
Abstract
Because aging is a multifactorial, pleiotropic process where many interacting mechanisms contribute to the organismal decline, the candidate gene approach rarely provides a clear message. This chapter discusses some of the inherent complexity, focusing on aspects that impinge upon protein homeostasis and maintain a healthy proteome. We discuss candidate genes that operate in these pathways, and compare their actions in invertebrates, mice and humans. We highlight several themes that emerge from recent research—the interconnections of pathways that regulate aging, the pleiotropic effects of mutations and other manipulations of the candidate proteins and the tissue specificity in these pleiotropic outcomes. This body of knowledge highlights the need for multiple specific readouts of manipulating longevity genes, beyond measuring lifespan, as well as the need to understand the integrated picture, beyond examining the immediate outputs of individual longevity pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Proteostasis (protein homeostasis)
- Lifespan and heathspan
- Proteotoxicity
- Stress responses
- Chaperone networks
- Caloric restriction
- Insulin/IGF-like signaling
- P53
- Sirtuins
- MTOR
Introduction
Aging is a multifactorial and pleiotropic process with many interacting mechanisms contributing to the decline. Because of this complexity, the candidate gene approach to understanding aging often relies on identifying human genes whose function during aging can be predicted based on animal models of aging. Then, association to longevity in humans is determined statistically if a variant of the candidate gene is prevalent among people who live healthy, long lives compared to people with average health span and lifespan . Many of the longevity genes identified thus far influence one of the following pathways: the insulin/insulin-like growth factor pathway , TOR (target of rapamycin) signaling, DNA repair, lipid metabolism, mitochondrial activity and nutrient intake. Understanding how these genetically distinct mechanisms interact at the molecular level to control longevity is a fundamental and fascinating problem, which has been subject to many reviews, e.g. [1–3]. In some cases, as for telomerase, the role in longevity is rather clear—maintaining telomere length [4, 5]. In others, such as mitochondrial activity, the connection is vague, because redox state and reactive oxygen species (ROS) encompass so many aspects of biology. In the case of other candidate genes, for example apolipoprotein E alleles that are prominently identified by genomic association studies [6], their mechanistic contribution to longevity is related to their different biochemical activities and in particular to their differential binding to damaged protein (reviewed recently by [7, 8]). Common to all these pathways is their role in cytoprotection against various cellular stresses. Clearly, maximal lifespan extension demands the co-activity of multiple cytoprotective pathways, as emphasized below. In this chapter we focus on the processes that guard the proteome during aging, an aspect that had been considered less than DNA repair or lipid metabolism.
Even at a simple-minded level, it is clear that the various types of stress impact or damage the proteome. Loss of enzymatic activity and translational fidelity, accumulation of somatic mutations, altered post-translational modification, are all ways in which the protein homeostasis network is functionally eroded upon alteration the conditions in either the cytosol, the mitochondria or the endoplasmic reticulum (ER) (Fig. 2.1). Damage to the proteome is intimately linked to the body’s stress responses , which deal with altered or damaged proteins by either attempting to repair the damage, dispose of the damaged molecules and/or compensate by increasing expression of the active one.

Proteotoxic stresses and stress responses in maintaining a healthy proteome. While heat shock and ER stress largely cause protein misfolding, mitochondrial and other redox stresses mostly cause damaged proteins indirectly, through oxidation and other post-translational modification (red arrows). Stress responses, located at the various cellular compartments, control protein misfolding either by refolding, sequestration or degradation (green arrows)
Cumulative Protein Damage and Aging
Cumulative damage is an often-discussed concept in the context of the genome, but it is clearly also important in the context of the proteome. Sustained exposure to environmental factors like UV radiation or free radicals causes incremental damage that is proportional to age and is problematic in particular with long-lived proteins. Even normal metabolism associated with daily activities, such as respiration and signaling, interface with stress responses and can cause cumulative protein damage (Fig. 2.1). The late onset of many degenerative diseases is commonly interpreted as reflecting cumulative damage, for example by accumulation of misfolded or aggregated proteins. In addition, proteome analysis by global methods often report on progressively damaged proteins through post-translational modifications that increase with age [9–11]. A remarkable example was provided by two species of bats, which generally live far longer than predicted by their size, relative to other organisms. The proteomes of these bats exhibit significantly lower protein carbonylation, resistance to protein oxidation under conditions of acute oxidative stress, low levels of ubiquitination and reduced proteasome activity [12]. Thus, diminished protein damage and removal are related to longevity, yet much more needs to be learned about the process of cumulative protein damage and how the thresholds are set so as to initiate disease late in life.
The accumulation of damaged proteins with aging is coupled to a progressively decreased ability of the normal quality control mechanisms, as discussed further in subsequent sections. The ability of quality control systems to detect and deal with damaged proteins is important at all times, but becomes particular critical when the quality control capacity is reduced. For example, the level of expression of molecular chaperones, which assist other proteins to attain or maintain a folded conformation, itself changes with age.
Perhaps the most consistently identified ‘aging gene’ in humans that affects both long lifespan and susceptibility to certain age-related diseases is the apolipoprotein E gene (ApoE) [7, 8]. The inheritance of the three alleles of ApoE correlates with protection or susceptibility to aspects of aging [7, 8, 13]. This correlation has been appreciated since the early 1990s, when a case-control studies found the ε4 allele, a previously known risk factor for cardio-vascular diseases and Alzheimer’s disease, to be significantly less frequent in centenarians than in controls. The ApoE allele most frequent in centenarians was ε2, suggesting that it was protective [14]. The nature of the protective effect of the ε2 allele is not entirely clear, as it has been associated with risk for certain types of ischemic heart disease and hyperlipoproteinemia [15], or found to have no association, positive or negative, with heart disease or stroke [16, 17].
Multiple mechanisms have been proposed to explain the effects of the ε4 isoform of the protein, which differs by 2 amino acids from the ε2 isoform (arginines instead of cysteines at positions 112 and 158). First, hyperlipidemia and hypercholesterolemia resulting from lipid transport abnormalities can directly contribute to heart disease, atherosclerosis, and stroke (reviewed in [7, 8]. In Alzheimer’s disease, ApoE is thought to be involved in binding and clearance of amyloid β (Aβ). ApoE ε4 has decreased interaction with Aβ [7, 8] and its deposition into senile plaques is higher in patients carrying the ε4 allele. Its direct role in developing the disease is still not clear, though, as increased deposition of amyloid is also seen in cognitively healthy ε4 carriers [18]. Soluble Aβ is another potential target of ApoE: in a mouse model, clearance of soluble Aβ from the brain depended on the isoform of human ApoE expressed [19]. Alternatively, ApoE ε4 may directly promote Aβ oligomer formation, more so than ε3 or ε2 [20]. Another hypothesis suggests a negative effect of ApoE ε4 on recycling of the ApoE receptor and thus on neural function [21]. Other possibilities include disruption of neural connectivity, independent of Aβ; increased tau hyperphosphorylation, cytoskeletal disruption and mitochondrial dysfunction by the proteolytic fragments of ApoE ε4, decreased protection from excitotoxicity, decreased synaptic function, and other deficiencies [22]. How all these phenotypic changes are caused by the two cys/arg substitutions is not known. One molecular consequence of the substitutions is decreased interaction of N-terminal and C-terminal domains of the ApoE protein, leading to decreased binding to the receptor and possibly to other interacting proteins [7].
It is worth noting that the association of specific alleles of ApoE with the detrimental cognitive phenotypes appears to be age-dependent [23]. For example, the presence of ε4 allele has been associated with better, not worse, memory and neural efficiency in young healthy people [24]. The differential effect of ε4 allele on memory performance depending on age was suggested to indicate antagonistic pleiotropy, a theme common for longevity genes: ApoE ε4 may be beneficial during development and early adulthood, at the expense of accelerated decline in cognitive function with ageing [25].
The action of ApoE exemplifies one type of coping with protein damage, systemic mechanisms that react to extracellular damaged proteins. The main focus of this chapter addresses a different type—intracellular mechanisms that monitor aging-dependent protein damage.
Proteostasis
Proteostasis , or protein homeostasis, is the state of a healthy proteome that is maintained by many sets of interactions among proteins and between proteins and metabolites, which enable the proteome to react to perturbations [26]. Like aging itself, proteostasis is highly complex and involves many hundreds of genes whose products engage in diverse protein complexes and function in many distinct metabolic pathways [26]. The interacting networks include protein synthesis, machineries of post-translational modifications, chaperone machines that assist in proper folding and various disposal systems. These protein networks are usually thought of as ‘stress responses’ (Fig. 2.1), but it is important to emphasize that they operate during normal physiology, even without external stress. Of course, since proteins are made and fold in different cellular compartments that present distinct environments, proteostasis networks in the cytosol, the endoplasmic reticulum and in other organelles have distinct functions and components, which evolved to address the distinct environmental conditions [27].
Also like aging, proteostasis is by definition tissue-specific, and while it has been studied in single cells for a long time, the molecular tools for studying it in whole organisms are still evolving. Here, we focus on some concepts and challenges linking these longevity pathways to the homeostasis of proteins .
Caloric Restriction
One of the most basic and general factors in determining lifespan is the diet. Much experimental work in animal models has shown that caloric restriction (CR) enables extension of lifespan , and studies in human populations often emphasize the inverse relationship between caloric intake and healthy aging [28]. Much of our understanding of the genetic factors that favor long lifespan also relates the function of these genes to their effects on the metabolism. A simplistic view of caloric restriction is that increased food intake leads to metabolic demands, which over time stress the organism and thus translates to shorter maximal lifespan or to poorer quality of aging. Clearly, sensing the intracellular nutrient and energy status involves many of the arms of the proteostasis signaling pathways in the cytosol, the secretory apparatus and the mitochondria. Caloric restriction has been shown to affect many genes involved in preserving cellular homeostasis during stressful conditions. Such genes, sometimes termed ‘vitagenes’ [29], encompass heat shock proteins (HSP), the thioredoxin and sirtuin protein systems, nutrient sensing systems like mTOR and redox systems. As discussed below, all these systems affect protein homeostasis.
Protein Misfolding During Aging
Many diseases whose molecular basis is misfolding of proteins are associated with aging and affect almost every tissue. A typical feature of diseases such as Huntignton disease, ALS, Alzheimer’s disease or systemic amyloidosis is their onset late in the life of adults. An illustrative example of such diseases is cataract, where most commonly progressive loss of function of the crystallins in the lens leads to visual degeneration. Lens cells lack the machinery for folding and degradation of proteins and are therefore thought to depend on “holdase” chaperone function to prevent protein aggregation. α-crystallin, one of the small heat shock proteins, is the built-in lens chaperone. It is present throughout life in the protected environment of the lens and over time it accumulates damage from oxidation, non-enzymatic modifications and proteolysis. This damage partially unfolds the crystallins and creates aggregation-prone intermediates. In the young lens, α-crystallin sequesters such intermediates effectively, but in an old lens α-crystallin’s capacity to bind polypeptides is saturated, and lens proteins are able to aggregate, promoting light scatter and loss of visual acuity [30].
The presence of insoluble proteins is a common feature of most aging-associated protein misfolding diseases (Fig. 2.1). Alzheimer’s and Parkinson’s diseases, type II diabetes and a host of lesser-known, but often equally serious conditions such as fatal familial insomnia are characterized by insoluble protein aggregates. These aggregates are found either in the cytoplasm, the nucleus or the endoplasmic reticulum (ER) and are often used in the diagnosis of the diseases [31]. Nonetheless, all too often the data only correlate the aggregates with aging ; relatively few studies address whether the misfolded protein itself causes accelerated aging through cellular dysfunction, or whether its misfolding and aggregation are consequences of the progressive decline of proteostasis [32–34]. The increased lifespan of C. elegans when aggregation-prone proteins are knocked down [35], or when proteostasis-stimulating compounds are used [36–38], supports the causative role of misfolding/aggregation. However, these approaches do not distinguish between the formation of protein aggregates themselves and accumulation of earlier misfolded intermediates. They also do not distinguish between genetic factors that change the aggregation behavior of the misfolded protein, and factors that change the organismal response to the misfolded protein; these can modify the lifespan independently of each other [39] and further work is needed to distinguish between these mechanisms. Furthermore, more data on the relation between protein aggregation and longevity in mammals is still required to demonstrate evolutionary conservation of the mechanisms that operate in the model genetic organisms [40].
Oxidative Damage to Proteins
Many candidate aging genes involved in the control of redox states and the response to oxidative stress have been highlighted and explored, no doubt because of the impact of the long-standing free radical theory of aging [41]. Oxidative damage to proteins, like damage to nucleic acids and lipids, is mediated by reactive oxygen species (ROS) and increases with age. ROS formation is a highly regulated process controlled by a complex network of intracellular signaling pathways . The intracellular nutrient and energy status of the cell, the functional state of mitochondria, and the concentration of ROS produced in mitochondria, in the cytosol and the ER are all sensed during normal physiology. CR appears to prolong life by reducing reactive oxygen species (ROS)-mediated oxidative damage. However, the correlation between ROS tolerance and lifespan is complex, which at the minimum contradict the existence of a simple relationship between ROS and aging [41].
Many long-lived mutants are resistant to treatment with ROS, and mutants selected for resistance to ROS are also long lived, but an exhaustive study on the effect of under- or over-expressing a wide variety of mouse genes coding for antioxidant enzymes showed that only the deletion of the Sod-1 gene affects lifespan [42]. Consistent with this, C. elegans devoid of all 5 SOD enzymes have normal lifespan under non-stressful conditions [43].
Numerous antioxidative enzymes (catalases, superoxide dismutases, glutathione enzymes, metal-binding proteins) have provided promising avenues for mechanistic analyses of ROS defenses, but inactivation of such candidate genes has shown at best a modest extension of lifespan. The current opinion is that ROS are not the sole determinant, but only a contributor to aging [41, 44].
Age-Related Decline in the Proteostasis Network
The proteostasis network is designed to react to the stresses that lead to misfolding, to the misfolded protein species, and to the protein aggregates that form [26]. Each of these processes can be countered by different arms of the network : Transcription and synthesis of anti-stress proteins such as folding enzymes or molecular chaperones are activated through the heat shock response (HSR) the unfolded protein response (UPR) in the ER and the mitochondrial UPR; Activities of chaperones are involved in recognition of misfolded molecules; Disposal systems such the proteasome or autophagy are activated to rid the tissues of the toxic misfolded species; Toxic proteins can be segregated to prevent cytotoxic effects.
The cytosolic stress-responsive machinery
Since misfolded proteins usually accumulate in the cytosol, even if they originate from the secreted proteome [31] they occupy and titrate cytosolic HSPs, and thereby activate HSF1, the main regulator of the HSR [45]. The activation of HSF1 involves hyper-phosphorylation and deacetylation, allowing HSF1 to trimerize, translocate into the nucleus, and bind to heat-shock elements in the promoter regions of the many HSR target genes. During aging, there is clearly a decline in the capacity of the HSR to counter stress. Up-regulation of the stress-inducible HSP70 by heat shock is reduced approximately 50 % in hepatocytes from old rats [46] and this decrease in HSP70 expression is also seen in other cells, such as human mononuclear cells and lymphocytes [47]. Similarly, expression of the other major cytosolic chaperone, HSP90, is reduced in fibroblasts from old rats [48]. The ability of HSF1 to bind the promoters of heat-shock genes in response to increased temperature was absent in the brains of 36 month old rats, while its levels were not affected [49]. In C. elegans, the inducibility of HSR becomes impaired in early adulthood [33], and this sharp transition seems to be mediated by the germline stem cell signaling at the onset of reproduction [50].
Expression of many HSPs differs significantly between tissues of ‘normal’ and long-lived Pit1(dw/dw) Snell dwarf mice. The magnitude and direction of the expression changes are tissue-specific and where HSP expression declines, it is often related to GH and/or IGF-I signaling [51]. These observations in mice parallel the role of IGF/insulin signaling in determining worm lifespan. On the other hand, forced overexpression of molecular chaperones is deleterious to growth of normal mammalian cells [52, 53], while the transformed phenotype of cancer cells depends on HSF1 and on an increased expression of chaperones [54–56]. A related example is the expression of the XBP-1 UPR transcription factor. Ectopic expression of the active form of XBP-1 specifically in C. elegans muscle reduced lifespan while expression in neurons prolonged longevity [57]. It appears then that the precise regulation of proteostasis networks is essential for the health of the organism, while accumulating evidence points to its dysregulation during aging as one of the main causes of cellular dysfunction and disease onset (Fig. 2.2).

Complexity of the effects of longevity pathways on aging. Examples of how intervention/regulation of transcription factors and chaperones either increase or decrease longevity and its quality
Molecular chaperones work in transient complexes with co-chaperones and catalytic factors and diminished chaperone capacity is brought about by down-regulation of these components, not only HSP70 and HSP90. For example, HSP110, which suppresses the important pro-apoptotic p38 MAPK stress signaling [58], is down-regulated in livers of three longevity mutants (Pit1(dw/dw), Prop1(df/df), Ghr−/−), and in both kidney and heart of Pit1(dw/dw) and Ghr−/− [51]. Hsph1 expression is likewise reduced in many tissues of mice exposed to the anti-aging effects of caloric restriction , including liver, heart, white adipose tissue, hypothalamus and colon [59].
Perhaps more importantly than the level of expression, the functionality of HSP70 from old rat livers was half that of HSP70 from young livers, as measured by in vitro chaperone assays [60]. The age-associated lower activity is important, because even if there is reduced level of HSP70 or HSP90, they are still highly abundant, accounting for more than 1 % of total cell protein even in aged tissues. Thus, reduced activity attests to reduced chaperone capacity, which may prevent repair of protein damage, leading to degeneration and cell death.
The ER stress-responsive machinery
Reduced inducibility of stress responses and reduced levels of chaperones with age are not restricted to the cytosolic proteostasis network. Several studies had documented age-related decline in UPR function, another important arm of the proteostatic network that reacts to stress in the ER. In C. elegans, the capacity to activate UPR signaling is strikingly reduced during early adulthood [50, 57] coinciding with the decline of HSR [33], and when the ability to respond is diminished, the extension of lifespan is also diminished.
Constitutive expression of ER chaperones and folding enzymes also declines with age. Hepatic expression of BiP, calnexin, ERp57 and ERp72 declined 30− 50 % in aged rats [61]. Liver expression of the ER gene dnajc3 (encoding p58IPK) was diminished by all three of the above murine longevity models and was also lower in kidney, muscle, and heart of the Pit1(dw/dw) mice. Dnajc3 is a stress-inducible co-chaperone for the UPR chaperone BiP critical for survival in response to ER stress [62], and there is evidence that this role of Dnajc3 in cellular stress response can have widespread physiological effects. For instance, mice that lack p58IPK have severely reduced body fat levels, low body weight and are diabetic, most likely due to apoptosis of pancreatic β cells [63]. Since 1/4 − 1/3 of the proteome is secretory and originates in the ER, and considering the preponderance of membrane and secreted protein among aging-related protein misfolding diseases, these data support the concept that diminished chaperone capacity contributes to the physiological decline associated with aging.
The UPR is implicated in many of these neurodegenerative and aging-related protein folding diseases. Activation of the UPR signaling by accumulation of misfolded proteins (not necessarily aggregates) is well established [64], but the outcomes of this activation are varied. For example, response to most of the misfolded mutants of α1 anti-trypsin results in induction of ER-associated proteasomal degradation (ERAD), but in response to accumulation of the PiZ mutant the UPR induces mostly the autophagy mechanism of disposal [65]. In addition, the effects of individual UPR pathways are quite complex and can be disease-specific. For example, deletion of XBP1, the central mediator of the IRE1 pathway, did not influence prion disease progression in animal models [66], but did delay onset and progression of ALS and HD in respective mouse models by stimulating autophagy [67].
Proteasomes
The proteasome is a multi-catalytic enzyme complex that mediates the degradation of both normal and damaged proteins. An age-related decline in proteasomal activity has been implicated in various age-related pathologies. Tomaru et al. created a transgenic mouse model with decreased proteasomal chymotrypsin-like activity, by over-expression of a thymus-specific subunit β5 that has weak chymotryptic activity [68]. These mice exhibited a shortened life span and developed age-related phenotypes. Poly-ubiquitinated and oxidized proteins accumulated in these mice, and the expression levels of cellular proteins such as Bcl-xL and RNase L were altered. When fed a high-fat diet, the β5 transgenic mice developed more pronounced obesity and hepatic steatosis than did wild-type mice. These results provide in vivo evidence that decreased proteasomal chymotrypsin-like activity affects longevity and aggravates age-related metabolic disorders [68]. In C. elegans, adaptation of peptide accessibility to the proteasome’s active site in response to environmental stress, through modifying its 19 S regulatory particle, is necessary for both resistance to proteotoxic insults and maintenance of animal life span under normal conditions [69].
Although proteasome activity is diminished when poly-ubiquitinated substrates accumulate, Hipp et al showed that an aggregated ubiquitinated substrate is not a direct inhibitor of proteasomes, but rather impairs degradation by saturating some as-yet-unidentified component [70].
At least one signaling pathway for proteasome dysfunction is through reactive oxygen species that originated from malfunctioning mitochondria and trigger an Nrf2-dependent up-regulation of the proteasomal subunits. This is a tissue- and age-specific regulatory circuit that adjusts the cellular proteasome activity according to temporal and/or spatial demands [71].
Proteasomes and aggresomes are likely complementary quality control pathways, since aggresome formation/growth is promoted when proteasomes are inhibited [72]. Each of these arms of the proteostasis network declines with age, thus diminishing the response of metabolic stress or to accumulating misfolded proteins and reducing the chances of return to homeostasis.
Autophagy
An independent way to dispose damaged proteins is the process of autophagy , where whole organelles can be engulfed by membrane, the contents of which then becomes an acidic degradative compartment. The formation of autophagosome requires a dedicated machinery that includes chaperones, and their relevance to aging is demonstrated by mutational inactivation of autophagy genes, which accelerates tissues aging in C. elegans and Drosophila. [73]. Inactivation of autophagy also suppresses life span extension by CR, aberrant insulin/IGF-1 or mTOR signaling , and lowers mitochondrial respiration. These findings suggest that the different longevity pathways in converge on the autophagy pathway to slow down aging and increase life span. Thus, autophagy is one central effector mechanism of animal aging [74].
Aggresomes
One way to cope with accumulated misfolded proteins is to segregate them in cytosolic protein inclusions, termed ‘aggresomes’, that are usually perinuclear, near the microtubule organizing center and whose formation and maintenance depends on microtubule-based protein traffic [75]. Aggresomes often contain aging-relevant misfolded proteins and were suggested as a coping mechanism to protect cells from the cytotoxic effects of misfolded proteins [31]. One line of evidence supporting this notion is the enhanced proteo-toxicity in a yeast poly-Q model when aggresome formation is inhibited [76]. Targeting misfolded proteins to aggresomes seem to depend on recognition signals, like proline-rich domains, present on some aggregation-prone proteins. Another signal that is likely involved in the formation of aggresomes is the ankyrin-like repeat in synphilin 1, a protein related to Parkinson Disease [77]. It is not currently known how the aggresome formation machinery changes with age.
Proteostasis in Somatic Tissues Vs. the Gonads
Hallmarks of somatic tissues of aging flies are gradual accumulation of ubiquitinated and carbonylated proteins and reduction of proteasome expression and activity. In contrast, gonads of aging flies were relatively free of proteome oxidative damage and maintained substantial and highly active proteasomes. Exposure of flies to oxidants induced higher proteasome activities in the gonads, which were, independently of age, more resistant than soma to oxidative challenge, based on reporter transgenes. Finally, inducible Nrf2 activation in transgenic flies promoted youthful proteasome expression levels in the aged soma, suggesting that age-dependent Nrf2 dysfunction leads to decreased somatic proteasome expression during aging [78]. The higher investment in maintenance of proteostasis in the gonads perhaps facilitates proteome stability across generations [78].
Like in Drosophila, C. elegans gonadal cells also display transcriptional and proteomic programs that endow them with stress-resistance, which renders them immortal and capable of regeneration on a scale not shared by somatic tissues. C. elegans mutants with increased longevity exhibit gene expression programs normally limited to the germ line. Decreased insulin-like signaling causes the somatic mis-expression of the germline-limited pie-1 and pgl family of genes in intestinal and ectodermal tissues. The FOXO transcription factor DAF-16, the major transcriptional effector of insulin-like signaling, directly regulates pie-1 [79]. Furthermore, Ruvkun et al. tested whether any of the other genes identified in screens for worm lifespan extension also displayed expression of germline genetic markers in somatic tissues, by tracking the expression of endogenous PGL-1. PGL-1 is normally restricted to the germline where it forms perinuclear punctate structures around mitotic and meiotic germ cells. However, RNAi inactivation of the cct-4 and cct-6 subunits of the cytosolic chaperonin complex, known to increase longevity , caused somatic pattern of expression of PGL-1—punctate perinuclear rings in hypodermal cells and cytoplasmic granules in intestinal cells, the same tissues where misexpression of pie-1 in the insulin-like signaling mutants was observed [79].
The ability of C. elegans to maintain proteostasis declines sharply after the onset of oogenesis [50]. Arrest of germline stem cell rescued protein quality control, resulting in maintenance of robust proteostasis in different somatic tissues of adult animals. Germline stem cell-dependent modulation of proteostasis requires several different signaling pathways, each affecting different aspects of proteostasis and unable to functionally complement the other pathways. Shemesh et al. propose that the decline in the maintenance of proteostasis in somatic tissues is linked in this fashion to reproduction [50].
Together, these disparate observations support a model where longevity-promoting mutations such as decreased insulin-like signaling or reduction of chaperone capacity causes mis-expression of germline specific transcription factors in somatic tissues and this change contributes to the increased survival and health of these animals.
Hormesis, Proteostasis and Aging
One of the well-known phenomena regarding stress responses is that low doses of stress are protective. Such phenomena, termed “pre-conditioning” have been well-known from studying heat shock response in flies (e.g. [80]) and are related to the concept of ‘hormesis’ that emerged from toxicology [81] and applied to aging research [29]. Many C. elegans longevity genes are associated with antagonistically pleiotropic effects on development, fertility and/or behavior [82]. One unifying principle is that life extension is generally associated with increased resistance to stress, but does not imply extension of good quality of life.
The C. elegans response to heat shock and mitochondrial dysfunction illustrates the role of hormesis in lifespan extension. Treatment of C. elegans with a brief but intense heat shock provides longevity increase, whereas sustained exposure is lethal, and subsequent treatments increasingly extend lifespan [83, 84]. This pattern is recapitulated in the response to inactivation of the mitochondrial ATPase atp-3 [85]. Both studies highlight the importance of low dose of stress for aging. They also underscore why functional inactivation of genes in the same pathway, e.g. RNAi, do not necessarily yield comparable longevity. Because knockdown experiments differ in the amount of residual protein activity, the longevity effects are either not seen or are found to contradict those of other studies.
An important and little understood feature of hormesis is that mild exposure to one stress improves tolerance to other deleterious treatments, like reduced mitochondrial function, oxidative stress, radiation and toxic compounds [86]. Together with increased stress tolerance, hormesis also increases lifespan [86]. Thus, maintenance of physiological processes and repair of damage are improved by low stress and increase the ability to buffer the damage when higher level of stress is encountered. It will be important to define in molecular terms to what extent the hormetic cross-protection is due to activation of multiple cytoprotective mechanisms or to unknown protection effects conferred by individual pathways. For example, the combination of mild ER stress and apoptotic signals triggers an autophagic response, which is necessary for the hormetic protection from neurodegeneration. This activation of autophagy inhibits caspase activation and apoptosis [87]. This is an example of how effects of different longevity promoting pathways can be integrated by autophagy to promote longevity.
Relations of Candidate Pathways of Longevity Genes to Proteostasis
The Insulin-like Pathway
Tom Johnson and colleagues discovered in the 1980s the first gene shown to limit lifespan in the C. elegans, named age-1 [88, 89], which was later shown to encode a PI3K protein [90]. The discovery and subsequent cloning of the daf-2 gene [91, 92], encoding a C. elegans insulin-IGF receptor , brought the insulin-like signaling pathway (ILS) into focus as the major lifespan regulator in worms. When daf-2 or age-1 expression was “silenced” the activity of the insulin/IGF-1 pathway also decreased and the worms lived longer. Since then, many other genes associated with the insulin-like pathway have been found to affect the lifespan of fruit flies and mice. Reducing the activity of the insulin-like signaling cascade through mutant receptors protects worms from proteotoxicity of various aggregative proteins, including the HD-linked peptide, polyQ40 [93] and the AD-associated peptide Aβ [94]. Inactivation of daf-16 or hsf-1, encoding a FOXO transcription factor in the ILS pathway and a heat-shock transcription factor, respectively, and of genes for small heat shock proteins (HSPs), accelerated the aggregation of polyQ40, supporting the idea that the ILS coordinately influences aging and protein aggregation through the action of DAF-16 and HSF1 [95, 96]. Thus, the hypothesis that the insulin-like signaling pathway plays an important role in the aging process is well supported by studies of aging in model organisms.
Echoing these studies in model genetic organisms, the ILS pathway is clearly important for aging in mammals. Reduction of IGF1 signaling (as in animals heterozygous for the IGF1 receptor) was reported to protect mice from oxidative stress [97] and from Aβ toxicity, and to extend their lifespan [98]. In humans, Suh et al. demonstrated a gender-specific over-representation of heterozygous mutations in the IGF1R gene among female centenarians, associated with reduced activity of the IGF1R, high serum IGF1 levels and smaller stature [99]. Variants of other genes involved in the ILS pathway predominate among long-lived individuals, like the FOXO3a transcription factor, which is linked to extreme longevity in several, ethnically-disparate populations [100–103]. The extreme example of decreased ILS signaling in humans is presented by Laron syndrome—IGF1 deficiency due to inactive GH receptor that results in dwarfism and obesity on the one hand, but apparently complete protection from cancer and relatively long life on the other [104]. The emerging picture is of evolutionary conservation for the action of the ILS pathway in conferring both protection from aggregation diseases and increased longevity [34, 105].
Why and how does the ILS pathway impact longevity? The answers are complex and not entirely coherent. In part, the answers are complicated because candidate proteins (e.g. FOXO3a, IGF1R) regulate multiple processes that are not necessarily linked mechanistically. This leads to pleiotropy, which cannot be simply related to longevity. For example, IGF1R+/− mice [106] and humans [107] displayed growth retardation and other skeletal defects not seen in some of the other studies. At least some of these phenotypic differences are due to the genetic background [108], but clearly the conclusion is not as simple as reducing IGF signaling increases lifespan in all cases, and the multiple effects on other morbidities should be taken into account. All these effects, due not only to amount of signaling but also to its timing and its tissue location, are integrated to yield the seemingly opposite effects of the ILS pathway on longevity (Fig. 2.2).
Another complicating aspect is that serum IGF-I levels peak during human puberty and declines thereafter. This is an important aspect of IGF signaling during aging that has been linked to cerebrovascular and brain aging and cognitive decline [109]. The congenital animal deficiency models do not mimic this age-related decline. A recent model where murine IGF-I production is ablated late in adulthood shows that an intact GH/IGF-1 axis is essential to maintain health span. Its disruption, even late in life, causes increased liver pathology, oxidative stress in liver and muscle and accelerated bone loss [110].
The observations that IGF-deficient humans have lower risk of developing diabetes or cancer [104, 111], provide an evolutionary driving force for maintaining mutations in ILS pathway components, even if they come at a cost to development. This realization spurred manipulation of aging as an emerging strategy aimed at postponing the manifestation of late-onset neurodegenerative disorders such as Alzheimer’s (AD) and Huntington’s diseases (HD) and slowing their progression once emerged. Indeed, a novel ILS inhibitor, NT219, mediates a long-lasting, inhibition of this signaling cascade by a dual mechanism; it reduces the auto- phosphorylation of IGF1R and directs the insulin receptor substrates 1 and 2 (IRS-1 and - 2) for degradation [38]. While NT219 treatment provides a proof-of-concept that ILS inhibitors can be useful against aging-onset diseases, it also highlights the complexity of the problem, because while decreased signaling is beneficial for an ALS worm model [112], increased IGF-I administration at disease onset slows the progression ALS in human subjects [113].
The ILS Pathway and Stress Responses
Insulin-like signaling and the stress responses in the cytosol (HSR), the ER (UPR) and the mitochondria are related mechanistically by proteostatic networks (Fig. 2.3a). One connection is through the C. elegans FOXO transcription factor daf-16, which appears to regulate HSP expression by interacting with HSF [95]. Unlike mammals, which have four FOXO genes with overlapping and different functions, C. elegans has to account for the same functions with a single FOXO-like protein, DAF-16. How then is its specificity achieved in biological regulation? Distinct isoforms are used to fine-tune the ILS-mediated processes in the context of a whole organism, and are expressed in distinct tissue patterns. One isoform, DAF-16a, is known to regulate longevity, stress response and dauer diapause while DAF-16b has only a minor role. A recently identified isoform, DAF-16d/f, is an additional isoform that is important for regulating longevity. DAF-16a and DAF-16d/f functionally cooperate to modulate IIS-mediated processes through differential tissue enrichment, preferential modulation by upstream kinases, and regulating distinct and overlapping target genes [114]. Another possible connection between HSR and ILS in C. elegans was recently proposed, by demonstration that ILS can directly control HSF activity by regulating two HSF-inhibitory proteins, DDL-1 and DDL-2. While both these proteins are evolutionarily conserved, it is not yet known whether this regulatory connection is also conserved [115].

Connections among proteostasis pathways affecting longevity. a Heat shock response (HSR) and the ER or mitochondrial unfolded protein responses (UPR, mtUPR respectively) up-regulate distinct chaperone networks . ILS can directly regulate chaperones through the FOXO transcription factor and it also interacts with the stress responses, via mechanisms that are still not fully elucidated. Chaperones, in turn, also impact ILS, for example by regulating IGF production. b Caloric Restriction increases sirtuin activity that, among other things, inhibits p53 activity in neuroendocrine cells with a consequent effect on insulin signaling. Red, representative components that have been defined as candidate longevity genes. Blue, a specific example from Drosophila shows how a dominant negative p53 (DN p53) reduces production of of insulin-like peptide 2 and thereby reduces downstream effectors and increases both stress resistance and lifespan [140]
The study of murine HSPs by [51] suggests important differences between the effects of GH/IGF-I signaling on HSP expression in mice, and the effects of ILS signaling on HSP expression in worms. The inhibition of ILS in the daf-2 mutant increases expression of HSP70 and HSP90 family members [116, 117] as well as small heat shock proteins [95, 118, 119], all outcomes that affect cellular protein homeostasis. All these effects of systemic ILS mutations in C. elegans on heat shock gene expression, still do not provide much insight into the cells and tissues that express HSP differentially. The effects of diminished GH and/or IGF-I signals on HSP gene expression in mice are more complicated than those described in C. elegans. When growth hormone receptor-deficient mice are compared to long-lived dwarf mice, the beneficial phenotypes like increased lifespan and lower incidence of kidney disease, cataracts, and joint disease [120] are not related in a simple manner to the level or type of HSP expression [51]. In humans, genetic association studies have correlated polymorphisms of HSPs genes with longevity and age-related disease [121], but the mechanisms underlying such associations are not clear. While HSP expression appears to enhance overall proteome stability [26], and may counteract age-related conditions resulting from accumulation of poly-Q or prion proteins [122, 123], CR in mice promotes widespread diminution of HSP expression [59]. HSP expression may therefore have both positive and negative effects, depending upon the tissues or cell types, or upon the developmental or physiological state of the organism.
Even though growth occurs in every tissue and IGF-1 receptors are found throughout the body, there is considerable difference in the contribution of distinct cells and tissues to the ILS-related effects on aging [124, 125]. In retrospect, this is not surprising given the hormonal nature of ILS, but nonetheless was only fully appreciated reasonably recently. The non-autonomous nature of ILS in C. elegans was proposed on the basis of both genetic [126] and microarray analyses [125] and followed by direct experimental demonstration that highlighted the role of neurons in secretion of insulin-like hormones [127].
The neuroendocrine nature of ILS signaling is also evident in extension of mouse lifespan through tissue-specific knockout of insulin receptor substrates [128, 129] and the IGF-1R+/− mice (see above). The reduced ILS as a result of targeting the brain, rather than in peripheral tissues, is particularly important as this sets the pattern for growth as well as lifespan [130]. IGF-1 feedback onto the hypothalamus during development plays a key role in determining the set point of the somatotropic axis throughout life. Thus, in both worms and mice neurons play a central role in ILS. Remarkably, as discussed below, the hypothalamus is also a central tissue for lifespan extension via another pathway, the sirtuins .
Another important connection between heat shock proteins and ILS is that during ER stress, IGF-I stimulates translational recovery and induces expression of the key molecular chaperone of the UPR, BiP/GRP78, thereby enhancing the folding capacity of the ER and promoting recovery from ER stress [131]. Conversely, BiP expression in CR mouse livers is reduced by 40 % and IGF1R signaling regulates BiP expression via the PI3K/AKT/mTORC1 axis, independent of the canonical UPR and FOXO1 [132]. The stress response and chaperone systems also impact ILS by controlling the production of hormones (Fig. 2.3a). An example is the dependence of IGF-I and –II themselves on GRP94. As shown in both cell culture and a mouse model, the level of IGFs produced is proportional to the activity of GRP94 [133–135]. Therefore, it is possible that the decline in IGF levels in humans with advanced age reflects a decline in chaperone activity. Indeed, preliminary indications suggest that hypomorphic alleles of GRP94 exist in human populations and affect ILS signaling (Argon et al. unpublished).
In a Drosophila model of muscle mitochondrial injury, mild muscle mitochondrial distress preserves mitochondrial function, impedes the age-dependent deterioration of muscle function and architecture, and prolongs lifespan. Strikingly, this effect is mediated by at least two pro-longevity signaling pathways: muscle-restricted, redox-dependent induction of genes that regulate the mitochondrial UPR and the transcriptional induction of the Drosophila ortholog of insulin-like growth factor-binding protein 7, which systemically antagonizes insulin signaling and facilitates mitophagy (autophagic degradation of mitochondria). Given that several secreted IGF-binding proteins (IGFBPs) exist in mammals, this work raises the possibility that muscle mitochondrial injury in humans may similarly result in the secretion of IGFBPs, with important ramifications for insulin-like signaling [136].
The P53 Pathway
p53 is often studied as either a tumor suppressor or as a guardian of genomic integrity in response to cellular damage and stress. Yet p53 plays a third role—a longevity regulator. Mouse models that expressed 2 truncated forms or a temperature-sensitive mutant of p53 [137, 138] were resistant to cancer, yet had significantly shortened longevity and a number of premature aging phenotypes. This effect on mouse longevity led Helfand et al. to investigate the role of the Drosophila p53 homologue, Dmp53, in longevity [139]. They found that null Dmp53 flies, while viable, are sick and have reduced lifespan, probably due to early negative effects on embryonic development. However, when two dominant-negative (DN) mutants of p53 that inhibit wild type p53 transactivation activity were expressed in neuronal cells of Drosophila, but not in other tissues, longevity was increased. Even more specifically, expression of the DN Dmp53 transgene in the 14 neurons of the brain that produce insulin-like peptides extends lifespan to a similar extent as pan-neuronal expression [140] (Fig. 2.2). The primary effect of DN Dmp53 in the insulin-producing neurons was on reducing production of insulin-like peptide 2 (dILP2), while other dILPs remained unaffected. This was sufficient to inhibit downstream insulin signaling, as evidenced by reduced PI-3 kinase function in fat bodies of both larval and adult flies (Fig. 2.3b). Moreover, increased dFoxO nuclear accumulation was observed in fat body cells, a key downstream readout for attenuated insulin signaling, as increased nuclear FoxO is associated with increased stress resistance [141]. Turning on expression of the DN Dmp53 construct in adult Drosophila brains increased lifespan while turning the expression off reduced lifespan extension, both in proportion to the age of turn-on/off [140].
Thus, alterations in p53 signaling in a specific subset of secretory neurons were shown to cause major effects on insulin signaling pathways in critical target organs such as the fat body. CR did not provide any additional lifespan extension beyond that observed in non-restricted neuronal DN Dmp53 flies [139], arguing that p53 signaling and CR operate in the same pathway (Fig. 2.3b). It is likely that this model applies also to mammalian systems, although with a more complicated network of interactors, whose unraveling should provide profound new insights into the genetics and biology of aging and longevity .
The Sirtuin Pathway
Sirtuins are evolutionarily conserved enzymes with histone deacetylases activity, which affect multiple pathways that are important for metabolic regulation and the overall health of organisms [142]. NAD(+) is a rate-limiting substrate for sirtuin deacylases and therefore an important cofactor regulating metabolic homeostasis. In the context of aging, an extra copy of the yeast Sir2 sirtuin gene was found in the 1990s to increase the lifespan of yeast and similar extension of lifespan has been replicated in other organisms, including flies and worms [143]. However, the role of sirtuins in promoting mammalian longevity has engendered a fierce controversy. Some studies suggested that sirtuins mediate the lifespan-extending effects of CR. Phenotypically, resveratrol supplementation of obese humans for 30 days induced metabolic changes that mimic the effects of CR on energy metabolism and behavior [144]; this resveratrol effect is thought to be mediated by activation of SIRT1. However, whole-body expression of Sirt1 (the homologue of yeast Sir2) was not sufficient to extend longevity in mice as it does in invertebrates, although it improved general health [145]. In fact, even the effects in worms and flies were called into question because longevity co-segregated with a second-site mutation affecting sensory neurons in the background of the experimental strains [146].
It has since been shown that a restricted diet boosts Sirt1 and neuronal activity in certain hypothalamic nuclei that control central autonomic nervous system functions in mice [147]. Like caloric restricted wild-type mice, BRASTO mice, which overexpress Sirt1 only in the brain, lived 11 % longer than their wild-type counterparts even on a normal diet [147, 148]. The adults were also more active physically and metabolically, just like young mice, suggesting that physiological aging slows down in BRASTO mice compared to controls. The benefit is at least partly due to more orderly sarcomeres and more normal mitochondria in their skeletal muscle [148]. Curiously, another BRASTO mouse with more uniform overexpression of Sirt1 throughout all nuclei in the hypothalamus exhibited none of these signs of youth, suggesting that Sirt1 expression specifically in the dorsomedial and lateral hypothalamus is important for the anti-aging effect [148]. This finding could help settle the controversy about the role of Sirt1 aging in mice as high Sirt1 expression throughout the body might suppress neural activity in the dorsomedial and lateral hypothalamus. It seems that in mammals, like in model organisms, the effects on longevity are due to the activity of certain neurons affecting certain target tissues, rather than to whole body, multi-tissue effects.
Another focus of controversy about sirtuins and aging has been whether the red-wine ingredient resveratrol exerts similar longevity effects to those of CR. Sinclair and colleagues discovered in 2003 that resveratrol sped up the normal deacetylase activity of SIR2 in vitro [149]. That discovery was challenged as pertaining only to in vitro assays and not to living cells. However, two new studies support that the fluorescent molecules used in vitro may have mimicked hydrophobic amino acids found in certain natural SIRT1 substrates. In fact, sirtuin activators appear to speed up SIRT1 only when it interacts with substrates that contain such hydrophobic residues [150, 151]. Importantly, the number of such substrates is limited and includes ones thought to help induce some of calorie restriction’s key health-promoting effects, such as FOXO3a. The SIRT1 protein includes a single amino acid, Glu320, which is not essential for its normal deacetylase activity, but is critical for boosting the enzyme’s activity and the mitochondrial effects by sirtuin activating compounds [151]. This implies that SIRT1 serves as a key channel for inducing the mitochondrial aspects of longevity.
Regarding sirtuin substrates, estrogen receptor (ER) signaling has a variety of neuroprotective effects, and several proteins involved in ERα signaling are acetylated, including ERα itself and Hsp90, a key chaperone in the functional regulation of ERα. The acetylation is important to the functions of these proteins, but deacetylases other than SIRT2 appear to be involved [152].
dSir2 overexpression in Drosophila and CR longevity extension were shown to operate in the same pathway [153]. Moreover, longevity extension via the sirtuins pathway is related mechanistically to the effects of the p53 pathway , because dSir2 overexpressing DN Dmp53 flies showed the same longevity as Sir2 overexpressing flies, indicating no additive effects. This suggests that CR, dSir2 and DN Dmp53 all act through similar pathways of longevity extension, through the inhibition of p53’s transcriptional activity by sirtuins, as discussed above (Fig. 2.3b).
A different pro-longevity mechanism for sirtuins was recently pointed out by Schmeisser et al., who found that sirtuin-mediated lifespan extension depends on methylation of nicotinamide [154]. This is an unexpected activity of sirtuins beyond histone deacetylation. All sirtuins convert NAD(+) into nicotinamide, and provision of either nicotinamide or its metabolite, 1-methylnicotinamide, extend C. elegans lifespan even in the absence of sir-2.1. A previously unknown C. elegans nicotinamide-N-methyltransferase, encoded by a gene now named anmt-1, generates 1-methylnicotinamide from nicotinamide. Disruption and overexpression of anmt-1 have opposing effects on lifespan independent of sirtuins, with loss of anmt-1 fully inhibiting sir-2.1-mediated lifespan extension. 1-methylnicotinamide is in turn a substrate for an aldehyde oxidase, GAD-3, that generates hydrogen peroxide, which acts as a mitohormetic reactive oxygen species signal promoting C. elegans longevity [154]. NAD(+) levels are reduced in aged mice and worms and decreasing NAD(+) levels results in a further reduction in worm lifespan. Conversely, genetic or pharmacological restoration of NAD(+) prevents age-associated metabolic decline and promotes longevity in worms [155]. These effects are dependent upon the protein deacetylase SIR-2.1 and involve the activation of stress signaling via the mitochondrial unfolded protein response (mtUPR) and the nuclear translocation and activation of FOXO transcription factor DAF-16. The implication is that augmenting mitochondrial stress signaling by modulating NAD(+) levels may be a way to prevent or treat age-related decline [155]. Acetylation by SIRT1 was also shown to directly regulate the DNA-binding activity of human HSF1, providing a mechanistic connection for the requirement of HSF1 in regulating life span [156].
The mitochondrial deacetylase Sirt3 is essential for antioxidant defense system [157] but its pro-aging effects are also becoming clearer at a biochemical level. In fasting mice, Sirt3 expression is decreased in skeletal muscle, resulting in increased mitochondrial protein acetylation. Deletion of Sirt3 led to impaired glucose oxidation in muscle, which was associated with decreased pyruvate dehydrogenase (PDH) activity, accumulation of pyruvate and lactate metabolites, and an inability of insulin to suppress fatty acid oxidation. Proteomic analysis showed that a major target of Sirt3 deacetylation is the E1α subunit of PDH (PDH E1α) and Sirt3 knockout or knockdown in myoblasts induced hyperacetylation of the PDH E1α, altering its phosphorylation and leading to lower PDH enzymatic activity. This inhibition of PDH activity switched skeletal muscle substrate utilization from carbohydrate oxidation toward lactate production and fatty acid utilization even in the fed state, contributing to a loss of metabolic flexibility. Thus, Sirt3 plays an important role in part by regulating PDH function through deacetylation, thus affecting mitochondrial substrate choice and metabolic flexibility [158].
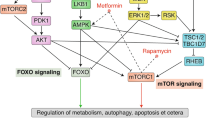
The Mtor Pathway
Much like sirtuins link genome quality control to a wide range of metabolic processes that impact aging, so do the mammalian (or mechanistic) target of rapamycin (mTOR) complexes. While the enzymatic activity of sirtuins is deacetylation, the activity of the TOR complexes is phosphorylation. Each is a common post-translation modification of enzymes, structural proteins or signaling proteins, and therefore affects either the level of activity or the stability of the protein and pathway.
The importance of mTOR for lifespan was first demonstrated by RNAi knockdown in C. elegans [159] and then extended by modulating the TOR homologues in flies, yeast and recently in mice. Lamming et al. demonstrated that female Mtor+/−Mlst8+/− mice have reduced mTORC1 activity and increased longevity [160], similar to the phenotype reported by Selman et al. for mice that lack S6K1, one of the principal substrates of mTOR [161]. Thus, the linkage between mTOR and lifespan is conserved in evolution.
Most commonly, the tool used to invoke mTOR complexes in longevity is the inhibitor rapamycin. Rapamycin treatment was shown to extend lifespan at blood levels that were 3X those of the typical therapeutic range for human immunosuppression [162]. However, the mechanism(s) accounting for the anti-aging effects of rapamycin is not yet clear. The effect on aging maybe through the anti-cancer effect, or by reducing protein synthesis, or by inducing autophagy, or by affecting stem cell, as it favors the retention of “stemness” and a more youthful phenotype in the adult stem cells types that have been studied (for details see review by [163]).
In vivo, the mTOR pathway receives inputs through a wide variety of signaling mechanisms and has roles in many aspects of physiology, which have been reviewed in depth [164]. These inputs are integrated by two mTOR complexes: mTORC1 responds to signals that include amino acids, glucose, WNT ligands, oxygen, cAMP, and insulin/IGF-1, namely to changes in metabolites or hormones that often signify metabolic stress. When activated, mTORC1 regulates protein synthesis and cell growth through phosphorylation of substrates that include S6 kinase (S6K) and eukaryotic initiation factor eIF4E binding protein (4E-BP). Thus, mTORC1 shapes the proteome in part through new protein synthesis [165]. mTORC2 (consisting of mTOR, rictor, mLST8/GβL, mSIN1, protor, DEPTOR) responds to less clear signals but may be activated by interaction with ribosomes [166], and thus affects the proteome at least at the level of protein synthesis. Nonetheless, mTORC2 activity regulates diverse kinases, including AKT, serum/glucocorticoid regulated kinase, and PKC-α, and thus indirectly regulates activity and or stability of many kinase substrates [166].
Acute treatment with rapamycin inhibits mTORC1 signaling, restricting growth and promoting longevity without reducing insulin sensitivity. On the other hand, chronic rapamycin treatment inhibits mTORC2 as well, impairing growth and insulin signaling, and perhaps promoting longevity [167, 168]. Interestingly, lifespan extension by disruption of mTORC1 in worms requires the worm’s homologues of NRF1/2 and FOXO, both transcription factors that control genes involved in stress defenses. Lifespan extension by rapamycin or by disruption of mTORC2, however, requires only the NRF1/2 homologue. Consistent with a role for general stress defenses in the benefits of rapamycin, both worms and flies with impaired TOR function are stress resistant, and induction of NRF1/2 and FOXO target genes has been detected in the livers of rapamycin-treated mice [167, 169] .
Despite the large number of studies with rapamycin, only recently was there genetic evidence that variation in any of the mTOR pathway genes plays a role in human longevity. Recently, using the Leiden Longevity cohort, seven of 40 mTOR pathway genes, and in particular Raptor, exhibited significant differential expression associated with longevity. This association was not explained by variation between the groups in the prevalence of type 2 diabetes, glucose levels or cancer [170]. It will be fascinating to discover which activities of mTORC1 are changed by these allelic variants.
It is important to emphasize that pathway cross-talks are important also to understanding mTOR activity towards longevity. There is an extensive cross talk between p53 and mTOR, one manifestation of which is that the activity of mTOR is increased in some but not all tissues of p53−/− mice, associated with the tendency to increased insulin and IGF-1 levels [171] (Fig. 2.3). At the same time, some of the endocrine and metabolic changes seen in diet-restricted mice were not seen in mice exposed to rapamycin, and the pattern of expression of hepatic genes involved in xenobiotic metabolism is also quite distinct in rapamycin-treated and diet-restricted mice [172]. These observations suggest that CR and mTOR inhibition extend mouse lifespan in different ways.
Conclusions
Many of the candidate longevity genes and so-called ‘vitagens’ affect longevity by impacting the health of the proteome. The major pathways of candidate longevity genes discussed here, meant only as examples, are all intimately connected with protein homeostasis: the mTOR pathway—though protein synthesis and activation of phosphorylation cascades; sirtuins—through changes in gene expression and protein modifications; ILS—through control of chaperone and anti-oxidant systems. These effects on the proteome lead to changes in the function and regulation of tissue and whole body metabolism. Even in the few major pathways used here as examples, aging is accompanied by reduced quantity of important components, or more importantly—reduced activity. These changes invariably degrade the ability of the protein networks to respond to stress, and even to normal life’s activities, and consequently, to restore the homeostasis. Understanding the impact of these and other pathways on the proteome is already beginning to yield chemical genetics approaches to improve protein homeostasis during aging.
A theme that emerges from multiple longevity pathways and several organisms is that the source of signals that matter for longevity is often neuroendocrine, either specific head neurons in the worm or nuclei in the mammalian hypothalamus. This feature is shared between the sirtuin and ILS pathways, stress responses including HSR and UPR, and perhaps many other longevity genes.
Another non-surprising theme to many longevity genes is the cross-talk between their action in one longevity pathway and other pathways. The intercalation of the longevity pathways is simply a mechanistic reiteration of the complexity of the aging process. As a consequence of the extensive cross-talk, another expected theme is that “there is no free lunch”. Almost any extension to lifespan or improvement of healthspan through manipulation of proteostasis genes comes at a cost of causing some morbidity. These considerations highlight the need for multiple specific readouts of manipulating longevity genes, beyond the simple measure of lifespan.
References
Kenyon CJ (2010) The genetics of ageing. Nature 464(7288):504–512
Lopez-Otin C et al (2013) The hallmarks of aging. Cell 153(6):1194–1217
Sahin E, Depinho RA (2010) Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature 464(7288):520–528
Aubert G, Lansdorp PM (2008) Telomeres and aging. Physiol Rev 88(2):557–579
Soerensen M (2012) Genetic variation and human longevity. Dan Med J 59(5):B4454
Murabito JM, Yuan R, Lunetta KL (2012) The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J Gerontol A Biol Sci Med Sci 67(5):470–479
Suri S et al (2013) The forgotten APOE allele: a review of the evidence and suggested mechanisms for the protective effect of APOE varepsilon2. Neurosci Biobehav Rev 37(10 Pt 2):2878–2886
Kulminski AM et al (2013) The role of lipid-related genes, aging-related processes, and environment in healthspan. Aging Cell 12(2):237–246
Baraibar MA, Ladouce R, Friguet B (2013) Proteomic quantification and identification of carbonylated proteins upon oxidative stress and during cellular aging. J Proteomics 92:63–70
Cabiscol E, Tamarit J, Ros J (2014) Protein carbonylation: proteomics, specificity and relevance to aging. Mass Spectrom Rev 33(1):21–48
Perluigi M, Swomley AM, Butterfield DA (2013) Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res Rev 13:75–89
Salmon AB et al (2009) The long lifespan of two bat species is correlated with resistance to protein oxidation and enhanced protein homeostasis. FASEB J 23(7):2317–2326
Beekman M et al (2013) Genome-wide linkage analysis for human longevity: genetics of healthy aging study. Aging Cell 12(2):184–193
Schachter F et al (1994) Genetic associations with human longevity at the APOE and ACE loci. Nat Genet 6(1):29–32
Breslow JL et al (1982) Studies of familial type III hyperlipoproteinemia using as a genetic marker the apoE phenotype E2/2. J Lipid Res 23(8):1224–1235
Song Y, Stampfer MJ, Liu S (2004) Meta-analysis: apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med 1411(2):137–147
Khan TA et al (2013) Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta-analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. Int J Epidemiol 42(2):475–492
Morris JC et al (2010) APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67(1):122–31
Castellano JM et al (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3(89):89ra57
Hashimoto T et al (2012) Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide. J Neurosci 32(43):15181–15192
Chen Y et al (2010) ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 107(26):12011–12016
Liu CC et al (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9(2):106–118
Jochemsen HM et al (2012) APOE epsilon4 differentially influences change in memory performance depending on age. The SMART-MR study. Neurobiol Aging 33(4):832 e15–22
Bloss CS et al (2008) Decreased cognition in children with risk factors for Alzheimer’s disease. Biol Psychiatry 64(10):904–906
Tuminello ER, Han SD (2011) The apolipoprotein e antagonistic pleiotropy hypothesis: review and recommendations. Int J Alzheimer’s Dis 2011:726197
Balch WE et al (2008) Adapting proteostasis for disease intervention. Science 319(5865):916–919
Gidalevitz T, Stevens F, Argon Y (2013) Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta 1833(11):2410–2424
Koubova J, Guarente L (2003) How does calorie restriction work? Genes Dev 17(3):313–321
Cornelius C et al (2013) Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: mitochondria as a “chi”. Immun Ageing 10(1):15
Moreau KL, King JA (2012) Protein misfolding and aggregation in cataract disease and prospects for prevention. Trends Mol Med 18(5):273–282
Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10(12):524–530
Gidalevitz T et al (2006) Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311(5766):1471–1474
Ben-Zvi A, Miller EA, Morimoto RI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A 106(35):14914–14919
Cohen E et al (2009) Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 139(6):1157–1169
David DC et al (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8(8):e1000450
Alavez S et al (2011) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472(7342):226–2269
Alavez S, Lithgow GJ (2012) Pharmacological maintenance of protein homeostasis could postpone age-related disease. Aging Cell 11(2):187–191
El-Ami T et al (2014) A novel inhibitor of the insulin/IGF signaling pathway protects from age-onset, neurodegeneration-linked proteotoxicity. Aging Cell 13, 165–174
Gidalevitz T et al (2013) Natural genetic variation determines susceptibility to aggregation or toxicity in a C. elegans model for polyglutamine disease. BMC Biol 11:100
Harrison DE et al (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253):392–395
Shore DE, Ruvkun G (2013) A cytoprotective perspective on longevity regulation. Trends Cell Biol 23(9):409–420
Perez VI et al (2009) Is the oxidative stress theory of aging dead? Biochim Biophys Acta 1790(10):1005–1014
Van Raamsdonk JM, Hekimi S (2012) Superoxide dismutase is dispensable for normal animal lifespan. Proc Natl Acad Sci U S A 109(15):5785–5790
Kikis EA, Gidalevitz T, Morimoto RI (2010) Protein homeostasis in models of aging and age-related conformational disease. Adv Exp Med Biol 694:138–159
Morimoto RI (2008) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22(11):1427–1438
Heydari AR et al (1994) Hsp70 and aging. Experientia 50(11–12):1092–1098
Singh R et al (2006) Reduced heat shock response in human mononuclear cells during aging and its association with polymorphisms in HSP70 genes. Cell Stress Chaperones 11(3):208–215
Liu AY et al (1989) Heat shock induction of HSP 89 is regulated in cellular aging. Biochem Biophys Res Commun 162(3):1302–1310
Shamovsky I, Gershon D (2004) Novel regulatory factors of HSF-1 activation: facts and perspectives regarding their involvement in the age-associated attenuation of the heat shock response. Mech Ageing Dev 125(10–11):767–775
Shemesh N, Shai N, Ben-Zvi A (2013) Germline stem cell arrest inhibits the collapse of somatic proteostasis early in Caenorhabditis elegans adulthood. Aging Cell 12(5):814–822
Swindell WR et al (2009) Endocrine regulation of heat shock protein mRNA levels in long-lived dwarf mice. Mech Ageing Dev 130(6):393–400
Elefant F, Palter KB (1999) Tissue-specific expression of dominant negative mutant Drosophila HSC70 causes developmental defects and lethality. Mol Biol Cell 10(7):2101–2117
Feder JH et al (1992) The consequences of expressing hsp70 in Drosophila cells at normal temperatures. Genes Dev 6(8):1402–1413
Nylandsted J, Brand K, Jaattela M (2000) Heat shock protein 70 is required for the survival of cancer cells. Ann N Y Acad Sci 926:122–125
Whitesell L et al (1994) Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A 91(18):8324–8328
Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer Nat Rev Cancer 5(10):761–72
Taylor RC, Dillin A (2013) XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell 153(7):1435–1447
Yamagishi N, Saito Y, Hatayama T (2008) Mammalian 105 kDa heat shock family proteins suppress hydrogen peroxide-induced apoptosis through a p38 MAPK-dependent mitochondrial pathway in HeLa cells. FEBS J 275(18):4558–4570
Swindell WR (2008) Comparative analysis of microarray data identifies common responses to caloric restriction among mouse tissues. Mech Ageing Dev 129(3):138–153
Shpund S, Gershon D (1997) Alterations in the chaperone activity of HSP70 in aging organisms. Arch Gerontol Geriatr 24(2):125–131
Erickson RR, Dunning LM, Holtzman JL (2006) The effect of aging on the chaperone concentrations in the hepatic, endoplasmic reticulum of male rats: the possible role of protein misfolding due to the loss of chaperones in the decline in physiological function seen with age. J Gerontol A Biol Sci Med Sci 61(5):435–443
Rutkowski DT et al (2007) The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol Biol Cell 18(9):3681–3691
Ladiges WC et al (2005) Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54(4):1074–1081
Hetz CA, Soto C (2006) Emerging roles of the unfolded protein response signaling in physiology and disease. Curr Mol Med 6(1):1
Pastore N et al (2013) Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med 5(3):397–412
Hetz C et al (2008) Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc Natl Acad Sci U S A 105(2):757–762
Hetz C et al (2009) XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev 23(19):2294–2306
Tomaru U et al (2012) Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol 180(3):963–972
Yun C et al (2008) Proteasomal adaptation to environmental stress links resistance to proteotoxicity with longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A 105(19):7094–7099
Hipp MS et al (2012) Indirect inhibition of 26 S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol 196(5):573–587
Tsakiri EN et al (2013) Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 12(5):802–813
Davis DP et al (2000) Inhibition of amyloid fiber assembly by both BiP and its target peptide. Immunity 13(4):433–442
Cuervo AM, Wong E (2014) Chaperone-mediated autophagy: roles in disease and aging. Cell Res 24(1):92–104
Toth ML et al (2008) Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4(3):330–338
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143(7):1883–1898
Wang Y et al (2009) Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 23(2):451–463
Zaarur N et al (2008) Triggering aggresome formation. Dissecting aggresome-targeting and aggregation signals in synphilin 1. J Biol Chem 283(41):27575–27584
Tsakiri EN et al (2013) Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J 27(6):2407–2420
Curran SP et al (2009) A soma-to-germline transformation in long-lived Caenorhabditis elegans mutants. Nature 459(7250):1079–1084
Karunanithi S et al (1999) Neuroprotection at Drosophila synapses conferred by prior heat shock. J Neurosci 19(11):4360–4369
Kaiser J (2003) Hormesis. Sipping from a poisoned chalice. Science 302(5644):376–379
Johnson TE et al (2000) Gerontogenes mediate health and longevity in nematodes through increasing resistance to environmental toxins and stressors. Exp Gerontol 35(6–7):687–94
Rea SL et al (2005) A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet 37(8):894–898
Wu D et al (2006) Visualizing hidden heterogeneity in isogenic populations of C. elegans. Exp Gerontol 41(3):261–270
Rea SL, Ventura N, Johnson TE (2007) Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol 5(10):e259
Cypser JR, Johnson TE (2002) Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. J Gerontol A Biol Sci Med Sci 57(3):B109–114
Fouillet A et al (2012) ER stress inhibits neuronal death by promoting autophagy. Autophagy 8(6):915–926
Johnson TE, Tedesco PM, Lithgow GJ (1993) Comparing mutants, selective breeding, and transgenics in the dissection of aging processes of Caenorhabditis elegans. Genetica 91(1–3):65–77
Friedman DB, Johnson TE (1988) A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118(1):75–86
Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382(6591):536–539
Kenyon C et al (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366(6454):461–464
Kimura KD et al (1997) daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277(5328):942–946
Morley JF et al (2002) The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99(16):10417–10422
Cohen E et al (2006) Opposing activities protect against age-onset proteotoxicity. Science 313(5793):1604–1610
Hsu AL, Murphy CT, Kenyon C (2003) Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300(5622):1142–1145
Morley JF, Morimoto RI (2004) Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell 15(2):657–664
Holzenberger M et al (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421(6919):182–187
Cohen E, Dillin A (2008) The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci 9(10):759–767
Suh Y et al (2008) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A 105(9):3438–3442
Tan Q et al (2013) A novel permutation test for case-only analysis identifies epistatic effects on human longevity in the FOXO gene family. Aging Cell 12(4):690–694
Anselmi CV et al (2009) Association of the FOXO3A locus with extreme longevity in a southern Italian centenarian study. Rejuvenation Res 12(2):95–104
Willcox BJ et al (2008) FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A 105(37):13987–13992
Flachsbart F et al (2009) Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci U S A 106(8):2700–2705
Laron Z (2008) The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones (Athens) 7(1):24–27
Deelen J et al (2013) Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age (Dordr) 35(1):235–249
Holzenberger M et al (2000) A targeted partial invalidation of the insulin-like growth factor I receptor gene in mice causes a postnatal growth deficit. Endocrinology 141(7):2557–2566
Abuzzahab MJ et al (2003) IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med 349(23):2211–2222
Xu J et al (2014) Longevity effect of IGF-1R(+/–) mutation depends on genetic background-specific receptor activation. Aging Cell 13(1):19–28
Sonntag WE et al (2013) Insulin-like growth factor-1 in CNS and cerebrovascular aging. Front Aging Neurosci 5:27
Gong Z et al (2014) Reductions in serum IGF-1 during aging impair health span. Aging Cell 13, 408–418
Yang J, Anzo M, Cohen P (2005) Control of aging and longevity by IGF-I signaling. Exp Gerontol 40(11):867–872
Boccitto M, Lamitina T, Kalb RG (2012) Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS One 7(3):e33494
Nagano I et al (2005) Beneficial effects of intrathecal IGF-1 administration in patients with amyotrophic lateral sclerosis. Neurol Res 27(7):768–772
Kwon ES et al (2010) A new DAF-16 isoform regulates longevity. Nature 466(7305):498–502
Chiang WC et al (2012) HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148(1–2):322–334
Halaschek-Wiener J et al (2005) Analysis of long-lived C. elegans daf-2 mutants using serial analysis of gene expression. Genome Res 15(5):603–615
Singh V, Aballay A (2006) Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci U S A 103(35):13092–13097
Walker GA, Lithgow GJ (2003) Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell 2(2):131–139
Lamitina ST, Strange K (2005) Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol 288(2):C467–474
Ikeno Y et al (2003) Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: correlation to extended longevity. J Gerontol A Biol Sci Med Sci 58(4):291–296
Altomare K et al (2003) The allele (A)(− 110) in the promoter region of the HSP70-1 gene is unfavorable to longevity in women. Biogerontology 4(4):215–220
Fujikake N et al (2008) Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem 283(38):26188–26197
Steele AD et al (2008) Heat shock factor 1 regulates lifespan as distinct from disease onset in prion disease. Proc Natl Acad Sci U S A 105(36):13626–13631
Dupont J, Holzenberger M (2003) Biology of insulin-like growth factors in development. Birth Defects Res Part C Embryo Today 69(4):257–271
Murphy CT et al (2003) Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424(6946):277–283
Apfeld J, Kenyon C (1998) Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell 95(2):199–210
Wolkow CA et al (2000) Regulation of C. elegans life-span by insulinlike signaling in the nervous system. Science 290(5489):147–150
Taguchi A, Wartschow LM, White MF (2007) Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 317(5836):369–372
Selman C et al (2008) Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J 22(3):807–818
Kappeler L et al (2008) Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol 6(10):e254
Novosyadlyy R et al (2008) Insulin-like growth factor-I protects cells from ER stress-induced apoptosis via enhancement of the adaptive capacity of endoplasmic reticulum. Cell Death Differ 15(8):1304–1317
Pfaffenbach KT et al (2012) GRP78/BiP is a novel downstream target of IGF-1 receptor mediated signaling. J Cell Physiol 227(12):3803–3811
Wanderling S et al (2007) GRP94 is essential for mesoderm induction and muscle development because it regulates IGF secretion. Mol Biol Cell 18(10):3764–3775
Ostrovsky O, Ahmed NT, Argon Y (2009) The chaperone activity of GRP94 toward insulin-like growth factor II is necessary for the stress response to serum deprivation. Mol Biol Cell 20(6):1855–1864
Barton E et al (2012) Deletion of muscle GRP94 impairs both muscle and body growth by inhibiting local IGF production. FASEB J 26(9):3691–3702
Owusu-Ansah E, Song W, Perrimon N (2013) Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155(3):699–712
Maier B et al (2004) Modulation of mammalian life span by the short isoform of p53. Genes Dev 18(3):306–319
Tyner SD et al (2002) p53 mutant mice that display early ageing-associated phenotypes. Nature 415(6867):45–53
Bauer JH et al (2009) dSir2 and Dmp53 interact to mediate aspects of CR-dependent lifespan extension in D. melanogaster. Aging (Albany NY) 1(1):38–48
Bauer JH et al (2007) Expression of dominant-negative Dmp53 in the adult fly brain inhibits insulin signaling. Proc Natl Acad Sci U S A 104(33):13355–13360
Salih DA, Brunet A (2008) FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 20(2):126–136
Haigis MC, Sinclair DA (2010) Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5:253–295
Blander G, Guarente L (2004) The Sir2 family of protein deacetylases. Annu Rev Biochem 73:417–435
Timmers S et al (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14(5):612–622
Herranz D et al (2010) Sirt1 improves healthy ageing and protects from metabolic syndrome-associated cancer. Nat Commun 1:3
Burnett C et al (2011) Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477(7365):482–485
Satoh A et al (2010) SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J Neurosci 30(30):10220–10232
Satoh A et al (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18(3):416–430
Howitz KT et al (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425(6954):191–196
Lakshminarasimhan M et al (2013) Sirt1 activation by resveratrol is substrate sequence-selective. Aging (Albany NY) 5(3):151–154
Hubbard BP et al (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339(6124):1216–1219
Suuronen T et al (2008) Regulation of ER alpha signaling pathway in neuronal HN10 cells: role of protein acetylation and Hsp90. Neurochem Res 33(9):1768–1775
Rogina B, Helfand SL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101(45):15998–16003
Schmeisser K et al (2013) Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat Chem Biol 9(11):693–700
Mouchiroud L et al (2013) The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154(2):430–441
Westerheide SD et al (2009) Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323(5917):1063–1066
Someya S et al (2010) Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143(5):802–812
Jing E et al (2013) Sirt3 regulates metabolic flexibility of skeletal muscle through reversible enzymatic deacetylation. Diabetes 62(10):3404–3417
Vellai T et al (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426(6967):620
Lamming DW et al (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335(6076):1638–1643
Selman C et al (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326(5949):140–144
Miller RA et al (2011) Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 66(2):191–201
Lamming DW et al (2013) Young and old genetically heterogeneous HET3 mice on a rapamycin diet are glucose intolerant but insulin sensitive. Aging Cell 12(4):712–718
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293
Hsu PP et al (2011) The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 332(6035):1317–1322
Zinzalla V et al (2011) Activation of mTORC2 by association with the ribosome. Cell 144(5):757–768
Robida-Stubbs S et al (2012) TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab 15(5):713–724
Soukas AA et al (2009) Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 23(4):496–511
Bjedov I et al (2010) Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11(1):35–46
Passtoors WM et al (2013) Gene expression analysis of mTOR pathway: association with human longevity. Aging Cell 12(1):24–31
Leontieva OV et al (2013) Dysregulation of the mTOR pathway in p53-deficient mice. Cancer Biol Ther 14(12):1182–1188
Miller RA et al (2014) Rapamycin-mediated lifespan increase in mice is dose and sex-dependent and appears metabolically distinct from dietary restriction. Aging Cell 13, 468–477
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Argon, Y., Gidalevitz, T. (2015). Candidate Genes That Affect Aging Through Protein Homeostasis. In: Atzmon, PhD, G. (eds) Longevity Genes. Advances in Experimental Medicine and Biology, vol 847. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2404-2_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2404-2_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2403-5
Online ISBN: 978-1-4939-2404-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)