
Abstract
Despite being one of the leading causes of maternal death and a major contributor of maternal and perinatal morbidity, the mechanisms responsible for the pathogenesis of preeclampsia (PE) have yet to be fully elucidated. PE affects the vasculature of many target maternal organs including the kidney, liver, and brain. Growing evidence supports the concept that the placenta plays a central role in the pathogenesis of PE and that reduced uteroplacental perfusion which develops as a result of abnormal cytotrophoblast invasion of spiral arterioles triggers the cascade of events leading to the maternal disorder. The hypoxic placenta in turn releases a variety of soluble factors which that generate widespread dysfunction of the maternal vascular endothelium. This dysfunction manifests as enhanced formation of factors such as endothelin, reactive oxygen species, and augmented vascular sensitivity to angiotensin II. In addition, PE is also associated with decreased formation of vasodilators such as nitric oxide and prostacyclin. The full elucidation of the molecular and cellular mechanisms involved in various stages of the disease process will hopefully lead to a more complete understanding of the etiology of PE and eventually lead to successful therapeutic intervention through the targeted disruption of novel pathways.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Preeclampsia (PE) is considered a pregnancy-specific syndrome that is diagnosed when new-onset hypertension and proteinuria occur after 20 weeks of gestation [1–3]. PE can progress rapidly to more severe complications such as seizures (eclampsia) and hemolysis, elevated liver enzymes, low platelet count (HELLP) syndrome, which can lead to cerebral hemorrhage, organ failure, and death [1–3]. PE is estimated to affect 5–7 % of all pregnancies. Despite being one of the leading causes of maternal death and a major contributor of maternal and perinatal morbidity, the mechanisms responsible for the pathogenesis of PE have not been fully elucidated. Hypertension associated with PE develops during pregnancy and remits after delivery, implicating the placenta as a central culprit in the pathogenic process [1–3]. PE affects the vasculature of many target organs including the brain, liver, and kidney [1–3]. Indeed, glomerular endotheliosis is considered an important characteristic lesion of women with PE [1–3].
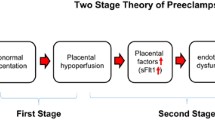
Although numerous factors including genetic, behavioral, and environmental factors have been implicated in the pathogenesis of PE, an important initiating event for the development of PE is thought to be placental ischemia/hypoxia ([1–3]; see Fig. 13.1). The hypoxic placenta, in turn, releases a variety of soluble factors that have profound effects on the peripheral vasculature and arterial pressure regulation. These factors include a host of molecules such as the soluble vascular endothelial growth factor (VEGF) receptor-1 (sFlt-1) , the angiotensin II type 1 receptor autoantibody (AT1-AA), and inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), which in turn generate widespread dysfunction of the maternal vascular endothelium. This dysfunction manifests as enhanced formation of factors such as endothelin , reactive oxygen species (ROS), and augmented vascular sensitivity to angiotensin II. In addition, PE is also associated with decreased formation of vasodilators such as nitric oxide (NO) . These alterations in vascular function not only lead to hypertension but multiorgan dysfunction including the brain, kidneys, and liver as well. Since PE remains to be one of the leading causes of maternal death and perinatal morbidity, identifying the mechanisms underlying abnormal placentation and the factors that link placental hypoxia and maternal cardiovascular and renal abnormalities remain important areas of investigation.

Hypothetical scheme depicting how abnormal cytotrophoblast invasion and subsequent reductions in spiral artery remodeling results in endothelial dysfunction and hypertension in PE
Abnormal Spiral Artery Remodeling and Placental Hypoxia in PE
During normal pregnancy, fetally derived cytotrophoblasts migrate to the uterine tissue in an orchestrated manner to invade and remodel the maternal uterine spiral arteries to ensure adequate oxygen and nutrient delivery to the developing uteroplacental unit [1–5]. This complex migration and invasion process results in the conversion of the high-resistance, small-diameter spiral arteries into high-capacitance, low-resistance vessels [4, 5]. It is believed that poor cytotrophoblast migration and/or vascular invasion during PE, leads to abnormal spiral artery remodeling and inadequate oxygen delivery to the developing uteroplacental unit [4, 5].
While the exact mechanisms responsible for the abnormal placental trophoblast migration/invasion and vascular remodeling in PE are unclear, results from a recent study by Hunkapiller et al. found that the absence of Notch2 in mice is associated with reduced vessel diameter and placental perfusion [6]. Additional findings that perivascular and endovascular cytotrophoblast often fail to express the Notch ligand, JAG1, in PE provides further evidence that defects in Notch signaling may be important in the pathogenesis of this pregnancy syndrome . Another recently described molecular pathway implicated in placental vascular development is the transcription factor storkhead box 1 (STOX1), a member of the winged helix transcription factor family [7]. Transgenic overexpression of STOX1 in the mouse leads to a phenotype that mimics PE in several key ways, most notably an increase in systolic blood pressure during gestation and elevated maternal circulating levels of soluble fms-like tyrosine kinase (sFlt)-1 and soluble endoglin [7]. While data from these recent studies are intriguing, much work remains to be done to elucidate the role of factors in mediating abnormal spiral artery remodeling in PE.
Factors Linking Placental Ischemia/Hypoxia with the Maternal Hypertension
Angiogenic Factors
One of the most intensely studied pathways in the pathophysiology of PE is that related to VEGF signaling [1–3, 8–11]. VEGF and the placental growth factor (PlGF-1) are also critically important in the maintenance of proper endothelial cell function in adult animals [8–11]. The VEGF signaling pathway came to prominence with the discovery of elevated circulating and placental levels of the soluble form of the VEGF receptor, fms-related tyrosine kinases-1 (sFlt-1) in preeclamptic women, especially in late gestation [8–11]. sFlt-1 is a circulating soluble receptor for both VEGF and PlGF, which when increased in maternal plasma leads to less circulating free-VEGF and free-PlGF, thus preventing their availability to maintain maternal endothelial integrity. In the kidney, this inactivation of free VEGF is believed to cause endotheliosis and proteinuria [12]. Subsequent studies of the regulation of sFlt-1 in cell culture and placental tissue in vitro have demonstrated that sFlt-1 is released from placental villi and trophoblast cells in response to reduced oxygen tensions similar to that seen in an ischemic placenta [1–3]. While sFlt-1 production appears to be regulated by hypoxia inducible factor-1, other factors such as TNF and the agonistic autoantibody to the AT1-AA also appear to be involved [1–3, 13] .
Several lines of evidence support a role for angiogenic factors in the pathogenesis of hypertension during PE. Several clinical studies have reported that sFlt-1 levels are strongly correlated with the severity of the PE [8–11]. In addition, chronic intravenous administration or adenovirus delivery of sFlt-1 to pregnant rats, to mimic plasma concentrations of sFlt-1 observed in preeclamptic women, decreases free VEGF and PlGF and produces hypertension and proteinuria [9, 14]. Moreover, a promising pilot study recently demonstrated that sFlt-1 could be removed from the maternal circulation of preeclamptic women by apheresis safely, and that this therapy reduced both blood pressure and proteinuria, with a trend toward increased gestational duration [15].
In addition to playing a pathogenic role in PE, angiogenic factors have been proposed as diagnostic markers for the syndrome. Several clinical studies were designed over the past decade to determine the potential of angiogenic factors as prediction tests in PE [1, 11]. While their accuracy fell short of sensitivities and likelihood ratios required for clinical use, prediction was much more reliable for early-onset PE. Ohkuchi et al. recently found that the sFlt-1 to PlGF ratio was a useful component for the prediction of PE when measured at 26–31 weeks of gestation [16]. Likewise, Perni et al. examined angiogenic factors in patients who had preexisting hypertension with superimposed PE, and found higher circulating levels of sFlt-1 prior to the 20th week of gestation in these patients versus pregnant women who had preexisting hypertension but did not develop PE [17]. These studies, along with other recent work, suggest that angiogenic balance could be a reliable marker of PE and allow detection prior to the onset of patient symptoms [11, 18]. Rana and colleagues recently suggested that angiogenic proteins alone account for the disease’s major phenotypes and therefore are extremely specific for both diagnosis and prognosis [11]. They also suggested that future screening studies should focus on prediction of the angiogenic form of PE rather than disease diagnosis based on nonspecific clinical criteria [11].
Immune Factors and Inflammation
The pathophysiology of PE is also thought to involve immune system abnormalities and inflammation [18–20]. Redman and colleagues proposed that fragments shed from the placental surface include pro-inflammatory proteins that may contribute to the systemic inflammatory response in normal pregnancy and the exaggerated inflammatory response in PE [19]. Supporting this concept are findings that proinflammatory cytokines, such as IL-6 and TNF-α, are elevated in preeclamptic women and placental ischemic rat models [18]. Moreover, infusion of proinflammatory cytokines into pregnant animals produces significant elevations in blood pressure [3, 18].
Maternal immune tolerance mechanisms are also implicated in the pathophysiology of PE. This maternal immune tolerance involves crucial interactions between regulatory CD4+ T cells and uterine natural killer cells recognizing and accepting the fetal antigens and facilitating placental growth. Partial failure of this interaction is thought to lead to poor placentation and dysfunctional placental perfusion and chronic immune activation originating from the placenta. Preeclamptic women have a decrease in circulating regulatory CD4+ T cells. Moreover, placental ischemic rats have a 47 % decrease in regulatory CD4+ T cells in the peripheral circulation when compared to normal pregnant rats [19]. T helper 17 cells, which are upregulated in a variety of autoimmune disorders, are also increased in preeclamptic women, and in placental ischemic rats [20]. While these data support the hypothesis that hypertension in response to placental ischemia represents a shift from the normal anti-inflammatory state of pregnancy to a pro-inflammatory state, the quantitative importance of CD4+ T cells and T helper 17 cells in the pathophysiology of PE remains to be determined [18].
A number of recent studies have also indicated that women with PE produce a novel agonistic autoantibody to the angiotensin II type I receptor [20–23]. Dechend and colleagues reported that sera from preeclamptic women contain an IgG (type 3) autoantibody that reacts with the AT1 receptor [22]. The binding of the AT1-AA to the seven amino acid stretch of the second extracellular loop of the angiotensin II type 1 receptor stimulates a chronotropic response from rat neonatal cardiomyocytes which can be attenuated with administration of an AT1 receptor antagonist. The is the basis of the bioassay primarily used for the detection of the autoantibody . These autoantibodies, isolated over a decade ago in preeclamptic women, have been studied more intensively recently, including their identification in the circulation of rats undergoing placental ischemia [3, 18, 24]. While infusion of the AT1-AA directly into pregnant animals results in moderate hypertension, the pathogenic importance of these antibodies remains to be fully elucidated, as their presence has been noted postpartum in a subset of preeclamptic patients even after the symptoms were resolved. Further studies are needed including determining how these unique antibodies are produced and how they interact with the other pathogenic agents in PE to produce the clinical phenotype .
Endothelin
There is growing evidence to suggest an important role for endothelin-1 (ET-1) in the pathophysiology of PE [25, 26]. Multiple studies have examined circulating levels of ET-1 in normal pregnant and preeclamptic cohorts, and found elevated levels of plasma ET-1 in the preeclamptic group, with some studies indicating that the level of circulating ET-1 correlates with the severity of the disease symptoms, though this is not a universal finding [25]. ET-1, however, is produced locally and plasma levels typically do not reflect tissue levels of the peptide. Animal studies have shown that a myriad of experimental models of PE (placental ischemia, sFlt-1 infusion, TNF-α infusion, and AT1-AA infusion) are associated with elevated tissue levels of ET-1 [2, 3, 25, 26]. A recent report also indicated increased vascular contractility to big ET-1 in the reduced uteroplacental perfusion pressure rat model of PE, an effect that was attributed to a greater contribution of matrix metalloproteinases to cleave bET-1 to ET-1 [27]. Finally, the fact that hypertension in pregnant rats, induced by placental ischemia or chronic infusion of sFlt-1, TNF-α, or AT1-AA [25, 26] can be completely attenuated by ETA receptor antagonism, strongly suggests that ET-1 is a final common pathway linking factors produced during placental ischemia to elevations in maternal blood pressure.
Nitric Oxide
Studies have suggested important roles for NO as a regulator of arterial pressure under various physiological and pathophysiological conditions [28–30]. NO is synthesized endogenously from L-arginine, oxygen, and nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) by various NO synthase (NOS) enzymes. NO production is elevated in normal pregnancy and these increments appear to play an important role in the vasodilatation that occurs in healthy pregnancy [28–30]. Thus, it was postulated that NO deficiency during PE might be involved in the disease process. Whether there is a reduction in NO production during PE is controversial. Much of the uncertainty originates from the difficulty in directly assessing the activity of the NO system in a clinical setting. Assessment of whole body NO production via measurement of 24-hour nitrate/nitrite excretion has yielded variable results, likely due to difficulties in controlling for factors such as nitrate intake and excretion [2–3]. Thus, the relative importance of NO deficiency in the pathogenesis of PE has yet to be fully elucidated.
In support of a role for NO deficiency in the pathogenesis of PE are reports from several laboratories that chronic NOS inhibition in pregnant rats produces hypertension associated with peripheral and renal vasoconstriction, proteinuria, intrauterine growth restriction, and increased fetal morbidity, a pattern resembling the findings of PE [28–30]. Placental ischemia has been reported to result in endothelial dysfunction and reduced NO production in some but not all vascular beds [29]. Moreover, L-arginine supplementation in animal models and in women with PE reduces blood pressure and improves pregnancy outcomes in some but not all studies [29]. Finally, hypertension induced by sFlt-1 in pregnant animal models is associated with significant reductions in NO synthesis [14].
Endoplasmic Reticulum and Oxidative Stress
Endoplasmic reticulum stress activates a number of signaling pathways aimed at restoring homeostasis. Burton and colleagues proposed that this mechanism to restore homeostasis fails and apoptotic pathways are activated to alter placental function in women who develop PE [31]. In addition, chronic, low levels of endoplasmic reticulum stress during the second and third trimesters may result in a growth-restricted phenotype. They also propose that higher levels of endoplasmic reticulum stress lead to activation of pro-inflammatory pathways that may contribute to maternal endothelial cell activation [31]. While endoplasmic reticulum stress is known to occur in PE, the importance of this abnormality in the pathophysiology has yet to be fully elucidated.
Oxidative stress has also been implicated in PE, as increased concentration of several oxidative stress markers have been reported systemically in preeclamptic women, among these peroxynitrite [32, 33]. Peroxynitrite concentrations in vascular endothelium were much higher in preeclamptic women versus normal gestation, concurrent with decreased levels of superoxide disumutase (SOD) and NOS [32, 33]. There is also evidence of increased oxidative stress during gestation in the placental ischemic rat hypertensive model, suggesting a link between placental ischemia/hypoxia and the production of reactive oxygen species [2, 3]. For example, the SOD-mimetic drug tempol, led to significant attenuation of the hypertensive response [2, 3]. In a related study, administration of the NADPH oxidase inhibitor apocynin also significantly attenuated placental ischemia-induced gestational hypertension, implicating the enzyme as an important source of pathogenic ROS in the reduced uterine perfusion pressure (RUPP) animal [2, 3]. Failure of the drug to fully normalize blood pressure, however, leaves open the possibility that alternative ROS production pathways are at work in the RUPP model. Further studies into the mechanism of ROS production in animal models of PE should help shed light into the importance of oxidative stress in the pathophysiology of PE and perhaps allow the identification of useful antioxidant strategies. It remains to be seen whether ROS production is a primary or secondary cause of PE pathophysiology, and how effective manipulation of the system will be in the search for effective therapies.
The Kidney and PE
PE is associated with decreases in renal blood flow and glomerular filtration rate and increases in protein excretion [1–3]. While the kidneys are an important target organ in PE, the pathophysiological mechanisms underlying the reduction in renal hemodynamics and proteinuria in PE has yet to be fully elucidated. VEGF is important to maintain endothelial cell function especially for the fenestrated endothelium found in glomeruli of the kidney, the brain, and liver. VEGF and VEGF receptors are highly expressed in the kidney. VEGF is expressed in podocytes in the glomerulus, and VEGF receptors are present on endothelial, mesangial, and peritubular capillary cells [1–3, 12]. Signaling between endothelial cells and podocytes is thought to be important for maintenance of the filtration function of the glomerulus and inhibitors of VEGF signaling have been shown to result in alterations in glomerular structure and function (see Fig. 13.2). In addition, ablation of VEGF-A from endothelial cells results in progressive endothelial degeneration and sudden death of mutant animals [12].

Potential mechanisms whereby placental ischemia leads to changes in renal hemodynamics and proteinuria
Because the endothelium is a major target organ for the actions of VEGF , it is likely that decreases in the production of endothelium-derived relaxing factors such as NO and enhanced production of vasoconstricting factors such as endothelin play a role in mediating the changes in renal hemodynamics in PE. (see Fig. 13.2) In support of this concept are studies demonstrating that inhibition of VEGF signaling with sFlt-1 or antibodies is associated with significant reductions in the expression of endothelial and neuronal NOS in the kidney [34]. Another potential mechanism whereby VEGF blockade could reduce renal hemodynamics and increase blood pressure is by enhancing ET-1 synthesis. Indeed, renal, endothelin levels are increased in placental ischemic rats and in pregnant rats infused with sFlt-1 [14]. In addition, ETA receptor antagonist significantly attenuates the vascular responses to placental ischemia or chronic infusion of sFlt-1 in pregnant animals [14].
The Brain and PE
Cerebrovascular abnormalities play a significant role in the pathogenesis of PE/eclampsia [35, 36]. Neurological symptoms such as headaches, blurred vision, nausea, drowsiness, and seizures are commonly reported in preeclamptic patients [1, 35–38]. Furthermore, the risk of developing a stroke during pregnancy and the postpartum year is increased in women with PE/eclampsia [39]. Approximately 40 % of all PE/eclampsia deaths are due to cerebrovascular events with cerebral hemorrhage contributing to 35 %, cerebral edema 3 %, and cerebral embolus 1 % of PE-related deaths [35–38].
Magnetic resonance imaging and computed tomography scans of the brain reveal abnormalities consistent with edema in preeclamptic patients [35–38]. Edema forms either from increased water transport into cells (cytotoxic edema) or through the disruption of the blood–brain barrier (BBB; vasogenic edema) [39]. The BBB, formed by the close association of endothelial cells, smooth muscle cells or pericytes (capillaries), and astrocytes, regulates the transport of substances between the blood and the brain tissue. Increased permeability of the cerebral vessels has been reported in both normal pregnancy and PE. For example, plasma from normal pregnant and even more so from preeclamptic women increases permeability of cerebral vessels in an ex vivo model [40]. These and other studies suggest that pregnancy itself induces changes in the cerebral vasculature, which may be exacerbated in the presence of increased arterial pressure, characteristic of PE.
Abnormalities in cerebral blood flow autoregulation may also contribute to cerebral dysfunction in PE (see Fig. 13.3). Cerebral blood flow is highly regulated and kept relatively constant even with fluctuations in blood pressure. Acute increases in blood pressure activate the vascular myogenic response, protecting neuronal tissue from damage. Women with severe PE have increased cerebral blood flow and perfusion pressure [41], and a recent study demonstrated impaired myogenic tone in the middle cerebral arteries and cerebral edema in placental ischemic rats [42]. Janzarik and colleagues recently reported that a history of previous PE is associated with poorer dynamic cerebral autoregulation values in subsequent pregnancies [43]. These conditions of increased cerebral blood flow and impaired myogenic reactivity render preeclamptic patients susceptible to neurological complications with acute increases in blood pressure. This concept is supported by studies demonstrating that during acute hypertension, pregnancy decreases vascular resistance and increases cerebral blood flow, resulting in a rightward shift in the autoregulatory curve, and cerebral edema.

Proposed mechanisms whereby placental ischemia leads to cerebrovascular abnormalities
Summary
Despite being one of the leading causes of maternal death and a major contributor of maternal and perinatal morbidity, the mechanisms responsible for the pathogenesis of PE have yet to be fully elucidated. PE affects the vasculature of many target maternal organs including the brain, liver, and kidney. Growing evidence supports the concept that the placenta plays a central role in the pathogenesis of PE and that reduced uteroplacental perfusion which develops as a result of abnormal cytotrophoblast invasion of spiral arterioles, triggers the cascade of events leading to the maternal disorder (see Fig. 13.1). The hypoxic placenta in turn releases a variety of soluble factors such as sFlt-1, AT1-AA, and inflammatory cytokines such as TNF-α which in turn generate widespread dysfunction of the maternal vascular endothelium. This dysfunction manifests as enhanced formation of factors such as endothelin, reactive oxygen species, and augmented vascular sensitivity to angiotensin II. In addition, PE is also associated with decreased formation of vasodilators such as NO . The full elucidation of the molecular and cellular mechanisms involved in various stages of the disease process will hopefully lead to a more complete understanding of the etiology of PE and eventually lead to successful therapeutic intervention through the targeted disruption of new and novel pathways.
References
Roberts JM, August PA, Bakris G, Barton JR, Bernstein IM, Druzin M, Gaiser RR, Granger JP, Jeyabalan A, Johnson DD, Karumanchi S, Lindheimer M, Owens MY, Saade GR, Sibai BM, Spong CY, Tsigas E, Joseph GF, O’Reilly N, Politzer A, Son S, Ngaiza K. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ task force on hypertension in pregnancy. Obstet Gynecol. 2013;122(5):1122–31.
Gilbert JS, Ryan MJ, Lamarca BB., Sedeek M, Murphy SR, Granger JP. Pathophysiology of hypertension during PE: linking placental ischemia with endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;294:H541–50.
Lamarca BD, Gilbert J, Granger JP. Recent progress toward the understanding of the pathophysiology of hypertension during PE. Hypertension. 2008;51:982–8.
Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of PE. Obstet Gynecol Ann. 1972;1:177–91.
Zhou Y, Fisher SJ, Janatpour M, Genbacev O, Dejana E, Wheelock M, Damsky CH. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J Clin Invest. 1997;99(9):2139–51.
Hunkapiller NM, Gasperowicz M, Kapidzic M, Plaks V, Maltepe E, Kitajewski J, Cross JC, Fisher SJ. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development. 2011;138:2987–98.
Doridot L, Passet B, Mehats C, Rigourd V, Barbaux S, Ducat A, Mondon F, Vilotte M, Castille J, Breuiller-Fouche M, Daniel N, Le Provost F, Bauchet AL, Baudrie V, Hertig A, Buffat C, Simeoni U, Germain G, Vilotte JL, Vaiman D. PE-like symptoms induced in mice by fetoplacental expression of STOX1 are reversed by aspirin treatment. Hypertension. 2013;61:662.
Levine RJ, Maynard SE, Qian C, et al. Circulating angiogenic factors and the risk of PE. N Engl J Med. 2004;350(7):672–83.
Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in PE. J Clin Invest. 2003;111(5):649–58.
Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of PE. N Engl J Med. 2004;350(7):672–83.
Rana S, Karumanchi SA, Lindheimer MD. Angiogenic factors in diagnosis, management, and research in PE. Hypertension. 2014;63(2):198–202.
Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111(5):707–16.
Zhou CC, Ahmad S, Mi T, Abbasi S, Xia L, Day MC, Ramin SM, Ahmed A, Kellems RE, Xia Y. Autoantibody from women with PE induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension. 2008;51:1010–9.
Murphy SR, Lamarca BB, CockrellL K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010;55:394–8.
Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, Schaarschmidt W, Jank A, Kribs A, Cornely OA, Kreyssig C, Hemphill L, Rigby AC, Khedkar S, Lindner TH, Mallmann P, Stepan H, Karumanchi SA, Benzing T. Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in PE. Circulation. 2011;124:940–50.
Ohkuchi A, Hirashima C, Matsubara S, Takahashi K, Matsuda Y, Suzuki M. Threshold of soluble fms-like tyrosine kinase 1/placental growth factor ratio for the imminent onset of PE. Hypertension. 2011;58(5):859–66.
Perni U, Sison C, Sharma V, Helseth G, Hawfield A, Suthanthiran M, et al. Angiogenic factors in superimposed PE: a longitudinal study of women with chronic hypertension during pregnancy. Hypertension. 2012;59(3):740–6.
Lamarca BD, Ryan MJ, Gilbert JS, Murphy SR, Granger JP. Inflammatory cytokines in the pathophysiology of hypertension during PE. Curr Hypertens Rep. 2007;9:480–5.
Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. 2010;63:534–43.
Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN, Jr, Lamarca B. CD4 + T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension. 2011;57:949–55.
Prins JR, Boelens HM, Heimweg J, Van Der Heide S, Dubois AE, Van Oosterhout AJ, Erwich JJ. PE is associated with lower percentages of regulatory T cells in maternal blood. Hypertens Pregnancy. 2009;28:300–11.
Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with PE develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–52.
Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–62.
LaMarca B, Wallace K, Herse F, Wallukat G, Martin JN, Weimer A, et al. Hypertension inresponse to placental ischemia during pregnancy: role of B lymphocytes. Hypertension. 2011;57(4):865–71.
George EM, Granger JP. Endothelin: key mediator of hypertension in PE. Am J Hypertens. 2011;24:964–9.
LaMarca B, Parrish M, Ray LF, et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin –1. Hypertension. 2009;54(4):905–9.
Abdalvand A, Morton JS, Bourque SL, Quon AL, Davidge ST. Matrix metalloproteinase enhances big-endothelin-1 constriction in mesenteric vessels of pregnant rats with reduced uterine blood flow. Hypertension. 2013;61:488–93.
Deng A, Engels K, Baylis C. Impact of nitric oxide deficiency on blood pressure and glomerular hemodynamic adaptations to pregnancy in the rat. Kidney Int. 1996;50(4):1132–8.
Alexander BT, Llinas MT, Kruckeberg WC, Granger JP. L-arginine attenuates hypertension in pregnant rats with reduced uterine perfusion pressure. Hypertension. 2004;43(4):832–6.
Conrad KP, Kerchner LJ, Mosher MD. Plasma and 24-h NO (x) and cGMP during normal pregnancy and PE in women on a reduced NO (x) diet. Am J Physiol. 1999;277(1 Part2):F48–57.
Burton GJ, Yung HW. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens. 2011;1:72–8.
Walsh SW. Maternal-placental interactions of oxidative stress and antioxidants in PE. Semin Reprod Endocrinol. 1998;16:93–104.
Roggensack AM, Zhang Y, Davidge ST. Evidence for peroxynitrite formation in the vasculature of women with PE. Hypertension. 1999;33:83–9.
Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54(3):652–8.
Demirtaş O, Gelal F, Vidinli BD, Demirtaş LO, Uluç E, Baloğlu A. Cranial MR imaging with clinical correlation in PE and eclampsia. Diagn Interv Radiol. 2005;11(4):189–94.
Manfredi M, Beltramello A, Bongiovanni LG, Polo A, Pistoia L, Rizzuto N. Eclamptic encephalopathy: imaging and pathogenetic considerations. Acta Neurol Scand. 1997;96(5):277–82.
Chakravarty A, Chakrabarti SD. The neurology of eclampsia: some observations. Neurol India. 2002;50(2):128–35.
Tang CH, Wu CS, Lee TH, Hung ST, Yang CY, Lee CH, et al. PE-eclampsia and the risk of stroke among peripartum in Taiwan. Stroke. 2009;40(4):1162–8.
Cipolla MJ, Sweet JG, Chan SL. Cerebral vascular adaptation to pregnancy and its role in the neurological complications of eclampsia. J Appl Physiol. 2011;110(2):329–39.
Amburgey OA, Chapman AC, May V, Bernstein IM, Cipolla MJ. Plasma from preeclamptic women increases blood-brain barrier permeability: role of vascular endothelial growth factor signaling. Hypertension. 2010;56(5):1003–8.
Choi SJ, Lee JE, Oh SY, Kim GM, Cho YS, Lee KH, et al. Maternal cerebral blood flow and glucose metabolism in pregnancies complicated by severe PE. Hypertens Pregnancy. 2012;31(1):177–88.
Ryan MJ, Gilbert EL, Glover PH, George EM, Masterson CW, McLemore GR, et al. Placental ischemia impairs middle cerebral artery myogenic responses in the pregnant rat. Hypertension. 2011;58(6):1126–31.
Janzarik WG, Ehlers E, Ehmann R, Gerds TA, Schork J, Mayer S, Gabriel B, Weiller C, Prömpeler H, Reinhard M. Dynamic cerebral autoregulation in pregnancy and the risk of PE. Hypertension. 2014;63:161–66.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Spradley, F., George, E., Palei, A., Warrington, J., Granger, J. (2015). Preeclampsia: Angiogenic Factors, Blood Pressure, and the Kidney. In: Weir, M., Lerma, E. (eds) Chronic Kidney Disease and Hypertension. Clinical Hypertension and Vascular Diseases. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-1982-6_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1982-6_13
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-1981-9
Online ISBN: 978-1-4939-1982-6
eBook Packages: MedicineMedicine (R0)