
Abstract
Once viewed as part of the “dark matter” of genome, long noncoding RNAs (lncRNAs), which are mRNA-like but lack open reading frames, have emerged as an integral part of the mammalian transcriptome. Recent work demonstrated that lncRNAs play multiple structural and functional roles, and their analysis has become a new frontier in biomedical research. In this chapter, we provide an overview of different lncRNA families, describe methodologies available to study lncRNA–protein and lncRNA–DNA interactions systematically, and use well-studied lncRNAs as examples to illustrate their functional importance during normal development and in disease states.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
In 2002, following high throughput sequencing of mouse cDNA libraries, Okazaki et al. revealed that a vast proportion of the mammalian transcriptome does not code for proteins and defined long noncoding RNAs (lncRNAs) as a significant transcript class (Okazaki et al. 2002). Ten years later, the ENCODE (Encyclopedia of DNA Elements) study reported the existence of over 9,640 lncRNA loci in the human genome, roughly half the number of protein-coding genes (Djebali et al. 2012). These studies have changed our view of the mammalian genome and highlighted the importance of understanding lncRNA function.
The biogenesis of lncRNAs is similar to that of mRNA in that many lncRNAs are transcribed by RNA polymerase II and have a 5′ cap and 3′ polyadenylation signal. Their size ranges widely from 200 bp to over 100 kb. lncRNAs can reside in either nuclear or cytoplasmic compartments and be soluble or chromatin-bound. Unlike small noncoding RNAs, such as microRNAs or piRNAs, lncRNAs function via diverse mechanisms and comprise different classes.
2 Systematic Discovery of Different lncRNA Classes
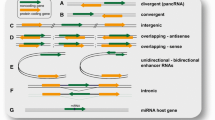
The completion of sequencing of the human and mouse genomes together with recent analyses of their transcriptional outputs have revealed that about 80 % of the mammalian genome is transcribed in a cell-specific manner, leading to a new understanding of transcriptional regulation, particularly of noncoding regions (Djebali et al. 2012; Dunham et al. 2012). Earlier studies exploiting the increased sensitivity of genome tiling arrays (Katayama et al. 2005; Kapranov et al. 2007; Preker et al. 2008), together with asymmetric strand-specific analysis of gene expression (ASSAGE) (He et al. 2008) and global run-on sequencing (GRO-seq) (Core et al. 2008), have revealed widespread antisense transcription (natural antisense transcripts, NATs) and promoter-associated transcripts (such as promoter-associated long RNAs, PALRs; promoter-upstream transcripts, PROMPTs) in mammalian cells (Fig. 4.1). More significant progress has been made in discovery of novel lncRNAs following improvement in RNA sequencing (RNA-seq) and application of integrated methodologies. Several studies have systematically identified lncRNAs in a variety of organisms by monitoring patterns of “K4-K36” chromatin modification (Khalil et al. 2009; Cabili et al. 2011; Ulitsky et al. 2011; Guttman and Rinn 2012). For example, it was determined that an intergenic transcript could be defined as a potential lncRNA if it exhibited histone 3 Lys 4 trimethylation (H3K4me3) in its promoter region followed by histone 3 Lys 36 trimethylation (H3K36me3) across its actively transcribed region. Other criteria have also been applied to evaluate whether transcripts are true lncRNAs, including the presence of a transcription initiation signal and polyA site, potential coding capacity, orthology features, and analysis of its expression pattern among tissue samples. Since many lncRNAs are transcribed from discrete loci in previously unannotated intergenic regions between protein-coding genes, they were termed large intergenic noncoding RNAs (lincRNAs) (Fig. 4.1). A survey of the entire mouse or human genome using these chromatin marks in numerous cell types or tissues revealed that 5,000–8,000 K4-K36 domains represented lincRNAs (Guttman and Rinn 2012; Cabili et al. 2011).

Pervasive transcription of various classes of lncRNA in mammalian genomes. PARLs promoter-associated long RNAs, NATs natural antisense transcripts, lincRNAs large intergenic noncoding RNAs, sno-lncRNAs snoRNA-related lncRNAs, circRNAs circular RNAs. See text for details
While a large proportion of the thousands of “K4-K36” lincRNAs identified by chromatin signature are polyadenylated (Cabili et al. 2011; Derrien et al. 2012), recent studies indicate that a number of Pol II-transcribed long noncoding RNAs are processed in alternative ways (Wilusz et al. 2008; Sunwoo et al. 2009; Burd et al. 2010; Yap et al. 2010; Hansen et al. 2011, 2013; Salzman et al. 2012; Yin et al. 2012; Jeck et al. 2013; Memczak et al. 2013). MALAT1 (also called NEAT2) and Menβ (also called NEAT1_2), which are both nuclear, are processed at their 3′ ends by RNase P (which processes the 5′ ends of tRNAs) (Wilusz et al. 2008; Sunwoo et al. 2009). RNase P cleavage leads to formation of the mature 3′ end of the lncRNA, which is protected by a highly conserved triple helical structures (Brown et al. 2012; Wilusz et al. 2012). Investigators using RNA-seq to define the population of non-polyadenylated “poly(A)-” transcripts in the human transcriptome report that both MALAT1 and Menβ and many previously unannotated intronic transcripts are enriched in poly(A)-transcriptomes (Yang et al. 2011a). Some of these intronic RNAs were further demonstrated to constitute a new family of lncRNAs derived from introns and capped by small nucleolar RNAs (snoRNAs) at both ends (sno-lncRNAs) (Yin et al. 2012) (Fig. 4.1). Additional non-polyadenylated lncRNAs from intronic regions were also found in various human cell lines (Derrien et al. 2012) and in Xenopus tropicalis (Gardner et al. 2012).
Circular RNAs (circRNAs) are a type of lncRNA that is protected from degradation by head-to-tail circularization. Several recent reports suggest that non-linearized RNAs are largely generated from back-spliced exons, in which splice junctions formed by an acceptor splice site at the 5′ end of an exon and a donor site at a downstream 3′ end (Burd et al. 2010; Yap et al. 2010; Hansen et al. 2011; Salzman et al. 2012). For example, the INK4a/ARF locus-associated lncRNA ANRIL participates directly in epigenetic transcriptional repression (Yap et al. 2010) (see Part VI for details). This locus also encodes heterogeneous species of RNA transcripts including a circular form of ANRIL (cANRIL) whose expression is correlated with INK4/ARF transcription (Burd et al. 2010). Moreover, sequencing of rRNA-depleted RNAs from human fibroblasts that had been digested with RNase R exonuclease identified numerous circRNAs containing non-colinear exons (Fig. 4.1). These RNAs are proposed to compete with endogenous RNAs in the cytoplasm (Jeck et al. 2013). Very recent studies demonstrate that many circRNAs form by back-spliced exons in animal cells (Memczak et al. 2013) and some of the most abundant ones function as efficient “sponges” to sequester microRNAs to regulate gene expression (Hansen et al. 2013; Memczak et al. 2013).
Pervasively transcribed lncRNAs exhibit several interesting features. First, although most are transcribed by RNA Pol II, many lncRNAs undergo maturation in ways different from mRNAs. Second, their expression is strikingly cell- or tissue-specific compared with coding genes and such RNAs are often coexpressed with neighboring genes (Cabili et al. 2011; Yin et al. 2012). Third, most show low primary sequence similarity with coding sequences (Cabili et al. 2011; Derrien et al. 2012; Zhu et al. 2013). Interestingly, however, loss- and gain-of-function studies of several lncRNAs in zebrafish demonstrate that they play key roles during embryonic development (Ulitsky et al. 2011), indicating the functional conservation despite their limited sequence conservation. Although detailed functions of lncRNAs are only beginning to be defined, several lines of evidence show that they participate in critical processes, such as X chromosome inactivation, genomic imprinting, maintenance of nuclear architecture, pluripotency, and developmental patterning (for reviews, see Wilusz et al. (2009), Chen and Carmichael (2010), Guttman and Rinn (2012), Rinn and Chang (2012)).
3 Novel High Throughput Approaches Enable Analysis of lncRNA–Protein Interactions
As a regulatory mechanism some lncRNAs partner with chromatin modifiers to either silence or activate genes epigenetically. A breakthrough in this field came from pioneering studies that identified the lncRNAs HOTAIR and Xist, which are transcribed from human HOX locus and mouse X-chromosome, respectively, and target chromatin repressor Polycomb proteins onto specific mammalian loci (Rinn et al. 2007; Zhao et al. 2008). Since then, lncRNAs from various genomic locations, including imprinted or cancer gene loci, have been shown to interact not only with chromatin repressors (Nagano et al. 2008; Pandey et al. 2008; Khalil et al. 2009; Yap et al. 2010; Zhao et al. 2010; Cabianca et al. 2012) and activators (Orom et al. 2010; Wang et al. 2011; Gomez et al. 2013; Lai et al. 2013) but with other types of proteins, including DNA methyltransferases (Schmitz et al. 2010), transcription factors (Yao et al. 2010; Jeon and Lee 2011), and splicing factors (Tripathi et al. 2010; Gong and Maquat 2011; Yin et al. 2012). Some lncRNAs, like HOTAIR and Xist, function to “guide” proteins to their targets (Rinn et al. 2007; Zhao et al. 2008). Others act as a scaffold to bridge protein complexes at specific genomic loci (Tsai et al. 2010), modify proteins allosterically to alter protein function (Shamovsky et al. 2006; Wang et al. 2008), or serve as a sponge to titrate away proteins in cells (Zhao et al. 2008; Tripathi et al. 2010; Yin et al. 2012).
Several technologies have been developed to allow unbiased identification of protein-interacting lncRNAs genome-wide by coupling RNA immunoprecipitation (RIP) with high throughput sequencing. Conceptually, RIP-seq is analogous to widely used technology ChIP-seq (chromatin IP coupled with high throughput sequencing), which was designed to identify transcription factor binding sites or histone modification patterns genome-wide. Both protocols rely on use of an antibody against the protein of interest to specifically pull-down either RNA or DNA associated with the protein. Major RIP-seq methods include native RIP-seq (nRIP-seq), cross-linked RIP-seq (CLIP-seq), photoactivatable-ribonucleoside-enhanced CLIP-seq (PAR-CLIP-seq), and individual-nucleotide resolution CLIP-seq (iCLIP-seq). The similarities and differences among these methods are enlisted in Table 4.1 and discussed below.
3.1 Native RNA Immunoprecipitation Coupled with High Throughput Sequencing (nRIP-seq)
Following our discovery that lncRNA Xist acts in cis to target Polycomb protein complex 2 (PRC2) onto X-chromosomes to establish the chromosome-wide heterochromatic mark trimethylated histone H3 residue lysine 27 (H3K27-3me) (Zhao et al. 2008), we predicted that other PRC2-interacting lncRNAs or mRNAs likely exist, as PRC2 occupies over 2,000 mammalian DNA loci through unknown mechanisms (Ku et al. 2008). To capture the PRC2 transcriptome in mouse embryonic stem cells (mESCs) we developed nRIP-seq (Zhao et al. 2010), a modification of previous RIP-ChIP strategies (Keene et al. 2006). Briefly, since PRC2 protein is primarily nuclear, an mESC nuclear extract is prepared, and then an antibody targeting Enhancer of zeste homolog 2 (Ezh2), the methyltransferase subunit of PRC2, was added for immunoprecipitation, followed by washing, RNA extraction, library construction, high throughput-sequencing and bioinformatics analysis. One advantage of the method is that it captures protein–RNA interaction in the native state since cross-linking reagents are generally not employed. Using nRIP-seq, we discovered over 9,000 Ezh2-interacting transcripts. Binding specificity was validated by generating a library under the same experimental conditions but made from an Ezh2-null cell line. Compared to the wild type library, we detected tenfold less RNA in the control library, suggesting that the Ezh2 transcriptome is highly enriched. Therefore, nRIP-seq is an excellent tool to study interactions between RNA binding proteins and their targets.
3.2 Cross-Linking and Immunoprecipitation (CLIP)-Seq
One limitation to nRIP-seq is that it cannot distinguish direct from indirect interactions. To do so requires application of techniques that utilize ultraviolet (UV) cross-linking of RNA to protein to precisely map protein binding sites in RNA. One of those methods, called cross-linking and immunoprecipitation (CLIP) (Ule et al. 2003), uses a short wave (254 nm) UV light to create a covalent bond between RNA and interacting proteins in living cells and allows stringent experimental manipulation in order to minimize capture of nonspecific lncRNAs. Following cross-linking, an antibody against a protein of interest is used to immunoprecipitate RNA, and then RNAses are used to digest unbound RNA fragments, leaving a 50–100 bp protein-interacting RNA fragment, which is then radiolabeled and then size-fractionated by SDS-PAGE. The cross-linked complex, usually slightly larger than the protein, is extracted and treated with proteinase K to remove RNA-bound protein. Recovered RNA is then ligated to adapters for reverse transcription and PCR amplification. Amplified cDNA libraries are then sequenced using multiple platforms, such as 454, Illumina, or SOLID, followed by bioinformatics data analysis.
CLIP and CLIP-seq have been used in diverse biological systems, including to identify splicing factor recognition sites, such as the RNA networks of splicing factor NOVA in mouse brain tissue (Ule et al. 2003; Licatalosi et al. 2008; Zhang et al. 2010), FOX2 binding sites in stem cells (Yeo et al. 2009), and SFRS1 interaction sites (Sanford et al. 2009) in human embryonic kidney cells. Multiple laboratories have also mapped mammalian microRNA–mRNA interaction sites through Argonaute CLIP-seq (Chi et al. 2009; Leung et al. 2011) [or Zisoulis et al. 2010 if including nematodes]. In addition, using this method, Guil et al. found nuclear protein hnRNPA1 binds a microRNA precursor and is required for microRNA-mediated repression (Guil and Caceres 2007), while Xu et al. showed that the germ cell-specific DNA/RNA-binding protein MSY2 binds small RNAs (Xu et al. 2009). These studies prove that this method is a powerful tool for identifying protein–RNA interaction in vivo. However, caveats include low cross-linking efficiency at short UV wavelengths (typically 1–5 % of RNA–protein complexes are cross-linked) and the inability to identify the precise nucleotide that binds to protein. Recently, a new method named PAR-CLIP-seq was developed based on CLIP-seq to provide solutions to these problems.
3.3 Photoactivatable-Ribonucleoside-Enhanced (PAR)-CLIP-Seq
The PAR-CLIP-seq method was developed in 2010 by Thomas Tuschl and colleagues (Hafner et al. 2010). Rather than cross-linking cells at 254 nm, Hafner et al. metabolically labeled cells with photoreactive nucleoside analogs, such as 4-thiouridine (4-SU) or 6-thioguanosine (6-SG), allowing more efficient cross-linking at 365 nm. Both nucleoside analogs are readily taken up by mammalian cells following their addition to cell culture medium and are relatively nontoxic, at least in human embryonic kidney (HEK) 293 cancer cells. Like CLIP-seq, cross-linked RNA–protein complexes are digested with RNases, followed by fractionation, isolation, proteinase K treatment, adaptor ligation, cDNA library construction, and high throughput sequencing. The advantage of this method is that cross-linking of 4-SU or 6-SG to proteins results in respective thymidine to cytidine and guanosine to adenosine transitions in cDNAs at 4-SU and 6-SG incorporation sites, making it possible to map interacting RNA nucleotides and distinguish true protein-binding RNA species from background.
Using this method, Tuschl and colleagues identified RNA interacting sites of several RNA- or microRNA-binding proteins, including PUM2, QK1, IGF2BP1-3, AGO/EIF2C1-4, and TNRC6A-C (Hafner et al. 2010). Another recent study reported ~26,000 HuR/ELAVL1 binding sites in HeLa cells (Lebedeva et al. 2011). Following comparison of the HuR/ELAVL1 and Argonaute 2 transcriptomes using CLIP-seq and PAR-CLIP-seq methods, Kishore et al reported small differences in accuracies of these methods in identifying binding sites of HuR and Ago2 proteins (Kishore et al. 2011). They also suggested that optimizing conditions used for RNases treatment are critical step for library bias (Kishore et al. 2011).
3.4 Individual-Nucleotide Resolution CLIP-Seq (iCLIP-Seq)
Both CLIP-seq and PAR-CLIP-seq require reverse transcription to pass through the amino acid covalently bound to RNA at the cross-linking site. Often, cDNAs are prematurely truncated immediately before that nucleotide (Urlaub et al. 2002). To resolve this problem König et al. developed iCLIP (Konig et al. 2010), in which cleavable adaptors are ligated after reverse transcription allowing RT products to be circularized. This step allows quantification of truncation sites and discrimination between unique cDNA products and PCR duplicates. The group has successfully applied quantitative iCLIP to predict dual splicing effects of T-cell intracellular antigen (TIA)-RNA interactions (Wang et al. 2010) in order to characterize RNA targets of the splicing factor TDP-43 in brain (Tollervey et al. 2011). Work from the same group suggests that direct competition between hnRNP C and U2AF65 protects the transcriptome from exonization of Alu elements (Zarnack et al. 2013). Two other groups have also used the method to define landscapes of the RNA splicing factors SRSF3 and SRSF4 (Anko et al. 2012), and U2AF65 (Schor et al. 2012).
Which method to choose for transcriptome analysis largely depends on the nature of RNA–protein interaction, and factors such as protein abundance, cellular location of protein–RNA complex, and specificity of the protein–RNA interaction. An important issue to be considered for any immunoprecipitation-based method is availability of a highly specific and sensitive antibody. Therefore, optimization for the immunoprecipitation should be carried out using different antibodies. Some studies also use epitope-tagged rather than endogenous proteins to isolate binding RNAs when a high quality antibody is not available. However, since overexpression of tagged proteins may alter protein function and protein–RNA interaction, this approach should be taken with additional validation experiments. Furthermore, a proper control dataset, including libraries produced using a nonspecific antibody such as IgG and/or control cells lacking the protein of interest, should be generated side-by-side with the target library in order to separate true sequences from background. With proper controls and experimental optimization, RIP-seq is a powerful tool for global analysis of subsets of mRNAs or lncRNAs bound to various RNA-binding proteins.
4 Use of lncRNA as Bait to Study lncRNA–Protein/DNA Interactions
LncRNAs are implicated in many important biological processes (for reviews, see Wilusz et al. (2009), Chen and Carmichael (2010), Guttman and Rinn (2012), Rinn and Chang (2012)). Although only a handful have been characterized mechanistically, evidence suggests that lncRNAs often function by recruiting, assembling, modifying, or scaffolding other cofactors, including proteins (Tripathi et al. 2010; Tsai et al. 2010; Yap et al. 2010; Yin et al. 2012), DNA (Martianov et al. 2007; Schmitz et al. 2010), and other factors (Cesana et al. 2011; Salmena et al. 2011; Hansen et al. 2013; Memczak et al. 2013). Clearly, identifying these cofactors is of key importance for understanding the function of a specific lncRNA.
4.1 Using lncRNA as a Bait to Investigate an lncRNA–Protein Complex
Although approaches exist to identify RNA binding proteins (RBPs), progress in this area has been hampered due to the instability of lncRNAs and the flexibility of their structures. On the other hand, like proteins, nucleic acids can be affinity-tagged, allowing one to decipher an lncRNA–protein complex using the RNA of interest as bait. A variety of tags, including RNA aptamers and chemical labels, have been developed to allow affinity purification of nucleic acids. Proteins captured using such tags are then systematically identified by mass spectrometry.
RNA aptamers or RNA affinity tags that bind to specific RNA molecules can also be used to identify binding proteins by affinity chromatography or visualize RNA trafficking in living cells (Zhou et al. 2002; Janicki et al. 2004; Mao et al. 2011; Vasudevan and Steitz 2007; Maenner et al. 2010a). Some RNA aptamers are naturally occurring, such as the MS2 coat protein-binding sequence, which is an RNA-hairpin structure that specifically binds to bacteriophage MS2 coat protein (Graveley and Maniatis 1998). Multiple copies of MS2 hairpins can be fused with a bait RNA using recombinant techniques, and then resultant tagged RNAs are obtained by in vitro or in vivo transcription. Affinity purification of the RNA–protein complex is then achieved by incubation of tagged RNAs with a fusion protein containing the MS2 coat protein and maltose-binding protein (MS2-MBP), followed by affinity selection by binding to amylose resin (Zhou et al. 2002; Vasudevan and Steitz 2007). This method has been used to isolate human spliceosomes assembled on a well-characterized model pre-mRNA (Zhou et al. 2002) and protein complexes associated with regulatory lncRNAs (Vasudevan and Steitz 2007; Maenner et al. 2010a). For example, it is known that the lncRNA Xist is required to maintain mammalian female X-chromosome inactivation (XCI) by recruiting silencing chromatin remodeling complexes (Penny et al. 1996; Zhao et al. 2008). Interestingly, the most conserved Xist RNA regions correspond to repeat elements, among which the A region is the most highly conserved and is critical for XCI initiation (Hoki et al. 2009). To identify proteins interacting with the mouse A Region, the entire A region as well as several fragments from it were fused with the MS2 hairpin followed by affinity purification and mass spectrometry (Maenner et al. 2010a). These analyses revealed components of PRC2 that directly bind A region, a discovery critical to our understanding of X inactivation. This MS2 hairpin and RNA fusion system together with the MS2 coat protein tagged with a fluorescent protein have also been used to track RNA localization in living cells (Janicki et al. 2004; Mao et al. 2011).
Other aptamers, such as streptavidin or Sephadex aptamers, which can bind small molecules as well as complex macromolecules, have been identified by screening synthetic libraries (Srisawat et al. 2001; Walker et al. 2008). Such aptamers have been fused to regulatory RNAs, usually short and abundant ncRNAs, to successfully pull down RNP complexes, including RPP1 RNA, the large RNA subunit of RNase P (Srisawat and Engelke 2001; Li and Altman 2002). However, as yet there is no generalized rule used to design bait RNAs or aptamers that can fold correctly and maintain a stable conformation; thus fusion of an aptamer to an lncRNA of interest may not always result in an ideal outcome, and synthetic aptamers are not yet widely used to study lncRNA–protein complexes. Recently, however, a streptavidin aptamer has been scaffolded to a tRNA backbone that can stabilize the aptamer RNA conformation (Iioka et al. 2011). This scaffold strategy achieved about a tenfold increase in affinity to efficiently pull down RNA–protein complexes from cell lysates (Iioka et al. 2011). Thus use of scaffolded aptamers remains a promising approach to study lncRNA–protein complexes.
Another alternative to RNA aptamers is chemical labeling. Incoporation of modified ribonucleotide triphosphates (rNTPs) containing compounds such as biotin into RNA during in vitro transcription has been used to isolate RNP complexes and applied to identify proteins specifically associated with lncRNAs (Huarte et al. 2010; Tsai et al. 2010; Yang et al. 2011b; Klattenhoff et al. 2013). In such assays, the biotin-incorporated lncRNA or a fragment of that sequence is first denatured/renatured with RNA structure buffer to maintain correct conformation. RNAs are then incubated with cellular/nuclear extracts and affinity beads, followed by washing and elution to collect associated proteins. Biotin labeling of different fragments of the PRC2-interacting lncRNA HOTAIR followed by affinity purification of associated RNP complexes surprisingly revealed that the HOTAIR 5′ or 3′ domain could retrieve either PRC2 or the LSD1/CoREST/REST complex (a histone demethylase complex that mediates H3K4me2 demethylation). This approach provided direct evidence that HOTAIR acts as a modular scaffold for at least two distinct histone modification complexes to coordinate specific combinations of histone modifications on target gene chromatin (Tsai et al. 2010). Recent studies have applied a similar strategy to reveal that distinct sets of proteins are associated with individual lncRNAs to modulate their function (Huarte et al. 2010; Yang et al. 2011b; Klattenhoff et al. 2013).
There are some limitations to the use of tagged RNAs to identify associated RNP complexes. First, proper folding is required for aptamer/RNA function as well as for RNA–protein interactions. Second, chemical modifications and fusion of affinity tags to RNA may lead to structural perturbations that inhibit RNA–protein complex formation. Third, chemically modified or aptamer-tagged RNAs made from in vitro transcription may not reflect the true nature of complexes formed in vivo. Finally, since cross-linking is rarely used (Huarte et al. 2010; Tsai et al. 2010; Yang et al. 2011b; Klattenhoff et al. 2013) in in vitro studies, transient lncRNA/protein interactions may not be captured, while nonspecific interactions may be unintentionally retrieved, leading to misinterpretation (Riley and Steitz 2013). However, it is worth noting that a recent study aimed at defining the mRNA interactome in human cells developed two complementary protocols for covalent UV cross-linking of RBPs to RNA. This study identified over 800 proteins associated with mRNAs by pull-down with oligo d(T) magnetic beads (Castello et al. 2012). In addition, two recent studies developed genome-wide assays to study both genomic binding sites of an lncRNA and lncRNA–protein complexes by incubating nuclear extracts with biotinylated antisense oligos targeting the lncRNA of interest (Chu et al. 2011; Simon et al. 2011) (see below for details). Thus, despite potential pitfalls inherent in these methods, accumulating evidence suggests that tagging an RNA bait with a reagent in order to “fish out” associated proteins is a reliable way to analyze lncRNA function.
4.2 Global Approaches to Study Genomic Binding Sites of an lncRNA
Many lncRNAs function at the level of chromatin by interacting with chromatin-modifying machinery or acting as scaffolds for multiple complexes (for reviews, see Guttman and Rinn (2012), Rinn and Chang (2012)). While some lncRNAs, such as Xist and Air, work in cis on neighboring genes (Nagano et al. 2008; Zhao et al. 2008), others, such as roX2 (in Drosophila) and HOTAIR, work in trans to regulate distant genes (Gelbart and Kuroda 2009; Rinn et al. 2007). In these cases, an lncRNA can interact directly or indirectly with a chromatin DNA through a specific RNA or protein. Thus, uncovering binding sites of these lncRNAs genome-wide is essential to understand their function. Recently, a combination of chemical tagging of RNAs and deep sequencing technology have allowed one to systematically identify those sites (Chu et al. 2011; Simon et al. 2011).
Two such approaches, named CHART (Capture Hybridization Analysis of RNA Targets) (Simon et al. 2011) and ChIRP (Chromatin Isolation by RNA Purification) (Chu et al. 2011), were independently developed by the Kingston and Chang laboratories, respectively. These approaches are similar in principle to ChIP-seq methodology. Briefly, chemically labeled oligos complementary to an lncRNA are incubated with cross-linked nuclear extracts, followed by affinity purification and elution of the precipitated materials, including DNAs and proteins. To obtain the genomic map of lncRNA occupancy, the reverse cross-linked DNAs are then subjected to deep sequencing and analyses (Fig. 4.2).
As lncRNAs are known to be highly structured, the key feature of these approaches is to design appropriate chemically labeled oligos that are specific and accessible to a structured lncRNA in cross-linked cells. Two principles are applied to design such oligos. In CHART, a mapping assay using RNase H, which hybrolyzes the RNA strand of a DNA–RNA hybrid, was adapted to probe sites on an lncRNA available to hybridization in extracts of cross-linked chromatin. The most sensitive or “accessible” regions in the lncRNA are then identified by incubating individual 20-mer complementary DNA oligos one at a time with the extracts. Identified regions are then used to design 24–25-mer desthiobiotin-conjugated C-oligos, which are 3′-modified by a single desthiobiotin and four oligoethyleneglycol spacers (Dejardin and Kingston 2009)), to allow biotin elution to achieve a lower background (Fig. 4.2) (Simon et al. 2011). In ChIRP, dozens of 20-mer biotinylated complementary DNA oligo probes that tile the entire length of an lncRNA are synthesized. These probes target all regions of an lncRNA equally, as there is no prior knowledge of its secondary structure or functional domains (Fig. 4.2) (Chu et al. 2011). Such unbiased design of oligonucleotides has been used successfully in single-molecule RNA fluorescent in situ hybridization and yielded highly specific signals (Raj et al. 2008).

Schematic overview of approaches used to analyze genomic binding sites of an lncRNA. See text for details

Schematic drawing of X-inactivation center (XIC) and Xist RNA. (a) A cluster of lncRNAs transcribed from the XIC is required for mammalian dosage compensation. lncRNAs include Xist (X-inactivation specific transcript), Tsix (antisense of Xist), Tsx (testis-specific X-linked gene), Xite (X-inactivation intergenic transcription element), RepA RNA, Jpx (also known as Enox (Expressed Neighbor of Xist)), and Ftx (Five prime to Xist). Ftx, JPX, and RepA lncRNAs promote Xist transcription, while Tsixn Xite and Tsx inhibit it. (b) Gene structure and repeat regions of Xist RNA. Exons are represented as boxes
To analyze global genomic binding sites of an lncRNA, principles used to design labeled DNA oligos can be slightly varied. In CHART, RNaseH mapping allows one to design labeled DNA oligos accessible to specific regions of an lncRNA (Simon et al. 2011). However, there are limitations to the method with lncRNAs such as HOTAIR, which harbors functional domains separate from each other (Tsai et al. 2010). Thus use of selected probes may result in potential loss of information due to failure to identify all functional domains of an lncRNA. This disadvantage potentially can be overcome by ChIRP, which uses unbiased biotinylated tiling oligos. However, ChIRP precipitation of nonspecific DNA fragments may occur due to off-target hybridization of pooled oligonucleotide probes. One way to eliminate this artifact is to split tiling oligos into “even” and “odd” probes based on their relative positions along the target RNA and then pooling them into two groups. Independent experiments run with “even” or “odd” probe groups and analyses then focus only on overlapping signals (Chu et al. 2011). As the two probe sets share no overlapping sequences, they target only the RNA of interest and its associated chromatin.
Both CHART and ChIRP are powerful in that they not only identify genomic maps showing lncRNA occupancy but provide a new insight into RNA–chromatin interaction at almost single nucleotide resolution. The lncRNA roX2 is known to localize to the X chromosome, where it acts together with the MSL complex to regulate X chromosome dosage compensation in Drosophila (Gelbart and Kuroda 2009). CHART has been successfully used to identify roX2 genomic binding (Simon et al. 2011). CHART-seq analysis of roX2 yielded the same preference for chromatin entry sites, namely, a GA-rich polypurine motif, as the MSL complex, consistent with the notion that roX2 is an integral subunit of the MSL complex bound to chromatin. ChIRP has also been successfully applied to several lncRNAs including TERC, roX2, and HOTAIR (Chu et al. 2011). Interestingly, motif analysis of HOTAIR ChIRP-seq-enriched sites revealed a novel GA-rich polypurine motif, suggesting that many lncRNAs function similarly to guide chromatin–lncRNA complexes such as PRC2-HOTAIR and MSL-roX to appropriate genomic loci. Finally, as both CHART and ChIRP utilize reversible cross-linking, enriched materials can be used to analyze proteins and even RNAs associated with the lncRNA of interest.
5 lncRNAs Function in X-Inactivation During Mammalian Development
The most extensively studied lncRNAs come from a region named the X-chromosome inactivation center (XIC) located on the X-chromosome (Rastan 1983; Rastan and Robertson 1985; Brown et al. 1991b). At least six different lncRNAs have been identified from this locus, and they function together to regulate the epigenetic process of X-chromosome inactivation (XCI) (Fig. 4.3a) (Brockdorff et al. 1991; Brown et al. 1991a; Lee et al. 1999; Lee and Lu 1999; Ogawa and Lee 2003; Tian et al. 2010; Anguera et al. 2011; Chureau et al. 2011). XCI failure results in female embryonic lethality; thus these lncRNAs play an essential role in female development. Their mechanisms and regulation are discussed below.
5.1 X-Chromosome Inactivation and Xist lncRNA
Indispensable for XCI is the 17Kb lncRNA named X-inactivation specific transcript (Xist) (Borsani et al. 1991; Brockdorff et al. 1991; Brown et al. 1991a). Other lncRNAs relevant to the locus likely function through regulating Xist expression. Xist is expressed at low levels from two active X-chromosomes in female embryos or female embryo-derived embryonic stem cells before XCI initiation and then is upregulated and, extraordinarily, starts to “coat” almost the entire X-chromosome. Xist spreading along the X transforms that allele into heterochromatin. Eventually, that in active allele (Xi) undergoes DNA methylation, repressing over a thousand genes. Xi remains silent during subsequent cell divisions, and Xist then becomes dispensable for chromosome-wide silencing, despite the observation that its expression remains high (Brown and Willard 1994; Csankovszki et al. 1999).
5.2 Xist Silencing Mechanisms
Xist is the only RNA reported so far capable of “spreading” along almost an entire chromosome and converting an active euchromatin to a condensed heterochromatin. An intensive area of investigation in the field of epigenetics is to understand how Xist achieves such silencing. Questions remain as to how it is tethered onto the inactive X and what silencing factors it interacts with.
5.2.1 Loading and Spreading of XIST on the Inactive X-Chromosome (Xi)
Currently, evidence suggests that specific DNA or RNA sequences facilitate Xist spreading. For example, naturally occurring or induced X:autosome translocations indicate that Xist spreads differently along autosomal DNA segments (Surralles and Natarajan 1998; Popova et al. 2006). A search for sequence differences between the X-chromosome and autosomes revealed that LINE (long interspersed elements) elements showed greater density on the X (Lyon 1998). Mary Lyon proposed that interspersed repetitive LINE elements act as booster elements to promote Xist spread. Indeed, a transgenic study in mouse embryonic stem cells (mESCs) showed that chromosome regions with greater LINE density are inactivated more efficiently by a Xist transgene (Popova et al. 2006). Recently, another study suggested that specific LINEs may participate in local propagation of XCI into regions that would otherwise escape it (Chow et al. 2010). However, the exact function of LINE elements in establishment of XCI remains to be investigated.
Trans-acting factors functioning in this process have been identified only recently. A landmark paper from Jeannie Lee’s group showed that the RNA/DNA binding protein Yin-Yang 1 (YY1) is required to tether Xist RNA to the inactive X (Jeon and Lee 2011). In YY1 knockdown cells, Xist RNA remained highly expressed but exhibited a diffuse localization pattern rather than forming a “cloud” on top of the Xi. However, since YY1 binding sites are widespread throughout the mammalian genome, it remains unclear why YY1 does not guide Xist RNA onto an entire autosome in the transgene studies. In addition to YY1, a screening study showed that the nuclear scaffold protein hnRNPU is essential for Xist loading and spreading (Hasegawa et al. 2010). Currently, it is not known whether YY1 and hnRNPU form a complex with Xist.
5.2.2 XIST Silences Genes Through Repressive Polycomb Proteins
Upon Xist spreading, the X-chromosome loses active chromatin marks, such as trimethylated histone H3 lysine 4 (H3K4-3me), and gains the heterochromatic marks H3K27-3me and mono ubiquinated H2A (H2AK119u1) (Plath et al. 2003; Silva et al. 2003; de Napoles et al. 2004; Fang et al. 2004; Okamoto et al. 2004). These observations suggest that Xist interacts with chromatin modifiers. Indeed, our work and that of others proved Xist to be a cofactor to target Polycomb group proteins onto X-chromosome during XCI.
First identified in Drosophila as repressing HOX loci (Alkema et al. 1995; Harding et al. 1995; Yu et al. 1995), Polycomb proteins are known to play critical roles in epigenetic gene regulation in almost all organisms, including mammals. Polycomb proteins comprise two major complexes, Polycomb Repressive Complex 1 (PRC1) and Polycomb Repressive Complex 2 (PRC2) (Simon and Kingston 2013). PRC2 bears histone methyltransferase activity through its catalytic subunit Ezh2 (Cao et al. 2002; Czermin et al. 2002; Kuzmichev et al. 2002). Once PRC2 establishes the H3K27-3me heterochromatin mark, protein with chromo-domain from PRC1 complex recognize that mark and guide PRC1 to specific genomic loci to establish ubiquinated histone H2A (H2AK119u1) (Fischle et al. 2003; Min et al. 2003; Wang et al. 2004). H2AK119u1 deposition is proposed to repress gene expression by facilitating chromatin compaction or inhibiting RNA polymerase II-dependent transcriptional elongation (Francis et al. 2004; Zhou et al. 2008). Both PRC1 and PRC2 complexes are highly enriched on the inactive X-chromosome during XCI establishment (Silva et al. 2003; de Napoles et al. 2004; Plath et al. 2003; Okamoto et al. 2004). Deletion of some Polycomb proteins, such as Eed from the PRC2 complex, reverses Xi in extra-embryonic tissues, suggesting an essential role for Polycomb proteins in maintaining XCI (Kalantry et al. 2006). To determine how PRC2 is recruited onto the inactive X-chromosome, we undertook studies showing that Xist directly binds to the Ezh2 subunit (Zhao et al. 2008). Interestingly, that interaction occurred at the Xist silencing domain.
Xist RNA contains six different repeat regions (A to F) (Fig. 4.3b), which likely represent distinct functional domains. The most well studied is the 5′ A region (Wutz et al. 2002), whose sequence is highly conserved among mouse and humans (Hong et al. 2000). A Xist transgene lacking the A region cannot initiate silencing (Wutz et al. 2002). Two independent silencing mechanisms are proposed for this region. One study suggests that the A region directly binds the splicing factor ASF/SF2 to ensure proper Xist splicing (Royce-Tolland et al. 2010). In another study, we assessed mESCs and discovered that a shorter transcript, named RepA RNA, is transcribed through the A region (Zhao et al. 2008). Both Xist and RepA RNA directly bind Ezh2 through A region repeats. Support for this observation came from the Reinberg group when they showed that Ezh2 phosphorylation enhances interaction between PRC2 and Xist/RepA RNAs (Kaneko et al. 2010). A different PRC2 subunit, Suz12, reportedly binds RepA RNA (Kanhere et al. 2010), although mechanisms underlying complex formation among lncRNAs and PRC2 subunits are not yet understood. We also found that after RepA RNA knockdown, Xist could not be upregulated; hence the H3K27-3me mark was not established and XCI was not initiated (Zhao et al. 2008). A similar phenotype was observed by Hoki et al. following genetic deletion of the A region in mouse (Hoki et al. 2009). These results suggest that the A region is required for Xist upregulation and such regulation requires the RepA transcript. Notably, a Xist mutant lacking A-repeats can target PRC2 and PRC1 onto Xi (Plath et al. 2003; Schoeftner et al. 2006), indicating that other Xist sequences also recruit Polycomb proteins. Such regions remain to be identified.
Several laboratories have studied the structure of the A region in order to understand the molecular basis for Xist and Polycomb interaction. Computational analysis revealed that the region contains 7.5–8.5 tandem repeats (variable among mouse and human) of a conserved ~26-mer sequence and predict a double stem loop structure within each repeat (Hendrich et al. 1993; Wutz et al. 2002; Duszczyk et al. 2011). That structure appears to be important for Rep A/PRC2 interaction (Zhao et al. 2008; Kanhere et al. 2010). Interestingly, NMR studies of an in vitro transcribed 26-mer showed that only the first predicted hairpin is formed internally, while the second mediates duplex formation among different repeats (Duszczyk et al. 2008; Duszczyk and Sattler 2012). FRET analysis using chemical and enzymatic probes to examine structure of the whole domain indicated that the entire A region contains two long stem-loop structures, each including four repeats (Maenner et al. 2010b). These studies highlight the importance of RNA structure for function.
Less is known about the structure and function of the other Xist repeats. Nevertheless, Repeat regions C and F have been shown to regulate Xist spreading, potentially through interaction with YY1 (Jeon and Lee 2011; Wutz et al. 2002; Beletskii et al. 2001; Sarma et al. 2010).
5.3 Xist Regulatory Mechanisms
5.3.1 Regulation by Other lncRNAs
Xist upregulation is controlled by cis-elements and trans-factors. Cis-elements include lncRNAs from the XIC, including the Xist antisense partner Tsix (Lee et al. 1999; Lee and Lu 1999), Xite (X-inactivation intergenic transcription elements) (Ogawa and Lee 2003), Jpx (also known as Enox (Expressed Neighbor of Xist) (Tian et al. 2010), Ftx (Five prime to Xist) (Chureau et al. 2011), and Tsx (Testes-specific X-linked) (Anguera et al. 2011) (Fig. 4.1). Tsix was identified as a Xist antagonist in 1999 in studies showing that its depletion promotes Xist transcriptional activation exclusively from the Tsix-disrupted allele (Lee et al. 1999; Lee and Lu 1999). During early development, Tsix and Xist display opposite expression patterns (Lee et al. 1999; Lee and Lu 1999). Tsix is expressed at ~10- to 100-fold higher levels than Xist and suppresses Xist expression before XCI (Shibata and Lee 2003). Tsix levels decrease during XCI initiation, allowing Xist upregulation (Lee et al. 1999; Lee and Lu 1999; Shibata and Lee 2003).
Several epigenetic mechanisms are proposed to explain Tsix activity. One is that Tsix modulates the chromatin state of the Xist promoter. This hypothesis is supported by two studies describing a gain of euchromatic marks at the Xist promoter following Tsix truncation in mESCs (Navarro et al. 2005; Sado et al. 2005), and another indicating an initial gain of heterochromatic marks at the Xist promoter followed by switching into euchromatic markers (Sun et al. 2006). These apparent differences in these results stem from use of male (Navarro et al. 2005; Sado et al. 2005) versus female mESCs (Sun et al. 2006). Others propose that Tsix forms an RNA duplex with Xist and regulates it through the RNAi pathway (Ogawa et al. 2008). Although this presents an attractive model for understanding sense and antisense RNA interactions, no microRNA has been identified from the Xist/Tsix locus, although Dicer and Tsix reportedly regulate Xist synergistically in mESCs (Ogawa et al. 2008). Recently, we reported that Tsix competes with Xist for PRC2 binding and interferes with PRC2 loading onto the inactive X at the early stage of XCI (Zhao et al. 2008).
Other lncRNAs functioning in XCI have not been extensively studied. However, it has been shown that deleting Xite from one of the Xs in female mESCs downregulates Tsix in cis and skews X-inactivation, suggesting that Xite promotes Tsix expression on the active X (Ogawa and Lee 2003). There are also reports that Jpx acts in trans to activate Xist and that Ftx is also a positive regulator of Xist (Tian et al. 2010; Chureau et al. 2011). X-inactivation is also mildly affected by loss of Tsix by mechanisms as yet uncharacterized (Anguera et al. 2011).
5.3.2 Regulation by Pluripotency Factors
It has long been postulated that XCI is coupled to the mESC pluripotency state, as XCI initiation is tightly associated with mESC differentiation. Furthermore, during reprogramming of mouse somatic cells, Xi reactivation accompanies the establishment of pluripotency (Maherali et al. 2007). Recent studies suggest pluripotent factors play a repressive role in Xist regulation. Xist upregulation occurs in Oct4 or Nanog conditional knockout male mESCs, and Xist coats both X chromosomes in Oct4 knockdown female mESCs as they undergo differentiation (Navarro et al. 2008; Donohoe et al. 2009). The Xist first intron is occupied by Oct4, Nanog, and Sox2 in undifferentiated mESCs (Navarro et al. 2008), suggesting that pluripotency factors directly repress Xist. However, intron1 deletion results in only a small increase in Xist expression, indicating the existence of unidentified cis-elements that interact with pluripotency factors (Nesterova et al. 2011). In addition to core pluripotency factors, other pluripotency genes, such as Prdm14, also inhibit Xist RNA expression (Ma et al. 2011). Interestingly, while one set of pluripotency factors represses Xist, a different set activates Tsix. Klf4, c-Myc, and Rex1 reportedly promote Tsix expression (Navarro et al. 2010), and Rex1 is required for efficient Tsix elongation. Factors such as RNF12 (Ring finger protein 12), an X-linked E3 ubiquitin ligase that targets Rex1 for degradation, are essential to initiate XCI and can activate Xist expression, even in male mESCs (Jonkers et al. 2009; Shin et al. 2010). Therefore, pluripotency factors are thought to block Xist expression directly or indirectly through Tsix activation.
6 ncRNA Function in Disease
In addition to their activity in normal physiological processes, just as other molecules, lncRNAs are also linked to human diseases, including a variety of human cancers and human genetic disorders. We will simply illustrate several lncRNAs and their involvement in human diseases below.
6.1 Xist, XCI and Cancer
Some female cancers, such as breast and ovarian cancers, exhibit loss of Xi and gain of an active X (Pageau et al. 2007), suggesting that XCI mis-regulation functions in tumorigenesis. Some X-linked oncogenes and tumors suppressors that are subject to XCI have been identified. Among them, Zuo et al. found that FOXP3 is an X-linked breast cancer tumor suppressor gene (Zuo et al. 2007). Additional evidence that Xist lncRNA drives cancer development came from a recent study reporting the occurrence of hematological cancer in Xist conditional knockout mice (Yildirim et al. 2013). Those authors deleted Xist in hematopoietic cells and found that only female mice developed myeloproliferative neoplasm and myelodysplastic syndrome, which is characterized by bone marrow fibrosis and chronic myelomonocytic leukemia. The study suggests that X reactivation perturbs maturation and longevity of hematopoietic stem cells, suggesting a novel role for Xist and XCI in adult stem cell and cancer biology.
6.2 Abnormal Expression of lncRNAs in Cancers
Abnormal expression of imprinted lncRNAs is seen in different types of cancer. For example, disruption of the lncRNA-related genomic imprinted Kcnq1 cluster is associated with human Beckwith–Wiedemann syndrome and Wilms’ tumor (O'Neill 2005). In addition, the paternally expressed antisense PEG8/IGF2AS, which is transcribed from the Igf2 locus, is significantly overexpressed in Wilms’ tumor samples compared to adjacent normal kidney tissue (Okutsu et al. 2000). Interestingly, although the lncRNA H19 has no obvious function in imprinted expression of Igf2 (Jones et al. 1998), it is essential for tumorigenesis (Matouk et al. 2007) and c-Myc directly induces H19 transcription during tumorigenesis (Barsyte-Lovejoy et al. 2006).
Many tumor suppressor genes are also associated with nearby antisense lncRNA transcripts. For example, the tumor suppressor gene p15, a cyclin-dependent kinase inhibitor implicated in leukemia, is regulated by its antisense transcript p15 ncRNA (p15AS) (Yu et al. 2008). p15AS and p15 sense expression is inversely correlated in leukemia progression, and p15 silencing is induced via both cis and trans mechanisms through heterochromatin formation, as shown by reduced H3K4 methylation and increased H3K9 methylation following introduction of p15AS into mammalian cells (Yu et al. 2008). Importantly, p15 silencing persists even after p15AS is downregulated (Yu et al. 2008). Another mode of such regulation is illustrated by ANRIL, the long antisense transcript of the INK4b/ARF/INK4a tumor suppressor locus (Yap et al. 2010). First, expression of ANRIL and its interacting protein chromobox 7 (CBX7), a subunit of the polycomb repressive complex 1(PRC1), is upregulated in prostate cancer tissues. Furthermore, ANRIL is associated with PRC1 complex recruitment to the INK4b/ARF/INK4a locus by specifically binding to CBX7. Interestingly, CBX7 contains structurally distinct modes to bind not only ANRIL, but also H3K37me, which is methylated by EZH2 of PRC2. Moreover, transfection of ANRIL antisense transcripts or expression vectors harboring CBX7 mutants that disrupt H3K27me modification or RNA binding promotes premature growth arrest (Yap et al. 2010). This study suggests that an lncRNA participates directly in cis-recruitment of PRC complexes to silence gene expression from a tumor suppressor locus.
Interestingly, the lncRNA HOTAIR functions in trans-recruitment of PRC2 complexes at non-HOX loci not only during developmental patterning (Rinn et al. 2007) but in cancer metastasis (Gupta et al. 2010). HOTAIR shows increased expression in primary breast tumor and metastases (Gupta et al. 2010) and in colorectal cancer (Kogo et al. 2011). HOTAIR loss decreases cancer invasiveness, while increased HOTAIR expression in epithelial cancer cells promotes metastasis by globally altering the chromatin state. Global changes include induction of genome-wide retargeting of PRC2 to an occupancy pattern, leading to PRC2-dependent altered histone H3 lysine 27 methylation and gene expression (Gupta et al. 2010). As noted, HOTAIR likely acts as a scaffold for at least two distinct complexes that mediate histone modification at targeted chromatin sites (Tsai et al. 2010), and it binds to many genomic sites through a GA-rich motif (Chu et al. 2011). Taken together, these studies strongly suggest that lncRNAs serve as important regulators in tumorigenesis, probably through direct targeting and recruitment of chromatin-modifying machinery at specific loci.
6.3 Abnormal Expression of lncRNAs in Human Genetic Disorders
lncRNAs also likely function in human genetic disorders, such as facioscapulohumeral muscular dystrophy (FSHD) (Cabianca et al. 2012) and Prader–Willi Syndrome (PWS) (Yin et al. 2012). Facioscapulohumeral muscular dystrophy (FSHD) is an autosomal-dominant disease associated with reduced copy number of a D4Z4 repeat sequences mapping to 4q35. D4Z4 deletion causes an epigenetic switch leading to de-repression of 4q35 genes (Cabianca and Gabellini 2010). The FSHD locus is normally a Polycomb repressive target; however, FSHD patients display loss of Polycomb silencing and a gain in Trithorax-dependent activation (a de-repression process). DBE-T is a chromatin-associated lncRNA originating from the FSHD-associated repetitive elements and is produced in FSHD patients. In FSHD patient, the expression of DBE-T is to coordinate the de-repression of 4q35 genes (Cabianca et al. 2012). DBE-T is thought to recruit the Trithorax group protein Ash1L to the FSHD locus, driving histone H3 lysine 36 dimethylation, chromatin remodeling, and 4q35 gene transcription (Cabianca et al. 2012).
While gain of DBE-T expression is likely involved in FSHD (Cabianca et al. 2012), loss of expression of sno-lncRNAs likely contributes to the pathogenesis of Prader–Willi Syndrome (PWS) (Yin et al. 2012). PWS is a multiple system human disorder characterized by global developmental delay, mental retardation, and morbid obesity is due to the absence of paternally expressed imprinted genes at 15q11.2-q13 (Cassidy et al. 2012). There are several coding and noncoding genes, including a cluster of 29 box C/D small nucleolar RNAs (snoRNAs) named SNORD116 expressed from this region (Cassidy et al. 2012). Although the role of each transcript in the pathogenesis of PWS remains largely unknown, lines of evidence in human patients suggest the SNORD116 snoRNA gene cluster plays a key role (Sahoo et al. 2008; de Smith et al. 2009; Duker et al. 2010). It is known that Box C/D snoRNAs usually function as guides for ribose methylation of an RNA target (Kiss 2001), however, no targets of SNORD116 snoRNAs has been validated so far (Cassidy et al. 2012). Thus, the function of these SNORD116 snoRNAs and the precise molecular cause of PWS still remain unknown.
Recently, investigators sequenced the repertoire of non-polyadenylated RNAs isolated from human embryonic stem cells (hESCs) (Yang et al. 2011a) and discovered a class of intron-derived and snoRNA-related lncRNAs (sno-lncRNAs). Interstingly, five sno-lncRNAs are produced from SNORD116 snoRNA gene cluster and are specifically deleted from the minimal deletion in PWS patients (Yin et al. 2012). In wild type hESCs, PWS region sno-lncRNAs are expressed at extremely high levels (similar in abundance to some housekeeping mRNAs) and accumulate near their sites of synthesis. Although these lncRNAs have little effect on local gene expression, surprisingly, each of them contains multiple specific binding sites for the alternative splicing regulator Fox2. Thus, PWS region sno-lncRNAs act as “molecular sponges” to strongly associate with Fox2 in the nucleus and alter patterns of Fox2-regulated alternative splicing in sno-lncRNA depleted cells (Yin et al. 2012). While in PWS patients, these PWS region sno-lncRNAs are not expressed, subsequently leading to altered nuclear distribution of Fox proteins and dysregulating splicing embryonically and in adults. These results implicate a new class of lncRNAs in PWS pathogenesis. Overall, all of these studies suggest that lncRNAs could serve as potential targets in treatment of diseases.
7 Conclusions
The number of non protein-coding transcripts identified over the past decade has increased exponentially, causing a dramatic shift in our perception of the mammalian genome from a focus on protein-coding genes to long noncoding elements. Emerging evidence suggests that lncRNAs play essential roles in a wide range of biological functions and various technologies have been developed to study their molecular mechanisms in vivo. These studies pave the way for exciting future discoveries relevant to lncRNAs.
References
Alkema MJ, van der Lugt NM, Bobeldijk RC, Berns A, van Lohuizen M (1995) Transformation of axial skeleton due to overexpression of bmi-1 in transgenic mice. Nature 374(6524):724–727
Anguera MC, Ma W, Clift D, Namekawa S, Kelleher RJ III, Lee JT (2011) Tsx produces a long noncoding RNA and has general functions in the germline, stem cells, and brain. PLoS Genet 7(9):e1002248
Anko ML, Muller-McNicoll M, Brandl H, Curk T, Gorup C, Henry I, Ule J, Neugebauer KM (2012) The RNA-binding landscapes of two SR proteins reveal unique functions and binding to diverse RNA classes. Genome Biol 13(3):R17
Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, Tsao MS, Penn LZ (2006) The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res 66(10):5330–5337
Beletskii A, Hong YK, Pehrson J, Egholm M, Strauss WM (2001) PNA interference mapping demonstrates functional domains in the noncoding RNA Xist. Proc Natl Acad Sci U S A 98(16):9215–9220
Borsani G, Tonlorenzi R, Simmler MC, Dandolo L, Arnaud D, Capra V, Grompe M, Pizzuti A, Muzny D, Lawrence C et al (1991) Characterization of a murine gene expressed from the inactive X chromosome. Nature 351(6324):325–329
Brockdorff N, Ashworth A, Kay GF, Cooper P, Smith S, McCabe VM, Norris DP, Penny GD, Patel D, Rastan S (1991) Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351(6324):329–331
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF (1991a) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349(6304):38–44
Brown CJ, Lafreniere RG, Powers VE, Sebastio G, Ballabio A, Pettigrew AL, Ledbetter DH, Levy E, Craig IW, Willard HF (1991b) Localization of the X inactivation centre on the human X chromosome in Xq13. Nature 349(6304):82–84
Brown CJ, Willard HF (1994) The human X-inactivation centre is not required for maintenance of X-chromosome inactivation. Nature 368(6467):154–156
Brown JA, Valenstein ML, Yario TA, Tycowski KT, Steitz JA (2012) Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc Natl Acad Sci U S A 109(47):19202–19207
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE (2010) Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 6(12):e1001233
Cabianca DS, Casa V, Bodega B, Xynos A, Ginelli E, Tanaka Y, Gabellini D (2012) A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 149(4):819–831
Cabianca DS, Gabellini D (2010) The cell biology of disease: FSHD: copy number variations on the theme of muscular dystrophy. J Cell Biol 191(6):1049–1060
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25(18):1915–1927
Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298(5595):1039–1043
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ (2012) Prader-Willi syndrome. Genet Med 14(1):10–26
Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM et al (2012) Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149(6):1393–1406
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147(2):358–369
Chen LL, Carmichael GG (2010) Long noncoding RNAs in mammalian cells: what, where, and why? Wiley Interdiscip Rev RNA 1(1):2–21
Chi SW, Zang JB, Mele A, Darnell RB (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460(7254):479–486
Chow JC, Ciaudo C, Fazzari MJ, Mise N, Servant N, Glass JL, Attreed M, Avner P, Wutz A, Barillot E et al (2010) LINE-1 activity in facultative heterochromatin formation during X chromosome inactivation. Cell 141(6):956–969
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44(4):667–678
Chureau C, Chantalat S, Romito A, Galvani A, Duret L, Avner P, Rougeulle C (2011) Ftx is a non-coding RNA which affects Xist expression and chromatin structure within the X-inactivation center region. Hum Mol Genet 20(4):705–718
Core LJ, Waterfall JJ, Lis JT (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322(5909):1845–1848
Csankovszki G, Panning B, Bates B, Pehrson JR, Jaenisch R (1999) Conditional deletion of Xist disrupts histone macroH2A localization but not maintenance of X inactivation. Nat Genet 22(4):323–324
Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111(2):185–196
de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M et al (2004) Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell 7(5):663–676
de Smith AJ, Purmann C, Walters RG, Ellis RJ, Holder SE, Van Haelst MM, Brady AF, Fairbrother UL, Dattani M, Keogh JM et al (2009) A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism. Hum Mol Genet 18(17):3257–3265
Dejardin J, Kingston RE (2009) Purification of proteins associated with specific genomic Loci. Cell 136(1):175–186
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22(9):1775–1789
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F et al (2012) Landscape of transcription in human cells. Nature 489(7414):101–108
Donohoe ME, Silva SS, Pinter SF, Xu N, Lee JT (2009) The pluripotency factor Oct4 interacts with Ctcf and also controls X-chromosome pairing and counting. Nature 460(7251):128–132
Duker AL, Ballif BC, Bawle EV, Person RE, Mahadevan S, Alliman S, Thompson R, Traylor R, Bejjani BA, Shaffer LG et al (2010) Paternally inherited microdeletion at 15q11.2 confirms a significant role for the SNORD116 C/D box snoRNA cluster in Prader-Willi syndrome. Eur J Hum Genet 18(11):1196–1201
Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R et al (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489(7414):57–74
Duszczyk MM, Sattler M (2012) (1)H, (1)(3)C, (1)(5)N and (3)(1)P chemical shift assignments of a human Xist RNA A-repeat tetraloop hairpin essential for X-chromosome inactivation. Biomol NMR Assign 6(1):75–77
Duszczyk MM, Wutz A, Rybin V, Sattler M (2011) The Xist RNA A-repeat comprises a novel AUCG tetraloop fold and a platform for multimerization. RNA 17(11):1973–1982
Duszczyk MM, Zanier K, Sattler M (2008) A NMR strategy to unambiguously distinguish nucleic acid hairpin and duplex conformations applied to a Xist RNA A-repeat. Nucleic Acids Res 36(22):7068–7077
Fang J, Chen T, Chadwick B, Li E, Zhang Y (2004) Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J Biol Chem 279(51):52812–52815
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17(15):1870–1881
Francis NJ, Kingston RE, Woodcock CL (2004) Chromatin compaction by a polycomb group protein complex. Science 306(5701):1574–1577
Gardner EJ, Nizami ZF, Talbot CC Jr, Gall JG (2012) Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev 26(22):2550–2559
Gelbart ME, Kuroda MI (2009) Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136(9):1399–1410
Gomez JA, Wapinski OL, Yang YW, Bureau JF, Gopinath S, Monack DM, Chang HY, Brahic M, Kirkegaard K (2013) The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 152(4):743–754
Gong C, Maquat LE (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470(7333):284–288
Graveley BR, Maniatis T (1998) Arginine/serine-rich domains of SR proteins can function as activators of pre-mRNA splicing. Mol Cell 1(5):765–771
Guil S, Caceres JF (2007) The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol 14(7):591–596
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL et al (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076
Guttman M, Rinn JL (2012) Modular regulatory principles of large non-coding RNAs. Nature 482(7385):339–346
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M et al (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141(1):129–141
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495(7441):384–388
Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, Kjems J (2011) miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J 30(21):4414–4422
Harding KW, Gellon G, McGinnis N, McGinnis W (1995) A screen for modifiers of deformed function in Drosophila. Genetics 140(4):1339–1352
Hasegawa Y, Brockdorff N, Kawano S, Tsutui K, Tsutui K, Nakagawa S (2010) The matrix protein hnRNP U is required for chromosomal localization of Xist RNA. Dev Cell 19(3):469–476
He Y, Vogelstein B, Velculescu VE, Papadopoulos N, Kinzler KW (2008) The antisense transcriptomes of human cells. Science 322(5909):1855–1857
Hendrich BD, Brown CJ, Willard HF (1993) Evolutionary conservation of possible functional domains of the human and murine XIST genes. Hum Mol Genet 2(6):663–672
Hoki Y, Kimura N, Kanbayashi M, Amakawa Y, Ohhata T, Sasaki H, Sado T (2009) A proximal conserved repeat in the Xist gene is essential as a genomic element for X-inactivation in mouse. Development 136(1):139–146
Hong YK, Ontiveros SD, Strauss WM (2000) A revision of the human XIST gene organization and structural comparison with mouse Xist. Mamm Genome 11(3):220–224
Huarte M, Guttman M, Feldser D, Garber M, Koziol MJ, Kenzelmann-Broz D, Khalil AM, Zuk O, Amit I, Rabani M et al (2010) A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142(3):409–419
Iioka H, Loiselle D, Haystead TA, Macara IG (2011) Efficient detection of RNA-protein interactions using tethered RNAs. Nucleic Acids Res 39(8):e53
Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH et al (2004) From silencing to gene expression: real-time analysis in single cells. Cell 116(5):683–698
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19(2):141–157
Jeon Y, Lee JT (2011) YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146(1):119–133
Jones BK, Levorse JM, Tilghman SM (1998) Igf2 imprinting does not require its own DNA methylation or H19 RNA. Genes Dev 12(14):2200–2207
Jonkers I, Barakat TS, Achame EM, Monkhorst K, Kenter A, Rentmeester E, Grosveld F, Grootegoed JA, Gribnau J (2009) RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell 139(5):999–1011
Kalantry S, Mills KC, Yee D, Otte AP, Panning B, Magnuson T (2006) The Polycomb group protein Eed protects the inactive X-chromosome from differentiation-induced reactivation. Nat Cell Biol 8(2):195–202
Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D (2010) Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev 24(23):2615–2620
Kanhere A, Viiri K, Araujo CC, Rasaiyaah J, Bouwman RD, Whyte WA, Pereira CF, Brookes E, Walker K, Bell GW et al (2010) Short RNAs are transcribed from repressed polycomb target genes and interact with polycomb repressive complex-2. Mol Cell 38(5):675–688
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL et al (2007) RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316(5830):1484–1488
Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J et al (2005) Antisense transcription in the mammalian transcriptome. Science 309(5740):1564–1566
Keene JD, Komisarow JM, Friedersdorf MB (2006) RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc 1(1):302–307
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A et al (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106(28):11667–11672
Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M (2011) A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods 8(7):559–564
Kiss T (2001) Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J 20(14):3617–3622
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S et al (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152(3):570–583
Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S et al (2011) Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res 71(20):6320–6326
Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17(7):909–915
Ku M, Koche RP, Rheinbay E, Mendenhall EM, Endoh M, Mikkelsen TS, Presser A, Nusbaum C, Xie X, Chi AS et al (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4(10):e1000242
Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16(22):2893–2905
Lai F, Orom UA, Cesaroni M, Beringer M, Taatjes DJ, Blobel GA, Shiekhattar R (2013) Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494(7438):497–501
Lebedeva S, Jens M, Theil K, Schwanhausser B, Selbach M, Landthaler M, Rajewsky N (2011) Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell 43(3):340–352
Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21(4):400–404
Lee JT, Lu N (1999) Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99(1):47–57
Leung AK, Young AG, Bhutkar A, Zheng GX, Bosson AD, Nielsen CB, Sharp PA (2011) Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs. Nat Struct Mol Biol 18(2):237–244
Li Y, Altman S (2002) Partial reconstitution of human RNase P in HeLa cells between its RNA subunit with an affinity tag and the intact protein components. Nucleic Acids Res 30(17):3706–3711
Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X et al (2008) HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456(7221):464–469
Lyon MF (1998) X-chromosome inactivation: a repeat hypothesis. Cytogenet Cell Genet 80(1–4):133–137
Ma Z, Swigut T, Valouev A, Rada-Iglesias A, Wysocka J (2011) Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol 18(2):120–127
Maenner S, Blaud M, Fouillen L, Savoye A, Marchand V, Dubois A, Sanglier-Cianferani S, Van Dorsselaer A, Clerc P, Avner P et al (2010a) 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol 8(1):e1000276
Maenner S, Blaud M, Fouillen L, Savoye A, Marchand V, Dubois A, Sanglier-Cianferani S, Van Dorsselaer A, Clerc P, Avner P et al (2010b) 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol 8(1):e1000276
Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R et al (2007) Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell 1(1):55–70
Mao YS, Sunwoo H, Zhang B, Spector DL (2011) Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat Cell Biol 13(1):95–101
Martianov I, Ramadass A, Serra Barros A, Chow N, Akoulitchev A (2007) Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 445(7128):666–670
Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, Galun E (2007) The H19 non-coding RNA is essential for human tumor growth. PLoS One 2(9):e845
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338
Min J, Zhang Y, Xu RM (2003) Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17(15):1823–1828
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P (2008) The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322(5908):1717–1720
Navarro P, Chambers I, Karwacki-Neisius V, Chureau C, Morey C, Rougeulle C, Avner P (2008) Molecular coupling of Xist regulation and pluripotency. Science 321(5896):1693–1695
Navarro P, Oldfield A, Legoupi J, Festuccia N, Dubois A, Attia M, Schoorlemmer J, Rougeulle C, Chambers I, Avner P (2010) Molecular coupling of Tsix regulation and pluripotency. Nature 468(7322):457–460
Navarro P, Pichard S, Ciaudo C, Avner P, Rougeulle C (2005) Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: implications for X-chromosome inactivation. Genes Dev 19(12):1474–1484
Nesterova TB, Senner CE, Schneider J, Alcayna-Stevens T, Tattermusch A, Hemberger M, Brockdorff N (2011) Pluripotency factor binding and Tsix expression act synergistically to repress Xist in undifferentiated embryonic stem cells. Epigenetics Chromatin 4(1):17
O’Neill MJ (2005) The influence of non-coding RNAs on allele-specific gene expression in mammals. Hum Mol Genet 14(Spec No 1):R113–R120
Ogawa Y, Lee JT (2003) Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell 11(3):731–743
Ogawa Y, Sun BK, Lee JT (2008) Intersection of the RNA interference and X-inactivation pathways. Science 320(5881):1336–1341
Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E (2004) Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303(5658):644–649
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H et al (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420(6915):563–573
Okutsu T, Kuroiwa Y, Kagitani F, Kai M, Aisaka K, Tsutsumi O, Kaneko Y, Yokomori K, Surani MA, Kohda T et al (2000) Expression and imprinting status of human PEG8/IGF2AS, a paternally expressed antisense transcript from the IGF2 locus, in Wilms’ tumors. J Biochem 127(3):475–483
Orom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q et al (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143(1):46–58
Pageau GJ, Hall LL, Ganesan S, Livingston DM, Lawrence JB (2007) The disappearing Barr body in breast and ovarian cancers. Nat Rev Cancer 7(8):628–633
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32(2):232–246
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N (1996) Requirement for Xist in X chromosome inactivation. Nature 379(6561):131–137
Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300(5616):131–135
Popova BC, Tada T, Takagi N, Brockdorff N, Nesterova TB (2006) Attenuated spread of X-inactivation in an X;autosome translocation. Proc Natl Acad Sci U S A 103(20):7706–7711
Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH (2008) RNA exosome depletion reveals transcription upstream of active human promoters. Science 322(5909):1851–1854
Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5(10):877–879
Rastan S (1983) Non-random X-chromosome inactivation in mouse X-autosome translocation embryos—location of the inactivation centre. J Embryol Exp Morphol 78:1–22
Rastan S, Robertson EJ (1985) X-chromosome deletions in embryo-derived (EK) cell lines associated with lack of X-chromosome inactivation. J Embryol Exp Morphol 90:379–388
Riley KJ, Steitz JA (2013) The “Observer Effect” in genome-wide surveys of protein-RNA interactions. Mol Cell 49(4):601–604
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129(7):1311–1323
Royce-Tolland ME, Andersen AA, Koyfman HR, Talbot DJ, Wutz A, Tonks ID, Kay GF, Panning B (2010) The A-repeat links ASF/SF2-dependent Xist RNA processing with random choice during X inactivation. Nat Struct Mol Biol 17(8):948–954
Sado T, Hoki Y, Sasaki H (2005) Tsix silences Xist through modification of chromatin structure. Dev Cell 9(1):159–165
Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaud AL, et al (2008) Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet 40(6):719–721
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146(3):353–358
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO (2012) Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7(2):e30733
Sanford JR, Wang X, Mort M, Vanduyn N, Cooper DN, Mooney SD, Edenberg HJ, Liu Y (2009) Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res 19(3):381–394
Sarma K, Levasseur P, Aristarkhov A, Lee JT (2010) Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc Natl Acad Sci U S A 107(51):22196–22201
Schmitz KM, Mayer C, Postepska A, Grummt I (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24(20):2264–2269
Schoeftner S, Sengupta AK, Kubicek S, Mechtler K, Spahn L, Koseki H, Jenuwein T, Wutz A (2006) Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J 25(13):3110–3122
Schor IE, Lleres D, Risso GJ, Pawellek A, Ule J, Lamond AI, Kornblihtt AR (2012) Perturbation of chromatin structure globally affects localization and recruitment of splicing factors. PLoS One 7(11):e48084
Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E (2006) RNA-mediated response to heat shock in mammalian cells. Nature 440(7083):556–560
Shibata S, Lee JT (2003) Characterization and quantitation of differential Tsix transcripts: implications for Tsix function. Hum Mol Genet 12(2):125–136
Shin J, Bossenz M, Chung Y, Ma H, Byron M, Taniguchi-Ishigaki N, Zhu X, Jiao B, Hall LL, Green MR et al (2010) Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature 467(7318):977–981
Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N (2003) Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4(4):481–495
Simon JA, Kingston RE (2013) Occupying chromatin: polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol Cell 49(5):808–824
Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE (2011) The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A 108(51):20497–20502
Srisawat C, Engelke DR (2001) Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA 7(4):632–641
Srisawat C, Goldstein IJ, Engelke DR (2001) Sephadex-binding RNA ligands: rapid affinity purification of RNA from complex RNA mixtures. Nucleic Acids Res 29(2):E4
Sun BK, Deaton AM, Lee JT (2006) A transient heterochromatic state in Xist preempts X inactivation choice without RNA stabilization. Mol Cell 21(5):617–628
Sunwoo H, Dinger ME, Wilusz JE, Amaral PP, Mattick JS, Spector DL (2009) MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19(3):347–359
Surralles J, Natarajan AT (1998) Position effect of translocations involving the inactive X chromosome: physical linkage to XIC/XIST does not lead to long-range de novo inactivation in human differentiated cells. Cytogenet Cell Genet 82(1–2):58–66
Tian D, Sun S, Lee JT (2010) The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143(3):390–403
Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V et al (2011) Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14(4):452–458
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA et al (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39(6):925–938
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329(5992):689–693
Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB (2003) CLIP identifies Nova-regulated RNA networks in the brain. Science 302(5648):1212–1215
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147(7):1537–1550
Urlaub H, Hartmuth K, Luhrmann R (2002) A two-tracked approach to analyze RNA-protein crosslinking sites in native, nonlabeled small nuclear ribonucleoprotein particles. Methods 26(2):170–181
Vasudevan S, Steitz JA (2007) AU-rich-element-mediated upregulation of translation by FXR1 and Argonaute 2. Cell 128(6):1105–1118
Walker SC, Scott FH, Srisawat C, Engelke DR (2008) RNA affinity tags for the rapid purification and investigation of RNAs and RNA-protein complexes. Methods Mol Biol 488:23–40
Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y (2004) Role of histone H2A ubiquitination in Polycomb silencing. Nature 431(7010):873–878
Wang KC, Yang YW, Liu B, Sanyal A, Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta RA et al (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472(7341):120–124
Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454(7200):126–130
Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, Ule J (2010) iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol 8(10):e1000530
Wilusz JE, Freier SM, Spector DL (2008) 3′ end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell 135(5):919–932
Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA (2012) A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26(21):2392–2407
Wilusz JE, Sunwoo H, Spector DL (2009) Long noncoding RNAs: functional surprises from the RNA world. Genes Dev 23:1494–1504
Wutz A, Rasmussen TP, Jaenisch R (2002) Chromosomal silencing and localization are mediated by different domains of Xist RNA. Nat Genet 30(2):167–174
Xu M, Medvedev S, Yang J, Hecht NB (2009) MIWI-independent small RNAs (MSY-RNAs) bind to the RNA-binding protein, MSY2, in male germ cells. Proc Natl Acad Sci U S A 106(30):12371–12376
Yang L, Duff MO, Graveley BR, Carmichael GG, Chen LL (2011a) Genomewide characterization of non-polyadenylated RNAs. Genome Biol 12(2):R16
Yang L, Lin C, Liu W, Zhang J, Ohgi KA, Grinstein JD, Dorrestein PC, Rosenfeld MG (2011b) ncRNA- and Pc2 methylation-dependent gene relocation between nuclear structures mediates gene activation programs. Cell 147(4):773–788
Yao H, Brick K, Evrard Y, Xiao T, Camerini-Otero RD, Felsenfeld G (2010) Mediation of CTCF transcriptional insulation by DEAD-box RNA-binding protein p68 and steroid receptor RNA activator SRA. Genes Dev 24(22):2543–2555
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM (2010) Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38(5):662–674
Yeo GW, Coufal NG, Liang TY, Peng GE, Fu XD, Gage FH (2009) An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16(2):130–137
Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT (2013) Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152(4):727–742
Yin QF, Yang L, Zhang Y, Xiang JF, Wu YW, Carmichael GG, Chen LL (2012) Long noncoding RNAs with snoRNA ends. Mol Cell 48(2):219–230
Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ (1995) Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378(6556):505–508
Yu W, Gius D, Onyango P, Muldoon-Jacobs K, Karp J, Feinberg AP, Cui H (2008) Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 451(7175):202–206
Zarnack K, Konig J, Tajnik M, Martincorena I, Eustermann S, Stevant I, Reyes A, Anders S, Luscombe NM, Ule J (2013) Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell 152(3):453–466
Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, Darnell RB (2010) Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329(5990):439–443
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT (2010) Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40(6):939–953
Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322(5902):750–756
Zhou W, Zhu P, Wang J, Pascual G, Ohgi KA, Lozach J, Glass CK, Rosenfeld MG (2008) Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29(1):69–80
Zhou Z, Licklider LJ, Gygi SP, Reed R (2002) Comprehensive proteomic analysis of the human spliceosome. Nature 419(6903):182–185
Zhu S, Zhang X-O, Yang L (2013) Panning for Long Noncoding RNAs. Biomolecules 3(1):226–241
Zuo T, Wang L, Morrison C, Chang X, Zhang H, Li W, Liu Y, Wang Y, Liu X, Chan MW et al (2007) FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 129(7):1275–1286
Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, Yeo GW (2010) Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol 17(2):173–179
Acknowledgements
We are grateful to Li Yang for critical reading of the manuscript. This work is supported by an AACR-Aflac, Inc., Career Development Award for Pediatric Cancer Research (J.C.Z.) and grants XDA01010206, 2012SSTP01 from CAS and grant 31271376 from NSFC (L.-L. Chen).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Chen, LL., Zhao, J.C. (2014). Functional Analysis of Long Noncoding RNAs in Development and Disease. In: Yeo, G. (eds) Systems Biology of RNA Binding Proteins. Advances in Experimental Medicine and Biology, vol 825. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1221-6_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1221-6_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1220-9
Online ISBN: 978-1-4939-1221-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)