
Abstract
Epilepsy is characterized by the periodic occurrence of seizures. Currently available anticonvulsant drugs and therapies are insufficient to controlling seizures in about one third of patients. Thus, there is an urgent need for new therapies that prevent generation of the disorder and improve seizure control in individuals already afflicted. The vast majority of epileptic cases are of idiopathic origin with their underlying mechanisms being unclear. Neurosurgical specimens from patients presenting with mesial temporal lobe epilepsy (MTLE) demonstrate marked reactive gliosis. Since recent studies have implicated astrocytes in important physiological roles in the central nervous system, such as synchronization of neuronal firing, it is plausible they may also have a role in seizure generation and/or seizure spread. In support of this view, various membrane channels, receptors and transporters in astrocytic membranes are altered in the epileptic brain. Excitingly, recent evidence suggests that in the course of the pathogenesis of MTLE, these glial changes alter homeostatic network functions and temporally precede the alterations in neurons. These findings might eventually classify MTLE as a glial rather than a neuronal disorder, and identify astrocytes as promising new targets for the development of more specific antiepileptic therapeutic strategies.
This chapter summarizes current evidence of astrocyte dysfunction in epilepsy and discusses presumed underlying mechanisms. Although research on astrocytes in epilepsy is still in its infancy, the review clearly demonstrate a critical role of astrocytes in the disturbance of K+ and transmitter homeostasis and its impact on seizure generation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Epilepsy
- Astrocyte
- Gap junction
- Connexin
- Pannexin
- K+ clearance
- Kir channel
- Gliotransmitter release
- Adenosine kinase
- Glutamine synthetase
- Inflammation
- AQP4 channel
8.1 Introduction
Astrocytes are active partners in neural information processing. Advanced electrophysiological and Ca2+ imaging techniques unraveled that these cells express a similar spectrum of ion channels and transmitter receptors as neurons, which allows them to sense and respond to neuronal activity. Despite the fact that the pathways enabling activation of astrocytes under physiological conditions are still ill-determined, evidence is emerging suggesting a critical role of astrocyte dysfunction in the pathogenesis of neurological disorders (Seifert et al. 2006) . In this review we will discuss recent work on specimens from patients with pharmacoresistant mesial temporal lobe epilepsy (MTLE) and corresponding animal models of epilepsy , which revealed alterations in expression, subcellular localization and function of astroglial K+ and water channels, resulting in impaired K+ buffering. Dysfunction of glutamate transporters and the astrocytic enzymes, glutamine synthetase (GS) and adenosine kinase (ADK) , as observed in epileptic tissue suggested that impaired astrocyte function is causative of hyperexcitation, seizure spread and neurotoxicity. Increasing evidence suggests that proinflammatory mediators cause dysfunctions in astrocytes, which individually orinconcert provoke neuronal hyperexcitability. Accordingly, astrocytes should be considered as promising targets for new therapeutic strategies. We will summarize current knowledge of astrocyte dysfunction in MTLE and discuss putative mechanisms underlying these alterations.
8.2 Impaired K+ Buffering in Epilepsy
8.2.1 K+ Uptake and K+ Spatial Buffering
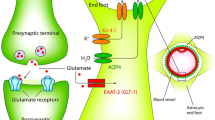
Intense neuronal activity elicits transient increases in the extracellular potassium concentration ([K+]o) which under pathological conditions like epilepsy can reach values of up to 10–12 mM (Heinemann and Lux 1977) . Even moderate rises in [K+]o have been shown to significantly increase neuronal excitability and synaptic transmission (Balestrino and Somjen 1986; Walz 2000) , underscoring the necessity of tight control of K+ homeostasis for normal brain function . This task is mainly accomplished by astrocytes which are characterized by a very negative resting potential and a high resting permeability for K+. Responsible for these glial membrane properties are essentially inwardly rectifying K+ channels of the Kir4.1 subtype (Seifert et al., 2009) . Astrocytes control [K+]o by two mechanisms: K+ uptake and spatial buffering (for review see (Kofuji and Newman 2004) . Net uptake of K+ is mainly mediated by Na+/K+ pumps and Na+/K+/Cl− cotransporters and to a minor extent by Kir4.1 channels (D’Ambrosio et al. 2002; Kofuji and Newman 2004; Ransom et al. 2000) . It is rather unlikely that this mechanism alone is sufficient for efficient clearance of excess [K+]o since intracellular K+ accumulation results in water influx and cell swelling. The spatial buffering model (Fig. 8.1) (Orkand et al. 1966) describes another, more effective mechanism for [K+]o clearance. It is based on the fact that astrocytes are electrically connected to each other via gap junction (GJ) channels to form a functional syncytium. According to the model, excessive extracellular K+ is taken up by astrocytes at sides of high neuronal activity redistributed through the astrocytic network to be released at regions of lower [K+]o. Here, uptake and release of K+ occur passively, via passive diffusion through weakly-rectifying Kir4.1 channels. These channels are particularly well suited for this task because they possess a high open probability at resting potential and their conductance increases at high [K+]o (Ransom and Sontheimer 1995) . Intercellular K +diffusion is also energy-independent, driven by the electrical gradient between the depolarized potential of glial cells at sides of K+ entry and the more negative membrane potential of the glial syncytium (Orkand et al. 1966; Walz 2000; Kofuji and Newman 2004) .

space-dependent spatial K+ buffering. Activity in a group of neurons has produced local increase in [K+]o to 12 mM (shaded area, left). This provokes a more positive membrane potential (Vm) that passively spreads through the coupled astrocytes. The positive shift of the K+ equilibrium potential (EK) is stronger than that of Vm of the cell exposed to high [K+]o because the latter is “clamped” by the neighboring, more negative astrocytes exposed to lower (normal) [K+]o (4 mM). The difference between EK and Vm drives K+ inward at the region where it is raised and outward at distant regions. The result is a net flux of K+ away from the region where it has accumulated extracellularly. Average [K+]i is not affected. The graph shows the distribution of EK and Vm as a function of distance along the astroglial syncytium. (Modified from Orkand and Ann (1986), reproduced with permission)
8.2.2 Kir4.1 Channels and K+ Buffering in Epilepsy
Increased [K+]o has been associated with the pathophysiology of epilepsy (Moody et al. 1974; Fisher et al. 1976; Lothman and Somjen 1976) , and it is known that high [K+]o is sufficient to trigger epileptiform activity in vitro (Traynelis and Dingledine 1988) . To assess the impact of Kir4.1 channels in K+ buffering , the effect of Ba2+-induced Kir channel block on stimulus-triggered rises in [K+]o or iontophoretically applied K+ was analyzed in sclerotic and non-sclerotic hippocampal slices from rat and man. It could be shown that Ba2+ significantly enhanced [K+]o accumulation under control conditions, but had no effect in sclerotic hippocampi. These findings provided evidence for the disturbance of Ba2+-sensitive K+-uptake in sclerosis (Heinemann et al. 2000; Jauch et al. 2002; Kivi et al. 2000) . Confirmation for this hypothesis came from patch clamp analysis, demonstrating significantly reduced Kir currents in the sclerotic CA1 region of neurosurgical specimens from patients presenting with MTLE (Hinterkeuser et al. 2000; Schröder et al. 2000; Bordey and Sontheimer 1998) . Moreover, Western blot revealed a 50 % down-regulation of Kir4.1 protein in human hippocampal sclerosis (HS) compared to post-mortem controls (Das et al. 2012) . In a recent study, (Heuser et al. 2012) used immunohistochemistry to examine the distribution of Kir4.1 in hippocampi from MTLE patients. They found significantly reduced astrocytic Kir4.1 immunoreactivity in patients with HS compared to non-sclerotic and autopsy controls. Interestingly, the reduction was most pronounced around vessels and presumably caused by disruption of the dystrophin-associated protein complex in astrocytic end-feet (Heuser et al. 2012). Together, these studies imply that impaired K+ clearance and increased seizure susceptibility in MTLE-HS result from reduced expression of Kir4.1 channels. However, it remains an open question whether this reduction represents cause, effect or adaptive response in TLE. In favor of a causative role for altered Kir channel expression in epilepsy , (David et al. 2009) showed in an albumin model of epilepsy that Kir4.1 down-regulation occurs before the onset of epileptic activity. Another study, however, reported no changes in astrocytic Kir currents 7–16 days following systemic injection of kainate, implying that Kir down-regulation represents a consequential event in epilepsy (Takahashi et al. 2010) .
Further support for the crucial role of Kir4.1 in glial K+ buffering emerged from the phenotype of Kir4.1 knockout mice (Djukic et al. 2007; Kofuji et al. 2000) . Global deletion of the Kir4.1 encoding gene, KCNJ10, resulted in marked motor impairments and premature death (postnatal day 8 (P8) to P24) (Neusch et al. 2001) . Mice with glia-specific deletion of Kir4.1 (cKir4.1−/− mice) displayed a similarly severe phenotype, including ataxia, seizures and early lethality before P30. At the cellular level, these mice showed substantial depolarization of gray matter astrocytes and consequently severely impaired astrocytic K+ and glutamate uptake (Djukic et al. 2007). Similar results were obtained after down-regulation of Kir4.1 by RNAi in cultured astrocytes (Kucheryavykh et al. 2007) . Follow-up studies performed on cKir4.1−/− animals substantiated the crucial role of Kir4.1 channels in K+ buffering and demonstrated that loss of Kir4.1 expression causes epilepsy (Chever et al. 2010; Haj-Yasein et al. 2011) .
Missense variations in the KCNJ10 gene have been linked to seizure susceptibility in man (Buono et al. 2004) . Loss-of-function mutations in KCNJ10 underlie an autosomal recessive disorder characterized by seizures , ataxia, sensorineural deafness, mental retardation and tubulopathy (EAST/SeSAME syndrome) (Bockenhauer et al. 2009; Scholl et al. 2009; Reichold et al. 2010; Williams et al. 2010) . Patients suffering from this disorder display focal and generalized tonic-clonic seizures since childhood. Another study reported that autism with seizures and intellectual disability is tentatively linked to gain-of-function mutations in KCNJ10 (Sicca et al. 2011) . Heuser et al. showed that a combination of three single nucleotide polymorphisms (SNPs) in the aquaporin 4 (AQP4) gene (encoding a water channel) together with two SNPs in the KCNJ10 gene was associated with MTLE (Heuser et al. 2010) . Association analysis in MTLE patients with a history of febrile seizures (FS) versus such without FS revealed that a combination of SNPs in KCNJ10, AQP4, and the area between KCNJ10 and KCNJ9 was significantly associated with MTLE-FS (Heuser et al. 2010).
8.3 GJ Communication in Epilepsy
8.3.1 Potential Roles of GJs in Epilepsy
Astrocytes in the adult brain are connected to each other via GJ channels composed of connexin43 (Cx43) and Cx30 (Nagy and Rash 2000) , allowing intercellular exchange of ions, second messengers, nutritional metabolites and amino acids. The astroglial syncytium has important functions, including spatial buffering of K+ ions (see above), trafficking and delivery of energetic metabolites to neurons (Giaume et al. 1997) , intercellular propagation of Ca2+waves (Scemes and Giaume 2006) , volume regulation (Scemes and Spray 1998), and adult neurogenesis (Kunze et al. 2009) ; reviewed by (Pannasch and Rouach 2013) .
The role of interastrocytic gap junctional coupling in the development and progression of epilepsy is still controversial (for review see (Nemani and Binder 2005; Carlen 2012; Steinhäuser et al. 2012)) . According to the spatial buffering concept (see above) the astroglial network is expected to possess antiepileptic function, since reduction of astrocytic coupling would result in accumulation of extracellular K+ and, consequently, to neuronal depolarization and a lowered threshold for seizure generation. In line with this hypothesis are results from transgenic mice with coupling-deficient astrocytes (Cx30−/− Cx43flox/flox hGFAP-Cre mice; dko mice). In these mice, clearance of K+ but also glutamate was disturbed. Consistently, these mice displayed spontaneous epileptiform events, a reduced threshold for the generating epileptic activity (Fig. 8.2), increased synaptic transmission and enhanced activity-induced astrocytic swelling (Wallraff et al. 2006; Pannasch et al. 2011) . Although these findings strongly support an anticonvulsive role of glial GJ networks, a potential seizure-promoting role emerged from the results by (Rouach et al. 2008). They elegantly demonstrated that astroglial GJs mediate activity-dependent intercellular trafficking of metabolites from blood vessels to sites of high energy demand, suggesting that this process is essential for the maintenance of synaptic activity under pathological conditions such as epilepsy . Additionally, involvement of GJ channels in the intercellular spread of Ca2+ waves favors a proconvulsive role of the astroglial syncytium, since alterations in the astrocytic coupling would influence the propagation of Ca2+ waves and, therefore, neuronal synchronization and spread of ictal activity (Gomez-Gonzalo et al. 2010) . Taken together, astroglial GJ networks might play a dual role in epilepsy, combining pro- and antiepileptic properties. Further work is needed to elucidate which of the mechanisms prevails under various circumstances.

Epileptiform field potentials (EFPs) in CA1 region of dko mice. a Spontaneous EFPs (CA1 region) occurred only in slices, bathed in artificial cerebrospinal fluid (ACSF), from dko mice but not in wild type (wt) mice (not shown). b Similarly, low-intensity Schaffer-collateral stimulation gave rise to EFPs only in dko slices. c EFPs induced by washout of Mg2+ (0 Mg2+) were more frequent in dko slices and occurred with shorter latency. Original traces from wt (top) and dko (bottom) mice. (From Wallraff et al. (2006), reproduced with permission)
8.3.2 Connexin Expression and Coupling in Epileptic Tissue
Seizure-induced changes in Cx expression have been investigated in several studies using a variety of animal models and human tissue (for reviews see (Nemani and Binder 2005; Giaume et al. 2010; Steinhäuser et al. 2012) . The results are conflicting and do not allow drawing definitive conclusions. In animals, increased (Gajda et al. 2003; Samoilova et al. 2003; Szente et al. 2002; Condorelli et al. 2002; Takahashi et al. 2010; Mylvaganam et al. 2010) , unchanged (Khurgel and Ivy 1996; Li et al. 2001; Söhl et al. 2000; Xu et al. 2009) and decreased (Elisevich et al. 1997a, 1998; Xu et al. 2009; David et al. 2009) Cx43 and/or Cx30 transcript and/or protein have been reported. This inconsistency might be explained by differences between animal models, seizure duration and investigated brain area. In human specimens, mainly up-regulation of Cx43 transcript and/or protein has been described (Aronica et al. 2001; Collignon et al. 2006; Fonseca et al. 2002; Naus et al. 1991) , although unchanged levels have also been reported in one study (Elisevich et al. 1997b). However, Cx expression does not necessarily reflect the extent of functional coupling, since post-translational modifications, such as phosphorylation, might alter GJ channel unitary conductance, open probability, trafficking or internalization. Hence, functional coupling analyses are indispensable to receive reliable results. Increased astrocytic coupling has been reported in a post-status epilepticus (SE) rat model of epilepsy (Takahashi et al. 2010), and in hippocampal slice cultures chronically exposed to bicuculline (Samoilova et al. 2003). In contrast, Xu et al. observed significantly reduced coupling in the hippocampal CA1 region in a genetic mouse model of tuberous sclerosis complex (Xu et al. 2009). Human coupling studies have so far only been performed on primary astrocyte cultures derived from epileptic specimens (Lee et al. 1995) . Using fluorescence recovery after photobleaching (FRAP), these authors found enhanced astrocyte coupling in cells from epileptic specimens.
Another approach to assess the role of GJ channels in epilepsy is pharmacological disruption of inter-astrocytic communication, i.e. GJ blockers, substances producing intracellular acidification or Cx mimetic peptides. Such experiments have been performed in a variety of in vivo and in vitro animal models of epilepsy (Bostanci and Bagirici 2006, 2007; Gajda et al. 2003; Gigout et al. 2006; Jahromi et al. 2002; Kohling et al. 2001; Medina-Ceja et al. 2008; Perez-Velazquez et al. 1994; Ross et al. 2000; Samoilova et al. 2003, 2008; Szente et al. 2002; Voss et al. 2009) . Most of these studies reported anticonvulsive effects of GJ inhibition although opposite effects were observed in the study by Voss et al. (2009). In neocortical slices from patients with MTLE or focal cortical dysplasia, GJ blockers attenuated spontaneous and evoked epileptiform activity (Gigout et al. 2006). Major problems with using GJ blockers are their significant side effects and poor Cx isoform-specificity.
In conclusion, Cx expression studies, functional coupling analyses and uncoupling experiments yield an inconsistent picture on the role of the astroglial network in the pathophysiology of epilepsy. Further work is needed to clarify this issue.
8.3.3 Role of Cx Hemichannels and Pannexin Channels in Epilepsy
In addition to inter-cellular communication, functional membrane-spanning Cx hemichannels (HCs) have been demonstrated in astrocytes. These channels are non-selective and permeable for large molecules, such as ATP, glutamate , glucose and glutathione. Under normal conditions these channels are closed, but the open probability increases upon depolarization, altered intra- and extracellular Ca2+ concentration, metabolic inhibition or proinflammatory cytokines (reviewed by (Theis and Giaume 2012; Orellana et al. 2009, 2013) . It has been suggested that activated HCs promote neuronal hyperactivity and synchronization through excessive release of ATP and glutamate, which in turn increases excitability and Ca2+ wave propagation (Bedner and Steinhauser 2013) . GJ channels and HCs are oppositely regulated by proinflammatory cytokines (Meme et al. 2006; Retamal et al. 2007) and may play differential roles in epilepsy . Indeed, Yoon et al. (2010) showed in hippocampal slice cultures exposed to bicuculline that selective inhibition of HCs by low concentrations of mimetic peptides had protective effects on seizure spread while blockade of both HCs and GJs by high doses of the peptide exacerbated the lesion.
In addition to the Cx HCs, another family of proteins, termedpannexins (Panx1–3), can form functional transmembrane channels (but not inter-cellular channels) in different cell types, including astrocytes and neurons. Like Cx HCs, pannexons possess a very low open probability at rest which, increases upon elevated [K+]o, depolarization, increased intracellular Ca2+ concentration ([Ca2+]i), mechanical stress and P2X7 receptor activation (Scemes and Spray 2012; Suadicani et al. 2012) . It has been hypothesized that Panxs contribute to seizures by releasing ATP (Santiago et al. 2011) . Indeed, increased Panx transcript levels have been found in an in vitro seizure model (Mylvaganam et al. 2010) . Moreover, pharmacological inhibition or genetic deletion of Panx1 resulted in reduced seizure activity during kainate-induced SE (Santiago et al. 2011).
8.4 Aquaporin-4 Dysfunction and Epilepsy
8.4.1 Role of Aquaporin-4 in Epilepsy
The aquaporins (AQPs) are a family of small (24–30 kDa) integral proteins that mediate transmembrane water movement in response to osmotic gradients. So far, fourteen AQPs have been identified in mammals. AQP4 is the predominant water channel in the brain, where it is mainly localized to astrocyte perivascular endfeet as well as perisynaptic processes (for review see (Binder et al. 2012; Papadopoulos and Verkman 2013)) . AQP4 has been implicated in the pathogenesis of epilepsy mainly due to their role in regulating extracellular fluid osmolarity and extracellular space (ECS) volume (Schwartzkroin et al. 1998) . Indeed, several studies have demonstrated that osmolarity-induced reduction in the ECS volume causes neuronal hyperexcitability (Dudek et al. 1990; Roper et al. 1992; Chebabo et al. 1995; Pan and Stringer 1996) , while increasing the ECS volume attenuates epileptiform activity (Traynelis and Dingledine 1989; Dudek et al. 1990; Pan and Stringer 1996; Haglund and Hochman 2005) . In addition to ECS volume regulation, the spatial overlap of AQP4 with Kir4.1 in glial endfeet gave rise to the hypothesis that AQP4 may be involved in K+ homeostasis (Binder et al. 2012).
Mechanistic insight into the role of AQP4 in ECS volume regulation, K+ clearance and neuronal excitability came from transgenic mice. AQP4-deficient (AQP4−/−) mice display mild ECS volume expansion as assessed by FRAP and the tetramethylammonium method (Binder et al. 2004b; Yao et al. 2008) . In acute seizure models, AQP4−/− mice exhibited elevated seizure threshold but prolonged seizure duration (Fig. 8.3) and enhanced frequency of spontaneous seizure during the early phase (Binder et al. 2004a; Binder and Steinhäuser 2006; Lee et al. 2012) . In addition, in vivo and in situ studies using K+ sensitive electrodes or a fluorescent K+ sensor revealed impaired stimulus-induced [K+]o clearance in AQP4-deficient mice (Binder and Steinhäuser 2006; Padmawar et al. 2005; Strohschein et al. 2011) . Interestingly, K+ spatial buffering was enhanced in AQP4−/− mice, probably due to improved GJ coupling (Strohschein et al. 2011). The expanded ECS volume found in AQP4-deficient mice offers an explanation for the elevated seizure threshold of these mice, while the impaired K+ uptake might account for the prolonged duration and increased frequency of seizures . As described in Sect. 8.2.1 above, insufficient [K+]o clearance would result in neuronal hyperexcitability. However, the mechanistic link between AQP4 expression and K+ homeostasis has not been resolved so far. The view of a functional interaction between the AQP4 and Kir4.1 (Nagelhus et al. 1999) is not supported by follow-up studies showing AQP4-independent Kir4.1 function (Ruiz-Ederra et al. 2007; Zhang and Verkman 2008; Strohschein et al. 2011) . A more reasonable hypothesis implies that astrocytic K+ uptake during neuronal activity triggers AQP4-dependent osmotic water uptake and, therefore, reduction of the ECS. ECS shrinkage, in turn, causes an increase of [K+]o and consequently further K+ uptake by astrocytes (Papadopoulos and Verkman 2013) . Recently, this hypothesis was supported by mathematical modeling (Jin et al. 2013) .

Electrographic seizure threshold and duration in wt vs. AQP4−/− mice. a Bipolar electrodes implanted in the right hippocampus, and sealed to the skull by an acrylic cap, were connected to a stimulator and an EEG acquisition system. Mice were awake and behaved normally at the onset of stimulation (inset). b EEG from wt and Aqp4 −/− mice. Baseline EEG prior to stimulation is similar (left). Hippocampal stimulation-induced electrographic seizures are shown for a wt (top) and an Aqp4 −/− mouse (bottom). Note the prolonged seizure of the Aqp4 −/− mouse. Behavioral arrest was observed in both animals during the seizure. Postictal depression is evident on the EEG in both mice. c Aqp4 −/− mice had a higher electrographic seizure threshold than wt controls. d Aqp4 −/− mice had remarkably longer stimulation-evoked seizures compared to wt controls. (From Binder et al. (2006), reproduced with permission)
8.4.2 AQP4 Expression and Regulation in Epileptic Tissue
AQP4 expression was investigated in hippocampi from MTLE patients using rtPCR, immunohistochemistry and gene chip analysis (Lee et al. 2004) . The authors found enhanced AQP4 levels in HS, but reduced expression of the dystrophin gene, which encodes the protein that is involved in anchoring AQP4 in perivascular endfeet, and speculated that polarity in astrocytic AQP4 distribution got lost. This finding was subsequently confirmed with immunogold electron microscopy and Western blot analysis (Eid et al. 2005) . This locally restricted reduction of AQP4 was accompanied by a loss of perivascular dystrophin, indicating that AQP4 mislocalization was caused by a disrupted dystrophin complex. Similar loss of perivascular AQP4 and dystrophin were found in tissue from patients with focal cortical dysplasia (Medici et al. 2011) . Further indication for the involvement of AQP4 in epilepsy and for the proposed interplay between AQP4 and Kir4.1 comes from genetic studies showing that several SNPs in the KCNJ10 and AQP4 genes are associated with MTLE (Heuser et al. 2010 ; see also paragraph 2.2).
In a recent study, Alvestad et al. (2013) explored, in the kainate model, whether the loss of perivascular AQP4 found in human MTLE is involved in epileptogenesis or merely represents a consequence of the condition. They could demonstrate that AQP4 mislocalization precedes the chronic phase of epilepsy, suggesting that astrocytic dysfunction is of pathophysiological relevance.
8.5 Altered Glutamate Homeostasis in Epilepsy
8.5.1 Extracellular Glutamate Levels in MTLE
Astrocytes modulate synaptic transmission and neuronal excitability by controlling extracellular neurotransmitter concentrations in the central nervous system (CNS) . Rapid clearance of excessive glutamate from the ECS as well as its recycling is essential for survival and normal brain function . To prevent excitotoxic accumulation of glutamate in the ECS, glutamate is taken up by astrocytes via specialized transporters, converted to glutamine by GS and shuttled back to neurons for re-synthesis of glutamate. Dysfunction of the glutamate metabolism has been proposed to be critically involved in the pathophysiology of epilepsy (Eid et al. 2008b; Coulter and Eid 2012) . This assumption is supported by the fact that glutamate and glutamate analogs cause seizures and neuronal loss in experimental epilepsy (Olney et al. 1972; Nadler and Cuthbertson 1980; Ben-Ari 1985; Fremeau et al. 2002) . Moreover, increased interictal glutamate levels and stronger seizure-induced glutamate transients were found in the hippocampi of MTLE patients (Cavus et al. 2005; During and Spencer 1993) . Among MTLE patients, those with HS displayed higher interictal extracellular glutamate levels (Petroff et al. 2003; Coulter and Eid 2012) . However, as discussed by Coulter and Eid (2012), the source of glutamate in HS is obscure, since one of the hallmarks of this pathology is loss of glutamatergic neurons in the hippocampal CA1 region.
8.5.2 Astrocytic Glutamate Uptake in Epilepsy
Astrocytes take up glutamate from the ECS via high-affinity glutamate transporters (excitatory amino acid transporters, EAATs), which utilize the electrochemical gradient of Na+ and K+ as a driving force. Five transporter isoforms have been identified and two of them, GLAST (in rodents; in human EAAT1) and GLT1 (in rodents; in human EAAT2), are preferentially expressed in astrocytes. The impact of astrocytic glutamate uptake became obvious from the phenotype of genetically engineered mice devoid of transporter proteins. GLT1 knockout mice displayed lethal spontaneous seizures, increased susceptibility to acute cortical injury and seizures after administration of subconvulsive doses of pentylenetetrazole (PTZ) (Tanaka et al. 1997) . Consistently, pharmacological inhibition of GLT1 in rat neocortex reduced the threshold for evoking epileptiform activity (Demarque et al. 2004; Campbell and Hablitz 2004) . In contrast, antisense knockdown of GLT1 in adult rats caused increased extracellular glutamate, but not seizures (Rothstein et al. 1996) . Mice deficient in GLAST showed no spontaneous seizures or electroencephalography (EEG) paroxysmal discharges, but amygdala kindling or PTZ-induced seizures were of longer duration, more severe and occurred after a shorter latency in mutant mice (Watanabe et al. 1999) .
In MTLE patients, inconsistent data on the regulation of EAAT1 and 2 have been published; no changes in transporter expression (Tessler et al. 1999; Eid et al. 2004) or decreased levels of EAAT 1/2 were found in epileptic hippocampi (Mathern et al. 1999; Proper et al. 2002) while found In kindled rats, unchanged GLT1 and GLAST levels were described (Akbar et al. 1997; Miller et al. 1997; Simantov et al. 1999) while decreased levels were found in the pilocarpine (Lopes et al. 2013) and albumin models (David et al. 2009) as well as in a tuberous sclerosis epilepsy model (Wong et al. 2003) . Finally, (Guo et al. 2010) observed decreased GLAST but unaffected GLT1 expression in the hippocampus of spontaneously epileptic rats.
Taken together, the data described so far indicate that glutamate uptake by astrocytes plays a crucial role in protecting neurons from hyperexcitability and excitotoxicity . However, whether this mechanism is disturbed in epilepsy is still under investigation.
8.5.3 Regulation of GS in Epilepsy
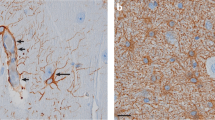
In the CNS, GSispredominantlyexpressed by astrocytes where it converts glutamate and ammonia to glutamine. Impaired GS activity has been hypothesized to play a crucial role in the pathogenesis of MTLE (Eid et al. 2013b). This hypothesis is supported by the following observations: (i) unilateral intrahippocampal infusion with the GS inhibitor methionine sulfoximine causes recurrent seizures and neuropathological changes similar to human MTLE (Eid et al. 2008a; Wang et al. 2009; Perez et al. 2012) ; (ii) reduced GS protein and enzyme activity have been found in MTLE-HS patients (Fig. 8.4) (Eid et al. 2004; van der Hel et al. 2005) ; interestingly, in a recent study no reduction of GS mRNA could be found in HS in the CA1 region, suggesting posttranscriptional modification of GS in epilepsy (Eid et al. 2013a); and (iii) mutations in the GS encoding gene, GLUL, are associated with reduced GS activity and epileptic seizures (Haberle et al. 2005, 2006, 2011) . One argument against a causative role of GS dysfunction in epileptogenesis, however, arises from the study performed in the kainate model (Hammer et al. 2008) . The authors demonstrated that although GS protein level was reduced during the chronic phase (confirming the findings in human tissue), increased GS levels were found in the latent period (prior to seizure onset). The mechanistic link between GS downregulation and seizure development is provided by the assumption that GS deficiency and the resulting decrease in glutamate to glutamine conversion causes intracellular glutamate accumulation. This accumulation, in turn, slows down glutamate uptake, leading to increased glutamate levels in the ECS (Coulter and Eid 2012; Eid et al. 2013b) . Support for this view emerges from recent work showing that GS inhibition causes indeed glutamate accumulation in hippocampal astrocytes (Perez et al. 2012).

Decreased GS immunoreactivity in the CA1 region of MTLE patients. There is dense and even distribution of GS-positive cells in the subiculum and CA1 region of autopsy (a) and non-MTLE hippocampi (d). High-power fields of subiculum in autopsy (b) and non-MTLE (e) hippocampi show that staining is confined to astrocytes. High-power fields of area CA1 in autopsy (c) and non-MTLE (f) hippocampi also show many GS-positive astrocytes. In MTLE hippocampus (g), there are many GS-positive cells in the subiculum but area CA1 is severely deficient in GS staining. High-power view of the subiculum in G (h) confirms presence of staining in astrocytes, which have fewer processes than GS-positive astrocytes in the corresponding areas of autopsy (b) and non-MTLE hippocampi (e). High-power view of area CA1 in G (i) confirms lack of GS staining in this region. Specificity controls with GS antiserum (j) and preimmune serum (k) on adjacent sections of the non-MTLE hippocampus shown in d–f reveal no staining in k. DG dentate gyrus. (Modified from Eid et al. (2004), reproduced with permission)
The glutamate-glutamine cycle is essential for replenishing the neurotransmitter pool and maintenance of synaptic activity. Interruption of the cycle through GS inhibition impairs inhibitory, i.e.γ-aminobutyric acid (GABA)-ergic transmission (Liang et al. 2006) , but had little effect on excitatory (glutamatergic) synaptic function (Kam and Nicoll 2007) . This phenomenon as well as its consequences on network excitability has been studied in detail in an in vitro model of astrocytic gliosis (Ortinski et al. 2010) . In this study, virus-induced gliosis in the hippocampus caused downregulation of GS expression and deficit in inhibitory, but not excitatory synaptic transmission. Employing voltage-sensitive dye imaging the authors showed that these inhibitory deficits entail network hyperexcitability, which could partially be reversed by exogenously supplied glutamine. These data emphasize the importance of proper GS function for inhibitory neurotransmission and prevention of seizure generation (Ortinski et al. 2010) .
8.6 Astrocyte Ca2+ Signaling and Gliotransmission in Epilepsy
8.6.1 Ca2+ Signaling
Already in the nineties of the past century it has been shown that astrocytes in culture (Cornell-Bell et al. 1990; Charles et al. 1991) and acute brain slices (Porter and McCarthy 1996) respond to glutamate with elevations in [Ca2+]i which propagate through the astroglial network. Increased astrocytic [Ca2+]i, in turn, induced release of gliotransmitter , including glutamate, ATP, D-serine and GABA (Halassa et al. 2007; Crunelli and Carmignoto 2013) . Hence, astrocytes not only sense, but also regulate synaptic transmission, neuronal excitability and plasticity. In human brain, bidirectional signaling between neurons and astrocytes has been demonstrated only recently (Navarrete et al. 2013) . However, the mechanism by which astrocytes release gliotransmitter is still controversially discussed .
Evidence for the involvement of astrocytic Ca2+ waves and concomitant release of transmitters in epilepsy emerge from studies in brain slices and in vivo, demonstrating increased Ca2+ oscillations in astrocytes during epileptiform activity, which could be suppressed by anti-epileptic drugs (Tian et al. 2005; Fellin et al. 2006) . In addition, metabotropic glutamate receptors , which mediate astrocytic Ca2+ signals, are up-regulated in experimental (Aronica et al. 2000; Ulas et al. 2000) and human (Tang and Lee 2001; Kandratavicius et al. 2013) epilepsy. Pilocarpine-induced SE caused long-lasting elevation in astrocytic [Ca2+]i and the resulting glutamate release contributed to neuronal excitotoxicity (Ding et al. 2007) .
8.6.2 Glutamate Release
Glutamate release from astrocytes synchronizes neuronal firing through activation of extrasynaptic NMDA receptors (Fellin et al. 2004; Angulo et al. 2004) . Epileptic discharges are characterized by excessive, hypersynchronous neuronal activity, leading to the suggestion that astrocytic glutamate release underlies the simultaneous activation of multiple neurons during such an event. Support for this view came from Tian et al. (2005) who studied paroxysmal depolarization shifts (PDSs) after inhibition of synaptic activity by tetrodotoxin (TTX) and Ca2+ channel blockers. PDSs, which underlie interictal activity, were largely insensitive to TTX but sensitive to α-amino-3-hydroxy-5-methyl-isoxazole propionate (AMPA) and N-methyl D-aspartate (NMDA) glutamate receptor antagonists, indicating that they were triggered by release of glutamate from extrasynaptic sources. Since photolysis of caged Ca2+ in individual astrocytes evoked local PDSs, the authors concluded that astrocytes are the primary source of glutamate in experimental seizure models (Tian et al. 2005) . This conclusion was challenged by a subsequent study showing that astrocytic glutamate is not required for the initiation of epileptiform activity, but might have amodulatory role (Fellin et al. 2006). This view is supported by an elegant study of Gomez-Gonzalo et al. (2010) , who combined patch clamp recording and Ca2+ imaging during in vitro seizures to assess the role of astrocytes in the generation of epileptiform activity. Their data revealed that ictal, but not interictal, discharges generate astrocytic [Ca2+]i elevations (Fig. 8.5). In a new in vitro model of focal seizures induced by local application of NMDA in the presence of 4-aminopyridine and low Mg2+, astrocytic Ca2+ signals preceded ictal discharges (Losi et al. 2010) . These early Ca2+ signals appeared to have a causative role in the generation of focal ictal discharges, since their inhibition prevented NMDA-stimulated ictal discharges, while their stimulation enhanced discharges. In contrast to the findings by Tian et al. (2005), no association between [Ca2+]i changes in astrocytes and interictal events could be found in this study. The authors concluded that bidirectional signaling between neurons and astrocytes in hyperexcitable networks generate a recurrent excitatory loop that promotes focal seizures (Gomez-Gonzalo et al. 2010) .

Astrocytes respond with Ca2+ elevations to ictal, but not interictal, discharges. a Ca2+ changes at basal activity (t 0 ) in neurons (n, black arrows) and astrocytes (a, white arrowheads), during an interictal (t 1 ) or an ictal (t 2 ) event after perfusion with picrotoxin/zero Mg2+. b Action potential bursts and Ca2+ change (green trace) of the patched neuron, and Ca2+ changes from other neurons and from astrocytes indicated in (a). Note the large Ca2+ rise in astrocytes upon ictal (t 2 ) but not interictal (t 1 ) events. c Ca2+ imaging in guinea pig entorhinal cortex before (t 0 ) and during (t 1 and t 2 ) ictal discharges induced by arterial perfusion with bicuculline. d Field potentials and Ca2+ changes from neuropil (dashed oval in c) and astrocytes (arrowheads in c) during ictal discharges. e Proportion of astrocytes activated during interictal and ictal events. Cell numbers are indicated above bars. (From Gomez-Gonzalo et al. 2010)
8.6.3 Astrocyte Release of ATP and Epilepsy
In addition to its role in propagating inter-astrocytic Ca2+ waves, ATP release from astrocytes directly modulates synaptic transmission (Kumaria et al. 2008) . Extracellular ATP is rapidly hydrolyzed to adenosine (Zimmermann and Braun 1996) . Adenosine, in turn, potently suppresses excitatory synaptic transmission through activation of presynaptic A1 receptors (A1Rs) (Pascual et al. 2005a; Fredholm et al. 2005) , indicating an anticonvulsive role of astrocytic ATP release (see Sect. 8.7).
In addition, extracellular ATP may activate purinergic receptors , which can have both pro- and anticonvulsant consequences. For instance, Torres et al. (2012) reported that the decrease in [Ca2+]o accompanying excitatory synaptic transmission triggers ATP release from astrocytes through Cx43 HCs that, in turn, enhances inhibitory transmission by activating P2Y1 receptors on interneurons. In contrast, ATP released through Panx1 channels aggravates seizures and prolongs SE (Santiago et al. 2011) . Astrocytic ATP has been proposed to possess seizure-promoting properties through activation of postsynaptic P2X receptors. The consequential elevation in neuronal Ca2+ levels promotes the insertion of AMPA receptors and synaptic strength (Gordon et al. 2005) .
Taken together, the role of gliotransmission in the pathophysiology of epilepsy is still unresolved. Clarification of the functional significance of the different astrocyte-to-neuron signaling pathways represents an intriguing challenge and attracts increasing interest for developing new, anti-epileptogenic therapies.
8.7 Adenosine Dysfunction in Epilepsy
8.7.1 Role of Astrocyte-derived Adenosine in Epilepsy
The anticonvulsive action of adenosine has been demonstrated already three decades ago (Dunwiddie 1980; Lee et al. 1984; Dragunow et al. 1985) . Since, various studies on experimental models and human epileptic tissue have confirmed and further elucidated these initial findings (Boison 2010, 2012) . Extracellular adenosine levels rapidly rise during seizures , a process that probably mediates seizure termination and postictal suppression (During and Spencer 1992). Mice intracranially implanted with adenosine-releasing polymers, encapsulated cells or embryonic stem cells showed profound reduction of seizure activity in the kindling model (Boison et al. 1999, 2002; Huber et al. 2001; Li et al. 2007b) . The anticonvulsive action of adenosine is mainly mediated via activation of G protein-coupled A1Rs (Boison 2010, 2012). Consistently, A1R agonists possess anticonvulsant and neuroprotective effects (Dunwiddie and Worth 1982; Barraco et al. 1984; Gouder et al. 2003; Li et al. 2013) , while antagonists promote seizures and aggravate neuronal damage (Dunwiddie 1990; Ault et al. 1987; Avsar and Empson 2004; Vianna et al. 2005) . Homozygous and heterozygous A1R knockout mice exhibit spontaneous seizures in the CA3 area of the hippocampus (Li et al. 2007a) . Furthermore, in the kainate model these animals displayed more severe SE and neuronal loss (Fedele et al. 2006) . In human MTLE-HS (Glass et al. 1996) and rodent HS (Cremer et al. 2009; Aden et al. 2004) A1Rs are downregulated.
Several studies have indicated that extracellular adenosine originates from hydrolyzed ATP released from astrocytes through Cx HCs, Panx channels (Kang et al. 2008; Santiago et al. 2011) or exocytosis (Pascual et al. 2005a) . The latter study investigated the role of astrocytes in regulating synaptic transmission using transgenic mice, which overexpressed a dominant-negative soluble N-ethylmaleimide-sensitive factor attachment protein receptor (dn-SNARE mice) specifically in astrocytes. In these mice adenosine-mediated heterosynaptic depression was absent, confirming the astrocytic origin of extracellular adenosine (Pascual et al. 2005b).
8.7.2 Role of Glial ADK in Epilepsy
The ambient adenosine level is controlled by the activity of the enzyme ADK, which is critically involved in adenosine metabolism by phosphorylating adenosine to 5′-AMP, and which in the adult brain is mainly expressed in astrocytes. Since cytoplasmic and extracellular adenosine levels are tightly balanced by nucleoside transporters, even small changes in ADK activity can influence extracellular adenosine concentrations and consequently neuronal excitability (Boison 2010; Aronica et al. 2013) . Overexpression of ADK was found in human MTLE-HS (Aronica et al. 2011) and in experimental epilepsy (Gouder et al. 2004; Aronica et al. 2011; Li et al. 2008, 2012) . Interestingly, in the intrahippocampal kainate model, Gouder et al. (2004) observed a transient reduction of ADK immunoreactivity during the first 24 h post SE, but pronounced overexpression during the later stages of epileptogenesis. The authors speculated that the initial reduction of ADK and the resulting increase in extracellular adenosine may contribute to termination of SE, while the enhanced ADK expression may contribute to chronic seizures by decreasing ambient adenosine. In experimental focal epilepsy, astrogliosis, ADK overexpression and spontaneous recurrent seizures are tightly associated (Li et al. 2008, 2007a, 2012).
In line with the expression studies, pharmacological inhibition of ADK potently inhibited seizures in experimental epilepsy (Kowaluk and Jarvis 2000; Gouder et al. 2004) . Moreover, transgenic or viral overexpression of ADK in mice resulted in spontaneous recurrent hippocampal seizures, while viral knockdown of ADK prevented seizures. Since ADK overexpression was not accompanied by astrogliosis , these studies indicate that ADK overexpression is sufficient to trigger seizures and cause chronic seizure activity (Li et al. 2007a, 2008; Theofilas et al. 2011; Boison 2010, 2012; Aronica et al. 2013) .
These above findings gave rise to the ADK hypothesis of epileptogenesis, which considers this enzyme both as a diagnostic marker as well as a potential therapeutic target to prevent epileptogenesis.
8.8 Astrocyte Immune Responses and Epilepsy
8.8.1 Astrocyte Changes Induced by Brain Inflammation
Brain inflammation has been implicated in the pathogenesis of epilepsy. Increased levels of inflammatory mediators , loss of blood-brain-barrier integrity and mono/lymphocyte infiltration were found in sclerotic tissue from MTLE patients as well as in animal models of TLE. There is growing evidence that inflammation is not only a consequence but also cause of epilepsy (Vezzani et al. 2011a) . This assumption is supported by the following set of findings. (i) Cytokines like interleukin-1beta (IL-1β) and damage associated molecular pattern (DAMPs) like high mobility group box 1 (HMGB1) promote seizures in experimental epilepsy (Maroso et al. 2011) . Preapplication of IL-1β or HMGB1 prolongs the duration of seizures induced by chemoconvulsant drugs (kainate, GABAA receptor antagonist bicuculline, while preapplication or overexpression of the corresponding antagonists or substances which inhibit IL-1β production attenuate seizure activity. Moreover, transgenic mice lacking functional receptors for the respective cytokines and DAMPs displayed reduced susceptibility for seizures (Vezzani et al. 2000; Maroso et al. 2010). (ii) Seizures arising during fever (febrile seizures) are the most frequent type of seizures in children (Dube et al. 2012) . (iii) Experimental induction of fever by lipopolysaccharide (LPS) injection or hyperthermia results in long-term enhancement of seizure susceptibility (Dube et al. 2012; Galic et al. 2008; Auvin et al. 2009, 2010) . However, the mechanism linking inflammation and epilepsy is still unclear. Proinflammatory cytokines enhance neuronal excitability by directly modifying the function of neuronal voltage- and receptor-gated ion channels (Vezzani et al. 2012). However, IL-1β and other cytokines may also indirectly influence neuronal excitability by altering astrocyte function. For instance, in culture or acute rat slices, IL-1β and tumor necrosis factor α (TNFα) inhibit glutamate reuptake and increase glial glutamate release (Bezzi et al. 2001; Hu et al. 2000; Ye and Sontheimer 1996) , which can be expected to produce hyperactivity. Furthermore, in cultured astrocytes cytokines inhibit Cx43 GJ channels, and open Cx43 HCs (Meme et al. 2006; Retamal et al. 2007) . Intriguingly, cytokine-induced uncoupling in culture could be rescued by treatment with the anti-epileptic drug levetiracetam (Keppra®) (Haghikia et al. 2008) . Whether astrocytic uncoupling and/or Cx HC activation is pro- or anticonvulsive needs to be clarified (see Sects. 8.3.2 and 3.3). Moreover, IL-1β and LPS increase ADK expression in human astrocyte cultures (Aronica et al. 2011) . As described above (Sect. 8.7.2), ADK overexpression is sufficient to trigger seizures. Of note, glial Kir4.1 expression in culture is down-regulated after exposure to IL-1β (Zurolo et al. 2012) . Given the essential role of Kir4.1 in K+ buffering (Sect. 8.2), the consequence of this down-regulation on neuronal excitability seems to be evident.
Collectively, these findings demonstrate that proinflammatory mediators cause several dysfunctions in astrocytes, which individually or in concert provoke neuronal hyperexcitability.
8.8.2 Involvement of Astrocytes in Brain Inflammation
Astrocytes contribute to inflammation in the CNS by producing a variety of chemokines and cytokines (Aronica et al. 2012). Cytokines like IL-1β and TNFα are overexpressed by astrocytes in both human and experimental epilepsy (Vezzani et al. 2008) . Importantly, during the latent period IL-1β was exclusively expressed by astrocytes in a rat model of epilepsy. In chronic experimental and human epilepsy about 10-fold more IL-1β-positive astrocytes than microglia were observed, indicating that astrocytes are the main source of IL-1β in the epileptic brain (Ravizza et al. 2008) .
An important role in the astrocyte immune response has been suggested for the IL-1 receptor/toll-like receptor (IL-1R/TLR) superfamily (Maroso et al. 2010, 2011) . In the human brain IL-1R and TLR4 are expressed at low levels in astrocytes under control conditions and are up-regulated in the sclerotic hippocampus from MTLE patients (Ravizza et al. 2008; Maroso et al. 2010). In the absence of pathogens, TLR signaling can be activated by DAMPs released by injured or dying cells. One member of the DAMP protein family, HMGB1, is released by cultured astrocytes upon IL-1β stimulation, and its nuclear to cytoplasmic translocation was observed in human and murine epileptic hippocampus (Maroso et al. 2010). Activation of IL-1R/TLR signaling induced Src kinase-dependent phosphorylation of the GluN2B subunit of the NMDA receptors, resulting in higher NMDA-dependent Ca2+ influx and neuronal hyperactivity (Vezzani et al. 2011b; Maroso et al. 2010). In addition, IL-1R/TLR signaling triggers, via nuclear factor kappa-light chain-enhancer of activated B cells (NF-κB) activation, transcription of several genes encoding downstream mediators of inflammation , like IL-6, TNFα or cyclooxygenase-2, which may further promote seizures (Vezzani et al. 2011b). Indeed, overexpression of NF-κB has been observed in the human sclerotic hippocampus (Crespel et al. 2002) .
These findings led to a model for the involvement of the IL-1R/TLR axis in epileptogenesis (Fig. 8.6). An inciting event, like infection, seizures or stroke, leads to cellular stress and/or injury and release of IL-1β (from microglia and astrocytes) and HMGB1 (from astrocytes and neurons) from their constitutive endogenous pools. Activation of IL-1R/TLR signaling in neurons and astrocytes by the released molecules results in several cellular changes (such as GluN2B phosphorylation), which contribute to the generation of the first seizure. Seizures, in turn, induce de novo synthesis of IL-1β, HMGB1 and other proinflammatory mediators, which contribute to seizure recurrence (Vezzani et al. 2011b; Maroso et al. 2010) . Thus, IL-1R/TLR signaling might represent promising targets for antiepileptic treatments.

Pathophysiological cascade mediated by IL-1R/TLR signaling in epilepsy. a Inciting events, initiated by local injuries or peripherally following infections, lead to activation of microglia , astrocytes and neurons. These cells release proinfammatory cytokines such as IL-1β, and danger signals such as HMGB1, thereby eliciting a cascade of inflammatory events in the target cells (i.e. neurons and glia) via activation of IL-1R1 and TLR4. This rapidly increases neuronal NMDA receptor Ca2+ conductance via ceramide/Src mediated phosphorylation of the GluN2B (NR2B) subunit, leading to hyperexcitability. Long-term decrease in seizure threshold results from activating transcription of genes, contributing to molecular and cellular changes involved in epileptogenesis and perpetuating inflammation. b Inflammation due to activation of IL1R/TLR signaling contributes to seizure generation by decreasing the threshold of neuronal excitability. Seizures recurrence, in turn, activates further inflammation, thereby establishing a vicious circle of events that contributes to the development of epilepsy. (From Vezzani et al. (2011), reproduced with permission)
8.9 Conclusions
The fact that astrocytes are now recognized as communication partners of neurons and do not merely represent “brain glue” has rekindled the question of the role of these cells in neurological disorders such as epilepsy . Compelling evidence is emerging demonstrating severe dysfunction of astrocytes in human and experimental epilepsy. However, several important questions still remain open. First, it is still unclear whether the reported alterations in astrocytes are causative of the condition or rather represent a compensatory phenomenon. Second, difficulties arise from the fact that the term “astrocyte” covers a heterogeneous group of cells, which complicates comparison of published work. Indeed, it is becoming increasingly clear that the molecular, functional and morphological properties of astrocytes do not only vary across brain areas but even within a given subregion. Nevertheless, the combination of molecular genetics with functional approaches will help clarifying the specific roles of astrocytes in epilepsy and enable developing novel therapeutic approaches to better treat this disorder.
References
Aden U, O’Connor WT, Berman RF (2004) Changes in purine levels and adenosine receptors in kindled seizures in the rat. Neuroreport 15:1585–1589
Akbar MT, Torp R, Danbolt NC, Levy LM, Meldrum BS, Ottersen OP (1997) Expression of glial glutamate transporters GLT-1 and GLAST is unchanged in the hippocampus in fully kindled rats. Neuroscience 78:351–359
Alvestad S, Hammer J, Hoddevik EH, Skare O, Sonnewald U, miry-Moghaddam M, Ottersen OP (2013) Mislocalization of AQP4 precedes chronic seizures in the kainate model of temporal lobe epilepsy. Epilepsy Res 105:30–41
Angulo MC, Kozlov AS, Charpak S, Audinat E (2004) Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–6927
Aronica E, Van Vliet EA, Mayboroda OA, Troost D, Da Silva FHL, Gorter JA (2000) Upregulation of metabotropic glutamate receptor subtype mGluR3 and mGluR5 in reactive astrocytes in a rat model of mesial temporal lobe epilepsy. Eur J Neurosci 12:2333–2344
Aronica E, Gorter JA, Jansen GH, Leenstra S, Yankaya B, Troost D (2001) Expression of connexin 43 and connexin 32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol 101:449–459
Aronica E, Zurolo E, Iyer A, de GM, Anink J, Carbonell C, Van Vliet EA, Baayen JC, Boison D, Gorter JA (2011) Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52:1645–1655
Aronica E, Ravizza T, Zurolo E, Vezzani A (2012) Astrocyte immune responses in epilepsy. Glia 60:1258–1268
Aronica E, Sandau US, Iyer A, Boison D (2013) Glial adenosine kinase-a neuropathological marker of the epileptic brain. Neurochem Int 63:688–695
Ault B, Olney MA, Joyner JL, Boyer CE, Notrica MA, Soroko FE, Wang CM (1987) Pro-convulsant actions of theophylline and caffeine in the hippocampus: implications for the management of temporal lobe epilepsy. Brain Res 426:93–102
Auvin S, Porta N, Nehlig A, Lecointe C, Vallee L, Bordet R (2009) Inflammation in rat pups subjected to short hyperthermic seizures enhances brain long-term excitability. Epilepsy Res 86:124–130
Auvin S, Shin D, Mazarati A, Sankar R (2010) Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia 51(Suppl 3):34–38
Avsar E, Empson RM (2004) Adenosine acting via A1 receptors, controls the transition to status epilepticus-like behaviour in an in vitro model of epilepsy. Neuropharmacology 47:427–437
Balestrino M, Somjen GG (1986) Chlorpromazine protects brain tissue in hypoxia by delaying spreading depression-mediated calcium influx. Brain Res 385:219–226
Barraco RA, Swanson TH, Phillis JW, Berman RF (1984) Anticonvulsant effects of adenosine analogues on amygdaloid-kindled seizures in rats. Neurosci Lett 46:317–322
Bedner P, Steinhauser C (2013) Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem Int 63:682–687
Ben-Ari Y (1985) Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14:375–403
Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A (2001) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710
Binder DK, Steinhäuser C (2006) Functional changes in astroglial cells in epilepsy. Glia 54:358–368
Binder DK, Oshio K, Ma T, Verkman AS, Manley GT (2004a) Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 15:259–262
Binder DK, Papadopoulos MC, Haggie PM, Verkman AS (2004b) In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci 24:8049–8056
Binder DK, Nagelhus EA, Ottersen OP (2012) Aquaporin-4 and epilepsy. Glia 60:1203–1214
Bockenhauer D et al (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360:1960–1970
Boison D (2010) Adenosine dysfunction and adenosine kinase in epileptogenesis. Open Neurosci J 4:93–101
Boison D (2012) Adenosine dysfunction in epilepsy. Glia 60:1234–1243
Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H (1999) Seizure suppression in kindled rats by intraventricular grafting of an adenosine releasing synthetic polymer. Exp Neurol 160:164–174
Boison D, Huber A, Padrun V, Deglon N, Aebischer P, Mohler H (2002) Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia 43:788–796
Bordey A, Sontheimer H (1998) Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res 32:286–303
Bostanci MO, Bagirici F (2006) The effects of octanol on penicillin induced epileptiform activity in rats: an in vivo study. Epilepsy Res 71:188–194
Bostanci MO, Bagirici F (2007) Anticonvulsive effects of carbenoxolone on penicillin-induced epileptiform activity: an in vivo study. Neuropharmacology 52:362–367
Buono RJ, Lohoff FW, Sander T, Sperling MR, O’Connor MJ, Dlugos DJ, Ryan SG, Golden GT, Zhao H, Scattergood TM, Berrettini WH, Ferraro TN (2004) Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res 58:175–183
Campbell SL, Hablitz JJ (2004) Glutamate transporters regulate excitability in local networks in rat neocortex. Neuroscience 127:625–635
Carlen PL (2012) Curious and contradictory roles of glial connexins and pannexins in epilepsy. Brain Res 1487:54–60
Cavus I, Kasoff WS, Cassaday MP, Jacob R, Gueorguieva R, Sherwin RS, Krystal JH, Spencer DD, bi-Saab WM (2005) Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann Neurol 57:226–235
Charles AC, Merrill JE, Dirksen ER, Sanderson MJ (1991) Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6:983–992
Chebabo SR, Hester MA, Aitken PG, Somjen GG (1995) Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695:203–216
Chever O, Djukic B, McCarthy KD, Amzica F (2010) Implication of kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional kir4.1 knock-out mice. J Neurosci 30:15769–15777
Collignon F, Wetjen NM, Cohen-Gadol AA, Cascino GD, Parisi J, Meyer FB, Marsh WR, Roche P, Weigand SD (2006) Altered expression of connexin subtypes in mesial temporal lobe epilepsy in humans. J Neurosurg 105:77–87
Condorelli DF, Mudo G, Trovato-Salinaro A, Mirone MB, Amato G, Belluardo N (2002) Connexin-30 mRNA is up-regulated in astrocytes and expressed in apoptotic neuronal cells of rat brain following kainate-induced seizures. Mol Cell Neurosci 21:94–113
Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ (1990) Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science 247:470–473
Coulter DA, Eid T (2012) Astrocytic regulation of glutamate homeostasis in epilepsy. Glia 60:1215–1226
Cremer CM, Palomero-Gallagher N, Bidmon HJ, Schleicher A, Speckmann EJ, Zilles K (2009) Pentylenetetrazole-induced seizures affect binding site densities for GABA, glutamate and adenosine receptors in the rat brain. Neuroscience 163:490–499
Crespel A, Coubes P, Rousset MC, Brana C, Rougier A, Rondouin G, Bockaert J, Baldy-Moulinier M, Lerner-Natoli M (2002) Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res 952:159–169
Crunelli V, Carmignoto G (2013) New vistas on astroglia in convulsive and non-convulsive epilepsy highlight novel astrocytic targets for treatment. J Physiol 591:775–785
D’Ambrosio R, Gordon DS, Winn HR (2002) Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol 87:87–102
Das A, Wallace GC, Holmes C, McDowell ML, Smith JA, Marshall JD, Bonilha L, Edwards JC, Glazier SS, Ray SK, Banik NL (2012) Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 220:237–246
David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A (2009) Astrocytic dysfunction in epileptogenesis: consequence of altered potassium and glutamate homeostasis? J Neurosci 29:10588–10599
Demarque M, Villeneuve N, Manent JB, Becq H, Represa A, Ben Ari Y, Aniksztejn L (2004) Glutamate transporters prevent the generation of seizures in the developing rat neocortex. J Neurosci 24:3289–3294
Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG (2007) Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci 27:10674–10684
Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD (2007) Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27:11354–11365
Dragunow M, Goddard GV, Laverty R (1985) Is adenosine an endogenous anticonvulsant? Epilepsia 26:480–487
Dube CM, McClelland S, Choy MK, Brewster AL, Noam Y, Baram TZ (2012) Fever, febrile seizures and epileptogenesis. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV (eds) Jasper’s basic mechanisms of the epilepsies. Oxford University, Betheseda, pp 343–352
Dudek FE, Obenaus A, Tasker JG (1990) Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett 120:267–270
Dunwiddie TV (1980) Endogenously released adenosine regulates excitability in the in vitro hippocampus. Epilepsia 21:541–548
Dunwiddie TV (1990) Electrophysiological aspects of adenosine receptor function. Adenosine Adenosine Recep 5:143–172
Dunwiddie TV, Worth T (1982) Sedative and anticonvulsant effects of adenosine analogs in mouse and rat. J Pharmacol Exp Ther 220:70–76
During MJ, Spencer DD (1992) Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 32:618–624
During MJ, Spencer DD (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341:1607–1610
Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, De Lanerolle NC (2004) Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363:28–37
Eid T, Lee TS, Thomas MJ, Amiry-Moghaddam M, Bjornsen LP, Spencer DD, Agre P, Ottersen OP, De Lanerolle NC (2005) Loss of perivascular aquaporin 4 may underlie deficient water and K+ homeostasis in the human epileptogenic hippocampus. Proc Natl Acad Sci U S A 102:1193–1198
Eid T, Ghosh A, Wang Y, Beckstrom H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, De Lanerolle NC (2008a) Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131:2061–2070
Eid T, Williamson A, Lee TS, Petroff OA, De Lanerolle NC (2008b) Glutamate and astrocytes-key players in human mesial temporal lobe epilepsy? Epilepsia 49(Suppl 2):42–52
Eid T, Lee TS, Wang Y, Perez E, Drummond J, Lauritzen F, Bergersen LH, Meador-Woodruff JH, Spencer DD, De Lanerolle NC, McCullumsmith RE (2013a) Gene expression of glutamate metabolizing enzymes in the hippocampal formation in human temporal lobe epilepsy. Epilepsia 54:228–238
Eid T, Tu N, Lee TS, Lai JC (2013b) Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int 63:670–681
Elisevich K, Rempel SA, Smith B, Allar N (1997a) Connexin 43 mRNA expression in two experimental models of epilepsy. Mol Chem Neuropathol 32:75–88
Elisevich K, Rempel SA, Smith BJ, Edvardsen K (1997b) Hippocampal connexin 43 expression in human complex partial seizure disorder. Exp Neurol 145:154–164
Elisevich K, Rempel SA, Smith B, Hirst K (1998) Temporal profile of connexin 43 mRNA expression in a tetanus toxin-induced seizure disorder. Mol Chem Neuropathol 35:23–37
Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D (2006) Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol 200:184–190
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743
Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG (2006) Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J Neurosci 26:9312–9322
Fisher RS, Pedley TA, Moody WJ Jr, Prince DA (1976) The role of extracellular potassium in hippocampal epilepsy. Arch Neurol 33:76–83
Fonseca CG, Green CR, Nicholson LF (2002) Upregulation in astrocytic connexin 43 gap junction levels may exacerbate generalized seizures in mesial temporal lobe epilepsy. Brain Res 929:105–116
Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM (2005) Adenosine and brain function. Int Rev Neurobiol 63:191–270
Fremeau RT Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D, Copenhagen DR, Storm-Mathisen J, Reimer RJ, Chaudhry FA, Edwards RH (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A 99:14488–14493
Gajda Z, Gyengesi E, Hermesz E, Ali KS, Szente M (2003) Involvement of gap junctions in the manifestation and control of the duration of seizures in rats in vivo. Epilepsia 44:1596–1600
Galic MA, Riazi K, Heida JG, Mouihate A, Fournier NM, Spencer SJ, Kalynchuk LE, Teskey GC, Pittman QJ (2008) Postnatal inflammation increases seizure susceptibility in adult rats. J Neurosci 28:6904–6913
Giaume C, Tabernero A, Medina JM (1997) Metabolic trafficking through astrocytic gap junctions. Glia 21:114–123
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Gigout S, Louvel J, Kawasaki H, D’Antuono M, Armand V, Kurcewicz I, Olivier A, Laschet J, Turak B, Devaux B, Pumain R, Avoli M (2006) Effects of gap junction blockers on human neocortical synchronization. Neurobiol Dis 22:496–508
Glass M, Faull RLM, Bullock JY, Jansen K, Mee EW, Walker EB, Synek BJL, Dragunow M (1996) Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res 710:56–68
Gomez-Gonzalo M, Losi G, Chiavegato A, Zonta M, Cammarota M, Brondi M, Vetri F, Uva L, Pozzan T, De Curtis M, Ratto GM, Carmignoto G (2010) An excitatory loop with astrocytes contributes to drive neurons to seizure threshold. PLoS Biol 8:e1000352
Gordon GR, Baimoukhametova DV, Hewitt SA, Rajapaksha WR, Fisher TE, Bains JS (2005) Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci 8:1078–1086
Gouder N, Fritschy JM, Boison D (2003) Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia 44:877–885
Gouder N, Scheurer L, Fritschy JM, Boison D (2004) Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci 24:692–701
Guo F, Sun F, Yu JL, Wang QH, Tu DY, Mao XY, Liu R, Wu KC, Xie N, Hao LY, Cai JQ (2010) Abnormal expressions of glutamate transporters and metabotropic glutamate receptor 1 in the spontaneously epileptic rat hippocampus. Brain Res Bull 81:510–516
Haberle J, Gorg B, Rutsch F, Schmidt E, Toutain A, Benoist JF, Gelot A, Suc AL, Hohne W, Schliess F, Haussinger D, Koch HG (2005) Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 353:1926–1933
Haberle J, Gorg B, Toutain A, Rutsch F, Benoist JF, Gelot A, Suc AL, Koch HG, Schliess F, Haussinger D (2006) Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J Inherit Metab Dis 29:352–358
Haberle J, Shahbeck N, Ibrahim K, Hoffmann GF, Ben-Omran T (2011) Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol Genet Metab 103:89–91
Haghikia A, Ladage K, Hinkerohe D, Vollmar P, Heupel K, Dermietzel R, Faustmann PM (2008) Implications of antiinflammatory properties of the anticonvulsant drug levetiracetam in astrocytes. J Neurosci Res 86:1781–1788
Haglund MM, Hochman DW (2005) Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol 94:907–918
Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA (2011) Evidence that compromised K(+) spatial buffering contributes to the epileptogenic effect of mutations in the human kir4.1 gene (KCNJ10). Glia 59:1635–1642
Halassa MM, Fellin T, Haydon PG (2007) The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med 13:54–63
Hammer J, Alvestad S, Osen KK, Skare O, Sonnewald U, Ottersen OP (2008) Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia 56:856–868
Heinemann U, Lux HD (1977) Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res 120:231–249
Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R, Lehmann TN (2000) Alterations of glial cell function in temporal lobe epilepsy. Epilepsia 41:S185–S189
Heuser K, Nagelhus EA, Tauboll E, Indahl U, Berg PR, Lien S, Nakken S, Gjerstad L, Ottersen OP (2010) Variants of the genes encoding AQP4 and Kir4.1 are associated with subgroups of patients with temporal lobe epilepsy. Epilepsy Res 88:55–64
Heuser K, Eid T, Lauritzen F, Thoren AE, Vindedal GF, Tauboll E, Gjerstad L, Spencer DD, Ottersen OP, Nagelhus EA, De Lanerolle NC (2012) Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J Neuropathol Exp Neurol 71:814–825
Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, Schramm J, Steinhäuser C (2000) Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci 12:2087–2096
Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC (2000) Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 7:153–159
Huber A, Padrun V, Deglon N, Aebischer P, Möhler H, Boison D (2001) Grafts of adenosine-releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci U S A 98:7611–7616
Jahromi SS, Wentlandt K, Piran S, Carlen PL (2002) Anticonvulsant actions of gap junctional blockers in an in vitro seizure model. J Neurophysiol 88:1893–1902
Jauch R, Windmuller O, Lehmann TN, Heinemann U, Gabriel S (2002) Effects of barium, furosemide, ouabaine and 4,4’-diisothiocyanatostilbene-2,2’-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res 925:18–27
Jin BJ, Zhang H, Binder DK, Verkman AS (2013) Aquaporin-4-dependent K(+) and water transport modeled in brain extracellular space following neuroexcitation. J Gen Physiol 141:119–132
Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192–9200
Kandratavicius L, Rosa-Neto P, Monteiro MR, Guiot MC, Assirati JA Jr, Carlotti CG Jr, Kobayashi E, Leite JP (2013) Distinct increased metabotropic glutamate receptor type 5 (mGluR5) in temporal lobe epilepsy with and without hippocampal sclerosis. Hippocampus 23:1212–1220
Kang J, Kang N, Lovatt D, Torres A, Zhao Z, Lin J, Nedergaard M (2008) Connexin 43 hemichannels are permeable to ATP. J Neurosci 28:4702–4711
Khurgel M, Ivy GO (1996) Astrocytes in kindling: relevance to epileptogenesis. Epilepsy Res 26:163–175
Kivi A, Lehmann TN, Kovacs R, Eilers A, Jauch R, Meencke HJ, Von Deimling A, Heinemann U, Gabriel S (2000) Effects of barium on stimulus-induced rises of [K+]o in human epileptic non-sclerotic and sclerotic hippocampal area CA1. Eur J Neurosci 12:2039–2048
Kofuji P, Newman EA (2004) Potassium buffering in the central nervous system. Neuroscience 129:1045–1056
Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA (2000) Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20:5733–5740
Kohling R, Gladwell SJ, Bracci E, Vreugdenhil M, Jefferys JG (2001) Prolonged epileptiform bursting induced by 0-Mg(2+) in rat hippocampal slices depends on gap junctional coupling. Neuroscience 105:579–587
Kowaluk EA, Jarvis MF (2000) Therapeutic potential of adenosine kinase inhibitors. Expert Opin Investig Drugs 9:551–564
Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ (2007) Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55:274–281
Kumaria A, Tolias CM, Burnstock G (2008) ATP signalling in epilepsy. Purinergic Signal 4:339–346
Kunze A, Congreso MR, Hartmann C, Wallraff-Beck A, Hüttmann K, Bedner P, Requardt R, Seifert G, Redecker C, Willecke K, Hofmann A, Pfeifer A, Theis M, Steinhäuser C (2009) Connexin expression by radial glia-like cells is required for neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A 106:11336–11341
Lee KS, Schubert P, Heinemann U (1984) The anticonvulsive action of adenosine: a postsynaptic, dendritic action by a possible endogenous anticonvulsant. Brain Res 321:160–164
Lee SH, Magge S, Spencer DD, Sontheimer H, Cornell-Bell AH (1995) Human epileptic astrocytes exhibit increased gap junction coupling. Glia 15:195–202
Lee TS, Eid T, Mane S, Kim JH, Spencer DD, Ottersen OP, De Lanerolle NC (2004) Aquaporin-4 is increased in the sclerotic hippocampus in human temporal lobe epilepsy. Acta Neuropathol 108:493–502
Lee DJ, Hsu MS, Seldin MM, Arellano JL, Binder DK (2012) Decreased expression of the glial water channel aquaporin-4 in the intrahippocampal kainic acid model of epileptogenesis. Exp Neurol 235:246–255
Li J, Shen H, Naus CC, Zhang L, Carlen PL (2001) Upregulation of gap junction connexin 32 with epileptiform activity in the isolated mouse hippocampus. Neuroscience 105:589–598
Li T, Quan LJ, Fredholm BB, Simon RP, Boison D (2007a) Adenosine dysfunction in astrogliosis: cause for seizure generation? Neuron Glia Biol 3:353–366
Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D (2007b) Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain 130:1276–1288
Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D (2008) Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 118:571–582
Li T, Lytle N, Lan JQ, Sandau US, Boison D (2012) Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia 60:83–95
Li M, Kang R, Shi J, Liu G, Zhang J (2013) Anticonvulsant activity of b2, an adenosine analog, on chemical convulsant-induced seizures. PLoS ONE 8:e67060
Liang SL, Carlson GC, Coulter DA (2006) Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J Neurosci 26:8537–8548
Lopes MW, Soares FM, de MN, Nunes JC, Cajado AG, de BD, de Cordova FM, da Cunha RM, Walz R, Leal RB (2013) Time-dependent modulation of AMPA receptor phosphorylation and mRNA expression of NMDA receptors and glial glutamate transporters in the rat hippocampus and cerebral cortex in a pilocarpine model of epilepsy. Exp Brain Res 226:153–163
Losi G, Cammarota M, Chiavegato A, Gomez-Gonzalo M, Carmignoto G (2010) A new experimental model of focal seizures in the entorhinal cortex. Epilepsia 51:1493–1502
Lothman EW, Somjen GG (1976) Functions of primary afferents and responses of extracellular K+ during spinal epileptiform seizures. Electroencephalogr Clin Neurophysiol 41:253–267
Maroso M, Balosso S, Ravizza T, Liu J, Aronica E, Iyer AM, Rossetti C, Molteni M, Casalgrandi M, Manfredi AA, Bianchi ME, Vezzani A (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat Med 16:413–419
Maroso M, Balosso S, Ravizza T, Liu J, Bianchi ME, Vezzani A (2011) Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Intern Med 270:319–326
Mathern GW, Mendoza D, Lozada A, Pretorius JK, Dehnes Y, Danbolt NC, Nelson N, Leite JP, Chimelli L (1999) Hippocampal GABA and glutamate transporter immunoreactivity in patients with temporal lobe epilepsy. Neurology 52:453–472
Medici V, Frassoni C, Tassi L, Spreafico R, Garbelli R (2011) Aquaporin 4 expression in control and epileptic human cerebral cortex. Brain Res 1367:330–339
Medina-Ceja L, Cordero-Romero A, Morales-Villagran A (2008) Antiepileptic effect of carbenoxolone on seizures induced by 4-aminopyridine: a study in the rat hippocampus and entorhinal cortex. Brain Res 1187:74–81
Meme W, Calvo CF, Froger N, Ezan P, Amigou E, Koulakoff A, Giaume C (2006) Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: potentiation by beta-amyloid. FASEB J 20:494–496
Miller HP, Levey AI, Rothstein JD, Tzingounis AV, Conn PJ (1997) Alterations in glutamate transporter protein levels in kindling- induced epilepsy. J Neurochem 68:1564–1570
Moody WJ, Futamachi KJ, Prince DA (1974) Extracellular potassium activity during epileptogenesis. Exp Neurol 42:248–263
Mylvaganam S, Zhang L, Wu C, Zhang ZJ, Samoilova M, Eubanks J, Carlen PL, Poulter MO (2010) Hippocampal seizures alter the expression of the pannexin and connexin transcriptome. J Neurochem 112:92–102
Nadler JV, Cuthbertson GJ (1980) Kainic acid neurotoxicity toward hippocampal formation: dependence on specific excitatory pathways. Brain Res 195:47–56
Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP (1999) Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia 26:47–54
Nagy JI, Rash JE (2000) Connexins and gap junctions of astrocytes and oligodendrocytes in the CNS. Brain Res Rev 32:29–44
Naus CCG, Bechberger JF, Paul DL (1991) Gap junction gene expression in human seizure disorder. Exp Neurol 111:198–203
Navarrete M, Perea G, Maglio L, Pastor J, Garcia de SR, Araque A (2013) Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb Cortex 23:1240–1246
Nemani VM, Binder DK (2005) Emerging role of gap junctions in epilepsy. Histol Histopathol 20:253–259
Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P (2001) Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci 21:5429–5438
Olney JW, Sharpe LG, Feigin RD (1972) Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol 31:464–488
Orellana JA, Saez PJ, Shoji KF, Schalper KA, Palacios-Prado N, Velarde V, Giaume C, Bennett MVL, Saez JC (2009) Modulation of brain hemichannels and gap junction channels by pro-inflammatory agents and their possible role in neurodegeneration. Antioxid Redox Signal 11:369–399
Orellana JA, Martinez AD, Retamal MA (2013) Gap junction channels and hemichannels in the CNS: regulation by signaling molecules. Neuropharmacology 75:567–582
Orkand RK, Nicholls JG, Kuffler SW (1966) Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol 29:788–806
Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA (2010) Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13:584–591
Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS (2005) K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Methods 2:825–827
Pan E, Stringer JL (1996) Influence of osmolality on seizure amplitude and propagation in the rat dentate gyrus. Neurosci Lett 207:9–12
Pannasch U, Rouach N (2013) Emerging role for astroglial networks in information processing: from synapse to behavior. Trends Neurosci 36:405–417
Pannasch U, Vargova L, Reingruber J, Ezan P, Holcman D, Giaume C, Sykova E, Rouach N (2011) Astroglial networks scale synaptic activity and plasticity. Proc Natl Acad Sci U S A 108:8467–8472
Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14:265–277
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG (2005a) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116
Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG (2005b) Astrocytic purinergic signaling coordinates synaptic networks. Science 310:113–116
Perez EL, Lauritzen F, Wang Y, Lee TS, Kang D, Zaveri HP, Chaudhry FA, Ottersen OP, Bergersen LH, Eid T (2012) Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis 47:331–337
Perez-Velazquez JL, Valiante TA, Carlen PL (1994) Modulation of gap junctional mechanisms during calcium-free induced field burst activity: a possible role for electrotonic coupling in epileptogenesis. J Neurosci 14:4308–4317
Petroff OA, Errante LD, Kim JH, Spencer DD (2003) N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology 60:1646–1651
Porter JT, McCarthy KD (1996) Hippocampal astrocytes in situ respond to glutamate released from synaptic terminals. J Neurosci 16:5073–5081
Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, De Graan PN (2002) Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125:32–43
Ransom CB, Sontheimer H (1995) Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol 73:333–346
Ransom CB, Ransom BR, Sontheimer H (2000) Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol (Lond) 522:427–442
Ravizza T, Gagliardi B, Noe F, Boer K, Aronica E, Vezzani A (2008) Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 29:142–160
Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R (2010) KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci U S A 107:14490–14495
Retamal MA, Froger N, Palacios-Prado N, Ezan P, Saez PJ, Saez JC, Giaume C (2007) Cx43 hemichannels and gap junction channels in astrocytes are regulated oppositely by proinflammatory cytokines released from activated microglia. J Neurosci 27:13781–13792
Roper SN, Obenaus A, Dudek FE (1992) Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann Neurol 31:81–85
Ross FM, Gwyn P, Spanswick D, Davies SN (2000) Carbenoxolone depresses spontaneous epileptiform activity in the CA1 region of rat hippocampal slices. Neuroscience 100:789–796
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang YF, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686
Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C (2008) Astroglial metabolic networks sustain hippocampal synaptic transmission. Science 322:1551–1555
Ruiz-Ederra J, Zhang H, Verkman AS (2007) Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 K+ channels in retinal muller cells. J Biol Chem 282:21866–21872
Samoilova M, Li J, Pelletier MR, Wentlandt K, Adamchik Y, Naus CC, Carlen PL (2003) Epileptiform activity in hippocampal slice cultures exposed chronically to bicuculline: increased gap junctional function and expression. J Neurochem 86:687–699
Samoilova M, Wentlandt K, Adamchik Y, Velumian AA, Carlen PL (2008) Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Exp Neurol 210:762–775
Santiago MF, Veliskova J, Patel NK, Lutz SE, Caille D, Charollais A, Meda P, Scemes E (2011) Targeting pannexin1 improves seizure outcome. PLoS ONE 6:e25178
Scemes E, Giaume C (2006) Astrocyte calcium waves: what they are and what they do. Glia 54:716–725
Scemes E, Spray DC (1998) Increased intercellular communication in mouse astrocytes exposed to hyposmotic shocks. Glia 24:74–84
Scemes E, Spray DC (2012) Extracellular K(+) and astrocyte signaling via connexin and pannexin channels Neurochem Res 37:2310–2316
Scholl UI, Choi M, Liu T, Ramaekers VT, Hausler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP (2009) Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A 106:5842–5847
Schröder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhäuser C (2000) Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41:S181–S184
Schwartzkroin PA, Baraban SC, Hochman DW (1998) Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 32:275–285
Seifert G, Schilling K, Steinhäuser C (2006) Astrocyte dysfunction in neurological disorders: a molecular perspective. Nat Rev Neurosci 7:194–206
Seifert G, Hüttmann K, Binder DK, Hartmann C, Wyczynski A, Neusch C, Steinhäuser C (2009) Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J Neurosci 29:7474–7488
Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, Grottesi A, Guerrini R, Masi G, Santorelli FM, Pessia M (2011) Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K(+) channel Kir4.1. Neurobiol Dis 43:239–247
Simantov R, Crispino M, Hoe W, Broutman G, Tocco G, Rothstein JD, Baudry M (1999) Changes in expression of neuronal and glial glutamate transporters in rat hippocampus following kainate-induced seizure activity. Brain Res Mol Brain Res 65:112–123
Söhl G, Güldenagel M, Beck H, Teubner B, Traub O, Gutierrez R, Heinemann U, Willecke K (2000) Expression of connexin genes in hippocampus of kainate-treated and kindled rats under conditions of experimental epilepsy. Mol Brain Res 83:44–51
Steinhäuser C, Seifert G, Bedner P (2012) Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60:1192–1202
Strohschein S, Hüttmann K, Gabriel S, Binder DK, Heinemann U, Steinhäuser C (2011) Impact of aquaporin-4 channels on K(+) buffering and gap junction coupling in the hippocampus. Glia 59:973–980
Suadicani SO, Iglesias R, Wang J, Dahl G, Spray DC, Scemes E (2012) ATP signaling is deficient in cultured Pannexin1-null mouse astrocytes. Glia 60:1106–1116
Szente M, Gajda Z, Said AK, Hermesz E (2002) Involvement of electrical coupling in the in vivo ictal epileptiform activity induced by 4-aminopyridine in the neocortex. Neuroscience 115:1067–1078
Takahashi DK, Vargas JR, Wilcox KS (2010) Increased coupling and altered glutamate transport currents in astrocytes following kainic-acid-induced status epilepticus. Neurobiol Dis 40:573–585
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699–1702
Tang FR, Lee WL (2001) Expression of the group II and III metabotropic glutamate receptors in the hippocampus of patients with mesial temporal lobe epilepsy. J Neurocytol 30:137–143
Tessler S, Danbolt NC, Faull RLM, Storm-Mathisen J, Emson PC (1999) Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience 88:1083–1091
Theis M, Giaume C (2012) Connexin-based intercellular communication and astrocyte heterogeneity. Brain Res 1487:88–98
Theofilas P, Brar S, Stewart KA, Shen HY, Sandau US, Poulsen D, Boison D (2011) Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 52:589–601
Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M (2005) An astrocytic basis of epilepsy. Nat Med 11:973–981
Torres A, Wang F, Xu Q, Fujita T, Dobrowolski R, Willecke K, Takano T, Nedergaard M (2012) Extracellular Ca(2)(+) acts as a mediator of communication from neurons to glia. Sci Signal 5:ra8
Traynelis SF, Dingledine R (1988) Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol 59:259–276
Traynelis SF, Dingledine R (1989) Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol 61:927–938
Ulas J, Satou T, Ivins KJ, Kesslak JP, Cotman CW, Balázs R (2000) Expression of metabotropic glutamate receptor 5 is increased astrocytes after kainate-induced epileptic seizures. Glia 30:352–361
van der Hel WS, Notenboom RG, Bos IW, van Rijen PC, van Veelen CW, De Graan PN (2005) Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64:326–333
Vezzani A, Moneta D, Conti M, Richichi C, Ravizza T, De Luigi A, De Simoni MG, Sperk G, Andell-Jonsson S, Lundkvist J, Iverfeldt K, Bartfai T (2000) Powerful anticonvulsant action of IL-1 receptor antagonist on intracerebral injection and astrocytic overexpression in mice. Proc Natl Acad Sci U S A 97:11534–11539
Vezzani A, Ravizza T, Balosso S, Aronica E (2008) Glia as a source of cytokines: implications for neuronal excitability and survival. Epilepsia 49(Suppl 2):24–32
Vezzani A, French J, Bartfai T, Baram TZ (2011a) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40
Vezzani A, Maroso M, Balosso S, Sanchez MA, Bartfai T (2011b) IL-1 receptor/Toll-like receptor signaling in infection, inflammation, stress and neurodegeneration couples hyperexcitability and seizures. Brain Behav Immun 25:1281–1289
Vezzani A, Friedman A, Dingledine RJ (2012) The role of inflammation in epileptogenesis. Neuropharmacology 69:16–24
Vianna EP, Ferreira AT, Dona F, Cavalheiro EA, da Silva Fernandes MJ (2005) Modulation of seizures and synaptic plasticity by adenosinergic receptors in an experimental model of temporal lobe epilepsy induced by pilocarpine in rats. Epilepsia 46(Suppl 5):166–173
Voss LJ, Jacobson G, Sleigh JW, Steyn-Ross A, Steyn-Ross M (2009) Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia 50:1971–1978
Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhäuser C (2006) The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26:5438–5447
Walz W (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36:291–300
Wang Y, Zaveri HP, Lee TS, Eid T (2009) The development of recurrent seizures after continuous intrahippocampal infusion of methionine sulfoximine in rats: a video-intracranial electroencephalographic study. Exp Neurol 220:293–302
Watanabe T, Morimoto K, Hirao T, Suwaki H, Watase K, Tanaka K (1999) Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res 845:92–96
Williams DM, Lopes CM, Rosenhouse-Dantsker A, Connelly HL, Matavel A, Uchi J, McBeath E, Gray DA (2010) Molecular basis of decreased Kir4.1 function in SeSAME/EAST syndrome. J Am Soc Nephrol 21:2117–2129
Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH (2003) Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol 54:251–256
Xu L, Zeng LH, Wong M (2009) Impaired astrocytic gap junction coupling and potassium buffering in a mouse model of tuberous sclerosis complex. Neurobiol Dis 34:291–299
Yao X, Hrabetova S, Nicholson C, Manley GT (2008) Aquaporin-4-deficient mice have increased extracellular space without tortuosity change. J Neurosci 28:5460–5464
Ye ZC, Sontheimer H (1996) Cytokine modulation of glial glutamate uptake: a possible involvement of nitric oxide. Neuroreport 7:2181–2185
Yoon JJ, Green CR, O’Carroll SJ, Nicholson LF (2010) Dose-dependent protective effect of connexin43 mimetic peptide against neurodegeneration in an ex vivo model of epileptiform lesion. Epilepsy Res 92:153–162
Zhang H, Verkman AS (2008) Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci 37:1–10
Zimmermann H, Braun N (1996) Extracellular metabolism of nucleotides in the nervous system. J Auton Pharmacol 16:397–400
Zurolo E, de Groot M, Iyer A, Anink J, Van Vliet EA, Heimans JJ, Reijneveld JC, Gorter JA, Aronica E (2012) Regulation of Kir4.1 expression in astrocytes and astrocytic tumors: a role for interleukin-1 beta. J Neuroinflamm 9:280
Acknowledgements
Supported by DFG (SFB/TR3, TP C1; SPP1172, SE774/3–2) and EU (FP7–202167 NeuroGLIA; ESF EuroEPINOMICS).
Conflict of Interest
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Bedner, P., Steinhäuser, C. (2014). Crucial Role for Astrocytes in Epilepsy. In: Parpura, V., Verkhratsky, A. (eds) Pathological Potential of Neuroglia. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0974-2_8
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0974-2_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0973-5
Online ISBN: 978-1-4939-0974-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)