pathways [57]. Lysophosphatidic acids are involved in regulating cell survival, apoptosis, motility, shape, differentiation, gene transcription, and malignant transformation, and are reportedly excellent substrates for the lipid phosphatase [58, 59]. However, to what extent this can affect physiology/pathophysiology needs to be determined.
It is interesting to note that even though most current sEH literature attributes the hydrolase domain to the cardiovascular effects seen in humans, the human sEH single nucleotide polymorphism most often associated with cardiovascular disease (R287Q) encodes a protein with significantly lower rather than elevated hydrolase activity [60]. Thus, solely incriminating the hydrolase domain for adverse cardio- and pulmonary-vascular effects seems premature and highlights the importance of further investigating the independent roles of the hydrolase and phosphatase domains. Indeed, some aspects of the phenotype of sEH−/− mice (e.g. pulmonary vascular muscularization) cannot be reproduced by chronic sEH inhibitor treatment, which may be an indirect indication of a physiological role for the phosphatase domain [61].
4 How do Lipid Epoxides Initiate Cellular Signaling?
Most is known about the actions of the epoxides or arachidonic acid or EETs for which several modes of signal initiation have been proposed. One of them involves the transactivation of the epidermal growth factor (EGF) receptor in endothelial cells, and the activation of this particular signaling pathway has been linked to cell proliferation and angiogenesis [62–64]. For actions other than angiogenesis, a separate mechanism has been proposed as a high-affinity EET binding site was reported to exist on monocytes and U937 cells [65–67]. Competition studies showed a specific high-affinity binding of 14,15- and 11,12-EET to a receptor that seems to be protein in nature [66, 68]. In addition, in isolated membranes, [3H]-14,15-EET binding was found to be specific, reversible, and saturable, and the ligand was not displaced by antagonists of the thromboxane, platelet-activating factor, or leukotriene receptors. However, binding was inhibited by 14,15- and 11,12-EETs, but not by inactive analogs of 14,15-EET or 15-HETE. Importantly, ligand binding was inhibited by GTPγS, indicating that the binding site or receptor is coupled to a G protein. Such findings are in agreement with other reports indicating the involvement of a G protein in the actions of the EETs [69, 70]. One characteristic of many EET-induced cellular responses such as gap junctional communication [71] and TRP channel translocation [72] is their ability to increase intracellular cyclic adenosine monophosphate (cAMP) levels and activate PKA [66, 73]. Moreover, an EET analog that is able to induce the complete relaxation of bovine coronary arteries also does so by increasing cAMP levels [74, 75]. Putting the evidence of a protein receptor on cell membranes together with that indicating a reliance on cAMP/PKA for EET-induced signaling, the existence of a Gαs-coupled EET receptor has been postulated [76]. However, to-date no specific EET receptor has been identified.
Many lipids also interact with intracellular fatty acid receptors such as the PPARs, and the EETs are no different. For example, ω-hydroxylated 14,15-EET and 14,15-DHET [77] are reported to bind with a high affinity to PPAR-α, while the EETs generated in endothelial cells in response to fluid shear stress increase PPAR-γ transcriptional activity [78]. Additional intracellular receptors for CYP products have not yet been identified, but one possibility is that these oxidized fatty acids bind to fatty acid binding proteins (FABPs) such as heart type (H) -FABP [79], which in turn mediate some of the physiologically relevant actions of these intermediates, possibly including the activation of PPARs [80].
A further proposed mechanism involves the incorporation of EETs into the plasma membrane, where they associate with effector molecules such as small G proteins [69] or change the lipid bilayer order, fluidity, and volume, and thereby regulate the flux of ions (e.g. Ca2+) across the membrane [81]. Certainly, the EETs can be esterified to phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositols [28]. Moreover, at least in the pancreas, it has been suggested that the long-chain acyl-coenzyme A (acyl-CoA) synthetase 4 activates EETs to form EET-CoAs that are incorporated into glycerophospholipids [82].
Given that enhanced EET production has frequently been correlated with an increase in intracellular cAMP levels, it is not entirely surprising that increased CYP expression and EET production are associated with activation of the cAMP-response element-binding protein (CREB) which underlies the EET-induced expression of COX-2 [83]. However, the first transcription factor reported to be regulated by CYP-derived EETs was nuclear factor κB (NF-κB) [84]. Indeed, the EET-dependent inhibition of the I-κB kinase led to classification of the EETs as anti-inflammatory mediators. However, this classification is complicated by the fact that some CYP epoxygenases can generate physiologically relevant levels of superoxide anions which tend to activate NF-κB, and thus functionally antagonize the inhibitory effects of EETs and promote the expression of adhesion molecules on endothelial cells [85]. The reason why CYP epoxygenases of the 2C family generate superoxide anions [85, 86] while the 2J enzymes do not [87, 88] is currently unclear but is probably related to substrate binding and metabolism. However, the differential ability to generate free radicals accounts for the disparate effects of these isozymes on vascular protection.
Other transcription factors that are reported to be modulated by EETs/DHETs are PPARα [77, 89, 90] and FOXO3a [91]. While the nuclear localization of FOXO3a is regulated by the EET-dependent activation of Akt [91], much less is known about the mechanisms involved in the EET-dependent activation of PPARα, or indeed the consequences of this effect.
5 CYP and Cardiovascular Function
5.1 Vascular Reactivity
The realization that EETs , especially 11,12- and 14,15-EET, can activate large conductance Ca2+-activated K+ channels (BKCa) on vascular smooth muscle cells to elicit hyperpolarization and relaxation led to their identification as a class of EDHF [2, 3]. The latter term is now recognized as an oversimplification as there are three principal mechanisms linked to the EDHF phenomenon: (i) an increase in endothelial [Ca2+]i following cell stimulation triggers the synthesis of a metabolite which is essential for the subsequent EDHF-mediated responses; (ii) K+, released from endothelial cells via Ca2+-dependent K+ (KCa) channels, induces smooth muscle hyperpolarization by activating inwardly rectifying K+ channels and/or the Na+/K+-ATPase on vascular smooth muscle cells; and (iii) endothelial cell hyperpolarization is transmitted to the vascular smooth muscle via gap junctions. The strengths and weaknesses of the arguments for each of these specific types of EDHF has been discussed at length [92] but each of them appears to be valid in certain vascular beds. Interestingly, all of these mechanisms can be modulated by EETs.
In endothelial cells, the activation of KCa channels by EETs is preceded by an increase in intracellular Ca2+ levels that can be accounted for by an increased open probability of nonselective cation channels of the TRP family. How this happens was initially attributed to the presence of an arachidonic acid-binding site in some of the TRP channels that can be activated by the parent lipid [93, 94] as well as the EETs [93, 95]. However, while relatively high concentrations of the EETs may affect TRP channels directly, more physiological concentrations activate TRP channels in a PKA-dependent manner that involves their translocation to caveolin-rich areas in the plasma membrane [72, 96]. There appear to be regioisomer-specific differences in EET-induced TRP channel translocation and activation as 5,6-EET, but not 11,12-EET, can activate TRPV4 in endothelial cells [93, 95], a phenomenon that underlies the EDHF-dependent, flow-induced vasodilatation [96]. On the other hand, 11,12-EET, but not 14,15-EET or 5,6-EET, enhance the bradykinin-induced capacitive Ca2+ influx in endothelial cells by stimulating the translocation of TRPC6 and TRPC3 to caveolin-rich areas in the plasma membrane [72].
5.2 Pulmonary Circulation
While increasing intracellular Ca2+ in endothelial cells elicits vasodilatation, the same process in vascular smooth muscle cells does exactly the opposite. This means that when EETs activate TRPC6 channels in pulmonary smooth muscle cells, an increase in pulmonary vascular tone would be expected. The fact that activation of TRPC6 channels plays a role in regulating hypoxic pulmonary vasoconstriction (a physiological mechanism by which pulmonary arteries constrict in hypoxic lung areas in order to redirect blood flow to areas with greater oxygen supply) was demonstrated using mice lacking the channel. Indeed, in pulmonary vascular smooth muscle cells from these animals, hypoxia completely failed to cause Ca2+ entry. It should be noted here that the TRPC6 is reported to primarily conduct Na+, and Ca2+ follows secondarily through voltage-gated Ca2+ channels or by the Na+/Ca2+ exchanger. In line with the disturbed Ca2+ entry, these animals completely lacked the initial, acute phase of hypoxia-induced pulmonary constriction [97]. Moreover, TRPC6−/− mice did not respond to 11,12-EET, although the eicosanoid induced a pronounced increase in pulmonary pressure in TRPC6+/− littermates. Furthermore, inhibition of the sEH potentiated the hypoxic pulmonary vasoconstriction in the heterozygous mice, but had no effect in the TRPC6−/− mice [48]. In line with the functional data, hypoxia and 11,12-EET caused the translocation of TRPC6 to caveolae in isolated pulmonary vascular smooth muscle cells. In addition, hypoxia-induced translocation of the channel could be prevented by pretreating the cells with an EET antagonist [48]. More recently [98], the site for pulmonary oxygen sensing was identified at alveolocapillary level, from which the hypoxic signal is propagated as endothelial membrane depolarization to upstream arterioles in an EET- and Cx40-dependent manner (Fig. 9.2; [99]).

Proposed mechanism for the differential consequences of epoxyeicosatrienoic acid (EET)-induced transient receptor potential (TRP) channel activation in the systemic and pulmonary circulations. a In the systemic circulation, EETs are generated in endothelial cells in response to stimulation (e.g. by bradykinin) following the activation of phospholipase A2 and cytochrome P450 (CYP)2C epoxygenases. EET-induced activation of protein kinase A (PKA) results in the translocation of TRP channels to the plasma membrane to potentiate the activation on KCa channels initiated by the phospholipase C (PLC)-induced conversion of phosphatidylinositol 4,5-bisphosphate (PIP 2 ) to diacylglycerol (DAG). The overall consequence is hyperpolarization and vasodilatation. b While activating TRPC6 in endothelial cells elicits vasodilatation, the same process in vascular smooth muscle cells does exactly the opposite as the TRPC6 channels in vascular smooth muscle cells (VSMC) primarily conduct Na+, Ca2+ follows secondarily through voltage-gated Ca2+ channels or by the Na+, Ca2+ exchanger (NCE). Reproduced from Loot and Fleming [99], with permission
The evidence for a vasodilator role of the arachidonic acid epoxides in humans is, needless to say, indirect and relies on the use of CYP inhibitors that cannot be guaranteed to be completely selective. That said, sulfaphenazole is one of the most selective inhibitors available for CYP2C9 [100], and while several studies failed to demonstrate any effects of sulfaphenazole on forearm vasodilatation in healthy subjects [86, 101, 102], a component of the flow-induced vasodilatation of skeletal muscle arterioles [103] and the radial artery [104–106], both of which have been shown to express CYP2C protein, is attenuated by the CYP inhibitor. Clearly, however, disease can affect responses as forearm vasodilator responses to acetylcholine could be blunted by CYP inhibitors in patients with hypercholesterolemia and reduced NO-dependent vasodilatation [107].
5.3 Hypertension and Atherosclerosis
Inhibition of the sEH increases intracellular levels of EETs, and thus prolongs their vasodilator and anti-inflammatory actions. Indeed, pharmacological inhibition of the sEH prevents angiotensin II-induced hypertension in rats and mice, and protects the kidney from hypertension-induced damage [44, 45, 52]. Furthermore, in humans increased sEH activity was associated with more advanced endothelial dysfunction and vascular inflammation [108].
Although the link between the sEH and cholesterol metabolism would make it logical to look at atherosclerosis , the situation is somehow less clear. Certainly, polymorphisms of the sEH have been linked with the risk of atherosclerosis and coronary heart disease [109–111]. Why this is the case is not known, but the initial report that sEH inhibitors can attenuate smooth muscle cell proliferation [112] most probably represented an off-target effect of the substance used [113]. In addition, some of the animal studies failed to deliver consistent results, and although inhibition of the sEH was reported to attenuate atherosclerosis , abdominal aortic aneurysm formation, and dyslipidemia by some researchers [114, 115], our group has been unable to detect clear effects. Furthermore, the effects on vascular remodeling are inconsistent with inhibition of the sEH preventing vascular remodeling in an inflammatory model but not in a blood flow-dependent model of neointima formation [116].
Most of the studies performed to date have focused on vascular smooth muscle cells, and the fact that monocytes express the sEH and a number of CYP enzymes has been largely overlooked. However, this area deserves much more attention as human and murine macrophages within atherosclerotic plaques express CYP2S1, a largely extrahepatic epoxygenase [117]. Interestingly, enzyme expression increased during monocyte differentiation to macrophages, and could be detected in classically activated or M1 macrophages and macrophages present in atherosclerotic plaques and inflamed tonsils, but not in macrophages polarized towards the M2 or alternatively-activated phenotype. Although the enzyme was able to accept several substrates and to generate bioactive epoxides from arachidonic acid, linoleic acid and EPA in an NADPH-dependent manner, perhaps from the macrophage polarization point of view the most relevant substrates seem to be prostaglandins G2 and H2 [117]. The resulting decrease in the immunomodulator prostaglandin E2 (PGE2) would certainly be expected to result in a macrophage subtype with attenuated angiogenic potential, but whether or not the CYP2S1 product 12(S)-hydroxyheptadeca-5Z,8E,10E-trienoic acid actively contributes to inflammation remains to be determined.
6 Angiogenesis and Cancer
Given the fact that the activation of KCa channels has been linked to endothelial cell proliferation [118–120], and EETs activate KCa channels, it would seem logical to assume that KCa activation would play a role in EET-induced proliferation. However, although the activation of KCa channels has been linked to endothelial cell proliferation induced by basic fibroblast growth factor [118], this mechanism appears to not be involved in the EET-induced proliferation of endothelial cells. The first hint that EETs may affect cell signaling and proliferation was obtained in renal epithelial cells [62, 121] and, soon afterwards, ‘authentic EDHF’ recovered from the luminal incubate of rhythmically stretched coronary arteries was found to activate a number of kinases, whose function was closely linked with endothelial cell proliferation [122]. Activation of these MAP kinases could be inhibited by treatment with CYP inhibitors, as well as by antisense oligonucleotides directed against CYP2C, and could be mimicked by the treatment of endothelial cells with 11,12-EET or by overexpression of CYP2C8 [122]. More detailed analysis of the mechanisms involved revealed that CYP epoxygenase-derived metabolites of arachidonic acid are able to transactivate the EGF receptor [123, 124]. 14,15-EET was initially suggested to act as a second messenger following activation of the EGF receptor; however, it appears that 14,15-EET can also elicit the release of heparin-binding EGF-like growth factor from a renal epithelial cell line via a process involving the activation of matrix metalloproteinases (MMP) [123]. Although the MMP involved has not yet been identified, a very similar mechanism seems to be responsible for the transactivation of the EGF receptor in endothelial cells [124]. The EET-mediated activation of the EGF receptor leads, in turn, to the activation of the kinase Akt and an enhanced expression of cyclin D1. All four EET regioisomers have been reported to elicit an increase in Akt phosphorylation and cell proliferation in murine endothelial cells, but only the proliferative effects of 5,6- and 14,15-EET are reportedly sensitive to a phosphatidylinositol 3-kinase (PI3-K) inhibitor, whereas the 8,9- and 11,12-EET-induced increase in [3H] thymidine incorporation seems to be dependent on the activation of the p38 MAP kinase [125]. In contrast, in bovine aortic endothelial cells 8,9-, 11,12-, and 14,15-EET-induced cell proliferation can be attenuated by MEK, ERK, and PI3-K inhibition [126]. Other signaling pathways also contribute to the increase in cyclin D1 expression, including the MAP kinase phosphatase-1 (MKP-1), which decreases c-Jun N-terminal kinase (JNK) activity [127]. Activation of Akt by EETs also induces phosphorylation and therefore inhibition of the forkhead factors FOXO1 and FOXO 3a, and subsequently a decrease in the expression of the cyclin-dependent kinase inhibitor p27kip1 [91]. The involvement of this mechanism in the CYP2C9-induced endothelial cell proliferation could be demonstrated by the transfection of CYP2C9-overexpressing cells with either a dominant negative Akt or a constitutively active FOXO3a, both of which inhibit CYP2C9-induced endothelial cell proliferation [91]. Although there is a precedent for the negative regulation of JNK after activation of Akt, inasmuch as Akt has been reported to phosphorylate and inactivate the kinase SEK1 and thus inactivate its substrate JNK [128], it remains unclear whether these pathways are linked to each other or are simply activated in parallel.
The first link between EETs and angiogenesis was obtained in co-cultures of astrocytes and endothelial cells. EETs released from astrocytes increased thymidine incorporation into endothelial cells and elicited the formation of capillary-like structures [129, 130]. Moreover, overexpression of CYP2C9 in, and/or the application of, 11,12- or 14,15-EET to monocultures of endothelial cells was associated with angiogenesis [124, 131]. In vivo data rapidly followed to support these in vitro findings, and EETs induced angiogenesis in the chick chorioallantoic membrane [124], as well as in EET-impregnated Matrigel plugs in adult rats [131] and an ischemic rat hindlimb model in which the overexpression of different CYP isozymes, including CYP2C11 and 2J2, was found to increase muscle capillary density [126]. Furthermore, tumor growth and metastasis can be increased by sEH inhibition in transgenic mice with high vascular EET levels, i.e. animals that overexpress either the human CYP2C8 or human CYP2J2 specifically in Tie-2-expressing cells, or that were treated with high concentrations of 14,15-EET [132]. All in all, such evidence indicated that activation of the CYP/sEH axis is linked with the promotion of angiogenesis; however, the latter models were somewhat artificial and focused on the products of arachidonic acid metabolism, largely ignoring the biological actions of other lipids that feed into the same CYP/sEH axis.
It was partly to assess the role of the sEH in angiogenesis under more physiological conditions that we determined the effects of the global and induced deletion of the sEH, as well as its pharmacological inhibition in vascular repair after ischemia and in the postnatal murine retina. We found that sEH deletion and inhibition resulted in markedly decreased angiogenesis [133] and vascular repair [47], and provided some of the first experimental data that linked the defect not to the accumulation of a PUFA epoxide but to the lack of diol production. To identify such lipids, liquid chromatography–tandem mass spectrometry (LC–MS/MS)-based lipid profiling approaches are used to screen for the PUFA epoxides or diols most affected by the deletion of the sEH, and, to date, biological activities have been attached to the DHA-derived diol 19,20-dihydroxydocosapentaenoic acid [133] and the linoleic acid-derived diol 12,13-dihydroxyoctadecenoic acid [47]. Interestingly, the signaling pathways targeted by the diols are distinct, as, while the defective vascular repair in sEH−/− mice could be attributed to altered Wnt signaling followed by attenuated progenitor cell proliferation and mobilization [47], defects in the retina could be linked to the translocation of presenilin 1 out of lipid rafts and the subsequent inhibition of the γ-secretase [133]. This means that the take-home message with respect to angiogenesis is that the ω-3/ω-6 profile of a particular tissue is likely to determine the overall effects on angiogenesis. Certainly, while EETs have been well-defined as angiogenic mediators [132, 134], a DHA-derived epoxide was recently reported to inhibit angiogenesis by preventing phosphorylation of the vascular endothelial growth factor receptor-2 (VEGFR2) [135]. This is of relevance since the lipids that feed into the CYP/sEH axis are largely provided by the diet, and regulating dietary intake of specific lipids, e.g. the fish oils EPA and DPA, has been linked with altered epoxide and diol profiles, as well as protection against vascular inflammation and cancer. On the other hand, increased dietary intake of linoleic acid is generally associated with inflammation and increased risk. It will therefore be interesting to determine to what extent diet can alter the influence of the CYP/sEH axis on angiogenesis and tumor growth [136, 137], as well as the development of the cardiovascular complications associated with the metabolic syndrome [136, 138].
To date, the CYP enzymes linked to angiogenesis have included the human 2C8/2C9 and 2J to enzymes, as well as the rat 2C11 and mouse 2c44 isoforms, all of which are epoxygenases. CYP1B1 is worth mentioning at this point, even though the enzyme is an estrogen-metabolizing CYP hydroxylase. CYP1B1 induction is an important factor in determining risk associated with hormone-mediated cancers, in particular as CYP1B1 is induced by hypoxia [139], probably because its expression is regulated by the AMP-activated protein kinase (AMPK) [140], and is involved in the metabolism of some clinically relevant anticancer agents [141]. In addition, CYP1B1 is tightly regulated by the angiogenic microRNA miR-27b [142–144]. The link to this particular microRNA is interesting as it has previously been described as a “regulator hub in lipid metabolism” [145]. Indeed, miR-27b levels are significantly upregulated by a high-fat diet and hepatic miR-27b and its target genes are inversely altered in a mouse model of dyslipidemia and atherosclerosis [145]. Whether or not CYP1B1 is involved in the latter is unclear but the enzyme has recently been linked with protection against angiotensin II-induced hypertension in female mice [146].
What makes CYP1B1 of interest in angiogenesis is that its deletion impaired revascularization in a model of oxygen-induced retinopathy in mice [147]. This effect was linked with a decrease in the expression of the endothelial NO synthase [148], as well as a corresponding increase in intracellular oxidative stress and increased production of thrombospondin-2, an endogenous inhibitor of angiogenesis [147, 149]. Interestingly, estrogen-induced angiogenesis has also been attributed to changes in endothelial NO synthase, thrombospondin, and free radical generation, making it tempting to speculate that CYP1B1 may actually mediate the effects of the hormone. Certainly, the CYP1B1-derived metabolites of β-estradiol promote angiogenesis in uterine artery endothelial cells [150]. Rather intriguingly, residues 41–48 of human CYP1B1 are part of a mitochondrial import signal, and the cleavage of CYP1B1 by serine proteases results in its targeting to mitochondria, which is associated with oxidative stress and mitochondrial dysfunction [151]. Given that angiogenic endothelial cells undergo changes in metabolism, the so-called Warburg effect [152], it will be interesting to determine whether or not CYP1B1 can also alter endothelial cell metabolism and mitochondrial function. Effects on CYP1B1 may also explain the antiangiogenic actions of the antidiabetic drug metformin, which prevents the tumor cell supernatant–induced upregulation of CYP1B1 in endothelial cells [140].
Abbreviations
- [Ca2+]i :
-
Intracellular Ca2+ concentration
- BKCa :
-
Large conductance Ca2+-activated K+ channels
- COX:
-
Cyclooxygenase
- CREB:
-
cAMP-response element-binding protein
- CYP:
-
Cytochrome P450
- DHA:
-
Docosahexenoic acid
- DHET:
-
Dihydroxyeicosatrienoic acid
- EDHFs:
-
Endothelium-derived hyperpolarizing factors
- EET:
-
Epoxyeicosatrienoic acid
- EGF:
-
Epidermal growth factor
- EPA:
-
Eicosapentaenoic acid
- FABPs:
-
Fatty acid-binding proteins
- HETE:
-
Hydroxyeicosatetraenoic acid
- KCa :
-
Ca2+-dependent K+ channels
- MKP-1:
-
MAP kinase phosphatase-1
- MMP:
-
Matrix metalloproteinase
- NFκB:
-
Nuclear factor κB
- NO:
-
Nitric oxide
- PI3-K:
-
Phosphatidylinositol 3-kinase
- PKA:
-
Protein kinase A
- PPAR:
-
Peroxisome proliferator-activated receptor
- PUFA:
-
Polyunsaturated fatty acid
- sEH:
-
Soluble epoxide hydrolase;
- TRP channels:
-
Transient receptor potential channels
References
Konkel A, Schunck WH. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta. 2011;1814:210–22.
Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–23.
Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–7.
Harder DR, Narayanan J, Gebremedhin D. Pressure-induced myogenic tone and role of 20-HETE in mediating autoregulation of cerebral blood flow. Am J Physiol Heart Circ Physiol. 2011;300:H1557–65.
Imig JD, Simpkins AN, Renic M, Harder DR. Cytochrome P450 eicosanoids and cerebral vascular function. Expert Rev Mol Med. 2011;13:e7.
Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther. 2006;111:584–95.
Chen L, Ackerman R, Guo AM. 20-HETE in neovascularization. Prostaglandins Other Lipid Mediat. 2012;98:63–8.
Guengerich FP. Human cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: structure, mechanism, and biochemistry. 2 ed. New York: Plenum Press; 1995. pp. 473–535.
Fleming I. The cytochrome P450 pathway in angiogenesis and endothelial cell biology. Cancer Metastasis Rev. 2011;30:541–55.
Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–30.
Oliw EH, Benthin G. On the metabolism of epoxyeicosatrienoic acids by ram seminal vesicles: isolation of 5(6)epoxy-prostaglandin F1a. Biochem Biophys Res Commun. 1985;126:1090–6.
Nüsing RM, Schweer H, Fleming I, Zeldin DC, Wegmann M. Epoxyeicosatrienoic acids affect electrolyte transport in renal tubular epithelial cells: dependence on cyclooxygenase and cell polarity. Am J Physiol Renal Physiol. 2007;293:F288–98.
Fisslthaler B, Hinsch N, Chataigneau T, Popp R, Kiss L, Busse R, Fleming I. Nifedipine increases cytochrome P4502C expression and EDHF-mediated responses in coronary arteries. Hypertension. 2000;36:270–5.
Fisslthaler B, Popp R, Michaelis UR, Kiss L, Fleming I, Busse R. Cyclic stretch enhances the expression and activity of coronary endothelium-derived hyperpolarizing factor synthase. Hypertension. 2001;38:1427–32.
Marden NY, Fiala-Beer E, Xiang SH, Murray M. Role of activator protein-1 in the down-regulation of the human CYP2J2 gene in hypoxia. Biochem J. 2003;373:669–80.
Michaelis UR, Fisslthaler B, Barbosa-Sicard E, Falck JR, Fleming I, Busse R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J Cell Sci. 2005;118:5489–98.
Alkayed NJ, Goyagi T, Joh HD, Klaus J, Harder DR, Traystman RJ, Hurn PD. Neuroprotection and P450 2C11 upregulation after experimental transient ischemic attack. Stroke. 2002;33:1677–84.
Earley S, Walker BR. Endothelium-dependent blunting of myogenic responsiveness after chronic hypoxia. Am J Physiol Heart Circ Physiol. 2002;283:H2202–9.
Earley S, Pastuszyn A, Walker BR. Cytochrome P-450 epoxygenase products contribute to attenuated vasoconstriction after chronic hypoxia. Am J Physiol Heart Circ Physiol. 2003;285:H127–36.
Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, Daujat M, Waechter F, Fabre JM, Carrere N, Maurel P. Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab Dispos. 2001;29:242–51.
Gerbal-Chaloin S, Daujat M, Pascussi JM, Pichard-Garcia L, Vilarem MJ, Maurel P. Transcriptional regulation of CYP2C9 gene. Role of glucocorticoid receptor and constitutive androstane receptor. J Biol Chem. 2002;277:209–17.
de Morais SMF, Schweikl H, Blaisdell J, Goldstein JA. Gene structure and upstream regulatory regions of human CYP2C9 and CYP2C18. Biochem Biophys Res Commun. 1993;194:194–201.
Ged C, Beaune P. Isolation of the human cytochrome P-450 IIC8 gene: multiple glucocorticoid responsive elements in the 5′ region. Biochim Biophys Acta. 1991;1088:433–5.
Bauersachs J, Christ M, Ertl G, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C expression and EDHF-mediated relaxation in porcine coronary arteries is increased by cortisol. Cardiovasc Res. 2002;54:669–75.
Transon C, Leemann T, Dayer P. In vitro comparative inhibition profiles of major human drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6 and CYP3A4) by HMG-CoA reductase inhibitors. Eur J Clin Pharmacol. 1996;50:209–15.
Fischer V, Johanson L, Heitz F, Tullman R, Graham E, Baldeck JP, Robinson WT. The 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor fluvastatin: effect on human cytochrome P-450 and implications for metabolic drug interactions. Drug Metab Dispos. 1999;27:410–6.
Qiu WP, Hu Q, Paolocci N, Ziegelstein RC, Kass DA. Differential effects of pulsatile versus steady flow on coronary endothelial membrane potential. Am J Physiol Heart Circ Physiol. 2003;285:H341–6.
Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–81.
Oesch-Bartlomowicz B, Richter B, Becker R, Vogel S, Padma PR, Hengstler JG, Oesch F. cAMP-dependent phosphorylation of CYP2B1 as a functional switch for cyclophosphamide activation and its hormonal control in vitro and in vivo. Int J Cancer. 2001;94:733–42.
Korsmeyer KK, Davoll S, Figueiredo-Pereira ME, Correia MA. Proteolytic degradation of heme-modified hepatic cytochromes P450: a role for phosphorylation, ubiquitination, and the 26S proteasome? Arch Biochem Biophys. 1999;365:31–44.
Anandatheerthavarada HK, Biswas G, Mullick J, Sepuri NB, Otvos L, Pain D, Avadhani NG. Dual targeting of cytochrome P4502B1 to endoplasmic reticulum and mitochondria involves a novel signal activation by cyclic AMP-dependent phosphorylation at Ser128. EMBO J. 1999;18:5494–504.
Eliasson E, Mkrtchian S, Ingelman-Sundberg M. Hormone- and substrate-regulated intracellular degradation of cytochrome P450 (2E1) involving MgATP-activated rapid proteolysis in the endoplasmic reticulum membranes. J Biol Chem. 1992;267:15765–9.
Minamiyama Y, Takemura S, Imaoka S, Funae Y, Tanimoto Y, Inoue M. Irreversible inhibition of cytochrome P450 by nitric oxide. J Pharmacol Exp Ther. 1997;283:1479–85.
Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxide attenuates the release of endothelium-derived hyperpolarizing factor. Circulation. 1996;94:3341–7.
Beetham JK, Tian T, Hammock BD. cDNA cloning and expression of a soluble epoxide hydrolase from human liver. Arch Biochem Biophys. 1993;305:197–201.
Newman JW, Morisseau C, Harris TR, Hammock BD. The soluble epoxide hydrolase encoded by EPXH2 is a bifunctional enzyme with novel lipid phosphate phosphatase activity. Proc Natl Acad Sci U S A. 2003;100:1558–63.
Cronin A, Mowbray S, Durk H, Homburg S, Fleming I, Fisslthaler B, Oesch F, Arand M. The N-terminal domain of mammalian soluble epoxide hydrolase is a phosphatase. Proc Natl Acad Sci U S A. 2003;100:1552–7.
Newman JW, Morisseau C, Hammock BD. Epoxide hydrolases: their roles and interactions with lipid metabolism. Prog Lipid Res. 2005;44:1–51.
Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, Hammock BD, Shyy JYJ, Zhu Y. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:9018–23.
Yamada T, Morisseau C, Maxwell JE, Argiriadi MA, Christianson DW, Hammock BD. Biochemical evidence for the involvement of tyrosine in epoxide activation during the catalytic cycle of epoxide hydrolase. J Biol Chem. 2000;275:23082–8.
Barbosa-Sicard E, Frömel T, Keserü B, Brandes RP, Morisseau C, Hammock BD, Braun T, Krüger M, Fleming I. Inhibition of the soluble epoxide hydrolase by tyrosine nitration. J Biol Chem. 2009;284:28156–63.
Ohtoshi K, Kaneto H, Node K, Nakamura Y, Shiraiwa T, Matsuhisa M, Yamasaki Y. Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem Biophys Res Commun. 2005;331:347–50.
Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–63.
Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–503.
Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–65.
Liu JY, Li N, Yang J, Li N, Qiu H, Ai D, Chiamvimonvat N, Zhu Y, Hammock BD. Metabolic profiling of murine plasma reveals an unexpected biomarker in rofecoxib-mediated cardiovascular events. Proc Natl Acad Sci U S A. 2010;107:17017–22.
Frömel T, Jungblut B, Hu J, Trouvain C, Barbosa-Sicard E, Popp R, Liebner S, Dimmeler S, Hammock BD, Fleming I. Soluble epoxide hydrolase regulates hematopoietic progenitor cell function via generation of fatty acid diols. Proc Natl Acad Sci U S A. 2012;109:9995–10000.
Keserü B, Barbosa-Sicard E, Popp R, Fisslthaler B, Dietrich A, Gudermann T, Hammock BD, Falck JR, Weissmann N, Busse R, Fleming I. Epoxyeicosatrienoic acids and the soluble epoxide hydrolase are determinants of pulmonary artery pressure and the acute hypoxic pulmonary vasoconstrictor response. FASEB J. 2008;22:4306–15.
Sinal CJ, Miyata M, Tohkin M, Nagata K, Bend JR, Gonzalez FJ. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J Biol Chem. 2000;275:40504–10.
Luria A, Weldon SM, Kabcenell AK, Ingraham RH, Matera D, Jiang H, Gill R, Morisseau C, Newman JW, Hammock BD. Compensatory mechanism for homeostatic blood pressure regulation in Ephx2 gene-disrupted mice. J Biol Chem. 2007;282:2891–8.
Seubert JM, Sinal CJ, Graves J, Degraff LM, Bradbury JA, Lee CR, Goralski K, Carey MA, Luria A, Newman JW, Hammock BD, Falck JR, Roberts H, Rockman HA, Murphy E, Zeldin DC. Role of soluble epoxide hydrolase in postischemic recovery of heart contractile function. Circ Res. 2006;99:442–50.
Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol. 2013;53:37–58.
Enayetallah AE, Luria A, Luo B, Tsai HJ, Sura P, Hammock BD, Grant DF. Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase. J Biol Chem. 2008;283:36592–8.
Luria A, Morisseau C, Tsai HJ, Yang J, Inceoglu B, De Taeye B, Watkins SM, Wiest MM, German JB, Hammock BD. Alteration in plasma testosterone levels in male mice lacking soluble epoxide hydrolase. Am J Physiol Endocrinol Metab. 2009;297:E375–83.
Enayetallah AE, Grant DF. Effects of human soluble epoxide hydrolase polymorphisms on isoprenoid phosphate hydrolysis. Biochem Biophys Res Commun. 2006;341:254–60.
Tran KL, Aronov PA, Tanaka H, Newman JW, Hammock BD, Morisseau C. Lipid sulfates and sulfonates are allosteric competitive inhibitors of the N-terminal phosphatase activity of the Mammalian soluble epoxide hydrolase. Biochemistry. 2005;44:12179–87.
Kovacs WJ, Olivier LM, Krisans SK. Central role of peroxisomes in isoprenoid biosynthesis. Prog Lipid Res. 2002;41:369–91.
Oguro A, Imaoka S. Lysophosphatidic acids are new substrates for the phosphatase domain of soluble epoxide hydrolase. J Lipid Res. 2012;53:505–12.
Morisseau C, Schebb NH, Dong H, Ulu A, Aronov PA, Hammock BD. Role of soluble epoxide hydrolase phosphatase activity in the metabolism of lysophosphatidic acids. Biochem Biophys Res Commun. 2012;419:796–800.
Przybyla-Zawislak BD, Srivastava PK, Vazquez-Matias J, Mohrenweiser HW, Maxwell JE, Hammock BD, Bradbury JA, Enayetallah AE, Zeldin DC, Grant DF. Polymorphisms in human soluble epoxide hydrolase. Mol Pharmacol. 2003;64:482–90.
Keserü B, Barbosa-Sicard E, Schermuly RT, Tanaka H, Hammock BD, Weissmann N, Fisslthaler B, Fleming I. Hypoxia-induced pulmonary hypertension: comparison of soluble epoxide hydrolase deletion vs. inhibition. Cardiovasc Res. 2010;85:232–40.
Chen JK, Wang D-W, Falck JR, Capdevila J, Harris RC. Transfection of an active cytochrome P450 arachidonic acid epoxygenase indicates that 14,15-epoxyeicosatrienoic acid functions as an intracellular messenger in response to epidermal growth factor. J Biol Chem. 1999;274:4764–9.
Chen J-K, Capdevila J, Harris RC. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:6029–34.
Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J. 2003;17:770–2.
Wong PY, Lin KT, Yan YT, Ahern D, Iles J, Shen SY, Bhatt RK, Falck JR. 14(R),15(S)-epoxyeicosatrienoic acid (14(R),15(S)-EET) receptor in guinea pig mononuclear cell membranes. J Lipid Mediat. 1993;6:199–208.
Wong PY, Lai PS, Falck JR. Mechanism and signal transduction of 14 (R), 15 (S)-epoxyeicosatrienoic acid (14,15-EET) binding in guinea pig monocytes. Prostaglandins Other Lipid Mediat. 2000;62:321–33.
Wong PY, Lai PS, Shen SY, Belosludtsev YY, Falck JR. Post-receptor signal transduction and regulation of 14(R),15(S)- epoxyeicosatrienoic acid (14,15-EET) binding in U-937 cells. J Lipid Mediat Cell Signal. 1997;16:155–69.
Snyder GD, Krishna UM, Falck JR, Spector AA. Evidence for a membrane site of action for 14,15-EET on expression of aromatase in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2002;283:H1936–42.
Li PL, Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res. 1997;80:877–84.
Node K, Ruan XL, Dai J, Yang SX, Graham L, Zeldin DC, Liao JK. Activation of Gas mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J Biol Chem. 2001;276:15983–9.
Popp R, Brandes RP, Ott G, Busse R, Fleming I. Dynamic modulation of interendothelial gap junctional communication by 11,12-epoxyeicosatrienoic acid. Circ Res. 2002;90:800–6.
Fleming I, Rueben A, Popp R, Fisslthaler B, Schrodt S, Sander A, Haendeler J, Falck JR, Morisseau C, Hammock BD, Busse R. Epoxyeicosatrienoic acids regulate Trp channel dependent Ca2+ signaling and hyperpolarization in endothelial cells. Arterioscler Thromb Vasc Biol. 2007;27:2612–8.
Fukao M, Mason HS, Kenyon JL, Horowitz B, Keef KD. Regulation of BKca channels expressed in human embryonic kidney 293 cells by epoxyeicosatrienoic acid. Mol Pharmacol. 2001;59:16–23.
Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, Deeter C, Sharma KK, Gauthier KM, Campbell WB. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–49.
Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB. Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20-125I-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharmacol Exp Ther. 2008;324:1019–27.
Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs). Prostaglandins Other Lipid Mediat. 2007;82:42–9.
Cowart LA, Wei S, Hsu MH, Johnson EF, Krishna MU, Falck JR, Capdevila JH. The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J Biol Chem. 2002;277:35105–12.
Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JY. The antiinflammatory effect of laminar flow: the role of PPARg, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci U S A. 2005;102:16747–52.
Widstrom RL, Norris AW, Spector AA. Binding of cytochrome P450 monooxygenase and lipoxygenase pathway products by heart fatty acid-binding protein. Biochemistry. 2001;40:1070–6.
Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci U S A. 2001;98:2323–8.
Capdevila JH, Falck JR, Harris RC. Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res. 2000;41:163–81.
Klett EL, Chen S, Edin ML, Li LO, Ilkayeva O, Zeldin DC, Newgard CB, Coleman RA. Diminished acyl-CoA synthetase isoform 4 activity in INS 832/13 cells reduces cellular epoxyeicosatrienoic acid levels and results in impaired glucose-stimulated insulin secretion. J Biol Chem. 2013;288:21618–29.
Michaelis UR, Falck JR, Schmidt R, Busse R, Fleming I. Cytochrome P4502C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler Thromb Vasc Biol. 2005;25:321–6.
Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285:1276–9.
Fleming I, Michaelis UR, Bredenkötter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51.
Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher AM, Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109:178–83.
Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem. 1996;271:3460–8.
Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC. Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res. 2004;95:506–14.
Fang X, Hu S, Watanabe T, Weintraub NL, Snyder GD, Yao J, Liu Y, Shyy JYJ, Hammock BD, Spector AA. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J Pharmacol Exp Ther. 2005;314:260–70.
Fang X, Hu S, Xu B, Snyder G, Harmon S, Yao J, Liu Y, Sangras B, Falck J, Weintraub NL, Spector AA. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator activated receptor alpha. Am J Physiol Heart Circ Physiol. 2006;290:H55–63.
Potente M, Fisslthaler B, Busse R, Fleming I. 11,12-Epoxyeicosatrienoic acid-induced inhibition of FOXO factors promotes endothelial proliferation by down-regulating p27Kip1. J Biol Chem. 2003;278:29619–25.
Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM, Weston AH. EDHF: bringing the concepts together. Trends Pharmacol Sci. 2002;23:374–80.
Watanabe H, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–8.
Zheng X, Zinkevich NS, Gebremedhin D, Gauthier KM, Nishijima Y, Fang J, Wilcox DA, Campbell WB, Gutterman DD, Zhang DX. Arachidonic acid-induced dilation in human coronary arterioles: convergence of signaling mechanisms on endothelial TRPV4-mediated Ca2+ entry. J Am Heart Assoc. 2013;2:e000080.
Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R, Nilius B. Modulation of the Ca2+ permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res. 2005;97:908–15.
Loot AE, Popp R, Fisslthaler B, Vriens J, Nilius B, Fleming I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc Res. 2008;80:445–52.
Weissmann N, Dietrich A, Fuchs B, Kalwa H, Ay M, Dumitrascu R, Olschewski A, Storch U, Schnitzler M, Ghofrani HA, Schermuly RT, Pinkenburg O, Seeger W, Grimminger F, Gudermann T. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc Natl Acad Sci U S A. 2006;103:19093–8.
Wang L, Yin J, Nickles HT, Ranke H, Tabuchi A, Hoffmann J, Tabeling C, Barbosa-Sicard E, Chanson M, Kwak BR, Shin HS, Wu S, Isakson BE, Witzenrath M, de Wit C, Fleming I, Kuppe H, Kuebler WM. Hypoxic pulmonary vasoconstriction requires connexin 40-mediated endothelial signal conduction. J Clin Invest. 2012;122:4218–30.
Loot AE, Fleming I. Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: central role of transient receptor potential (TRP) C6 channels. J Cardiovasc Pharmacol. 2011;57:140–7.
Khojasteh S, Prabhu S, Kenny J, Halladay J, Lu A. Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet. 2011;36:1–16.
Passauer J, Büssemaker E, Lassig G, Pistrosch F, Fauler J, Gross P, Fleming I. Baseline blood flow and bradykinin-induced vasodilator responses in the human forearm are insensitive to the CYP 2C9 inhibitor sulfaphenazole. Clin Sci (Lond). 2003;105:513–8.
Passauer J, Pistrosch F, Lässig G, Herbrig K, Büssemaker E, Gross P, Fleming I. Nitric oxide- and EDHF-mediated arteriolar tone in uremia is unaffected by selective inhibition of vascular cytochrome P450 2C9. Kidney Int. 2005;67:1907–12.
Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol (Lond). 2003;546:307–14.
Bellien J, Iacob M, Gutierrez L, Isabelle M, Lahary A, Thuillez C, Joannides R. Crucial role of NO and endothelium-derived hyperpolarizing factor in human sustained conduit artery flow-mediated dilatation. Hypertension. 2006;48:1088–94.
Bellien J, Joannides R, Iacob M, Arnaud P, Thuillez C. Evidence for a basal release of a cytochrome-related endothelium-derived hyperpolarizing factor in the radial artery in humans. Am J Physiol Heart Circ Physiol. 2006;290:H1347–52.
Fischer D, Landmesser U, Spiekermann S, Hilfiker-Kleiner D, Hospely M, Muller M, Busse R, Fleming I, Drexler H. Cytochrome P450 2C9 is involved in flow-dependent vasodilation of peripheral conduit arteries in healthy subjects and in patients with chronic heart failure. Eur J Heart Fail. 2007;9:770–5.
Ozkor MA, Murrow JR, Rahman AM, Kavtaradze N, Lin J, Manatunga A, Quyyumi AA. Endothelium-derived hyperpolarizing factor determines resting and stimulated forearm vasodilator tone in health and in disease. Circulation. 2011;123:2244–53.
Schuck RN, Theken KN, Edin ML, Caughey M, Bass A, Ellis K, Tran B, Steele S, Simmons BP, Lih FB, Tomer KB, Wu MC, Hinderliter AL, Stouffer GA, Zeldin DC, Lee CR. Cytochrome P450-derived eicosanoids and vascular dysfunction in coronary artery disease patients. Atherosclerosis. 2013;227:442–8.
Fornage M, Hinojos CA, Nurowska BW, Boerwinkle E, Hammock BD, Morisseau CH, Doris PA. Polymorphism in soluble epoxide hydrolase and blood pressure in spontaneously hypertensive rats. Hypertension. 2002;40:485–90.
Wei Q, Doris PA, Pollizotto MV, Boerwinkle E, Jacobs J, Siscovick DS, Fornage M. Sequence variation in the soluble epoxide hydrolase gene and subclinical coronary atherosclerosis: interaction with cigarette smoking. Atherosclerosis. 2007;190:26–34.
Lee CR, North KE, Bray MS, Fornage M, Seubert JM, Newman JW, Hammock BD, Couper DJ, Heiss G, Zeldin DC. Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet. 2006;15:1640–9.
Davis BB, Thompson DA, Howard LL, Morisseau C, Hammock BD, Weiss RH. Inhibitors of soluble epoxide hydrolase attenuate vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2002;99:2222–7.
Davis BB, Morisseau C, Newman JW, Pedersen TL, Hammock BD, Weiss RH. Attenuation of vascular smooth muscle cell proliferation by 1-cyclohexyl-3-dodecyl urea is independent of soluble epoxide hydrolase inhibition. J Pharmacol Exp Ther. 2006;316:815–21.
Ulu A, Davis BB, Tsai HJ, Kim IH, Morisseau C, Inceoglu B, Fiehn O, Hammock BD, Weiss RH. Soluble epoxide hydrolase inhibitors reduce the development of atherosclerosis in apolipoprotein E-knockout mouse model. J Cardiovasc Pharmacol. 2008;52:314–23.
Zhang LN, Vincelette J, Cheng Y, Mehra U, Chen D, Anandan SK, Gless R, Webb HK, Wang YX. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol. 2009;29:1265–70.
Revermann M, Schloss M, Barbosa-Sicard E, Mieth A, Liebner S, Morisseau C, Geisslinger G, Schermuly RT, Fleming I, Hammock BD, Brandes RP. Soluble epoxide hydrolase deficiency attenuates neointima formation in the femoral cuff model of hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2010;30:909–14.
Frömel T, Kohlstedt K, Popp R, Yin X, Barbosa-Sicard E, Thomas AC, Liebertz R, Mayr M, Fleming I. Cytochrome P4502S1: a novel monocyte/macrophage fatty acid epoxygenase in human atherosclerotic plaques. Basic Res Cardiol. 2013;108:1–12.
Wiecha J, Munz B, Wu Y, Noll T, Tillmanns H, Waldecker B. Blockade of Ca2+-activated K+ channels inhibits proliferation of human endothelial cells induced by basic fibroblast growth factor. J Vasc Res. 1998;35:363–71.
Faehling M, Koch ED, Raithel J, Trischler G, Waltenberger J. Vascular endothelial growth factor-A activates Ca2+-activated K+ channels in human endothelial cells in culture. Int J Biochem Cell Biol. 2001;33:337–46.
Wolfram Kuhlmann CR, Wiebke LD, Schaefer CA, Kerstin MA, Backenkohler U, Neumann T, Tillmanns H, Erdogan A. Lysophosphatidylcholine-induced modulation of Ca2+-activated K+ channels contributes to ROS-dependent proliferation of cultured human endothelial cells. J Mol Cell Cardiol. 2004;36:675–82.
Chen JK, Falck JR, Reddy KM, Capdevila J, Harris RC. Epoxyeicosatrienoic acids and their sulfonimide derivatives stimulate tyrosine phosphorylation and induce mitogenesis in renal epithelial cells. J Biol Chem. 1998;273:29254–61.
Fleming I, Fisslthaler B, Michaelis UR, Kiss L, Popp R, Busse R. The coronary endothelium-derived hyperpolarizing factor (EDHF) stimulates multiple signalling pathways and proliferation in vascular cells. Pflugers Arch Eur J Physiol. 2001;442:511–8.
Chen J-K, Capdevila J, Harris RC. Heparin-binding EGF-like growth factor mediates the biological effects of P450 arachidonate epoxygenase metabolites in epithelial cells. Proc Natl Acad Sci U S A. 2002;99:6029–34.
Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J. 2003;17:770–2.
Pozzi A, Macias-Perez I, Abair T, Wey S, Su Y, Zent R, Falk JR, Capdevila JH. Charaterization of 5,6-and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–46.
Wang Y, Wei X, Xiao X, Hui R, Card JW, Carey MA, Wang DW, Zeldin DC. Arachidonic acid epoxygenase metabolites stimulate endothelial cell growth and angiogenesis via mitogen-activated protein kinase and phosphatidylinositol 3-kinase/Akt signaling pathways. J Pharmacol Exp Ther. 2005;314:522–32.
Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–6.
Park HS, Kim MS, Huh SH, Park J, Chung J, Kang SS, Choi EJ. Akt (protein kinase B) negatively regulates SEK1 by means of protein phosphorylation. J Biol Chem. 2002;277:2573–8.
Munzenmaier DH, Harder DR. Cerebral microvascular endothelial cell tube formation: role of astrocytic epoxyeicosatrienoic acid release. Am J Physiol Heart Circ Physiol. 2000;278:H1163–7.
Zhang C, Harder DR. Cerebral capillary endothelial cell mitogenesis and morphogenesis induced by astrocytic epoxyeicosatrienoic acid. Stroke. 2002;33:2957–64.
Medhora M, Daniels J, Mundey K, Fisslthaler B, Busse R, Jacobs ER, Harder DR. Epoxygenase-driven angiogenesis in human lung microvascular endothelial cells. Am J Physiol Heart Circ Physiol. 2003;284:H215–24.
Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnés CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122:178–91.
Hu J, Popp R, Frömel T, Ehling M, Awwad K, Adams RH, Hammes HP, Fleming I. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J Exp Med. 2014;211:281–95.
Grocott-Mason R, Fort S, Lewis MJ, Shah AM. Myocardial relaxant effect of endogenous nitric oxide in isolated ejecting hearts. Am J Physiol. 1994;266:H1699–705.
Zhang G, Panigrahy D, Mahakian LM, Yang J, Liu JY, Stephen Lee KS, Wettersten HI, Ulu A, Hu X, Tam S, Hwang SH, Ingham ES, Kieran MW, Weiss RH, Ferrara KW, Hammock BD. Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc Natl Acad Sci U S A. 2013;110:6530–5.
Pauwels EK. The protective effect of the Mediterranean diet: focus on cancer and cardiovascular risk. Med Princ Pract. 2011;20:103–11.
Gerber M. Omega-3 fatty acids and cancers: a systematic update review of epidemiological studies. Br J Nutr. 2012;107(Suppl 2):S228–39.
Robinson L, Mazurak V. N-3 polyunsaturated fatty acids: relationship to inflammation in healthy adults and adults exhibiting features of metabolic syndrome. Lipids. 2013;48:319–32.
Kirwan RP, Felice L, Clark AF, O’Brien CJ, Leonard MO. Hypoxia regulated gene transcription in human optic nerve lamina cribrosa cells in culture. Invest Ophthalmol Vis Sci. 2012;53:2243–55.
Dallaglio K, Bruno A, Cantelmo AR, Esposito AI, Ruggiero L, Orecchioni S, Calleri A, Bertolini F, Pfeffer U, Noonan DM, Albini A. Paradoxic effects of metformin on endothelial cells and angiogenesis. Carcinogenesis. 2014;doi:10.1093/carcin/bgu 001.
Sissung TM, Price DK, Sparreboom A, Figg WD. Pharmacogenetics and regulation of human cytochrome P450 1B1: implications in hormone-mediated tumor metabolism and a novel target for therapeutic intervention. Mol Cancer Res. 2006;4:135–50.
Tsuchiya Y, Nakajima M, Takagi S, Taniya T, Yokoi T. MicroRNA regulates the expression of human cytochrome P450 1B1. Cancer Res. 2006;66:9090–8.
Devlin AH, Thompson P, Robson T, McKeown SR. Cytochrome P450 1B1 mRNA untranslated regions interact to inhibit protein translation. Mol Carcinog. 2010;49:190–9.
Chuturgoon AA, Phulukdaree A, Moodley D. Fumonisin B1 modulates expression of human cytochrome P450 1b1 in human hepatoma (Hepg2) cells by repressing Mir-27b. Toxicol Lett. 2014;227:50–5.
Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, Collins FS, Remaley AT, Sethupathy P. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology. 2013;57:533–42.
Jennings B, George L, Pingili A, Khan N, Estes A, Fang X, Gonzalez F, Malik K. Estrogen metabolism by cytochrome P450 1B1 modulates the hypertensive effect of angiotensin II in female mice. Hypertension. 2014;64:134–40.
Tang Y, Scheef EA, Wang S, Sorenson CM, Marcus CB, Jefcoate CR, Sheibani N. CYP1B1 expression promotes the proangiogenic phenotype of endothelium through decreased intracellular oxidative stress and thrombospondin-2 expression. Blood. 2009;113:744–54.
Tang Y, Scheef EA, Gurel Z, Sorenson CM, Jefcoate CR, Sheibani N. CYP1B1 and endothelial nitric oxide synthase combine to sustain proangiogenic functions of endothelial cells under hyperoxic stress. Am J Physiol Cell Physiol. 2010;298:C665–78.
Palenski TL, Gurel Z, Sorenson CM, Hankenson KD, Sheibani N. Cyp1B1 expression promotes angiogenesis by suppressing NF-kB activity. Am J Physiol Cell Physiol. 2013;305:C1170–84.
Jobe SO, Ramadoss J, Koch JM, Jiang Y, Zheng J, Magness RR. Estradiol-17b and its cytochrome P450- and catechol-O-methyltransferase-derived metabolites stimulate proliferation in uterine artery endothelial cells: role of estrogen receptor-alpha versus estrogen receptor-beta. Hypertension. 2010;55:1005–11.
Bansal S, Leu A, Gonzalez FJ, Guengerich FP, Roy Chowdhury A, Anandathheerthavarada HK, Avadhani NG. Mitochondrial targeting of cytochrome P450 (CYP) 1B1 and its role in polycyclic aromatic hydrocarbon-induced mitochondrial dysfunction. J Biol Chem. 2014;289:9936–51.
Polet F, Feron O. Endothelial cell metabolism and tumour angiogenesis: glucose and glutamine as essential fuels and lactate as the driving force. J Intern Med. 2013;273:156–65.
Acknowledgments
The author acknowledges the work of the many groups whose work it has not been possible to cite here because of space limitations. Work performed in the author’s own laboratory was supported by the Deutsche Forschungsgemeinschaft (SFB-TR 23, A6 and Exzellenzcluster 147 ‘Cardio-Pulmonary System’).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Fleming, I. (2015). Cytochrome P450-Derived Lipid Mediators and Vascular Responses. In: Schmidt, M., Liebner, S. (eds) Endothelial Signaling in Development and Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2907-8_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2907-8_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2906-1
Online ISBN: 978-1-4939-2907-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)