
Abstract
In the nervous system, various unique glycans not found in other tissues are expressed on glycoproteins, and their expression/functions have been studied using specific antibodies/lectins. Among brain-specific glycans in mammals, we focus on human natural killer-1 (HNK-1) and related Cat-315 epitopes, which can be detected using specific antibodies. It is known that the HNK-1 epitope is expressed on N- and O-mannosylated glycans and that Cat-315 mAb preferentially recognizes the HNK-1 epitope on brain-specific “branched O-mannose glycan.” The β1,6-branched O-mannose structure is synthesized by a brain-specific glycosyltransferase, N-acetylglucosaminyltransferase-IX (GnT-IX, also designated as GnT-Vb). Using GnT-IX gene-deficient mice and specific antibodies/lectins, the function of GnT-IX was found to be quite different from that of its ubiquitous homologue, GnT-V. Using Cat-315 mAb, the receptor protein tyrosine phosphatase-beta (RPTPβ) was identified as an in vivo target glycoprotein for GnT-IX. Analysis of the function of branched O-mannose glycan on RPTPβ indicated that its loss promoted the recovery process after myelin injury (called remyelination) in brain and that this phenomenon is probably caused in vivo by reduced activation of astrocytes in GnT-IX-deficient brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
6.1 Introduction
The mammalian nervous system is composed of various cell types, including neurons, astrocytes, myelinating cells (oligodendrocytes and Schwann cells), and microglia. Each type of cell communicates with the others to elaborate a complicated neural network. In the nervous system, unique glycan structures are expressed which are quite different from those in other tissues and are exemplified by the human natural killer-1 (HNK-1) epitope and polysialic acid (PSA) (Kleene and Schachner 2004). Accumulating evidence has shown that brain-specific glycans are essential for maintaining brain functions, such as learning/memory, cognition, and behavior (Weinhold et al. 2005; Yamamoto et al. 2002). Specific lectins and antibodies are useful for both carbohydrate detection and expression/functional studies. For instance, good monoclonal antibodies for detecting HNK-1 and PSA, anti-HNK-1 and 12E3 mAbs, respectively, are now widely used (Yamamoto et al. 2000; 2002). In addition to the use of these antibodies for the identification of target glycoproteins (e.g., neural cell adhesion molecule), they are used for elucidation of the biosynthetic pathways of glycosylation.
Lectins, proteins which recognize specific glycan structures, are now widely used to detect glycans. Plant lectins such as conA (concanavalin A), WGA (wheat germ agglutinin), PHA (phytohemagglutinin), etc. (Sharon and Lis 2004), are often used to characterize glycans in cells or tissues. Meanwhile, in mammals, the functions of animal lectins have been well studied in the immune system. For instance, some Siglecs (sialic acid-binding immunoglobulin type lectins) or C-type lectins are specifically expressed in subsets of immune cells and involved in regulation of immune cell activity or recognition of pathogens (Crocker et al. 2007; Hardison and Brown 2012). Recent advances in lectin biology allowed us to perform cellular glycomic analyses using lectin microarrays (Tateno et al. 2010). Although lectins are a useful tool, their affinity and specificity for their ligand glycans are often lower than that of anti-carbohydrate antibodies, which sometimes makes it difficult to use them to determine glycan structure.
This chapter focuses on a brain-specific glycan, the HNK-1 epitope. We describe its expression on branched O-mannose glycans and the function of a brain-specific glycosyltransferase, N-acetylglucosaminyltransferase-IX (GnT-IX) (Inamori et al. 2006), in its synthesis. Using specific antibodies/lectins, an in vivo target glycoprotein expressing this modification was identified. Using a mouse model, we also found that branched O-mannose glycans play key roles in the remyelination process after myelin injury by regulating astrocyte activation (Kanekiyo et al. 2013).
6.2 HNK-1 Epitope
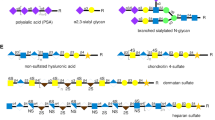
The HNK-1 epitope was first found as an antigen on the surface of human natural killer cells in 1981 (Abo and Balch 1981). However, subsequent studies revealed that the HNK-1 glycan is almost exclusively found in the nervous system (Kruse et al. 1984). The glycan structure of HNK-1 was identified as HSO3-3GlcAβ1-3Galβ1-4GlcNAc- (Fig. 6.1) (Ariga et al. 1987; Chou et al. 1986; Tokuda et al. 1998). The terminal sulfated glucuronic acid residue is a rare structure in mammalian N- and O-glycans other than glycosaminoglycans. A key step in its biosynthesis is the addition of GlcA, and the major enzyme responsible for this step, glucuronyltransferase-P (GlcAT-P), is specifically expressed in the nervous system (Terayama et al. 1997). Later, by expression cloning and use of an HNK-1 mAb, the sulfotransferase designated HNK-1ST was cloned (Bakker et al. 1997).

Putative structure of the Cat-315 epitope and unique O-mannose glycan structure on αDG. Cat-315-reactive glycan, which possesses terminal HNK-1 epitope(s), is specifically expressed in brain. The Cat-315-reactive glycan is expressed on RPTPβ and other molecules expressed in brain. In contrast, unique phosphorylated O-mannose glycans that have never been identified on other glycoproteins are expressed on αDG. Phosphorylated O-mannose glycan is essential for binding to ligands or IIH6 mAb
Function of the HNK-1 glycan was examined using GlcAT-P-deficient mice in which HNK-1 mAb reactivity almost disappears. GlcAT-P-deficient mice have reduced synaptic plasticity and impaired learning/memory functions (Yamamoto et al. 2002). Spine (postsynapse) structure in primary neurons from GlcAT-P-deficient brains was consistently abnormally immature (Morita et al. 2009b). To explore the molecular mechanism by which the HNK-1 glycan regulates spine maturation and learning, HNK-1 mAb was used to identify glycoproteins expressing the HNK-1 glycan. This resulted in identification of the glutamate receptor subunit GluA2 (Morita et al. 2009a). Compared with other HNK-1-carrying glycoproteins, GluA2 is highly enriched in the postsynaptic density fraction, and among glutamate receptor subunits was the one selectively modified with HNK-1. GluA2 is also known to be an essential molecule for spine maturation and synaptic plasticity (Isaac et al. 2007), and the loss of HNK-1 in GlcAT-P-deficient neurons causes instability of GluA2 at the synaptic membrane. These findings strongly suggest that the HNK-1 glycan on GluA2 plays a key role in learning and memory function in vivo.
HNK-1 glycan is expressed on both N-glycan and O-mannose glycans in brain. Although the glycan structure and carrier glycoprotein for N-linked HNK-1 are well characterized (Kruse et al. 1984; Morita et al. 2009a; Voshol et al. 1996), the expression pattern and function of O-mannose-linked HNK-1 are not. In the following sections, the expression and function of O-mannose glycans in brain are described prior to focusing on brain-specific “branched” O-mannose glycans carrying a terminal HNK-1 modification.
6.3 O-Mannose Glycans in the Brain
Although O-mannose glycans are abundantly expressed in mammalian brain, with evidence that one third of brain O-glycans are O-mannosylated (Chai et al. 1999), their functions remain to be clarified. The most well-studied O-mannosylated glycoprotein in mammals is α-dystroglycan (αDG), and a number of studies have demonstrated that O-mannosylation of αDG is essential for skeletal muscle function in mice and humans (Godfrey et al. 2011). Previous reports have shown that αDG binds to ligands such as laminin or pikachurin via its unusual phosphorylated O-mannose glycan, which was detected using the IIH6 mAb (Fig. 6.1) (Chiba et al. 1997; Inamori et al. 2012; Sato et al. 2008; Yoshida-Moriguchi et al. 2010). However, in brain, the content of O-mannose glycan on αDG is almost negligible (Stalnaker et al. 2011), indicating that the O-mannose glycan in brain modulates functions of glycoproteins other than αDG. To date, several neural glycoproteins such as neurocan, neurofascin 186, and CD24, enriched and purified using specific lectins or antibodies, have been shown to be modified by O-mannose glycans (Bleckmann et al. 2009; Pacharra et al. 2012; 2013); however, the functions of their O-mannose glycans are still unclear. Meanwhile, a neural phosphatase, receptor protein tyrosine phosphatase-beta (RPTPβ), was found to be modified by branched O-mannose glycans with terminal HNK-1 epitope(s) (Abbott et al. 2008; Kanekiyo et al. 2013). These glycans are detected by the HNK-1 mAb as well as the Cat-315 mAb. Although the epitope for the Cat-315 mAb has not been accurately determined, it binds preferentially to O-mannosylated HNK-1 (6.1) (Dwyer et al. 2012). Moreover, the major glycoprotein carrying the Cat-315 epitope changes from RPTPβ to aggrecan during mouse brain development (Dino et al. 2006). Using these specific mAbs, the biological function of O-mannose modifications in brain was studied as described below.
Expression in brain of a “branched” O-mannose glycan not found in O-mannosylated glycans from other tissues (Figs. 6.1 and 6.2) (Chai et al. 1999; Yuen et al. 1997) indicated the unique activity of a branching enzyme present specifically in brain. We and others identified the brain-specific enzyme as GnT-IX (Vb).

Structures of a branched O-mannose glycan and an N-glycan. GnT-IX catalyzes formation of the β1,6-branch of an O-mannose glycan. GnT-V catalyzes β1,6-branch formation on the α6-mannose arm in an N-glycan. GnT-IX also shows modest activity toward N-glycans in vitro
6.4 GnT-IX Is a Brain-Specific O-Mannose Branching Enzyme
GnT-IX was originally identified in silico as a homologue of GnT-V by both our laboratory and that of Dr. Pierce (Inamori et al. 2003; Kaneko et al. 2003), and for that reason, GnT-IX is also designated as GnT-Vb. GnT-V is ubiquitously expressed and is responsible for N-glycan branching at the α6-mannose arm (Fig. 6.2). Many studies have shown that GnT-V is involved in cancer progression and growth factor receptor signaling (Granovsky et al. 2000; Lau et al. 2007). In brain, the GnT-V product, which can be detected with the L4-PHA lectin, is involved in depression-like behavior (Soleimani et al. 2008). Although GnT-IX shows high sequence identity to GnT-V (42 % in the case of humans), its expression and function are different from those of GnT-V. GnT-IX is highly specific to brain (Kizuka et al. 2011) and exhibits weak activity toward N-glycans in vitro. We and others have reported that GnT-IX has preferential branching activity toward O-mannose glycans (Fig. 6.2) (Alvarez-Manilla et al. 2010; Inamori et al. 2004). As described above, branched O-mannosyl glycans terminally modified with sialic acid, Lewis epitope, or HNK-1 are found only in the brain (Chai et al. 1999; Yuen et al. 1997). To date, no lectin/antibody has been developed that can specifically detect all O-mannose glycans or distinguish linear O-mannose glycans from branched ones. However, using some specific detection probes and mouse models, the functions and expression patterns of O-mannose glycans in brain have been gradually identified.
6.5 In Vivo Enzymatic Functions of GnT-IX and GnT-V
Our group and Dr. Pierce’s group independently generated GnT-IX (Vb) gene-deficient mice (Kanekiyo et al. 2013; Lee et al. 2012). These mutant mice showed no overt phenotype in terms of brain morphology and fertility. However, they showed reduced reactivity to both Cat-315 and HNK-1 mAbs, supporting the idea that Cat-315 recognizes O-mannosylated glycans with terminal HNK-1 modification(s) in brain. Using immunoprecipitation, we identified RPTPβ as one of the major target proteins for GnT-IX in brain, consistent with previous findings in cultured cells (Abbott et al. 2008). Dr. Pierce’s group analyzed glycan structures from GnT-V- or GnT-IX-deficient brain and found that GnT-IX acts on O-mannose glycans but not on N-glycans and that GnT-IX cannot compensate for GnT-V’s function in the biosynthesis of N-glycans in vivo (Lee et al. 2012). In addition, lectin blot analysis using L4-PHA showed that loss of lectin reactivity is solely caused by knockout of GnT-V but not by knockout of GnT-IX, indicating that GnT-V acts on N-glycans, while GnT-IX acts on O-mannose glycans in vivo (Kanekiyo et al. 2013). These findings indicate that the in vivo function of GnT-IX is different from that of its homologue GnT-V. Moreover, Dr. Pierce’s group observed no apparent change in αDG binding to IIH6 mAb or laminin in GnT-IX-deficient mouse brain tissue, indicating that the target glycoprotein for GnT-IX function is probably not αDG.
6.6 GnT-IX-Deficient Mice Show Enhanced Recovery from Demyelinating Damage
Based on the fact that a target of GnT-IX, RPTPβ, is known to be critical for recovery from demyelinating damage in vivo (Harroch et al. 2002), we hypothesized that O-mannose glycans on RPTPβ produced by GnT-IX are also involved in the demyelination/remyelination process. Demyelination is found in many pathological conditions including multiple sclerosis, and promoting remyelination is a rational strategy for treating demyelinating diseases (Fancy et al. 2011). We induced demyelination in wild-type and GnT-IX-deficient mice using the copper chelator “cuprizone,” which induces oligodendrocyte damage through oxidative stress (Torkildsen et al. 2008). In wild-type mouse brain, myelin is progressively damaged by cuprizone. In contrast, in GnT-IX-deficient mouse brain, once early-phase demyelination has occurred as in the case of wild-type mice, significant recovery of myelin (remyelination) was observed (Fig. 6.3) (Kanekiyo et al. 2013).

Recovery from myelin injury is promoted in GnT-IX-deficient mice. Myelin in the corpus callosum was stained with anti-myelin basic protein antibody. Cuprizone feeding for 4 weeks causes demyelination in both wild-type and GnT-IX-deficient mice. Further treatment with cuprizone causes more severe demyelination in wild-type mice, while GnT-IX-deficient mice show recovery from myelin damage (remyelination). Figure is modified from that published previously by Kanekiyo et al. (2013)
As a potential mechanism for this effect, we found that astrocyte activation was reduced in the damaged region in GnT-IX-deficient brain (Kanekiyo et al. 2013). In wild-type brain, Cat-315-positive astrocytes accumulated in demyelinated corpus callosum, and astrocyte activation is one of the mechanisms underlying suppression of the remyelination process by inhibiting oligodendrocyte differentiation from oligodendrocyte precursor cells (OPCs) (Wang et al. 2011). Concomitantly, oligodendrocyte lineage analysis revealed that oligodendrocyte differentiation is actually enhanced in GnT-IX-deficient mice (Kanekiyo et al. 2013). Based on these data, we suggest that the branched O-mannose glycans on RPTPβ are involved in astrocyte activation, which probably suppresses oligodendrocyte differentiation and remyelination. A schematic model for the role of branched O-mannosyl glycans on astrocytic RPTPβ in remyelination is shown in Fig. 6.4. Cultured primary astrocytes isolated from nascent pups also showed decreased activation in vitro (Kanekiyo et al. 2013), strongly suggesting that GnT-IX can regulate the astrocyte activation process with or without cuprizone treatment. Further analysis of the role of GnT-IX is needed to clarify how branched O-mannose glycans are involved in astrocyte activation. On the basis of these findings, we suggest that targeting of protein glycosylation may be a novel therapeutic strategy for demyelinating disorders.

Putative model for the regulatory role of branched O-mannosyl glycans on astrocytic RPTPβ in remyelination after myelin injury. Cuprizone administration causes reversible demyelination. Activated astrocytes are known to inhibit differentiation of oligodendrocyte precursor cells, leading to impairment of remyelination. In the absence of branched O-mannosyl glycans (GnT-IX knockout), astrocyte activation is suppressed, and remyelination is enhanced. Figure modified from the paper by Kanekiyo et al. (2013)
No conflict of interest is declared.
References
Abbott KL, Matthews RT, Pierce M. Receptor tyrosine phosphatase beta (RPTPbeta) activity and signaling are attenuated by glycosylation and subsequent cell surface galectin-1 binding. J Biol Chem. 2008;283:33026–35.
Abo T, Balch CM. A differentiation antigen of human NK and K cells identified by a monoclonal antibody (HNK-1). J Immunol. 1981;127:1024–9.
Alvarez-Manilla G, Troupe K, Fleming M, Martinez-Uribe E, Pierce M. Comparison of the substrate specificities and catalytic properties of the sister N-acetylglucosaminyltransferases, GnT-V and GnT-Vb (IX). Glycobiology. 2010;20:166–74.
Ariga T, et al. Characterization of sulfated glucuronic acid containing glycolipids reacting with IgM M-proteins in patients with neuropathy. J Biol Chem. 1987;262:848–53.
Bakker H, Friedmann I, Oka S, Kawasaki T, Nifant'ev N, Schachner M, Mantei N. Expression cloning of a cDNA encoding a sulfotransferase involved in the biosynthesis of the HNK-1 carbohydrate epitope. J Biol Chem. 1997;272:29942–6.
Bleckmann C, et al. O-glycosylation pattern of CD24 from mouse brain. Biol Chem. 2009;390:627–45.
Chai W, Yuen CT, Kogelberg H, Carruthers RA, Margolis RU, Feizi T, Lawson AM. High prevalence of 2-mono- and 2,6-di-substituted manol-terminating sequences among O-glycans released from brain glycopeptides by reductive alkaline hydrolysis. Eur J Biochem. 1999;263:879–88.
Chiba A, Matsumura K, Yamada H, Inazu T, Shimizu T, Kusunoki S, Kanazawa I, Kobata A, Endo T. Structures of sialylated O-linked oligosaccharides of bovine peripheral nerve alpha-dystroglycan. The role of a novel O-mannosyl-type oligosaccharide in the binding of alpha-dystroglycan with laminin. J Biol Chem. 1997;272:2156–62.
Chou DK, Ilyas AA, Evans JE, Costello C, Quarles RH, Jungalwala FB. Structure of sulfated glucuronyl glycolipids in the nervous system reacting with HNK-1 antibody and some IgM paraproteins in neuropathy. J Biol Chem. 1986;261:11717–25.
Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–66.
Dino MR, Harroch S, Hockfield S, Matthews RT. Monoclonal antibody Cat-315 detects a glycoform of receptor protein tyrosine phosphatase beta/phosphacan early in CNS development that localizes to extrasynaptic sites prior to synapse formation. Neuroscience. 2006;142:1055–69.
Dwyer CA, Baker E, Hu H, Matthews RT. RPTPzeta/phosphacan is abnormally glycosylated in a model of muscle-eye-brain disease lacking functional POMGnT1. Neuroscience. 2012;220:47–61.
Fancy SP, Chan JR, Baranzini SE, Franklin RJ, Rowitch DH. Myelin regeneration: a recapitulation of development? Annu Rev Neurosci. 2011;34:21–43.
Godfrey C, Foley AR, Clement E, Muntoni F. Dystroglycanopathies: coming into focus. Curr Opin Genet Dev. 2011;21:278–85.
Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med. 2000;6:306–12.
Hardison SE, Brown GD. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol. 2012;13:817–22.
Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, Schlessinger J. A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat Genet. 2002;32:411–4.
Inamori K, Endo T, Ide Y, Fujii S, Gu J, Honke K, Taniguchi N. Molecular cloning and characterization of human GnT-IX, a novel beta1,6-N-acetylglucosaminyltransferase that is specifically expressed in the brain. J Biol Chem. 2003;278:43102–9.
Inamori K, et al. N-Acetylglucosaminyltransferase IX acts on the GlcNAc beta 1,2-Man alpha 1-Ser/Thr moiety, forming a 2,6-branched structure in brain O-mannosyl glycan. J Biol Chem. 2004;279:2337–40.
Inamori K, Mita S, Gu J, Mizuno-Horikawa Y, Miyoshi E, Dennis JW, Taniguchi N. Demonstration of the expression and the enzymatic activity of N-acetylglucosaminyltransferase IX in the mouse brain. Biochim Biophys Acta. 2006;1760:678–84.
Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, Campbell KP. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–6.
Isaac JT, Ashby MC, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–71.
Kanekiyo K, et al. Loss of branched o-mannosyl glycans in astrocytes accelerates remyelination. J Neurosci. 2013;33:10037–47.
Kaneko M, Alvarez-Manilla G, Kamar M, Lee I, Lee JK, Troupe K, Zhang W, Osawa M, Pierce M. A novel beta(1,6)-N-acetylglucosaminyltransferase V (GnT-VB)(1). FEBS Lett. 2003;554:515–9.
Kizuka Y, Kitazume S, Yoshida M, Taniguchi N. Brain-specific expression of N-acetylglucosaminyltransferase IX (GnT-IX) is regulated by epigenetic histone modifications. J Biol Chem. 2011;286:31875–84.
Kleene R, Schachner M. Glycans and neural cell interactions. Nat Rev Neurosci. 2004;5:195–208.
Kruse J, Mailhammer R, Wernecke H, Faissner A, Sommer I, Goridis C, Schachner M. Neural cell adhesion molecules and myelin-associated glycoprotein share a common carbohydrate moiety recognized by monoclonal antibodies L2 and HNK-1. Nature. 1984;311:153–5.
Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, Dennis JW. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–34.
Lee JK, Matthews RT, Lim JM, Swanier K, Wells L, Pierce JM. Developmental expression of the neuron-specific N-acetylglucosaminyltransferase Vb (GnT-Vb/IX) and identification of its in vivo glycan products in comparison with those of its paralog, GnT-V. J Biol Chem. 2012;287:28526–36.
Morita I, Kakuda S, Takeuchi Y, Itoh S, Kawasaki N, Kizuka Y, Kawasaki T, Oka S. HNK-1 glyco-epitope regulates the stability of the glutamate receptor subunit GluR2 on the neuronal cell surface. J Biol Chem. 2009a;284:30209–17.
Morita I, Kakuda S, Takeuchi Y, Kawasaki T, Oka S. HNK-1 (human natural killer-1) glyco-epitope is essential for normal spine morphogenesis in developing hippocampal neurons. Neuroscience. 2009b;164:1685–94.
Pacharra S, Hanisch FG, Breloy I. Neurofascin 186 is O-mannosylated within and outside of the mucin domain. J Proteome Res. 2012;11:3955–64.
Pacharra S, Hanisch FG, Muhlenhoff M, Faissner A, Rauch U, Breloy I (2013) The lecticans of mammalian brain perineural net are O-mannosylated. J Proteome Res. 12(4):1764-71
Sato S, et al. Pikachurin, a dystroglycan ligand, is essential for photoreceptor ribbon synapse formation. Nat Neurosci. 2008;11:923–31.
Sharon N, Lis H. History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology. 2004;14:53R–62.
Soleimani L, Roder JC, Dennis JW, Lipina T. Beta N-acetylglucosaminyltransferase V (Mgat5) deficiency reduces the depression-like phenotype in mice. Genes Brain Behav. 2008;7:334–43.
Stalnaker SH, et al. Glycomic analyses of mouse models of congenital muscular dystrophy. J Biol Chem. 2011;286:21180–90.
Tateno H, Kuno A, Itakura Y, Hirabayashi J. A versatile technology for cellular glycomics using lectin microarray. Methods Enzymol. 2010;478:181–95.
Terayama K, Oka S, Seiki T, Miki Y, Nakamura A, Kozutsumi Y, Takio K, Kawasaki T. Cloning and functional expression of a novel glucuronyltransferase involved in the biosynthesis of the carbohydrate epitope HNK-1. Proc Natl Acad Sci U S A. 1997;94:6093–8.
Tokuda A, Ariga T, Isogai Y, Komba S, Kiso M, Hasegawa A, Tai T, Yu RK. On the specificity of anti-sulfoglucuronosyl glycolipid antibodies. J Carbohydr Chem. 1998;17:535–46.
Torkildsen O, Brunborg LA, Myhr KM, Bo L. The cuprizone model for demyelination. Acta Neurol Scand Suppl. 2008;188:72–6.
Voshol H, van Zuylen CW, Orberger G, Vliegenthart JF, Schachner M. Structure of the HNK-1 carbohydrate epitope on bovine peripheral myelin glycoprotein P0. J Biol Chem. 1996;271:22957–60.
Wang Y, Cheng X, He Q, Zheng Y, Kim DH, Whittemore SR, Cao QL. Astrocytes from the contused spinal cord inhibit oligodendrocyte differentiation of adult oligodendrocyte precursor cells by increasing the expression of bone morphogenetic proteins. J Neurosci. 2011;31:6053–8.
Weinhold B, Seidenfaden R, Rockle I, Muhlenhoff M, Schertzinger F, Conzelmann S, Marth JD, Gerardy-Schahn R, Hildebrandt H. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem. 2005;280:42971–7.
Yamamoto N, Inui K, Matsuyama Y, Harada A, Hanamura K, Murakami F, Ruthazer ES, Rutishauser U, Seki T. Inhibitory mechanism by polysialic acid for lamina-specific branch formation of thalamocortical axons. J Neurosci. 2000;20:9145–51.
Yamamoto S, et al. Mice deficient in nervous system-specific carbohydrate epitope HNK-1 exhibit impaired synaptic plasticity and spatial learning. J Biol Chem. 2002;277:27227–31.
Yoshida-Moriguchi T, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92.
Yuen CT, Chai W, Loveless RW, Lawson AM, Margolis RU, Feizi T. Brain contains HNK-1 immunoreactive O-glycans of the sulfoglucuronyl lactosamine series that terminate in 2-linked or 2,6-linked hexose (mannose). J Biol Chem. 1997;272:8924–31.
Conflict of Interest
The author declares that he has no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kizuka, Y., Kanekiyo, K., Kitazume, S., Taniguchi, N. (2014). Use of Glycan-Targeted Antibodies/Lectins to Study the Expression/Function of Glycosyltransferases in the Nervous System. In: Yu, R., Schengrund, CL. (eds) Glycobiology of the Nervous System. Advances in Neurobiology, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1154-7_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1154-7_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1153-0
Online ISBN: 978-1-4939-1154-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)