
Abstract
Major advancements in the treatment of cancer and immune-mediated diseases have been made over the past few decades with the introduction of a variety of new biologic and chemotherapeutic agents. Their use, however, has been associated with a variety of adverse cutaneous reactions, some of which may even lead to the discontinuation of the inciting therapy. In this chapter, the clinical and histologic features of the following reactions are discussed: granuloma annulare/interstitial granulomatous dermatitis and urticaria/cellulitis associated with tumor necrosis factor-alpha inhibitors, neutrophilic dermatoses associated with granulocyte colony-stimulating factor, injection site reactions of interferon, hand-foot skin reaction of kinase inhibitor (sorafenib, sunitinib), urticaria and infusion reactions of CD20 inhibitors, Richter’s syndrome associated with CD52 inhibitors, and pseudodermatomyositis associated with chronic hydroxyurea.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Chronic Lymphocytic Leukemia
- Pyoderma Gangrenosum
- Injection Site Reaction
- Certolizumab Pegol
- Neutrophilic Dermatosis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Over the past few decades, the introduction of new biologic agents, such as tumor necrosis factor-alpha inhibitors, has resulted in potent disease-modifying effects in a variety of immune-mediated diseases. There have also been major advancements in cancer chemotherapy with the recent addition of granulocyte colony-stimulating factor, interferon, kinase inhibitors, and CD20 and CD52 inhibitors for the treatment of hematologic malignancies as well as solid tumors. However, a variety of toxicities, including adverse cutaneous reactions, are seen in association with these agents. Awareness of these cutaneous toxicities and recognition of corresponding histologic features is of diagnostic and therapeutic importance.
TNF-α Inhibitors
Tumor necrosis factor-α (TNF-α) is a powerful proinflammatory cytokine that is a central player to the activation of inflammation (Parameswaran and Patial 2010). Lymphocytes, macrophages, endothelial cells, Langerhans cells, as well as keratinocytes, melanocytes, and many others are capable of producing TNF-α (Deng et al. 2006). It is rapidly released in response to trauma, infection, or exposure to toxins wherein it functions as the “master regulator” of proinflammatory cytokine production (Parameswaran and Patial 2010). Despite its role in the normal host defense response, TNF-α is also central to the pathogenesis of several chronic inflammatory conditions and autoimmune disorders, including rheumatoid arthritis, psoriasis, inflammatory bowel disease, and ankylosing spondylitis (Kollias et al. 1999; Moustou et al. 2009).
Biologic antibody inhibitors of TNF-α, including infliximab (Remicade, Centocor, USA), etanercept (Enbrel, Immunex, USA), adalimumab (Humira), and certolizumab pegol (Cimzia), have been used with success in the treatment of these disorders. Nevertheless, their use has been associated with class-wide adverse reactions events, including opportunistic infections (Kim and Solomon 2010), reactivation of latent tuberculosis (Keane et al. 2001), lupus-like syndrome (Williams and Cohen 2011), demyelinating disease (Omair et al. 2012), exacerbation of heart failure (Colombel et al. 2004), and lymphoma (Adams et al. 2004). Cutaneous adverse events of antitumor necrosis factor therapy include infusion and injection site reactions, psoriasiform eruptions, granulomatous dermatitis, lupus-like disorders, vasculitis, cutaneous infections, and cutaneous neoplasms (Moustou et al. 2009; Bovenschen et al. 2006; Esser et al. 2004; Deng et al. 2006) (Table 15.1).
Granuloma Annulare/Interstitial Granulomatous Dermatitis
TNF-α inhibitor-associated granulomatous reactions, including interstitial granulomatous dermatitis and granuloma annulare (GA), are rare cutaneous lymphoid reactions that have only recently been reported in the literature (Deng et al. 2006; Voulgari et al. 2008).
Clinical Presentation
Granulomatous adverse cutaneous reactions may occur anywhere from 1 month to over 1 year after initiating anti-TNF-α therapy and may occur after treatment with any of the aforementioned agents (Deng et al. 2006; Voulgari et al. 2008). Cases of interstitial granulomatous dermatitis presented with rapid-onset, asymptomatic to mildly pruritic, 1–4 cm macules or indurated papules or plaques, many of which are annular in morphology, that occur on the trunk, shoulders, and upper extremities (Deng et al. 2006). In the first reported series, this reaction led to the discontinuation of treatment in four of five patients (Deng et al. 2006). Of these four patients, the eruption cleared or improved within 2 months of discontinuation (Deng et al. 2006). One patient had to be maintained on therapy due to the severity of underlying rheumatoid arthritis, and the lesions persisted at 3-month follow-up (Deng et al. 2006).
TNF-α inhibitor-induced GA presented with erythematous skin eruptions covering the fingers, hands, and forearms (Voulgari et al. 2008). In the largest report in the literature, nine such cases were reported among 197 patients (incidence, 4.5 %) undergoing anti-TNF therapy for the treatment of rheumatoid arthritis (Voulgari et al. 2008). Biologic therapy was maintained on seven of nine of these cases (Voulgari et al. 2008). Lesions were treated with topical corticosteroids with resolution of eruption after 3–4 weeks (Voulgari et al. 2008).
Histology Features
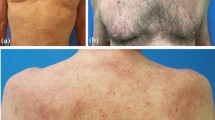
Histopathologic examination of TNF-α inhibitor-associated GA is characterized by palisading granulomas surrounding an area of necrobiosis and mucin deposition (Voulgari et al. 2008; Stewart et al. 2011) (Fig. 15.1). The histology of interstitial granulomatous dermatitis is characterized by a diffuse interstitial infiltrate of histiocytes and lymphocytes that palisade around partially degenerated or “piecemeal”-fragmented collagen, resulting in granulomatous foci in the mid and deep dermis (Deng et al. 2006). Eosinophils, neutrophils, and multinucleated giant cells may be present in scant numbers (Deng et al. 2006).

A 50-year-old female on Humira for Crohn’s disease presented with a pruritic and indurated plaques and papules on her left leg. A skin biopsy shows an interstitial and perivascular infiltrate of histiocytes and lymphocytes consistent with an interstitial granulomatous dermatitis
Pathogenesis
The precise pathomechanism underlying TNF-α inhibitor-induced GA is not known, but evidence suggests that anti-TNF therapy may induce autoreactive T cells (Sfikakis and Kollias 2003). Paradoxically, infliximab has been reported to have led to the rapid improvement of disseminated GA (Hertl et al. 2005). Chu et al. (1994) postulated that the granulomatous infiltrate in interstitial granulomatous dermatitis is a secondary reaction to dermal collagen damage induced initially by the deposition of immune complexes in dermal vessels and subsequent ischemia, followed by complement and neutrophil activation. It is known that interstitial granulomatous dermatitis can present in association with rheumatoid arthritis, lupus erythematosus, and other systemic disorders (Long et al. 1996) and may also be induced by other medications, including calcium channel blockers, lipid-lowering agents, and others (Magro et al. 1998).
Enbrel (Etanercept): Urticaria and Cellulitis
Urticaria and cellulitis are the most commonly reported cutaneous toxicity associated with Enbrel/etanercept, although it is not frequently biopsied.
Clinical Presentation
Etanercept-induced urticaria or urticaria-like reactions typically present 3–4 months after receiving twice-weekly subcutaneous etanercept injections (Skytta et al. 2000; Borrás-Blasco et al. 2009). After the inciting injection, an erythematous, macular, papular, or plaque-like urticarial lesion with or without central clearing develops initially on the extensor surfaces of the elbows and, subsequently, on the extensor surfaces of the knees (Borrás-Blasco et al. 2009). Lesions may also appear elsewhere on the body, such as the buttocks, trunk, proximal limbs, or ears (Skytta et al. 2000; Borrás-Blasco et al. 2009). Laboratory abnormalities, including eosinophilia, are typically absent (Skytta et al. 2000). These lesions have been reported to resolve after cessation of etanercept treatment (Skytta et al. 2000) or a course of steroid (Borrás-Blasco et al. 2009).
Reactions at the injection site can be urticarial reactions (Batycka-Baran et al. 2012) or eosinophilic cellulitis-like reaction (Winfield et al. 2006). Typically within 24 h of receiving the first dose, the patient developed evanescent, pruritic, slightly indurated, and erythematous plaques surrounding the injection site. These lesions resolved upon treatment with antihistamines and prednisone (Winfield et al. 2006) or topical steroids (Batycka-Baran et al. 2012).
Histologic Features
Histopathologic examination of the etanercept injection site urticarial lesions reveals a perivascular inflammatory infiltrate composed predominantly of neutrophils without eosinophils (Batycka-Baran et al. 2012). Immunophenotypic study may reveal either CD8+ (Zeltser et al. 2001) or CD4+ (González-López et al. 2007) predominance of T lymphocytes within the dermal infiltrate. Histology of the eosinophilic cellulitis-like injection site reaction to etanercept demonstrates an unremarkable epidermis and underling perivascular and interstitial lymphocytic infiltrates with numerous eosinophils with associated edema in the dermis (Winfield et al. 2006) (Fig. 15.2). Other notable features are multiple foci of hypereosinophilic collagen surrounded by eosinophils in the dermis as well as flame figures (Winfield et al. 2006).

A 47-year-old male with a history of psoriasis and on Enbrel treatment developed a lesion at the injection site on his right upper arm. A skin biopsy revealed an interstitial and perivascular infiltrate (a) of lymphocytes, histiocytes, and many eosinophils (b) in the dermis consistent with a hypersensitivity reaction
Pathogenesis
Batycka-Baran et al. (2012) proposed that the kinetics of the reaction, as revealed by the time course between the injection and onset of cutaneous eruption, may provide insight into the mechanism. It is thought that injection site reactions occurring rapidly after the first injection are due to an irritative mechanism and may not require discontinuation of treatment (Batycka-Baran et al. 2012). Urticarial lesions that occur within hours of the second or later injection with etanercept may reflect an immediate type I, IgE-mediated hypersensitivity reaction that would require stopping the medication and close monitoring for possible anaphylaxis (Batycka-Baran et al. 2012). Finally, reactions that occur hours after the injection and persist for several days may reflect an Arthus-like type III hypersensitivity reaction, mediated in part by the recruitment of neutrophilic granulocytes (Batycka-Baran et al. 2012).
G-CSF: Neutrophilic Dermatoses
Granulocyte colony-stimulating factor (G-CSF) is a neutrophil-specific growth factor that promotes the survival, proliferation, and maturation of neutrophil progenitors (Kaushansky 2006). This cytokine also enhances their phagocytic capacity, superoxide anion production, and bacterial killing mechanisms (Spiekermann et al. 1997). In contrast, granulocyte-macrophage colony-stimulating factor (GM-CSF) stimulates the production and activation of eosinophils, basophils, monocytes, and dendritic cells in addition to neutrophils.
G-CSF is used clinically to treat neutropenia associated with chemotherapy, myelodysplasia, or aplastic anemia as well as to reduce infections in patients with congenital, idiopathic, or cyclic neutropenia (Prendiville et al. 2001). Mild adverse effects include transient bone pain, edema, arthralgias, and myalgias as well as elevations in serum lactate dehydrogenase (LDH), alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (AP) levels (Paydas et al. 1993). It has also been found to increase the risk of treatment-related acute myelogenous leukemia, particularly in those with congenital neutropenia, wherein approximately 10 % will develop a malignant myeloid disorder (Kaushansky 2006; Dong et al. 1995).
Adverse cutaneous effects of G-CSF (Table 15.2) include diffuse eruption or reaction at injection site (Alvarez-Ruiz et al. 2004; Valks et al. 1998). Skin biopsies show an increase in the number of dermal macrophages, often enlarged in size (Alvarez-Ruiz et al. 2004; Valks et al. 1998). Neutrophilic dermatoses (Prendiville et al. 2001; Johnson and Grimwood 1994), including pyoderma gangrenosum (Ross et al. 1991) and Sweet syndrome (Paydas et al. 1993; Fukutoku et al. 1994), rare cases of vasculitis (Jain 1994; Couderc et al. 1995), and folliculitis (Ostlere et al. 1992) have been reported in association with G-CSF.
Sweet Syndrome Associated with G-CSF
Sweet syndrome, first documented by RD Sweet in (1964), is characterized by a pentad of findings, including fever, neutrophilia, multiple erythematous and painful cutaneous papules or nodules, dense dermal infiltrate of neutrophils, and rapid response to steroid therapy (Paydas et al. 1993). Cases of Sweet syndrome are categorized based on etiology into classical/idiopathic, malignancy-associated, and drug-induced (Cohen 2007). Classical Sweet syndrome is associated with upper respiratory tract and gastrointestinal infections, inflammatory bowel disease, and pregnancy (Cohen 2007). Malignancy-associated Sweet syndrome is most commonly caused by hematologic malignancies (especially acute myelogenous leukemia), but solid tumors of the genitourinary and gastrointestinal tracts and breast have also been associated. A wide variety of medications have been associated with drug-induced Sweet syndrome, including antibiotics such as trimethoprim-sulfamethoxazole, all-trans retinoic acid, and antineoplastic agents such as Imatinib (Cohen 2007). However, G-CSF is the most frequently and widely implicated medications in causing Sweet syndrome (White et al. 2006).
Walker and Cohen (1996) put forth the following diagnostic criteria for drug-induced Sweet syndrome: (1) abrupt onset of painful erythematous plaques or nodules, (2) histopathologic evidence of a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis, (3) fever >38 °C, (4) temporal relationship between drug ingestion and clinical presentation or temporally related recurrence after reintroduction of drug, and (5) temporally related resolution of lesions after drug withdrawal or treatment with systemic corticosteroids. Drug-induced Sweet syndrome typically affects patients between 29 and 68 years of age (mean, 45 years) and presents anywhere from days to weeks (mean, 7.5 days) after initiation of drug therapy. It is characterized by the development of fever, arthralgias, and diffuse, erythematous, painful papules, plaques, or nodules found most commonly in the upper extremities (71 %) but may also affect the chest (50 %), head and neck (43 %), lower extremities (36 %), or oral mucous membranes (7 %) (Walker and Cohen 1996). Lesions on the face may follow a periorbital or periauricular distribution (Bidyasar et al. 2008). Cutaneous lesions in a photodistributed pattern (Walker and Cohen 1996) as well as lesions that develop clinical features of necrosis (Fukutoku et al. 1994) have also been described. Ocular manifestations, characterized by “bilateral conjunctivitis” or “conjunctival lesions,” have also been documented in case reports (Bidyasar et al. 2008; Paydas et al. 1993).
Histopathologic examination of these cutaneous lesions reveals superficial edema and an underlying dense infiltrate of neutrophils in the dermis (Walker and Cohen 1996) (Fig. 15.3). Nuclear features of apoptosis such as karyorrhexis and an absence of vasculitis and pathogenic microbes are typical features (Bidyasar et al. 2008; Walker and Cohen 1996). The histologic differential diagnosis for drug-induced Sweet syndrome includes Sweet syndrome proper, abscess/cellulitis, granuloma faciale, leukemia cutis, lobular neutrophilic panniculitis, rheumatoid neutrophilic dermatitis, and pyoderma gangrenosum (Cohen et al. 2007).

A 51-year-old female on granulocyte colony-stimulating factor presented with erythematous plaques on her left upper arm. A skin biopsy showed marked papillary dermal edema and underlying band-like infiltrate of neutrophils consistent with Sweet syndrome
Laboratory abnormalities include neutrophilia, although this is much less common with drug-induced Sweet syndrome, especially that associated with G-CSF, than it is with Sweet syndrome proper (Walker and Cohen 1996). The cutaneous lesions in G-CSF associated Sweet syndrome may appear with the rise or normalization of neutrophil counts (Walker and Cohen 1996) and may disappear when they fall (van Kamp et al. 1994). Elevations in the erythrocyte sedimentation rate, anemia, and thrombocytosis have also been documented (Walker and Cohen 1996).
The mainstay of treatment for all forms of drug-induced Sweet syndrome is removal of the offending agent, after which the lesions typically resolve within 1–2 weeks (Walker and Cohen 1996). Clinical improvements in both cutaneous lesions and the overall condition have been achieved with low-dose systemic steroids alone (Bidyasar et al. 2008) as well as in combination with colchicine (van Kamp et al. 1994). Remission of the myeloid disorder, such as acute myeloid leukemia or myelodysplastic syndrome, also results in resolution of Sweet syndrome.
The precise etiology and pathogenic mechanism underlying all forms of Sweet syndrome is currently unknown. Elevated levels of G-CSF and interleukin-6 have been documented in a G-CSF-naïve patient with myelodysplastic syndrome-associated Sweet syndrome (Reuss-Borst et al. 1993). G-CSF, a cytokine, likely plays a direct pathogenic role, given that serum levels appear to directly correlate with dermatosis activity (Kawakami et al. 2004).
Pyoderma Gangrenosum Associated with G-CSF
The first case of biopsy-proven neutrophilic dermatosis without vasculitis was reported by Ross et al. (1991) in a 50-year-old woman who underwent G-CSF therapy for neutropenia related to chemotherapy for metastatic small cell lung cancer. Since this initial report, few additional cases of pyoderma gangrenosum associated with G-CSF and pegylated G-CSF therapy have been reported in the literature (Lewerin et al. 1997; Miall et al. 2006). G-CSF-associated pyoderma gangrenosum typically presents days to weeks after starting G-CSF therapy (Lewerin et al. 1997). It is characterized by the simultaneous onset of fever and erythematous plaques that can appear anywhere on the body, such as the face, trunk, upper, and lower extremities (Miall et al. 2006). The lesions rapidly progresses in size, swiftly develop blue or black edematous central regions of necrosis, and may appear clinically as a hemorrhagic blister, earning the name “bullous pyoderma gangrenosum” (Lewerin et al. 1997). There does not appear to be a correlation between the dosage or duration of therapy and the severity of this adverse cutaneous reaction (Ross et al. 1991).
Histopathologic examination reveals typical features of pyoderma gangrenosum characterized by a dense neutrophilic dermal infiltrate (Lewerin et al. 1997). Features of vasculitis are typically absent but have been described in association with this lesion (Miall et al. 2005; Lewerin et al. 1997). Studies for acid-fast bacilli, bacteria, and fungi are typically negative (Ross et al. 1991).
It appears that this adverse cutaneous reaction may not resolve with simple withdrawal of G-CSF (Lewerin et al. 1997). Treatment success has been reported with cyclosporin A (Lewerin et al. 1997). Others have had success with high-dose steroids in combination with dapsone (an anti-neutrophil agent) and topical tacrolimus (Miall et al. 2005). Interestingly, there was also a recent report of a case of Crohn’s-associated pyoderma gangrenosum that was successfully treated with perilesional granulocyte-macrophage colony-stimulating factor (GM-CSF) (Shpiro et al. 1998).
Classic pyoderma gangrenosum is most commonly associated with inflammatory bowel disease, rheumatologic disorders, or malignancy. The pathogenesis of this condition is poorly understood but thought to be related to loss of altered neutrophil trafficking and chemotaxis, genetic variations, and dysregulation of the innate immune system (Ahronowitz et al. 2012).
Interferon: Lymphocutaneous Injection Site Reactions
Interferon is a complex family of host-derived glycoproteins produced and secreted by eukaryotic cells in response to viruses, antigens, and mitogens (Baron et al. 1991). During their study of cellular viral infections, Isaacs and Lindemann (1957) identified a protein produced by cells infected by the virus that interfered with the virus’ ability to infect other cells. It is now known that interferons, of which there are three main groups (IFN-α, IFN-β, IFN-γ), have intrinsic antiviral, antimicrobial, immunomodulatory, and antiproliferative properties (Baron et al. 1991). Thus, today they are used to treat a variety of malignancies, viral infections, and inflammatory conditions, including hairy cell leukemia (IFN-α2b), chronic hepatitis C infection (IFN-α2b), multiple sclerosis (IFN-β1a), and chronic granulomatous disease (IFN-γ1b).
Despite their therapeutic yield, the doses required are frequently associated with significant side effects, including flu-like symptoms, weight loss, depression, as well as neurologic toxicity (Krainick et al. 1998). Moreover, interferon is believed to precipitate autoimmune disorders such as type I diabetes, thyroid disease, and systemic lupus erythematosus and may also worsen certain granulomatous diseases (Selmi et al. 2006).
Psoriasis was one of the early cutaneous reactions (Table 15.3) developed in association with interferon treatment (Quesada and Gutterman 1986), and there have been multiple subsequent reports in the literature (Ketikoglou et al. 2005). Eczematous drug eruptions associated with interferon, which often present as ill-defined clusters of coalescing, erythematous, blanchable, pruritic papules often on the extremities and trunk, have also been described (Dereure et al. 2002; Moore et al. 2004). Sarcoidosis has been described in patients receiving interferon treatment for hepatitis C as well as in a patient receiving adjuvant interferon-alpha immunotherapy for melanoma (Fantini et al. 2009; Alonso-Perez et al. 2006).
Alopecia has been reported in up to 19 % of patients treated with combination interferon and ribavirin, including alopecia areata (Agesta et al. 2002) and alopecia universalis (Demirturk et al. 2006). Vitiligo has been reported in a number of patients who were treated for hepatitis C with interferon (Tomasiewicz et al. 2006). Cases of cutaneous necrosis (Dalmau et al. 2005), fixed drug eruption (Tai and Tam 2005), lichenoid eruption (Pinto et al. 2003), lupus erythematosus (Fukuyama et al. 2000), indurated erythema (Detmar et al. 1989), leukocytoclastic vasculitis (Christian et al. 1997), as well as pyoderma gangrenosum (Montoto et al. 1998) have been reported in association with interferon.
Delivered through the intramuscular or subcutaneous route, interferon has also been associated with local injection site reactions in just under 5 % of patients, including local necrotizing (Krainick et al. 1998), granulomatous and suppurative (Sanders et al. 2002), lupus erythematosus-like (Arrue et al. 2007), embolia cutis medicamentosa (Koontz and Alshekhlee 2007), and local alopecia (Lang et al. 1999) lesions. The pathogenesis of these reactions remains uncertain. Multiple mechanisms have been proposed, and these, in addition to the basic clinical presentation and histopathology, are summarized by each pattern of reaction below. Whether there is a common underlying mode of pathogenesis underlying these reactions is yet to be determined, but interferon’s role in modulating multiple inflammatory pathways is likely central to the explanation.
Local Cutaneous Necrotizing Lesions
Krainick et al. (1998) described seven cases of local, painful cutaneous necrotizing lesions that arose in patients undergoing intramuscular or subcutaneous interferon therapy for Philadelphia chromosome-positive chronic myelogenous leukemia (CML). They occurred without correlation to specific stage of disease, course of illness, or interferon type. The lesions arose anywhere from 3 to 108 months after initiating interferon therapy. The lesions were described as necrotizing ulcers or abscess with surrounding erythematous, painful induration. Low-grade fever was commonly present. Microbiologic culture of the lesion was most commonly sterile and ultrasound findings were consistent with cellulitis.
Histopathologic examination revealed ulceration with underlying inflamed granulation tissue with scattered eosinophils in the dermis. One case was managed with reconstructive surgery, another with local surgical debridement and topical antibiotics, while the others generally resolved with more conservative measures even while maintained on therapy. The pathogenesis of these ulcers remains uncertain, but it is believed that local interferon-mediated keratinocyte toxicity, immune complex formation, and/or ischemia through multiple mechanisms, including interferon-mediated inhibition of angiogenesis, endothelial cell disruption, vasospasm, and intravascular thrombosis are involved (Sanders et al. 2002).
Granulomatous and Suppurative Dermatitis
Sanders et al. (2002) describe two cases wherein dusky, violaceous, multilobulated papules or indurated plaques and nodules with central ulceration developed at interferon injection sites in one patient undergoing treatment for non-remitting metastatic renal cell carcinoma (RCC) and another for metastatic melanoma. Histopathologic examination revealed suppurative and granulomatous dermatitis involving the superficial and deep dermis. The inflammatory cells were composed of neutrophils and histiocytes. Noninflammatory intravascular thrombosis was noted in the second case. Special stains for acid-fast bacilli, fungi, and bacteria were negative. Culture of lesions in one case was sterile and cultures from the second case contained surface colonization of bacteria that were not known to cause granulomas. Both patients’ injection site reactions resolved with conservative management, and one was maintained on therapy.
The authors proposed that a pathogenic mechanism involving tissue ischemia and intravascular thrombi may play a role in interferon-induced granulomatous and suppurative dermatitis, citing morphologic and biochemical similarities with pyoderma gangrenosum and cutaneous Crohn’s disease (Sanders et al. 2002).
Lupus Erythematosus-Like Reaction at Injection Site
Arrue et al. (2007) described five cases of cutaneous lesions at the interferon inoculation site with a lupus erythematosus-like histopathologic pattern. Three patients underwent treatment for metastatic melanoma and two patients for multiple sclerosis. The lesions generally occurred anywhere from days to years after inoculation with interferon and were described as painful erythematous nodules or exudative, infiltrated plaques that resolved spontaneously or with minimal treatment within 2–3 weeks. These patients had no history of prior lupus erythematosus, and serum studies conducted after the development of the lesions generally revealed negative antinuclear and deoxyribonucleic acid (DNA) antibodies and normal complement levels.
Histopathologic examination of biopsy specimens revealed prominent dermal mucin deposition highlighted by colloidal iron stain, perivascular and periadnexal infiltrate, and interface changes at the dermal follicular epithelium junction, while the epidermis exhibited only mild acanthosis and parakeratosis. In three of the cases, there was complete destruction of the folliculosebaceous unit, which was replaced by an inflammatory reaction or mucin pools.
While the pathogenesis of this adverse cutaneous reaction is uncertain, the authors of the case report postulate that abnormalities in platelet activation as well as the ability of interferon to transform fibroblast growth factor-β1, a cytokine that stimulates fibroblasts and, thus, increases the dermal mucin production, may play a role (Arrue et al. 2007).
Embolia Cutis Medicamentosa
Koontz and Alshekhlee (2007) reported a case of a 55-year-old man undergoing treatment with subcutaneous IFN-β1a who subsequently developed multiple painful necrotic skin lesions at the injection sites in various stages of healing. The diagnosis of embolia cutis medicamentosa was made based on the histologic findings of a thrombotic vasculopathy characterized by thrombotic occlusion of numerous capillaries. The authors hypothesized that patient self-administration of the medication resulted in inadvertent, deeper administration of the medication, leading to accidental arterial or periarterial injection of the agent. Their hypothesis is supported by the observation that after the patient was switched to an auto-injector, which controlled needle depth, no new lesions developed (Koontz and Alsshekhlee 2007).
Embolia cutis medicamentosa (Nicolau syndrome) was first described by Nicolau and Freudenthal in 1924 when they observed this phenomenon after an injection of bismuth salts in patients being treated for syphilis (Koontz and Alsshekhlee 2007). This adverse cutaneous effect is thought to be due to ischemic necrosis secondary to occlusion of the microvasculature (Luton et al. 2006). Proposed pathogenic mechanisms include intra-arterial, periarterial, or perivenous phenomenon wherein embolic or crystal occlusion by the injected agent, pain and sympathetically mediated vasospasm, or vascular trauma-induced thrombosis disrupts the microcirculation (Luton et al. 2006).
Alopecia
Lang et al. (1999) described three cases of transient, localized hair loss at IFN-α2b injection sites in patients undergoing treatment for chronic hepatitis C infection. The lesions were described as patches of non-scarring alopecia with or without perifollicular erythema or papules. Histopathologic examination revealed a non-scarring alopecia with perifollicular and perivascular lymphohistiocytic infiltrate at the infundibulum and isthmus. It is believed that multiple mechanisms may be at play in this reaction, including the formation of autoantibodies directed against follicular epithelium through interferon-mediated induction of previously masked surface antigens, and direct replicative inhibition of germinative hair cells (Lang et al. 1999).
Kinase Inhibitor (Sorafenib, Sunitinib): Hand-Foot Skin Reaction
Sorafenib and sunitinib are two novel, rationally designed, orally available, multikinase inhibitors (MKIs) used in the treatment of various malignancies for their anti-angiogenic and antiproliferative properties. Initially identified as a Raf-kinase inhibitor, sorafenib has also been shown to inhibit vascular endothelial growth factor receptor (VEGFR) 2–3, platelet-derived growth factor receptor (PDGFR)-β, Fms-like tyrosine kinase 3 (Flt-3), RET, c-KIT, p38α (member of the MAP-kinase family), and B-Raf (Wilhelm and Chien 2002). Sunitinib targets VEGFR 1–3, PDGFR-α, c-Kit, Flt-3, colony-stimulating factor receptor-1, and the glial cell line-derived neurotrophic factor receptor (Chow and Eckhardt 2007). Sorafenib is currently approved for the treatment of metastatic renal cell carcinoma (RCC) (Singer et al. 2012) but has also been shown to be effective in treating hepatocellular carcinoma (HCC) (Kokudo et al. 2012). Sunitinib has been approved for the treatment of gastrointestinal stromal tumor (GIST) as well as advanced RCC but has also been shown to be affective against neuroendocrine, colon, and breast cancers in phase II studies (Chow and Eckhardt 2007).
Significant cutaneous adverse events are present in up to 80 % of patients undergoing treatment with kinase inhibitors, of which the hand-foot skin reaction (HFSR) may be the most significant cutaneous toxicity. In a retrospective review of 109 patients on sorafenib for RCC or HCC and 119 patients receiving sunitinib for RCC or GIST, Lee et al. (2009) identified that HFSR was the most commonly observed cutaneous toxicity, seen in 48 % of patients on sorafenib and 36 % of patients on sunitinib. Moreover, HFSR is regarded as the most clinically significant dermatologic toxicity of sorafenib and sunitinib therapy, given the severity of symptoms and the potential for consequent chemotherapeutic dose modification (Chu et al. 2008; Lacouture et al. 2008).
Besides HFSR, a variety of other cutaneous reactions and neoplasms has been reported in association with sorafenib and sunitinib (Table 15.4) (Robert et al. 2005, 2011; Smith et al. 2009; Raymond et al. 2010; Bovenschen et al. 2006; Kong et al. 2008; Rosenbaum et al. 2008; Chu et al. 2008; Lacouture et al. 2008). Smith et al. (2009) reported 15 patients who developed keratoacanthoma-type squamous cell carcinomas associated with sorafenib treatment. Regional squamous cell carcinoma has also been recently reported to develop following limb infusion of sorafenib (Raymond et al. 2010). Rapid development of pigmented lesions has been reported in association with blistering disorders, immunodeficiency, and chemotherapeutic agents (Bovenschen et al. 2006). Kong et al. (2008) reported eruptive melanocytic nevi in two patients receiving sorafenib for treatment of metastatic RCC. Other skin changes associated with kinase inhibitors include nonspecific rash, skin discoloration, xerosis, hair color changes, alopecia, and nail changes (Rosenbaum et al. 2008).
Clinical Presentation
The hand-foot skin reaction (HFSR) is a distinct set of palmoplantar lesions in areas of frequent trauma or friction. They can develop anytime from 3 to 56 days (median, 18.4 days) after or within 4 weeks (83 %) of initiating therapy with sorafenib or sunitinib (Lee et al. 2009). These lesions are characterized by multiple, scaly, keratotic plaques on the knuckles of the dorsal hand, hyperkeratotic callus-like lesions on pressure areas of the sole, and/or yellow-colored blisters surrounded by an erythematous halo on fingers and palm (Lee et al. 2009). They are commonly associated with significant tenderness and paresthesias and result in significant alterations in quality of life (Lacouture et al. 2008).
Histologic Features
Skin biopsies from seven patients with HFSR associated with sorafenib or sunitinib showed band-like foci of necrotic keratinocytes in all seven, with two resulting in blister formation (Lacouture et al. 2008). All specimens showed superficial telangiectasia, sparse perivascular lymphocytic infiltrates, and subtle dilatation and cystic degeneration of eccrine glands (Lacouture et al. 2008). Dermal eosinophils are not prominent. The extent of this epidermal change appears to correlate with the duration of therapy with the multikinase inhibitor; less than 30 days of exposure was associated with only intraepidermal changes, while longer exposure times led to changes in the outermost stratum corneum, evident as hyperkeratosis and parakeratosis (Lacouture et al. 2008). In a case series by Lee et al. (2009), similar changes were noted in the skin biopsies from six patients. The general histopathologic pattern is an interface dermatitis with some degree of epidermal necrosis or vesiculation, as noted in some cases (Fig. 15.4) (Lacouture et al. 2008; Lee et al. 2009).

A 51-year-old woman with hepatitis C and hepatocellular carcinoma on sorafenib treatment presented with bilateral vesicles and bullae on her feet. These blisters resolved after cessation of sorafenib treatment. A skin biopsy from her left dorsal foot showed focal epidermal necrosis and necrotic keratinocyte within the epidermis. There is papillary dermal edema and a perivascular lymphocytic infiltrate in the dermis
Differential Diagnosis/Pathogenesis
The differential diagnosis for hand-foot skin reaction (HFSR) includes hand-foot syndrome (HFS), palmoplantar keratoderma, radiation recall dermatitis, localized epidermal necrolysis, generalized epidermal necrolysis, and acute graft-versus-host disease (Lacouture et al. 2008; Nagore et al. 2000).
The pathogenesis of this adverse cutaneous reaction is uncertain. The most widely accepted theory is direct toxicity of chemotherapeutic agents against acral epithelium, which may occur through inhibition of the keratinocyte c-Kit receptor (Chu et al. 2008). Other direct cytotoxic agents have been hypothesized to inflict direct keratinocyte toxicity through eccrine sweat gland excretion (Jacobi et al. 2005). Chu et al. (2008) has suggested that direct effect of sorafenib on eccrine gland receptors may also play a pathogenic role. The histologic findings of apoptotic keratinocytes with satellitosis of lymphocytes led Beard et al. (1993) to propose the host-versus-altered-host response. Despite these suggestions, sorafenib and sunitinib’s inhibitory effect on vascular endothelial growth factor (VEGF) and platelet-derived growth factor at the capillary and vascular endothelial level is likely central to the explanation, given that this reaction occurs in areas exposed to repeated trauma such as the palms and soles (Lacouture et al. 2008). Alas, direct VEGF inhibitor (bevacizumab) use does appear to associate with more severe sorafenib-associated HFSR (Azad et al. 2009). However, given that HFSR incidence is rare with isolated bevacizumab use (Chu et al. 2008), sorafenib and sunitinib’s inhibitory effect on multiple kinase pathways suggests that there are multiple conspiring mechanisms underlying this reaction.
Treatment
There have been no randomized trials comparing the efficacy of various agents in the treatment of hand-foot skin reaction (HFSR) to multikinase inhibitors. The literature to date supports the use of supportive podiatric measures, including a variety of moisturizers, cotton socks, shock absorbers, and soft shoes that minimize pressure points (Chu et al. 2008). Lacouture et al. (2008) obtained significant clinical improvement in treating sorafenib- and sunitinib-induced HFSR with topical urea (keratolytic that supports skin integrity) alongside tazarotene (reduces epidermal proliferation and dermal inflammation) and/or topical fluorouracil.
CD20 Inhibitors: Urticaria and Infusion Reactions
In 1997, rituximab (Rituxan, IDEC-C2B8) became the first monoclonal antibody product approved in the United States for the treatment of a malignancy (Dillman 1999). Rituximab is a chimeric monoclonal antibody composed of human immunoglobulin G1 heavy-chain sequences and murine immunoglobulin variable regions that targets the human CD20 antigen (Maloney 2012). CD20 is a multimeric, cell-surface complex antigen that is specific to B cells (Maloney 2012). Physiologically, CD20 regulates transcellular calcium transport and is involved in the regulation of B-cell activation and proliferation (Maloney 2012). Anti-CD20 antibodies such as rituximab are thought to contribute to B-cell-specific destruction through a combination of synergistic mechanisms including complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, direct antiproliferative effect, and the induction of apoptosis (Maloney 2012).
Originally approved by the Food and Drug Administration for the treatment of follicular B-cell lymphoma, rituximab is one of the most widely used anticancer drugs in the United States. It is currently used to treat an array of malignancies and several inflammatory conditions, including chronic lymphocytic leukemia (Bauer et al. 2012), B-cell lymphomas (Maloney 2012), as well as rheumatoid arthritis (Moots and Naisbett-Groet 2012), systemic lupus erythematosus (Coca and Sanz 2012), and refractory nephrotic syndrome (van Husen and Kemper 2011). Additional anti-CD20 antibodies, including ofatumumab (human antibody), tositumomab (murine antibody), and obinutuzumab (human antibody), have been developed, but their use has been less well documented (Maloney 2012).
Rituximab is typically administered by intravenous infusion (Maloney 2012). Infusion-related reactions occur at an incidence rate of up to 14 %, as documented in clinical trials (Maloney 2012). To date, there have been three case reports of rituximab infusion-related urticarial reactions in patients undergoing treatment for follicular B-cell lymphoma, despite premedication with diphenhydramine, acetaminophen, and steroids (Heinzerling et al. 2000; Errante et al. 2006; Rey et al. 2009). This cutaneous reaction typically occurs 1 h after the first infusion and is characterized by a pruritic, erythematous to violaceous, well-demarcated, irregularly shaped plaques that develop at the site of existing or previously excised tumors (Errante et al. 2006). The lesions spontaneously regress one to two hours after stopping the infusion (Rey et al. 2009; Errante et al. 2006). Formal histopathologic descriptions of these lesions have yet to be described in the literature, as biopsies of this rapidly resolving reaction may not be readily taken.
Infusion-related localized urticaria is thought to be an epiphenomenon of malignant B-cell destruction as opposed to a true hypersensitivity reaction (Errante et al. 2006). Because it occurs within an hour after the very first infusion further supports this hypothesis (Rey et al. 2009). Van der Kolk et al. (2001) have previously demonstrated that complement activation and subsequent release of tumor necrosis factor-α, interleukin (IL)-6, and IL-8 correlate with the severity of rituximab-related side effects. As such, cytokine release may be involved in the pathomechanism of infusion-related urticaria (Errante et al. 2006). Interestingly, all three reported cases of rituximab infusion-induced urticaria have been in patients undergoing treatment for follicular B-cell lymphoma, which is known to be sensitive to rituximab (Rey et al. 2009).
Other infusion-related adverse effects include fever, chills, nausea, and headache (Maloney 2012; Errante et al. 2006). More serious adverse reactions, including cytokine-release syndrome associated with lymphopenia, thrombocytopenia, and hepatotoxicity (Winkler et al. 1999), potentially life-threatening noninfectious pulmonary toxicity (Hadjinicolaou et al. 2012), and cardiac arrhythmias and acute coronary syndromes (Lee and Kukreti 2012), have been described in the literature. A single case report of Stevens-Johnson syndrome associated with rituximab therapy has been described (Lowndes et al. 2002). Otherwise, cutaneous adverse reactions are characterized predominantly by lesion-localized urticaria.
Alemtuzumab (Campath-1H): Richter’s Syndrome
Alemtuzumab (Campath-1H) is a humanized, monoclonal antibody that targets CD52, a cell-surface peptide antigen expressed on normal and malignant B and T lymphocytes (Hale et al. 1985). Approved as salvage therapy for patients with chronic lymphocytic leukemia (CLL) and cutaneous T-cell lymphoma who have failed purine analogue treatment, alemtuzumab induces more prolonged depletion of CD4 and CD8 T-cell subpopulations (Keating et al. 2002). Adverse events associated with alemtuzumab include infusion-related rigors, fever, nausea, vomiting, and rash (Keating et al. 2002). Opportunistic infections, cardiac toxicity, and neutropenia may also occur (Keating et al. 2002; Lenihan et al. 2004).
Richter’s syndrome (RS) refers to the development of a second, more aggressive lymphoma in a patient with CLL (Richter 1928). The secondary lymphoma is usually a diffuse large B-cell lymphoma (DLBCL), but the development of Hodgkin’s lymphoma (Jamroziak et al. 2012) and T-cell lymphoma (Lee et al. 1995) after CLL has also been described. Studies to date have estimated that Richter’s syndrome occurs in anywhere from 1 to 10 % of patients with CLL (Robertson et al. 1993). Clinically, RS is characterized by sudden clinical deterioration with rapid increase in lymphadenopathy or extranodal involvement and less commonly the presence of a monoclonal gammopathy, lytic bone lesions, and multiple cytogenetic changes (Robertson et al. 1993). Molecular analyses of immunoglobulin heavy- and light-chain rearrangements suggest that the secondary lymphoma generally arises from a transformation of the underlying CLL cell (Robertson et al. 1993). However, biclonal origin in RS has been described in the literature (Tohda et al. 1990).
To date, there have been two reported cases in the literature that describe Richter’s or Richter’s-like transformation associated primarily with alemtuzumab therapy (Janssens et al. 2006; Faguer et al. 2007). A long-term follow-up, retrospective study of 38 patients with CLL who underwent alemtuzumab therapy found that there was an increased but statistically insignificant risk of developing Richter’s syndrome in patients on alemtuzumab therapy (16 %, 6/38) compared with consecutive historical controls who underwent alternative salvage therapy (12 %, 9/75) (Karlsson et al. 2006). In their report on 39 patients who developed Richter’s syndrome, Robertson et al. (1993) noted that most of those that had developed the transformation had active and more advanced disease. Thus, whether alemtuzumab therapy poses a true risk for the development of Richter’s syndrome is still to be determined. Nonetheless, a summary of the reported clinical and pathologic features and a discussion of proposed pathogenic mechanisms of alemtuzumab-induced Richter’s syndrome are noted below.
Clinical Presentation/Histologic Features
Janssens et al. (2006) report the case of a patient with B-CLL who after failing treatment with chlorambucil was started on alemtuzumab. Three months after initiating alemtuzumab therapy, the patient developed a fever and rapidly increasing lactate dehydrogenase (LDH), with recurrent thrombocytopenia and autoimmune hemolytic anemia, which had previously precluded her from fludarabine. Bone marrow biopsy led to a diagnosis of large B-cell lymphoma, revealing the Richter’s transformation. Molecular microbiologic studies documented a negative Epstein-Barr virus (EBV) status. Molecular analysis of the immunoglobulin heavy chain confirmed that the CLL and the RS were of different clonal origin, and further analyses revealed that, in fact, the RS cells had evolved from a clone that was present 2 years before the clinical appearance of RS.
Faguer et al. (2007) report the case of a patient with cutaneous T-cell lymphoma (CTCL) who, 2 months after initiating alemtuzumab therapy, developed high fever, night sweats, and dramatic diffuse enlargement of lymph nodes and left temporal cutaneous tumor. A month prior to this presentation, the patient had developed febrile CMV viremia, which after treatment with ganciclovir and prophylaxis with valganciclovir, became and remained negative. Biopsy disclosed a transformed CTCL with CD30+ T cells, revealing a Richter’s syndrome-like phenomenon. Rising titers of EBV virus were documented at this time, but in situ hybridization with EBV-specific EBER probes on the biopsy specimen were negative. The patient was treated with a separate course of chemotherapy with no obvious clinical response.
Pathogenesis
It is known that in typical RS, acquisition of TP53 mutations and/or 17p deletions are frequent molecular events (Rossi and Gaidano 2009). However, whether these mutations are sufficient to lead to the disease remains to be determined. The authors of both case reports above are in agreement that alemtuzumab may have promoted the uncontrolled growth of a novel, Richter’s clone by eradicating the initial B-CLL or CTCL efficiently and by inducing T-cell depletion with consequent impairment of immunosurveillance (Janssens et al. 2006; Faguer et al. 2007). However, the relationship between chemotherapy-induced immunosuppression and the development of RS remains a controversial issue (Cohen et al. 2002).
Previous studies have emphasized the role of circulating natural killer lymphocytes in the cytotoxic response against Sézary cells (Bouaziz et al. 2005). The transformation of CLL into diffuse large B-cell lymphoma has also been reported after rituximab (Cohen et al. 2002). A very recent case was also reported by Salihoglu et al. (2013 in press) of a CLL patient who developed diffuse large B-cell lymphoma after treatment with rituximab, alemtuzumab, and reduced-intensity conditioning allogenic stem cell transplantation. However, considering the advanced stage of disease, it is still possible that this transformation reflects the natural history of the disease process itself as opposed to being a direct result of chemotherapeutics (Cohen et al. 2002). Further study and experience with alemtuzumab may reveal further insight.
Chronic Hydroxyurea: Pseudodermatomyositis
Hydroxyurea is a cytostatic, antineoplastic agent that interrupts the formation of deoxyribonucleotides from ribonucleotides by inhibiting the enzyme ribonucleotide diphosphate reductase. It is used to treat myeloproliferative disorders such as chronic myelogenous leukemia (CML), essential thrombocythemia, polycythemia vera, as well as sickle cell anemia and severe, refractory psoriasis (Richard et al. 1989; Halverstam et al. 2008).
Major adverse reactions include bone marrow suppression (Oskay et al. 2002) and life-threatening alveolitis (Dacey and Callen 2003; Senet et al. 1995) as well as an elevated risk of multiple actinic keratoses and non-melanoma skin cancers, which is thought to be related to the inhibition of DNA damage repair mechanisms (Rocamora et al. 2000; Saraceno et al. 2008; Papi et al. 1993; Stasi et al. 1992). Documented adverse cutaneous effects associated with long-term hydroxyurea therapy include xerosis, hyperpigmentation, atrophy, lower extremity ulcerations, partial alopecia, and chromonychia (Hagen et al. 2012; Richard et al. 1989) (Table 15.5), but these effects are also common with use of other chemotherapeutic agents such as cytarabine, doxorubicin, busulfan, and bleomycin (Senet et al. 1995).
Pseudodermatomyositis is a cutaneous reaction unique to long-term hydroxyurea therapy that was first described in 1975 by Kennedy et al. who documented three patients undergoing said treatment for chronic myelogenous leukemia that developed erythematous lesions, violaceous papules, and atrophy involving the dorsa of the metacarpophalangeal and interphalangeal joints. Sigal et al. (1984) later described the eruption as dermatomyositis-like but ultimately eliminated the diagnosis because of the absence of muscular abnormalities. Interestingly, this rare but characteristic cutaneous side effect has not been documented in psoriatic patients treated with hydroxyurea, but this may be related to the shorter duration of therapy (Senet et al. 1995).
Clinical Presentation
Pseudodermatomyositis typically presents after years (2–10) of hydroxyurea therapy with scaly, linear, and erythematous plaques on the dorsa of the hands (Kennedy et al. 1975) that may or may not be tender (Suehiro et al. 1998). These lesions may strongly resemble the Gottron’s sign of dermatomyositis proper (Rocamora et al. 2000). Associated findings include localized atrophy, nails telangiectatic changes, scaly poikilodermatous plaques, or violaceous papules on the feet, elbows, palms, or face (Hagen et al. 2012). The violaceous and erythematous lesions, which can appear around the eyelids and elsewhere on the face, resemble the heliotrope rash of dermatomyositis (Oskay et al. 2002). However, there are typically no signs of proximal muscle weakness or muscle enzymes and electromyography abnormalities (Senet et al. 1995). Furthermore, laboratory evaluation typically reveals negative antinuclear antibody as well as normal aldolase creatine kinase levels (Dacey and Callen 2003). Unlike in dermatomyositis proper, there is little data to suggest that pseudodermatomyositis is associated with any underlying malignancy (Robinson and Reed 2011).
Histologic Features
Histopathologic examination of skin biopsies shows hyperkeratosis, epidermal atrophy, vacuolar changes in the basal layer, and perivascular lymphohistiocytic infiltrate in the superficial dermis (Bahadoran et al. 1996; Senet et al. 1995). Variations on this common theme include focal lichenoid reactions, dyskeratotic cells, dermal mucin deposition, and dermal telangiectasia (Dacey and Callen 2003). Direct immunofluorescence studies have been reportedly negative (Senet et al. 1995).
Differential Diagnosis/Pathogenesis
The histologic differential diagnosis includes dermatomyositis, poikiloderma, lichenoid dermatitis, and graft-versus-host disease (Hagen et al. 2012). The mechanism underlying this reaction is not known at this time. Chronic, cumulative cytologic and selective DNA damage to the basal layer and epidermis is likely involved and may be related to the accumulation of the drug or one of its metabolites (Richard et al. 1989; Senet et al. 1995; Dacey and Callen 2003). Given that leg ulceration is another common adverse event associated with long-term hydroxyurea use, Suehiro et al. (1998) have postulated that chronic cumulative toxicity of the drug on the dividing cells of the basal epidermal layer may play a pathogenic role.
Treatment
Pseudodermatomyositis is a mild eruption that resolves without treatment between 10 days and 18 months after discontinuation of hydroxyurea but runs a benign course even if the medication is continued (Senet et al. 1995).
References
Adams AE, Zwicker J, Curiel C, Kadin ME, Falchuk KR, Drews R, Kupper TS. Aggressive cutaneous T-cell lymphomas after TNF-alpha blockade. J Am Acad Dermatol. 2004;51(4):660–2.
Agesta N, Zabala R, Diaz-Perez JL. Alopecia areata during interferon alpha-2b/ribavirin therapy. Dermatology. 2002;205(3):300–1.
Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum: a comprehensive review. Am J Clin Dermatol. 2012;13(3):191–211. doi:10.2165/11595240-000000000-00000.
Alonso-Perez A, Ballestero-Diez M, Fraga J, Garcia-Diez A, Fernandez-Herrera J. Cutaneous sarcoidosis by interferon therapy in a patient with melanoma. J Eur Acad Dermatol Venereol. 2006;20(10):1328–9.
Alvarez-Ruiz S, Penas PF, Fernandez-Herrera J, Sanchez-Perez J, Fraga J, Garcia-Diez A. Maculopapular eruption with enlarged macrophages in eight patients receiving G-CSF or GM-CSF. J Eur Acad Dermatol Venereol. 2004;18(3):310–3.
Arrue I, Saiz A, Ortiz-Romero PL, Rodriguez-Peralto JL. Lupus-like reaction to interferon at the injection site: report of five cases. J Cutan Pathol. 2007;34 Suppl 1:18–21.
Azad NS, Aragon-Ching JB, Dahut WL, Gutierrez M, Figg WD, Jain L, et al. Hand-foot skin reaction increases with cumulative sorafenib dose and with combination anti-vascular endothelial growth factor therapy. Clin Cancer Res. 2009;15(4):1411–6. doi:10.1158/1078-0432.CCR-08-1141.
Bahadoran P, Castanet J, Lacour JP, Perrin C, Del Giudice P, Mannocci N, et al. Pseudo-dermatomyositis induced by long-term hydroxyurea therapy: report of two cases. Br J Dermatol. 1996;134(6):1161–3.
Baron S, Tyring SK, Fleischmann Jr WR, Coppenhaver DH, Niesel DW, Klimpel GR, et al. The interferons. Mechanisms of action and clinical applications. JAMA. 1991;266(10):1375–83.
Batycka-Baran A, Flaig M, Molin S, Ruzicka T, Prinz JC. Etanercept-induced injection site reactions: potential pathomechanisms and clinical assessment. Expert Opin Drug Saf. 2012;11(6):911–21. doi:10.1517/14740338.2012.727796.
Bauer K, Rancea M, Roloff V, Elter T, Hallek M, Engert A, Skoetz N. Rituximab, ofatumumab and other monoclonal anti-CD20 antibodies for chronic lymphocytic leukaemia. Cochrane Database Syst Rev. 2012;11:CD008079. doi:10.1002/14651858.CD008079.pub2.
Beard JS, Smith KJ, Skelton HG. Combination chemotherapy with 5-fluorouracil, folinic acid, and alpha-interferon producing histologic feature of graft-versus-host disease. J Am Acad Dermatol. 1993;29(2 Pt 2):325–30.
Bidyasar S, Montoya M, Suleman K, Markowitz AB. Sweet syndrome associated with granulocyte colony-stimulating factor. J Clin Oncol. 2008;26(26):4355–6. doi:10.1200/JCO.2008.16.2933.
Borrás-Blasco J, Gracia-Perez A, Rosique-Robles JD, Nuñez-Cornejo C, Casterá MD, Abad FJ. Urticaria due to etanercept in a patient with psoriatic arthritis. South Med J. 2009;102(3):304–5. doi:10.1097/SMJ.0b013e31819450e7.
Bouaziz JD, Ortonne N, Giustiniani J, Schiavon V, Huet D, Bagot M, Bensussan A. Circulating natural killer lymphocytes are potential cytotoxic effectors against autologous malignant cells in sezary syndrome patients. J Invest Dermatol. 2005;125(6):1273–8.
Bovenschen HJ, Tjioe M, Vermaat H, de Hoop D, Witteman BM, Janssens RW, et al. Induction of eruptive benign melanocytic naevi by immune suppressive agents, including biologicals. Br J Dermatol. 2006;154:880–4.
Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25(7):884–96.
Christian MM, Diven DG, Sanchez RL, Soloway RD. Injection site vasculitis in a patient receiving interferon alfa for chronic hepatitis C. J Am Acad Dermatol. 1997;37(1):118–20.
Chu D, Lacouture ME, Fillos T, Wu S. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol. 2008;47(2):176–86. doi:10.1080/02841860701765675.
Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130(10):1278–83.
Coca A, Sanz I. Updates on B-cell immunotherapies for systemic lupus erythematosus and Sjogren’s syndrome. Curr Opin Rheumatol. 2012;24(5):451–6. doi:10.1097/BOR.0b013e32835707e4.
Cohen PR. Sweet’s syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
Cohen Y, Da’as N, Libster D, Amir G, Berrebi A, Polliack A. Large – cell transformation of chronic lymphocytic leukemia and follicular lymphoma during or soon after treatment with fludarabine-rituximab-containing regimens: natural history- or therapy-related complication? Eur J Haematol. 2002;68(2):80–3.
Colombel JF, Loftus Jr EV, Tremaine WJ, Egan LJ, Harmsen WS, Schleck CD, et al. The safety profile of infliximab in patients with Crohn’s disease: the Mayo clinic experience in 500 patients. Gastroenterology. 2004;126(1):19–31.
Couderc LJ, Philippe B, Franck N, Balloul-Delclaux E, Lessana-Leibowitch M. Necrotizing vasculitis and exacerbation of psoriasis after granulocyte colony-stimulating factor for small cell lung carcinoma. Respir Med. 1995;89(3):237–8.
Dacey MJ, Callen JP. Hydroxyurea-induced dermatomyositis-like eruption. J Am Acad Dermatol. 2003;48(3):439–41.
Dalmau J, Pimentel CL, Puig L, Peramiquel L, Roe E, Alomar A. Cutaneous necrosis after injection of polyethylene glycol-modified interferon alfa. J Am Acad Dermatol. 2005;53(1):62–6.
Demirturk N, Aykin N, Demirdal T, Cevik F. Alopecia universalis: a rare side effect seen on chronic hepatitis C treatment with peg-INF and ribavirin. Eur J Dermatol. 2006;16(5):579–80.
Deng A, Harvey V, Sina B, Strobel D, Badros A, Junkins-Hopkins JM, et al. Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Dermatol. 2006;142(2):198–202.
Dereure O, Rason-Peyron N, Larrey D, Blanc F, Guilhou JJ. Diffuse inflammatory lesions in patients treated with interferon alpha and ribavirin for hepatitis C: a series of 20 patients. Br J Dermatol. 2002;147(6):1142–6.
Detmar U, Agathos M, Nerl C. Allergy of delayed type to recombinant interferon alpha 2c. Contact Dermatitis. 1989;20(2):149–50.
Dillman RO. Infusion reactions associated with the therapeutic use of monoclonal antibodies in the treatment of malignancy. Cancer Metastasis Rev. 1999;18(4):465–71.
Dong F, Brynes RK, Tidow N, Welte K, Löwenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med. 1995;333(8):487–93.
Errante D, Bernardi D, Bianco A, De Nardi S, Salvagno L. Rituximab-related urticarial reaction in a patient treated for primary cutaneous B-cell lymphoma. Ann Oncol. 2006;17(11):1720–1.
Esser AC, Abril A, Fayne S, Doyle JA. Acute development of multiple keratoacanthomas and squamous cell carcinomas after treatment with infliximab. J Am Acad Dermatol. 2004;50(5 Suppl):S75–7.
Faguer S, Launay F, Ysebaert L, Mailhol C, Estines-Chartier O, Lamant L, Paul C. Acute cutaneous T-cell lymphoma transformation during treatment with alemtuzumab. Br J Dermatol. 2007;157(4):841–2.
Fantini F, Padalino C, Gualdi G, Monari P, Giannetti A. Cutaneous lesions as initial signs of interferon alpha-induced sarcoidosis: report of three new cases and review of the literature. Dermatol Ther. 2009;22 Suppl 1:S1–7. doi:10.1111/j.1529-8019.2009.01263.x.
Fukutoku M, Shimizu S, Ogawa Y, Takeshita S, Masaki Y, Arai T, et al. Sweet’s syndrome during therapy with granulocyte colony-stimulating factor in a patient with aplastic anaemia. Br J Haematol. 1994;86(3):645–8.
Fukuyama S, Kajiwara E, Suzuki N, Miyazaki N, Sadoshima S, Onoyama K. Systemic lupus erythematosus after alpha-interferon therapy for chronic hepatitis C: a case report and review of the literature. Am J Gastroenterol. 2000;95(1):310–2.
González-López MA, Martínez-Taboada VM, González-Vela MC, Blanco R, Fernández-Llaca H, Rodríguez-Valverde V, Val-Bernal JF. Recall injection-site reactions associated with etanercept therapy: report of two new cases with immunohistochemical analysis. Clin Exp Dermatol. 2007;32(6):672–4.
Hadjinicolaou AV, Nisar MK, Parfrey H, Chilvers ER, Ostör AJ. Non-infectious pulmonary toxicity of rituximab: a systematic review. Rheumatology (Oxford). 2012;51(4):653–62. doi:10.1093/rheumatology/ker290.
Hagen JW, Magro CM, Crowson AN. Emerging adverse cutaneous drug reactions. Dermatol Clin. 2012;30(4):695–730. doi:10.1016/j.det.2012.06.016.
Hale G, Swirsky D, Waldmann H, Chan LC. Reactivity of rat monoclonal antibody CAMPATH-1 with human leukaemia cells and its possible application for autologous bone marrow transplantation. Br J Haematol. 1985;60(1):41–8.
Halverstam CP, Lebwohl M. Nonstandard and off-label therapies for psoriasis. Clin Dermatol. 2008;26(5):546–53. doi:10.1016/j.clindermatol.2007.10.023.
Heinzerling LM, Urbanek M, Funk JO, Peker S, Bleck O, Neuber K, et al. Reduction of tumor burden and stabilization of disease by systemic therapy with anti-CD20 antibody (rituximab) in patients with primary cutaneous B-cell lymphoma. Cancer. 2000;89(8):1835–44.
Hertl MS, Haendle I, Schuler G, Hertl M. Rapid improvement of recalcitrant disseminated granuloma annulare upon treatment with the tumour necrosis factor-alpha inhibitor, infliximab. Br J Dermatol. 2005;152(3):552–5.
Isaacs A, Lindemann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):258–67.
Jacobi U, Waibler E, Schulze P, Sehouli J, Oskay-Ozcelik G, Schmook T, et al. Release of doxorubicin in sweat: first step to induce the palmar-plantar erythrodysesthesia syndrome? Ann Oncol. 2005;16(7):1210–1.
Jain KK. Cutaneous vasculitis associated with granulocyte colony-stimulating factor. J Am Acad Dermatol. 1994;31(2 Pt 1):213–5.
Jamroziak K, Grzybowska-Izydorczyk O, Jesionek-Kupnicka D, Gora-Tybor J, Robak T. Poor prognosis of Hodgkin variant of Richter transformation in chronic lymphocytic leukemia treated with cladribine. Br J Haematol. 2012;158(2):286–8; author reply 289. doi:10.1111/j.1365-2141.2012.09127.x.
Janssens A, Berth M, De Paepe P, Verhasselt B, Van Roy N, Noens L, et al. EBV negative Richter’s syndrome from a coexistent clone after savage treatment with alemtuzumab in a CLL patient. Am J Hematol. 2006;81(9):706–12.
Johnson ML, Grimwood RE. Leukocyte colony-stimulating factors. A review of associated neutrophilic dermatoses and vasculitides. Arch Dermatol. 1994;130(1):77–81.
Karlsson C, Norin S, Kimby E, Sander B, Porwit Macdonald A, et al. Alemtuzumab as first-line therapy for B-cell chronic lymphocytic leukemia: long-term follow-up of clinical effects, infectious complications and risk of Richter transformation. Leukemia. 2006;20(12):2204–7.
Kaushansky K. Lineage-specific hematopoietic growth factors. N Engl J Med. 2006;354(19):2034–45.
Kawakami T, Ohashi S, Kawa Y, Takahama H, Ito M, Soma Y, Mizoguchi M. Elevated serum granulocyte colony-stimulating factor levels in patients with active phase of sweet syndrome and patients with active behcet disease: implication in neutrophil apoptosis dysfunction. Arch Dermatol. 2004;140(5):570–4.
Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345(15):1098–104.
Keating MJ, Flinn I, Jain V, Binet JL, Hillmen P, Byrd J, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood. 2002;99(10):3554–61.
Kennedy BJ, Smith LR, Goltz RW. Skin changes secondary to hydroxyurea therapy. Arch Dermatol. 1975;111(2):183–7.
Ketikoglou I, Karatapanis S, Elefsiniotis I, Kafiri G, Moulakakis A. Extensive psoriasis induced by pegylated interferon alpha-2b treatment for Hepatitis B. Eur J Dermatol. 2005;15(2):107–9.
Kim SY, Solomon DH. Tumor necrosis factor blockade and the risk of viral infection. Nat Rev Rheumatol. 2010;6(3):165–74. doi:10.1038/nrrheum.2009.279.
Kokudo N, Nakajima J, Hatano E, Numata K. Current status of hepatocellular carcinoma treatment in Japan: practical use of sorafenib (Nexavar®). Clin Drug Investig. 2012;32 Suppl 2:25–35. doi:10.2165/1163023-S0-000000000-00000.
Kollias G, Douni E, Kassiotis G, Kontoyiannis D. The function of tumour necrosis factor and receptors in models of multi-organ inflammation, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Ann Rheum Dis. 1999;58 Suppl 1:I32–9.
Kong HH, Sibaud V, Chanco Turner ML, Fojo T, Hornyak TJ, Chevreau C. Sorafenib-induced eruptive melanocytic lesions. Arch Dermatol. 2008;144(6):820–2. doi:10.1001/archderm.144.6.820.
Koontz D, Alshekhlee A. Embolia cutis medicamentosa following interferon beta injection. Mult Scler. 2007;13(9):1203–4.
Krainick U, Kantarjian H, Broussard S, Talpaz M. Local cutaneous necrotizing lesions associated with interferon injections. J Interferon Cytokine Res. 1998;18(10):823–7.
Lacouture ME, Reilly LM, Gerami P, Guitart J. Hand foot skin reaction in cancer patients treated with the multikinase inhibitors sorafenib and sunitinib. Ann Oncol. 2008;19(11):1955–61. doi:10.1093/annonc/mdn389.
Lang AM, Norland AM, Schuneman RL, Tope WD. Localized interferon alfa-2b-induced alopecia. Arch Dermatol. 1999;135(9):1126–8.
Lee L, Kukreti V. Rituximab-induced coronary vasospasm. Case Rep Hematol. 2012;2012:984986. doi:10.1155/2012/984986.
Lee A, Skelly ME, Kingma DW, Medeiros LJ. B-cell chronic lymphocytic leukemia followed by high grade T-cell lymphoma. An unusual variant of Richter’s syndrome. Am J Clin Pathol. 1995;103(3):348–52.
Lee WJ, Lee JL, Chang SE, Lee MW, Kang YK, Choi JH, et al. Cutaneous adverse effects in patients treated with the multitargeted kinase inhibitors sorafenib and sunitinib. Br J Dermatol. 2009;161(5):1045–51. doi:10.1111/j.1365-2133.2009.09290.x.
Lenihan DJ, Alencar AJ, Yang D, Kurzrock R, Keating MJ, Duvic M. Cardiac toxicity of alemtuzumab in patients with mycosis fungoides/Sézary syndrome. Blood. 2004;104(3):655–8.
Lewerin C, Mobacken H, Nilsson-Ehle H, Swolin B. Bullous pyoderma gangrenosum in a patient with myelodysplastic syndrome during granulocyte colony-stimulating factor therapy. Leuk Lymphoma. 1997;26(5–6):629–32.
Long D, Thiboutot DM, Majeski JT, Vasily DB, Helm KF. Interstitial granulomatous dermatitis with arthritis. J Am Acad Dermatol. 1996;34(6):957–61.
Lowndes S, Darby A, Mead G, Lister A. Stevens-Johnson syndrome after treatment with rituximab. Ann Oncol. 2002;13(12):1948–50.
Luton K, Garcia C, Poletti E, Koester G. Nicolau syndrome: three cases and review. Int J Dermatol. 2006;45(11):1326–8.
Magro CM, Crowson AN, Schapiro BL. The interstitial granulomatous drug reaction: a distinctive clinical and pathological entity. J Cutan Pathol. 1998;25(2):72–8.
Maloney DG. Anti-CD20 antibody therapy for B-cell lymphomas. N Engl J Med. 2012;366(21):2008–16. doi:10.1056/NEJMct1114348.
Miall FM, Harman K, Kennedy B, Dyer MJ. Pyoderma gangrenosum complicating pegylated granulocyte colony-stimulating factor in Hodgkin lymphoma. Br J Haematol. 2006;132(1):115–6.
Montoto S, Bosch F, Estrach T, Blade J, Nomdedeu B, Nontserrat E. Pyoderma gangrenosum triggered by alpha2b-interferron in a patient with chronic granulocytic leukemia. Leuk Lymphoma. 1998;30(1–2):199–202.
Moore MM, Elpern DJ, Carter DJ. Severe, generalized nummular eczema secondary to interferon alfa-2b plus ribavirin combination therapy in a patient with chronic hepatitis C virus infection. Arch Dermatol. 2004;140(2):215–7.
Moots RJ, Naisbett-Groet B. The efficacy of biologic agents in patients with rheumatoid arthritis and an inadequate response to tumour necrosis factor inhibitors: a systematic review. Rheumatology (Oxford). 2012;51(12):2252–61. doi:10.1093/rheumatology/kes217.
Moustou AE, Matelovits A, Dessinioti C, Antoniou C, Sfikakis PP, Stratigos AJ. Cutaneous side effects of anti-tumor necrosis factor biologic therapy: a clinical review. J Am Acad Dermatol. 2009;61(3):486–504. doi:10.1016/j.jaad.2008.10.060.
Nagore E, Insa A, Sanmartin O. Antineoplastic therapy-induced palmar plantar erythrodysesthesia (‘hand-foot’) syndrome. Incidence, recognition and management. Am J Clin Dermatol. 2000;1(4):225–34.
Omair MA, Alnaqbi KA, Lee P. Rituximab in a patient with ankylosing spondylitis with demyelinating disease: a case report and review of the literature. Clin Rheumatol. 2012;31(8):1259–61. doi:10.1007/s10067-012-2002-8.
Oskay T, Kutluay L, Ozyilkan O. Dermatomyositis-like eruption after long-term hydroxyurea therapy for polycythemia vera. Eur J Dermatol. 2002;12(6):586–8.
Ostlere LS, Harris D, Prentice HG, Rustin MHA. Widespread folliculitis induced by human granulocyte-colony stimulating factor therapy [Letter]. Br J Dermatol. 1992;127:193.
Papi M, Didona B, DePita O, Abruzzese E, Stasi R, Papa G, Cavalieri R. Multiple skin tumors on light-exposed areas during long-term treatment with hydroxyurea. J Am Acad Dermatol. 1993;28(3):485–6.
Parameswaran N, Patial S. Tumor necrosis factor-α signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20(2):87–103.
Paydas S, Sahin B, Seyrek E, Soylu M, Gonlusen G, Acar A, Tuncer I. Sweet syndrome associated with G-CSF. Br J Dermatol. 1993;85(1):191–2.
Pinto JM, Marques MS, Correia TE. Lichen planus and leukocytoclastic vasculitis induced by interferon alpha-2b in a subject with HCV-related chronic active hepatitis. J Eur Acad Dermatol. 2003;17(2):193–5.
Prendiville J, Thiessen P, Mallory SB. Neutrophilic dermatoses in two children with idiopathic neutropenia: association with granulocyte colony-stimulating factor (G-CSF) therapy. Pediatr Dermatol. 2001;18(5):417–21.
Quesada JR, Gutterman JU. Psoriasis and alpha-interferon. Lancet. 1986;1(8496):1466–8.
Raymond AK, Puri PK, Selim MA, Tyler DS, Nelson KC. Regional squamous cell carcinomas following sorafenib therapy and isolated limb infusion for regionally advanced metastatic melanoma of the limb. Arch Dermatol. 2010;146(12):1438–9. doi:10.1001/archdermatol.2010.367.
Reuss-Borst MA, Pawelec G, Saal JG, Horny HP, Müller CA, Waller HD. Sweet’s syndrome associated with myelodysplasia: possible role of cytokines in the pathogenesis of the disease. Br J Haematol. 1993;84(2):356–8.
Rey J, Wickenhauser S, Ivanov V, Coso D, Gastaut JA, Bouabdallah R. A case of rituximab-related urticarial reaction in cutaneous B-cell lymphoma. J Eur Acad Dermatol Venereol. 2009;23(2):210. doi:10.1111/j.1468-3083.2008.02792.x.
Richard M, Truchetet F, Friedel J, Leclech C, Heid E. Skin lesions simulating chronic dermatomyositis during long-term hydroxyurea therapy. J Am Acad Dermatol. 1989;21(4 Pt 1):797–9.
Richter MN. Generalized reticular cell sarcoma of lymph nodes associated with lymphatic leukemia. Am J Pathol. 1928;4(4):285–92.7.
Robert C, Soria JC, Spatz A, Le Cesne A, Malka D, Pautier P, et al. Cutaneous side-effects of kinase inhibitors and blocking antibodies. Lancet Oncol. 2005;6(7):491–500.
Robert C, Arnault JP, Mateus C. RAF inhibition and induction of cutaneous squamous cell carcinoma. Curr Opin Oncol. 2011;23(2):177–82. doi:10.1097/CCO.0b013e3283436e8c.
Robertson LE, Pugh W, O’Brien S, Kantarjian H, Hirsch-Ginsberg C, Cork A, et al. Richter’s syndrome: a report on 39 patients. J Clin Oncol. 1993;11(10):1985–9.
Robinson AB, Reed AM. Clinical features, pathogenesis and treatment of juvenile and adult dermatomyositis. Nat Rev Rheumatol. 2011;7(11):664–75. doi:10.1038/nrrheum.2011.139.
Rocamora V, Puig L, Alomar A. Dermatomyositis-like eruption following hydroxyurea therapy. J Eur Acad Dermatol Venereol. 2000;14(3):227–8.
Rosenbaum SE, Wu S, Newman MA, West DP, Kuzel T, Lacouture ME. Dermatological reactions to the multitargeted tyrosine kinase inhibitor sunitinib. Support Care Cancer. 2008;16(6):557–66. doi:10.1007/s00520-008-0409-1.
Ross HJ, Moy LA, Kaplan R, Figlin RA. Bullous pyoderma gangrenosum after granulocyte colony-stimulating factor treatment. Cancer. 1991;68(2):441–3.
Rossi D, Gaidano G. Richter syndrome: molecular insights and clinical perspectives. Hematol Oncol. 2009;27(1):1–10. doi:10.1002/hon.880.
Salihoglu A, Ozbalak M, Keskin D, Tecimer T, Soysal T, Ferhanoglu B. An unusual presentation of a chronic lymphocytic leukemia patient with 17p deletion after reduced-intensity transplantation: Richter syndrome and concomitant graft-versus-host disease-case report. Transplant Proc. 2013;S0041-1345(12):1304–8. doi:10.1016/j.transproceed.2012.12.001.
Sanders S, Busam K, Tahan SR, Johnson RA, Sachs D. Granulomatous and suppurative dermatitis at interferon alfa injection sites: report of 2 cases. J Am Acad Dermatol. 2002;46(4):611–6.
Saraceno R, Teoli M, Chimenti S. Hydroxyurea associated with concomitant occurrence of diffuse longitudinal melanonychia and multiple squamous cell carcinomas in an elderly subject. Clin Ther. 2008;30(7):1324–9.
Selmi C, Lleo A, Zuin M, Podda M, Rossaro L, Gershwin ME. Interferon alpha and its contribution to autoimmunity. Curr Opin Investig Drugs. 2006;7(5):451–6.
Senet P, Aractingi S, Porneuf M, Perrin P, Duterque M. Hydroxyurea-induced dermatomyositis-like eruption. Br J Dermatol. 1995;133(3):455–9.
Sfikakis PP, Kollias G. Tumor necrosis factor biology in experimental and clinical arthritis. Curr Opin Rheumatol. 2003;15(4):380–6.
Shpiro D, Gilat D, Fisher-Feld L, Shemer A, Gold I, Trau H. Pyoderma gangrenosum successfully treated with perilesional granulocyte-macrophage colony stimulating factor. Br J Dermatol. 1998;138(2):368–9.
Sigal M, Crickx B, Blanchet P, Perron J, Simony J, Belaïch S. Cutaneous lesions induced by long-term use of hydroxyurea. Ann Dermatol Venereol. 1984;111(10):895–900 [Article in French].
Singer EA, Gupta GN, Srinivasan R. Targeted therapeutic strategies for the management of renal cell carcinoma. Curr Opin Oncol. 2012;24(3):284–90. doi:10.1097/CCO.0b013e328351c646.
Skytta E, Phjankoski H, Savolainen A. Etanercept and urticaria in patients with juvenile idiopathic arthritis. Clin Exp Rheumatol. 2000;18:533–4.
Smith KJ, Haley H, Hamza S, Skelton HG. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35(11):1766–70. doi:10.1111/j.1524-4725.2009.01289.x.
Spiekermann K, Roesler J, Emmendoerffer A, Elsner J, Welte K. Functional features of neutrophils induced by G-CSF and GM-CSF treatment: differential effects and clinical implications. Leukemia. 1997;11(4):466–78.
Stasi R, Cantonetti M, Abruzzesse E, Papi M, Didona B, Cavalieri R, Papa G. Multiple skin tumors in long-term treatment with hydroxyurea. Eur J Dermatol. 1992;48(2):121–2.
Stewart LR, George S, Hamacher KL, Hsu S. Granuloma annulare of the palms. Dermatol Online J. 2011;17(5):7.
Suehiro M, Kishimoto S, Wakabayashi T, Ikeuchi A, Miyake H, Takenaka H, et al. Hydroxyurea dermopathy with a dermatomyositis-like eruption and a large leg ulcer. Br J Dermatol. 1998;139(4):748–9.
Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349–56.
Tai YJ, Tam M. Fixed drug eruption with interferon-beta-1b. Australas J Dermatol. 2005;46(3):154–7.
Tohda S, Morio T, Suzuki T, Nagata K, Kamiyama T, Imai Y, et al. Richter syndrome with two B cell clones possessing different surface immunoglobulins and immunoglobulin gene rearrangements. Am J Hematol. 1990;35(1):32–6.
Tomasiewicz K, Modzewska R, Semczuk G. Vitiligo associated with pegylated interferon and ribavirin treatment of patients with chronic hepatitis C: a case report. Adv Ther. 2006;23(1):139–42.
Valks R, Vargas E, Munoz E, Fernandez-Herrera J, Garcia-Diez A, Fraga J. Dermal infiltrate of enlarged macrophages in patients receiving chemotherapy. J Cutan Pathol. 1998;25(5):259–64.
Van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115(4):807–11.
van Husen M, Kemper MJ. New therapies in steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome. Pediatr Nephrol. 2011;26(6):881–92. doi:10.1007/s00467-010-1717-5. Epub 2011 Jan 13.
van Kamp H, van den Berg E, Timens W, Kraaijenbrink RA, Halie MR, Daenen SM. Sweet’s syndrome in myeloid malignancy: a report of two cases. Br J Haematol. 1994;86(2):415–7.
Voulgari PV, Markatseli TE, Exarchou SA, Zioga A, Drosos AA. Granuloma annulare induced by anti-tumour necrosis factor therapy. Ann Rheum Dis. 2008;67(4):567–70.
Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5 Pt 2):918–23.
White JM, Mufti GJ, Salisbury JR, du Vivier AW. Cutaneous manifestations of granulocyte colony-stimulating factor. Clin Exp Dermatol. 2006;31(2):206–7.
Wilhelm S, Chien DS. Bay 43-9006: preclinical data. Curr Pharm Des. 2002;8(25):2255–7.
Williams VL, Cohen PR. TNF alpha antagonist-induced lupus-like syndrome: report and review of the literature with implications for treatment with alternative TNF alpha antagonists. Int J Dermatol. 2011;50(5):619–25. doi:10.1111/j.1365-4632.2011.04871.x.
Winfield H, Lain E, Horn T, Hoskyn J. Eosinophilic cellulitis-like reaction to subcutaneous etanercept injection. Arch Dermatol. 2006;142:218–20.
Winkler U, Jensen M, Manzke O, Schulz H, Diehl V, Engert A. Cytokine-release syndrome in patients with B-cell chronic lymphocytic leukemia and high lymphocyte counts after treatment with an anti-CD20 monoclonal antibody (rituximab, IDEC-C2B8). Blood. 1999;94(7):2217–24.
Zeltser R, Valle L, Tanck C, Holyst M, Ritchlin C, Gaspari AA. Clinical, histological, and immunophenotypic characteristics of injection site reactions associated with etanercept: a recombinant tumor necrosis factor alpha receptor: Fc fusion protein. Arch Dermatol. 2001;137(7):893–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Lee, J.J., Hoang, M.P. (2014). Cutaneous Lymphoid Infiltrates in Patients Receiving Biologic Modifiers. In: Cualing, H., Kadin, M., Hoang, M., Morgan, M. (eds) Cutaneous Hematopathology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0950-6_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0950-6_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0949-0
Online ISBN: 978-1-4939-0950-6
eBook Packages: MedicineMedicine (R0)