
Abstract
The luminal-mucosal interface of the intestinal tract is the first relevant location where microorganism-derived antigens and all other potentially immunogenic particles face the scrutiny of the powerful mammalian immune system. Upon regular functioning conditions, the intestinal barrier is able to effectively prevent most environmental and external antigens to interact openly with the numerous and versatile elements that compose the mucosal-associated immune system. This evolutionary super system is capable of processing an astonishing amount of antigens and non-immunogenic particles, approximately 100 tons in one individual lifetime, only considering food-derived components. Most important, to develop oral tolerance and proper active immune responses needed to prevent disease and inflammation, this giant immunogenic load has to be managed in a way that physiological inflammatory balance is constantly preserved. Adequate functioning of the intestinal barrier involves local and distant regulatory networks integrating the so-called brain-gut axis. Along this complex axis both brain and gut structures participate in the processing and execution of response signals to external and internal changes coming from the digestive tract, using multidirectional pathways to communicate. Dysfunction of brain-gut axis facilitates malfunctioning of the intestinal barrier, and vice versa, increasing the risk of uncontrolled immunological reactions that may trigger mucosal and brain low-grade inflammation, a putative first step to the initiation of more permanent gut disorders. In this chapter, we describe the structure, function and interactions of intestinal barrier, microbiota and brain-gut axis in both healthy and pathological conditions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The survival of living organisms greatly depends on the ability of species and individuals to constantly provide a series of complex and dynamic repository responses to counteract internal and environmental threats. This functional equilibrium, named homeostasis, relies upon the adequate integration of every generated response to a threat. At the gastrointestinal level, the mucosal surfaces are the first location where immunogenic particles, environmental toxins and microorganism-derived antigens gain access to the immune system [1]. The luminal side of the mucosa of the ileum and jejunum is coated with hundreds of tiny finger-like structures called villi, which in turn are composed by myriads of microvilli, rendering a final physical contact area of about 400 m2. This enormous epithelial surface area favours nutrient absorption and water and electrolyte transport across. However, it also designed to select which luminal antigens should face the components of the mucosal-associated immune system. This selection process is aimed at preventing the generation of inadequate pro-inflammatory signals [2]. Mucosal processing of antigens and non-immunogenic molecules will at the end, determine whether tolerogenic or non-tolerogenic immune responses are raised to keep homeostasis [3].
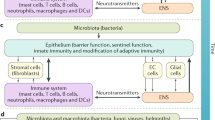
The intestinal mucosal barrier consists of different consecutive layers, including the intestinal flora and external mucus, the columnar epithelium and extracellular matrix below, and the innermost lamina propria. Within the lamina propria we can find blood and lymph vessels, a plethora of resident immune cells (plasma cells, lymphocytes, macrophages, eosinophils, mast cells, dendritic cells, etc.), and a significant number of intrinsic and extrinsic nerve terminals (Fig. 4.1). All of these components may display effector and modulatory functions relevant to the control of inflammation, absorption and secretion, transport of macromolecules and metabolic processes [4]. Considerable evidence now supports the existence of multidirectional communication between the components of this local regulatory network [5, 6]. Communication is driven by the release of chemical mediators, such as neuropeptides, neurohormones, neurotransmitters, cytokines, chemokines, growth factors, and other regulatory molecules.

Intestinal barrier function. The intestinal barrier has evolved to guarantee homeostasis through the execution of basic weeping off functions, such as water secretion and intestinal peristaltism, and by the development of immunological surveillance. This barrier is composed by several levels of protection aimed at preventing and selecting toxin and antigen penetration across. The most external laters harbours mucus, enzymes, antimicrobial peptides and the intestinal microbiota. Just below, a single-cell layer of epithelial cells, sealed by intercellular junctions, regulates the transcellular and paracellular passage of substances. Intermingled goblet cells secrete mucins that dissolve in water to form mucus, a major contributor to the retention of secretions containing antibacterial peptides and digestive enzymes, and to keep epithelial hydration. The epithelium also displays microbial recognition receptors and is able to release immune mediators. Lamina propria leukocytes produce proteases and cytokines to modify epithelial secretory activity and permeability range of the epithelium. M cells are found in the follicle-associated epithelium of the Peyer’s patches and transport antigens from the luminal side to immune cells across the epithelial barrier. IgA is produced by plasma cells, and transported through, and secreted by, the epithelium to the luminal side. Both, the central and the enteric nervous system, interact with the immune system, the smooth muscle and the epithelium to regulate immune responses, absorption and secretion, motility, and also visceral sensitivity. Note: IEL intraepithelial lymphocyte
The regulation of gut physiology is also achieved through the activity of both the enteric nervous system (ENS) and the central nervous system (CNS). ENS is an extensive neural network, also known as the second brain, containing approximately 100 million neurons embedded in the gastrointestinal lining, similar number to the spinal cord [7]. The ENS contains sensory neurons, inter-neurons, and motor neurons, which primarily control motility, absorption and secretion, but also visceral sensitivity. In addition, the ENS is wired with multiple terminals from ascending and descending CNS pathways that help to control gut function. To understand gut physiology and pathology, it is of particular importance to consider the role of the autonomic nervous system, and the hypothalamic pituitary-adrenal axis (HPA) because both systems also establish a vast and complex array of integrative and bidirectional interactions between the brain and the gut, the brain-gut axis.
The Intestinal Barrier
The intestinal barrier has evolved to guarantee homeostasis through the execution of basic weeping off functions, such as water secretion, to wash off harmful substances that may be present in the intestinal lumen, and by the development of a programme, that includes active immunological surveillance. One of the first steps to fight unwanted or harmful stimuli involves the release of mucus, defensins, secretory-immunoglobulin A, and other chemical mediators to the lumen [8]. In addition, the importance of maintaining epithelial permeability tight to prevent the passage of noxious substances, was emphasized in the early 1990s [9], and reiterated by many authors thereafter.
Structure and Function of Intestinal Barrier
Mucus
The entire intestinal mucosal surface is covered by a layer of mucus gel, thicker than 100 μm secreted by goblet cells (GCs). Mucus protects the epithelial lining from luminal sheer forces, adhesion and invasion by microorganisms, dietary, chemical and radiation toxins, and other antigens present in the intestinal lumen [10]. The mucus layer also contributes to the retention of mucosal secretions containing antibacterial peptides and digestive enzymes [11, 12] and keeps epithelial hydration. Mucus seems to participate in epithelial renewal, differentiation and integrity, and relates to other biological processes [13]. More recently, mucus has also been shown to enhance oral tolerance by imprinting dendritic cells with anti-inflammatory properties through the assembly of a galectin-3-dectin-1-FcγRIIB receptor complex that activated β-catenin, interfering with the expression of inflammatory, but not tolerogenic cytokines by dendritic cells [14].
Components of mucus include water, phospholipids, the negatively charged mucins (MUC), which provide a chemical barrier to protect the underlying epithelium, and a variety of trefoil factors and other antimicrobials such as secretory IgA [15], cathelicidins and defensins that provide the physical and immune protection against luminal agents [16]. Mucus secreted at the apical brush border binds the glycocalyx to form a viscoelastic gel with hydrophobic and surfactant properties, dependent on the presence of phospholipids at the most apical part. Hydrophobicity helps to fight enteric bacteria and to regulate gut permeability [17].
MUC represent the most abundant component of the mucus gel. MUC are huge glycoproteins composed of a central protein backbone rich in serine, threonine and proline. These glycoproteins are highly glycosylated by attached oligosaccharides, which contain blood group structures and are initiated by N-acetyl-galactosamine that is O-linked onto serine or threonine at the protein core [18–20]. These O-linked oligosaccharides are responsible for MUC properties. Up to 20 different MUC genes have been identified to date (MUC1 to MUC20) [21], with site and cell-specific expression. Several secreted mucins (MUC2, MUC5AC, MUC5B, and MUC6) function as extracellular viscous secretions whereas others appear as membrane-associated mucins (MUC1, MUC3 and MUC4) in the glycocalyx [22]. MUC1–4 represent the most abundant secreted mucins in the human intestine. The first identified human secretory mucin was MUC2 that is also the principal secreted MUC [23], and is normally restricted to GCs [24]. In mice, it has been shown that colonic mucus consists of two layers with similar protein composition, being MUC2 the major structural component. The inner layer is firmly attached to the epithelium and functions as a barrier to prevent bacterial invasion while the outer layer is a loose matrix usually colonized by bacteria [25]. Thickness of the inner mucus layer varies down along the intestine according to luminal concentration of bacteria, being thicker at the highly colonized colonic segment, and thinner at the less colonized small intestine [26]. Baseline secretion of MUC is a constitutive pathway where small vesicles transport MUC directly to the cell surface where immediate and full exocytosis of their contents takes place. The release and secretion of packaged MUC is a different pathway regulated by specific stimuli including microbes and their products, and neuroendocrine and inflammatory/immune mediators. Mucus production is tightly regulated by different protein families, such as MUC and protein O-fucosyltransferase 1 (POFUT1) family members. Dysfunction of mucus secretion can lead to the development of intestinal inflammation as shown by the susceptibility of MUC2 KO mice to develop spontaneous colitis, and by a more severe intestinal response to the administration of dextran sulphate sodium (DSS) [27]. These mice also display impaired host resistance to parasitic infection [28], and over-enhanced susceptibility to Salmonella enterica serovar typhimurium [29]. Decreased production and alteration of the O-glycosylation profile of MUC2 has been associated with increased inflammation in ulcerative colitis [30, 31]. Moreover, increased susceptibility to ulcerative colitis [32] and Crohn’s disease [33] has been linked to a rare variable number of tandem repeat alleles of the MUC3 gene. Mice defective in intestinal POFUT1 exhibit chronic intestinal inflammation in association with an alteration of mucus-associated flora, goblet cell hyperplasia and hypertrophy and elevated production of mucus [34].
Resistin-like molecule (RELM)-β is a cysteine-rich protein also present in the mucus layer and specifically produced by intestinal GCs. RELM-β upregulates MUC2 and M1/MUC5AC gene expression in the human colonic HT29 cell line. Pretreatment of murine colon with RELM-β significantly attenuates trinitrobenzene sulphonic acid (TNBS)-induced colitis [35] while RELM-β deficient mice show increased susceptibility to T-cell-dependent TNBS-induced colitis. Therefore, available evidence suggests that RELM-β plays an important role in colonic inflammation [36].
Trefoil factors, a group of small cysteine-rich peptides, are also essential protective components of the mucus layer and contribute to mucosal repair, particularly, the trefoil factor 3 synthesized and secreted by intestinal GCs [37, 38]. Trefoil factor 3 deficient mice are highly susceptible to DSS, chemotherapy and radiation-induced colitis [39, 40], and display prominent hypoxia-elicited increases in intestinal permeability [41].
Epithelial Lining
The intestinal epithelium is a single polarized continuous layer of columnar cells of only 20 μm thick that covers the intestinal surface and separates the intestinal lumen from the internal milieu. Although it functions primarily as a physical barrier, it also regulates the absorption of dietary nutrients, water and electrolytes. The passage of molecules from the intestinal lumen to the lamina propria takes place mainly through two different routes: (1) The paracellular pathway, which allows small molecules (<600 Da) diffuse through tight junctions (TJs) located between adjacent intestinal epithelial cells; and, (2) The transcellular pathway, which allows the passage of larger particles through the epithelial cells via endocytosis or exocytosis processes [42].
The intestinal epithelium contains several stem cell-derived cellular types, such as absorptive enterocytes, GCs, Paneth cells, enteroendocrine cells, and M cells, as shown in panel 1 of Fig. 4.2. This epithelial population renews every 3–5 days from pluripotential stem cells located in the intestinal crypts to ensure cellular integrity all along the intestinal epithelium. Pluripotential stem cells migrate to the tip of the villus where final differentiation takes place [43]. Signalling cascades such as the wnt and the Notch pathway are involved in epithelial proliferation and differentiation, essential processes to regulate homeostasis in the intestinal epithelium [44].

Ultrastructure of the intestinal mucosa. Transmission electron micrographs of the intestinal epithelium and the lamina propria of the human jejunum. The intestinal mucosa is responsible for nutrient absorption and water secretion, which require a selectively permeable barrier. Panel 1—Intestinal epithelium. The epithelium functions primarily as a physical barrier between the external environment and the internal milieu. It is composed by enterocytes, secretory cells and immune cells, all supported on the basal side by a basement membrane underneath which the lamina propria harbors blood and lymph vessels, resident immune cells and nerve terminals. GC goblet cell, IEL intraepithelial lymphocyte, EC enterochromaffin cell, Ep epithelial cell. Panel 2—Intercellular junctions. The epithelial cells are polarized cells bound together through specific junctions. The apical junctional complex delineates the apical and the basal regions of the epithelial cells. It limits the uptake of microbial and food-derived antigens and prevents the passage of cellular elements across. TJs are located at the most apical site of the epithelium followed by the subjacent adherens junction and the desmosomes. TJ tight junction, AJ adherens junction, D Desmosome. Panel 3—Lamina propria. Most of the immune elements of the intestinal barrier are located in the lamina propria, where they develop innate and adaptive responses in coordination with the nervous system and the epithelium. Eo eosinophil, NE nerve endings, PC plasma cell, MC mast cell, L lymphocyte, Ep epithelial cell
Enterocytes
Enterocytes are key elements of the epithelial lining. Although, the most important endeavour of these cells is to maintain the integrity of the intestinal physical barrier, enterocytes reinforce barrier strength by also developing immunologic activity. Enterocytes express innate immune receptors [45], act as non-professional antigen presenting cells, and release several chemokines and cytokines such as fractalkine [46] or thymic stromal lymphopoietin [47] involved in leukocyte recruitment and in dendritic cell regulation.
Enterocytes are tightly bonded to each other through the apical junctional complex that separates the apical membrane from the basolateral membrane. This apical junctional complex is composed TJs, adherens junctions, and desmosomes, as shown in panel 2 of Fig. 4.2. The junctional complex limits the uptake of microbial and food derived antigens and prevents the passage of cellular elements across. TJs are located at the most apical site of the epithelium and composed of intracellular and surface-membrane proteins. Intracellular proteins are zonula occludens (ZO)-1, ZO-2 and ZO-3, as well as cingulin. Surface-membrane or transmembrane proteins include occludin, claudins, and junctional adhesion molecules (JAMs). TJs seal the intercellular space and regulate intestinal permeability. Adherens junctions are located below TJs and mainly composed by e-cadherin, catenin, and actin filaments. These protein complexes provide the necessary strength to hold the cells together.
Occludin was the first TJ transmembrane protein identified. It belongs to the TJ-associated MAL and related proteins for vesicle trafficking and membrane link (MARVEL) proteins, and contains a MARVEL domain. The function of occludin remains to be elucidated. On one hand, occludin deficient mice do not show alterations in TJ assembly and permeability [48], but, on the other hand, occludin seems to play a role in the regulation of integrity rather than in the de novo assembly of the TJs [21]. Furthermore, in vitro observations suggest that occludin localization to the TJ complex is regulated by phosphorylation [49]. Regulation of occludin phosphorylation implicates several kinases including protein kinases C, mitogen-activated protein kinases (MAPKs), Rho kinases, and the Src-Family kinases [50]. When occludin is highly phosphorylated on serine and threonine residues, it is selectively located at the TJ. In contrast, occludin dephosphorylation at those residues by protein phosphatases, results in redistribution of the protein to the cytoplasm [24].
The claudin family of transmembrane proteins consists of 24 members with a molecular weight ranging from 20 to 27 kDa. Each member shows a specific organ and tissue distribution. This protein family plays a central role in the regulation of barrier function. Some claudins make up pores that allow preferential passage of specific ions, while others reduce the transit of specific ions. The strength, size, and ion selectivity of TJs is determined by claudins, as reflected by massive trans-epidermal water loss and death of mice within one day of birth affecting claudin-1 deficient mice [51]. Moreover, segmental barrier properties along the crypt-villus axis and throughout the length of the intestine do correlate with the disposition of claudins [52, 53]. In the human intestine, both ileal and colonic mucosa express tightening claudins-1, -3, -4, -5 and -7 [54, 55]. However, the expression of the permeability mediator claudin-2 is restricted to the crypt, in the colon [30, 56], yet detected in the crypt and the villus, in the small bowel [31]. Differences in the expression and distribution of claudins may reflect adaptation to specific physiological functions carried out by the different segments down the intestinal tract.
A third group of transmembrane receptors found at TJs is the family of JAMs. JAMs have been implicated in the construction and assembly of TJs [57], and in the regulation of intestinal permeability and inflammation [58]. JAM-A deficient mice display increased intestinal permeability and inflammatory cytokine production, and marked epithelial apoptosis to DSS-induced colitis [59]. More recently, reduced intestinal JAM-A expression has been described in irritable bowel syndrome (IBS) patients, possibly contributing to intestinal barrier dysfunction in these patients [60]. JAMs are also present on blood cells, such as leukocytes, thereby contributing to the process of trans-endothelial migration [61].
The TJ transmembrane proteins, claudins, occludin, and JAMs are linked to the actomyosin fibers of the cytoskeleton by members of the ZO family [62]. This association to the peri-junctional actomyosin ring seems crucial for the dynamic regulation of permeability at paracellular spaces. Interestingly, only ZO-1 and ZO-2 are relevant for claudin recruitment, TJ formation and for epithelial barrier function [63].
Far from being static, TJs are quite mobile structures that readily adapt to changing conditions and challenging stimuli. Regulation of intestinal permeability involves different functional pathways. Fast changes in permeability occur usually via myosin light chain kinase (MLCK)-mediated cytoskeleton contraction, and by endocytosis of TJ proteins [64, 65]. In contrast, lasting permeability disturbances involve the transcriptional modulation of TJ proteins, epithelial cell apoptosis and ultrastructural alterations in the epithelium [66].
Phosphorylation of myosin II regulatory light chain (MLC) induces actomyosin cytoskeleton contraction and increased TJ junction permeability. Rho GTPases have been shown to regulate TJs through redistribution of ZO-1, and reorganization of JAM-1 away from the TJ membrane [67]. Up-regulation of zonulin expression increased intestinal permeability to bacterial and gliadin exposure. In fact, this zonulin-mediated intestinal barrier defect has been advocated to play a central role in the origin of celiac disease [68] and type 1 diabetes [69].
Secretory Cells
The intestinal epithelium also houses different types of specialized epithelial called secretory cells that contribute to the reinforcement of the intestinal epithelial barrier, mainly goblet cells, Paneth cells and enteroendocrine cells.
GCs are scattered through the epithelial lining. GCs that mainly secrete mucins, but also trefoil peptides, RELM-β and Fc-γ binding protein. GC distribution varies throughout the gastrointestinal tract, the number increasing from the duodenum to the distal colon. The number of GCs is probably regulated by the intestinal microbiota because germ-free mice have less and smaller GCs than regular mice [70].
Paneth cells are located at the base of the crypts of Lieberkühn. Similar to the other intestinal epithelial cell types, they evolve from stem cells at the bottom of the crypt. Contrary to other cell types, Paneth cells migrate downwards, to the bottom of the crypt, where they synthesize and secrete antimicrobial peptides and other proteins to the intestinal lumen. Among them, lysozyme, α-defensins, TNF-α, and secretory phospholipase A2 type IIA, contribute to maintain host-microbe homeostasis and to protect stem cells from pathogens [71, 72]. Certain defects in Paneth may be linked to the pathogenesis of Crohn’s disease [73, 74] and necrotizing enterocolitis [75, 76].
Gut enteroendocrine cells spread all along the intestinal epithelium where they function as highly specialized chemoreceptors sensing changes in luminal osmolarity, pH and nutrient composition. Although they represent less than 1 % of the entire gut epithelial population, enteroendocrine cells constitute the largest endocrine organ of the human body. Products released by enteroendocrine cells include hormones, such as ghrelin, somatostatin, cholecystokinin, gastric inhibitory polypeptide, glucagon-like peptides and peptide YY, and neurotransmitters such as serotonin [77]. Enteroendocrine cells inform the brain-gut axis mostly through the activation of neural pathways [78].
The Intestinal Immune System
Mucosa-associated lymphoid tissue is a diverse and diffuse defence system found at most mucosal surfaces of the body, such as the respiratory system and the eye conjunctiva. The immune response generated by this system provides generalized immunization at all mucosal surfaces [79]. About 70 % of whole body’s immune cells reside within the gastrointestinal tract shaping the gut-associated lymphoid tissue (GALT), which is conformed in two different compartments: the organized immune inductive sites, and the diffuse effector sites.
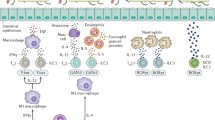
Diffuse GALT is composed of two lymphocyte populations distributed at both sides of the basal lamina. Intraepithelial lymphocytes are found between epithelial cells, above the basal lamina. Lamina propria lymphocytes reside in lamina propria along with many other types of immune cell, such as eosinophils, dendritic cells, mast cells, macrophages or plasma cells (panel 3 of Figs. 4.2 and 4.3). The majority of intraepithelial lymphocytes are CD8+ T cells that function as surface gatekeepers of the intestinal barrier because the constantly monitor and respond against luminal bacteria and other antigens. Lamina propria lymphocytes constitute a much more heterogeneous population, approximately 50 % of which correspond to plasma cells, 30 % to T lymphocytes, and the remaining 20 % to macrophages, dendritic cells, mast cells and eosinophils. Resident B lymphocytes complete their maturation into plasma cells, mostly producing IgA, but IgM and IgG. Activated T and B-lymphocytes express α4β7 integrin and mucosal endothelial cells of Peyer’s patches, mesenteric lymph nodes and lamina propria of the small and large intestine constitutively express the mucosal addressin cell adhesion molecule-1 that interacts with α4β7 integrin to recirculate lymphocytes between the blood and the gastrointestinal tract [80].

Resident immunocytes in the intestinal mucosa. The majority of the immune cells within the body reside in the gastrointestinal tract (Gut-associated lymphoid tissue, GALT), and are distributed in two different compartments: the organized inductive sites, and the diffuse effector sites. The diffuse GALT is composed of intraepithelial lymphocytes, between the epithelial cells, and the lamina propria lymphocytes, which reside below the basal lamina, along with other immune cells. The figure shows intestinal micrographs (×400 magnification) processed for H&E staining to identify mucosal eosinophils (1), and immunohistochemistry for T-lymphocytes (2, CD3+), B-lymphocytes (3, CD20+), macrophages (4, CD68+), plasma cells (5, CD138+), and mast cells (6, CD117+)
Inductive sites of the GALT include organized lymphoid structures in the small intestine such as Peyer’s patches, mesenteric lymph nodes, isolated lymphoid follicles, and lymphocytes and antigen-presenting cells. Peyer’s patches are macroscopic lymphoid aggregates found at the submucosal levels in the antimesenteric border of the intestine. The follicle-associated epithelium covering Peyer’s patches contains M cells, another special cell type that plays a role in monitoring the gut lumen and maintaining intestinal barrier function. M cells display several unique properties including apical microfolds instead of microvilli, no mucus layer, and a reduced glycocalyx, which facilitate the capture of luminal antigens and microorganisms and their transport to contact underlying immune cells [81]. Peyer’s patches also contain antigen-presenting cells, mainly dendritic cells, but also macrophages. These antigen-presenting cells capture luminal antigens (taken up by M cells in the Peyer’s patch dome), to further process and present them to immunocompetent cells in association with the major histocompatibility complex.
Innate immunity is present in both animals and plants [82]. It serves the host defence via immediate, but non-specific, responses to a wide variety of pathogens. The main components of innate immune response include pattern recognition receptors (PRRs), and antimicrobial peptides.
PPRs are a class protein that responds to small molecular sequences consistently found on pathogens, named pathogen-associated molecular patterns (PAMP). PRRs include Toll-like receptors (TLRs) and Nod-like receptors (NLRs).
The TLR family consists of at least 13 transmembrane receptors containing a large leucine-rich repeats extracellular domain that recognizes different bacterial, viral, parasite or self-derived ligands, such as lipopolysaccharide, peptidoglycan, muramyl dipeptide, lipoteichoic acids, and bacterial DNA. After activation upon PAMP recognition, TLRs initiate downstream signalling cascades, leading to transcriptional responses and to the initiation of both innate immune responses (macrophage activation and induction of antimicrobial peptides for various cell types) and the adaptive immune response (induction of T cell responses and maturation of dendritic cells) [83]. In many tissues, mast cells, dendritic cells, monocytes/macrophages and B cells express TLRs [84]. Healthy intestinal epithelial cells express relatively low levels of TLRs, such as TLR-4, perhaps explaining why lipopolysaccharide does not induce a potent inflammatory response in normal intestine [85]. By contrast, and consistent with the idea that chronic intestinal inflammation may be the result of uncontrolled responses to components of the intestinal bacterial flora, the intestinal epithelium of patients with inflammatory bowel disease (IBD) shows increased expression of TLR-4 [86]. The cellular localization of TLRs is also influenced by the polarized epithelial cell organization. TLR5 is expressed on the basolateral surface of intestinal epithelia only, where if becomes stimulated by luminal flagellin exposure when disruption of the epithelial barrier. Therefore, its localization prevents inappropriate stimulation by flagellin, but allows recognition of invasive pathogens [87]. Similarly, TLR9 activation through apical and basolateral surface domains induces distinct transcriptional responses. Whereas basolateral TLR9 strongly stimulates proinflammatory chemokine secretion, through NF-kappaB activation, apical TLR9 stimulation invokes a unique response in which ubiquitinated IkappaB accumulates in the cytoplasm preventing NF-kappaB activation conferring mucosal tolerance towards microbial exposure [88].
NLR constitutes a large family of 23 intracellular PRRs, being nucleotide-binding oligomerization domain (NOD)1, NOD2 and NALP3 the most extensively described. NOD1 and NOD2 recognize intracellular bacterial cell products, and NALP3 responds to multiple stimuli to form a multi-protein complex termed the NALP3 inflammasome, which promotes the release of the IL-1 family of cytokines. Most NLRs share a similar structure consisting of a centrally located NOD, a C-terminal leucine-rich repeat that detects PAMPs, and a variable N-terminal domain that is critical for downstream signalling through the recruitment of adaptors or effector molecules [89]. NOD1 recognizes γ-d-glutamyl-meso-diaminopimelic acid, which is found in the peptidoglycan structures of all gram-negative as well as in several gram-positive bacteria [90]. In contrast, NOD2 recognizes muramyl dipeptide, which is found in nearly all gram-positive and gram-negative organisms [91]. Upon ligand recognition, NOD1 and NOD2 induce the activation of NF-kappaB and MAPKs pathways leading to the activation of both innate and adaptive immune responses. In contrast, other NLRs such as Ipaf and cryopyrin respond to microbial components through the assembly of multiprotein complexes termed “inflammasomes” that promote caspase-1 activation to generate the proinflammatory cytokines IL-1β and IL-18 [92]. NOD1 is expressed by intestinal epithelial cells [93] while NOD2 expression is predominantly found in monocytes and Paneth cells [73]. Both NOD1 and NOD2 have been shown to modulate inflammation and mediate efficient clearance of bacteria from the mucosal tissue during Salmonella colitis [94]. In addition, NOD2-deficient mice display an increased load of commensal resident bacteria, and a diminished ability to prevent intestinal colonization by pathogenic bacteria [95]. NOD-2 mutations have been identified in Crohn’s disease patients and could be related to an impaired release of antimicrobial peptides from Paneth cells [96].
Antimicrobial peptides are endogenous antibiotics that are constitutively expressed in intestinal epithelial cells, yet may be also inducible in immune cells and Paneth cells [97]. They include compounds such as lactoferrin, hepcidin, bactericidal/permeability increasing protein, lysozyme and overall, defensins and cathelicidins.
Defensins are a family of small cationic peptides (29–45 amino acids) that exhibit a wide and potent antimicrobial activity spectrum against gram-negative, and gram-positive bacteria, fungal and yeast, parasites, viruses, and even tumor cells [98]. Defensins have been identified in both prokaryotes and eukaryotes. Although structurally different, most defensins display cationic and amphiphilic properties which confer them the capacity to permeabilize the bacterial cell membrane. In mammals, these peptides are expressed in mucosal epithelial cells and phagocytes, but also are released into the intestinal lumen, several grams daily, by Paneth cells [99]. Defensins act as effector and regulatory molecules of the innate immune response. In addition, defensins also enhance adaptive response acting on phagocytic cells and mast cells to induce the release of inflammatory mediators and to regulate the complement system. Defensins also interact with dendritic cells and T cells to increase antigen-specific immune response [100].
These peptides are classified as α and β-defensins according to their disulphide bond pairing pattern. The human α-defensins 1–4, conventionally referred as to neutrophil defensin (human neutrophil peptide, HNP), although defensins HNP1-3 are also expressed in epithelial cells of inflammed mucosa [101]. In contrast, human α-defensin 5 and 6 (HD5 and HD6) are only expressed in Paneth cells of the small intestine [102]. HD5 has been shown to induce IL-8 expression on intestinal epithelial cells [103], and to protect mice from DSS colitis and Salmonella infection [104]. More recently, HD6 has been shown to form fibrils and nanonets that surround and entangle bacteria to protect the small intestine against invasion by diverse enteric pathogens [105].
Human β-defensin-1 is constitutively expressed in the small intestine and the colon. In contrast, Human β-defensins-2-4 expression is inducible [106] in inflammatory conditions such as IBD [107, 108] or infection by enteroinvasive bacteria [109].
The other major class of antimicrobial peptides is the cathelicidin group. In mammals, about 35 members have been identified, but only one in humans: hCAP18/LL37 [110]. Although regarded as neutrophil specific, hCAP18/LL37is also expressed in other leukocytes, keratinocytes and epithelial cells of the respiratory, genitourinary and gastrointestinal tract [111], and in human breast milk [112].
Expression of hCAP18/LL37 in human colonic epithelial cells has been related to cell differentiation [113]. Infection of intestinal epithelial cells by Shigella spp. inhibits the expression of hCAP18/LL37 [114], while bacterial components such as sodium butyrate [115] or TLR-ligands such as bacterial DNA [116] induce its expression.
Acquired immunity is restricted to vertebrates and constitutes a second line of defence against pathogens. It is driven by B and T lymphocytes through specific receptors and confers protection against re-exposure to the same antigen. Antigen binding to these receptors results in clonal expansion of these cells and the initiation of a directed immune response. Functionally speaking, within the adaptive immunity, we can distinguish inductive and effector compartments. Antigen presentation and naive T and B-lymphocytes activation occurs in the inductive compartment. In the effector compartment sensitized cells against different antigens extravasate and differentiate to carry out the destruction of pathogens. IgA secretion has been shown to be regulated through TLR-signalling [117] but also by changes in the composition of intestinal Microbiota [118].
Intestinal Barrier Dysfunction
Stress, Hormones and Neurotransmitters
Stress represents a threat to the internal homeostasis. In response to stress, a coordinated response is initiated to maintain stability through the autonomic, endocrine, and immune systems. The main systems activated during the stress response are the sympatho-adrenomedullary, a component of the sympathetic division of the autonomic nervous system, and the HPA axis. The autonomic nervous system provides, through its sympathetic and parasympathetic arms, the fastest response to stressor exposure, leading to rapid alterations in physiological state through neural innervation of end organs. Stress activation of the HPA axis stimulates the parvocellular neurons in the paraventricular nucleus of the hypothalamus to secrete corticotropin-releasing-factor (CRF), which in turn travels to the anterior pituitary to promote the synthesis of corticotropin (ACTH) [119]. ACTH, when released into the systemic circulation, activates the adrenal cortex to induce cortisol and corticosterone secretion that circulate through the bloodstream to reach every tissue [120]. Adaptation to stress through the activation of the sympatho-adrenomedullary system and the HPA axis to maintain homeostasis is called “allostasis”. However, excessive stress exposure impairs this adaptive response, eventually predisposing these subjects to the development of disease or to exacerbation of previous existing ones [121], specially in stress-sensitive disorders, like IBS.
At the experimental level, different type of stresses, acute and chronic, physical or psychological, have been shown to influence properties of the intestinal barrier function, including as ion and water secretion, intestinal permeability, mucus secretion, and also intestinal flora. Ion and water secretion allows the intestine to wash away noxious substances present in the intestinal lumen, preventing adhesion to the mucosal surfaces and penetration to the lamina propria. The jejunum of rats submitted to restraint stress or cold restraint stress was found to show an increase its baseline short-circuit current, indicative of enhanced anion secretion [122]. Later, it was observed that peripheral CRF and repetitive exposure to water avoidance stress reproduced stress-induced rat jejunal and colonic epithelial barrier dysfunction via cholinergic and adrenergic nerves and mast cells [123, 124]. More recently, it has been shown that chronic psychosocial stress also activates mucosal mast cells and increases baseline short-circuit current in both the jejunum and the colon [125]. In humans, studies using jejunal segmental perfusion techniques reveal that acute physical or psychological stress either reduce net water absorption or increase secretion in healthy subjects and in patients with food allergy [126, 127] through the parasympathetic nervous system and mast cell activation [128]. More recently, we have extended these observations to show that in healthy female volunteers that intestinal water secretion during cold pain stress was significantly reduced in those with moderate background stress compared to those with low stress [129]. This observation could indicate a loss of regulatory mechanisms in subjects suffering from continuous life stress.
Both paracellular and transcellular permeability to small and large molecules increased in response to acute and chronic stress in the rodent jejunum and colon [130–133]. Several mechanisms, including mast cells, CRF [134], MLCK, and cytokines like interferon gamma, and interleukin-4 [135] have been implicated. In humans, it is known that surgery, trauma, and gastrointestinal infections [136] increase intestinal permeability. CRF has been shown to enhance transcellular uptake of macromolecules in human colonic mucosa via CRF-R1 and CRF-R2 receptors, located on subepithelial mast cells [137]. Unpublished observations from our group indicate that intravenous CRF increased intestinal permeability in healthy subjects and in IBS patients [138]. Acute psychological stress also increases small intestinal permeability in humans and peripheral CRF reproduces the effect of stress and mast cell stabilization blocks the effect of both stress and CRF, suggesting the involvement of mast cells [139]. Cold pain stress also increased intestinal permeability in female healthy subjects, although this response was larger in women with moderate background stress. Increased intestinal permeability has been found in diarrhoea prone IBS patients [140]. These findings provide new insight into the complex interplay between the central nervous system and gastrointestinal function in man.
Acute stress causes mucin release in the rat colon, along with enhanced secretion of rat mast cell protease II and prostaglandin 2. These changes were reproduced by intravenous or intracerebral injection of CRF in non-stressed rats, and were inhibited by the administration of a CRF antagonist or a mast cell stabilizer [141]. In addition, stress-induced release of mucin was abolished in mast-cell deficient mice, highlighting a key role of mast cells in stress-mediated mucin release [142]. In contrast, rats submitted to chronic stress displayed mucus depletion along with increased bacterial adhesion and penetration into enterocytes [143].
Stress can also induce microbiological changes in the intestinal flora. Maternal separation in infant rhesus monkeys decreased faecal bacteria, especially Lactobacilli, and increased their susceptibility to opportunistic bacterial infections [144]. Similarly, prenatal stress reduced the overall numbers of Bifidobacteria and Lactobacilli in the newborn infants [145]. Interestingly probiotic treatment ameliorates stress-induced changes in the gastrointestinal tract [146] and attenuates the observed Lactobacilli reduction in maternally-deprived rat pups [147]. In addition to these microbiological changes, dexamethasone administration in rats enhanced bacterial adherence to the mucosa, decreased secretory-immunoglobulin A secretion, and increased intestinal permeability [148]. More recently, Söderholm et al. showed that chronic psychological stress in rats, leads to an increased antigen and bacterial uptake in follicle associated epithelium from Peyer’s patches [149] as well as in the villous ileal and colonic epithelium. Emotional stress during take-off in cosmonauts induced changes in faecal Bifidobacteria and Lactobacillus, as well as an increase in Escherichia coli, whereas a substantial increase in Enterobacteria and Clostridia was found after the flight [150]. These stress-induced changes in the faecal flora have been related to catecholamine release into the intestinal lumen and/or into the systemic circulation, as the addition of various catecholamines to cultures of gram negative bacteria resulted in dramatic increases in growth of E. coli, Yersinia enterocolitica and Pseudomonas aeruginosa [151].
Mast cells are known to modulate stress-mediated responses of the epithelial barrier function, to orchestrate the mucosal immune function and to participate in the defence against bacteria [152, 153]. To exert these functions, enteric mast cells are strategically located within the gastrointestinal tract, developing an optimal sensory and effector interaction within the local regulatory neuroendocrine networks. Upon activation, mast cells act as effector cells, through the selective (piecemeal degranulation) (Fig. 4.4) or massive release (anaphylactic degranulation) of preformed or newly produced biological mediators. More relevant to stress-mediated inflammation is their ability to communicate, bidirectionally, with both the enteric, autonomic and central nervous systems. Anatomical contacts between mast cells and enteric nerve fibres have been demonstrated in the human gastrointestinal mucosa and these contacts increase, when inflammation is present [154]. An increase in the nerve-to-mast cell proximity in the colonic mucosa of IBS patients has been positively correlated with the severity and frequency of abdominal pain [155]. This mast cell-enteric nerve interaction provides a physical substrate for bidirectional communication between the CNS and the gut, by which stress might influence gastrointestinal physiology. This is reflected in vivo by the release of mast cell products into the lumen of the human small intestine after cold stress, which is accompanied by increased epithelial secretion [128]. Mast cell mediators released after degranulation can sensitize mesenteric afferents and nociceptive receptors [156]. Among the potential mast cell mediators involved, both histamine and serotonin induce intestinal secretion of water, electrolytes and mucus. In addition, mast cells from IBS patients release more histamine and tryptase than intestinal mast cells from normal subjects [157] a fact that has been linked to the generation of visceral hypersensitivity, through the activation of proteinase-activated receptors type 2. These receptors can modulate enteric neurotransmission, secretion, motility, epithelial permeability, and visceral sensitivity, and are also known to regulate intestinal inflammation [158]. However, altered expression of histamine H1 and H2 receptor subtypes has recently been reported in mucosal biopsies from distal gut of IBS patients, suggesting that these receptors could also play a role in these processes [159].

Intestinal mast cells. Enteric mast cells are known to modulate the epithelial barrier function, to orchestrate the mucosal immunity and to participate in the defence against bacteria. They are strategically located within the gastrointestinal tract, developing sensory and effector interactions within the local regulatory neuroendocrine networks. Upon activation, mast cells act as effector cells, through the selective (piecemeal degranulation) or massive release (anaphylactic degranulation) of preformed or newly produced mediators. The figure shows transmission electron micrographs of ultrastructural characteristics of mucosal mast cells: a resting mast cell in health (H), with granules filled (white arrows) and no signs of degranulation; piecemeal degranulation in a mast cell from a patient with irritable bowel syndrome (IBS), identified by partial or total emptiness of granules content (black arrows) and intact granules (white arrow); and anaphylactic degranulation in a mast cell from a food allergy patient (FA), identified by fusion of granule membranes devoid of content (black arrow). Barr indicates 2 μm
CRF and related peptides are the most important neuroendocrine factors mediating the effects of stress, both at the central and peripheral level. CRF urocortin (Ucn) 1, Ucn 2 and Ucn 3 exert their effects after binding to G protein-coupled receptor subtypes, CRF-R1 and CRF-R2, signalling through cAMP [160]. After physical or psychological stress, neural or immune release of CRF and urocortins mediate autonomic, hormonal, and behavioural responses to stress and stimulate the ENS to modulate gastrointestinal motility and secretion [161–163]. Increased CRF and urocortin expression has been demonstrated in the colonic mucosa of IBD patients [164, 165].
Vasoactive intestinal peptide is also involved in the regulation of chloride secretion, mucin release, paracellular permeability and epithelial cell proliferation [166, 167]. Psychological stress increases vasoactive intestinal peptide levels in the small intestine of mice [168] and vasoactive intestinal peptide has been implicated in the regulation of the intestinal barrier function, through its direct effect on tight junction-associated protein, ZO-1, in epithelial cells [169].
Substance P participates in gut inflammation by interacting mainly with the neurokinin-1 receptor, expressed on nerves, epithelial, endothelial and smooth muscle cells, and immune cells, such as mast cells, macrophages, and T cells [170]. This neuropeptide has been found to stimulate macrophage and eosinophil secretion of pro-inflammatory cytokines, to increase NK cell activity and migration, and to activate the release of chemokines from leukocytes. It also induces the release of vasoactive mediators from mast cells, contributing to chloride secretion, intestinal permeability, vascular leakiness and oedema at sites of inflammation, modulating diarrhoea, inflammation, and motility [171]. Substance P mediates stress-induced CRF expression in mice eosinophils, and eosinophil-derived CRF is responsible for mast cell activation and consequently, epithelial barrier dysfunction [172].
Nerve growth factor (NGF) has been involved in the development of stress-induced barrier dysfunction [173] and hyperalgesia during inflammation [174, 175]. These effects seem to be mediated by CRF and mast cells [176, 177]. Maternal deprivation has been shown to induce hyperalgesia to rectal distension and to enhance colon permeability in association with elevated NGF expression [173]. A subsequent study from the same group showed that CRF, acting through its receptor CRF-R1, stimulated NGF release from mast cells, which in turn increased gut paracellular permeability [178]. More recently, norepinephrine has been shown to induce visceral sensitivity to colorectal distension by increasing the expression of NGF in the rat colon wall [179]. These findings support the importance of NGF in stress-induced visceral hypersensitivity, but also in stress-induced barrier dysfunction.
Sex steroids also play a role in modulating intestinal barrier, although conflicting results have been described. Estrogen can bind to two different receptors named estrogen receptor-α and β. Estrogen receptor-α mediates estrogen signalling in the development of secondary sex characteristics, and the regulation of the menstrual cycle and sperm maturation [180]. In contrast, estrogen receptor-β is mainly expressed in epithelial cells and is the most abundant estrogen receptor in the colon [181]. Both progesterone and estradiol have been shown to reduce chloride secretion in intestinal epithelial cells [182, 183], whereas estradiol has also been found to reinforce epithelial permeability [184], and to up-regulate JAM-A and occludin expression [185].
Other hormones have been involved in the regulation of intestinal barrier function (Table 4.1).
Infections
Intestinal pathogens have developed specific strategies to gain access to the lamina propria. Strategies include direct TJ disruption, the production of toxins that induce fluid and electrolyte secretion, and the activation of the inflammatory cascade [186]. Vibrio cholerae can directly alter TJs through its cytotoxin hemagglutinin protease, a metalloproteinase that disrupts occludin-ZO-1 interactions leading to TJ and cytoskeleton anchorage destabilization [187]. In addition, other toxins have been involved in TJ disruption by V. cholerae such as the RTX toxin, that crosslinks actin inducing cell rounding and increased permeability [188], or the ZO toxin, that fragments ZO-1 and occludin and disrupts the actin cytoskeleton [189, 190]. Clostridium difficile infection produces two distinct exotoxins, Toxin A and B (TcdA and TcdB), that through RhoA GTPases inactivation cause actin filament disaggregation and cell rounding, resulting in increased paracellular permeability [191, 192]. Recent findings suggest that toxin A could even disrupt directly TJ proteins [193]. Clostridium perfringens enterotoxin utilizes claudin-3 and 4 as receptors [194] to bind the enterocyte surface where it forms small protein complexes in the plasma membrane that interact with other proteins forming a large complex, that at the end triggers massive permeability changes [195]. Enteropathogenic E. coli infection directly disrupts TJ through occludin dephosphorylation and dissociation from TJs to the cytoplasma [196] and MLC phosphorylation [197] enhancing intestinal permeability.
Intestinal Microbiota
Intestinal microbiota has been shown to influence intestinal barrier function and the brain-gut axis [198, 199]. Intestinal microflora displays several important functions to maintain gut homeostasis, such as nutrient digestion, vitamin and hormone production and most importantly, protection from microbial colonization, achieved through competition for intestinal nutrients and for attachment sites [200]. Probiotics are live microorganisms which, when consumed in adequate amounts, confer a health benefit on the host. Increasing evidence suggests that probiotics implement intestinal epithelial homeostasis and enhance barrier tightness and integrity. In contrast with pathogens, probiotics have been shown to increase occludin expression [201], and to enhance ZO-2 expression in parallel to its redistribution towards the cell boundaries via silencing of PKCζ [202] thereby leading to TJ stabilization and the restoration of the epithelial barrier. Specific Lactobacilus salivarus strains prevent hydrogen peroxide-induced reduction in transepithelial resistance when added to Caco-2 cell monolayers [203]. Similarly, Lactobacillus rhamnosus GG improves intestinal barrier function in the immature murine gut through the induction of claudin 3 expression [204], the regulation of apoptosis and the promotion of cytoprotective responses [205]. Interestingly, probiotics have also demonstrated beneficial effects in other tissues such as the skin barrier [206] or the respiratory tract [207, 208].
There is a significant body of evidence indicating that probiotics can also prevent intestinal barrier damage in conditions such as IBD or experimental stress. In rats, DSS-induced colitis was ameliorated by Lactobacillus reuteri decreasing the bacterial translocation from the intestine to mesenteric lymph nodes [209]. E. coli Nissle 1917 has been shown to confer protection against murine DSS colitis-associated increase in mucosal permeability through up-regulation of ZO-1 expression [210]. Moreover, a probiotic mixture of Lactobacillus acidophilus, Bifidobacterium lactis, Lactobacillus plantarum and Bifidobacterium breve helped to maintain the integrity of colonic mucosal barrier in the DSS model by down-regulating macrophage nitric oxide production and by enhancing mucus production [211]. In this model, the administration of a probiotic mixture prevented not only the decrease in TJ proteins expression, but also the increase of epithelial apoptotic ratio induced by acute colitis [212]. Furthermore, in patients with severe pouchitis, probiotics were able to restore the mucosal barrier, as they decreased E. coli K12 passage through the intestinal epithelium in Ussing chambers [213].
Probiotics also play a role in stress-induced intestinal damage and psychiatric comorbidity. Lactobacillus farciminis has been shown to suppress stress-induced hyperpermeability and endotoxemia, and to prevent HPA axis response and neuroinflammation in rats submitted to partial restraint stress [214]. Probiotic administration to mice submitted to food and mobility restriction increased IgA producing cells, CD4+ cells in the lamina propria of the small intestine, and secretory IgA in the lumen and also reduced the levels of IFN-γ [215]. Bifidobacterium lactis CNCM I-2494 has been shown to suppress gut hypersensitivity and colonic barrier disruption induced by partial restraint stress in rats [216, 217]. In the last years, attention has been also pointed to the potential role of microbiota in the pathophysiology of psychiatric disorders such as depression and anxiety [218] and neurodevelopmental disorders such as autism. Interestingly, treatment with the human commensal Bacteroides fragilis restores gut permeability, alters microbial composition, and ameliorates defects in communicative, stereotypic, anxiety-like and sensorimotor behaviors in a mouse model of the autism spectrum disorder [219]. Since psychiatric comorbidities are highly common in functional gastrointestinal disorders, the emerging role of microbiota and probiotics in the regulation of intestinal and brain barrier function and its implication in behavioral changes in the host certainly will boost investigations in this field in the years ahead.
Inflammatory Mediators
Several inflammatory mediators have been involved in intestinal barrier regulation. In vitro experiments with epithelial cell monolayers demonstrated that interferon-γ and TNF-α induce epithelial barrier dysfunction through MLCK up-regulation and MLC phosphorylation [220, 221], although they can also disrupt intestinal permeability through down-regulation of occludin transcription [222] and up-regulation of the channel-forming TJ protein claudin-2 expression. In addition, TNF-α but also IL-1β have been shown to inhibit electrogenic sodium absorption in the rat distal colon [223], and mice injected with TNF-α present diarrhoea as a consequence of Na+/H+ exchange inhibition [224].
Similarly, IL-13 and IL-4 increased paracellular permeability in a dose- and time-dependent fashion and IL-4, but not IL-13, stimulated chloride secretion in T84 cells [225] through a PI3K pathway [226]. In contrast, IL-10 has been identified as a protector cytokine in barrier function as the addition of this cytokine to T84 cells prevents interferon-γ-induced disruption of T84 monolayer barrier integrity and limits chloride secretion [227]. Moreover, IL-10 deficient mice display increased intestinal permeability [228] and most importantly, develop spontaneous colitis [229], suggesting that increased permeability predisposes to intestinal inflammation.
Although, beyond the limits of this chapter. It is to know that many other cytokines have been involved in barrier function such as IL-17A, IL-17F, IL-22, and IL-26, interferon-α, interferon-β, transforming growth factor-α, and -β [230, 231].
Nutritional Factors
Some dietary compounds are able to induce intestinal barrier dysfunction in susceptible individuals such as in celiac disease and food allergy. The gliadin fraction of wheat gluten is the environmental triggering of celiac disease. In genetically predisposed subjects gluten exposure may lead to increased intestinal permeability and inflammation. Recent evidence has shown that the increase in intestinal permeability occurs through the activation of the zonulin pathway in a MyD88-dependent fashion [232]. The protein zonulin is the target of the Zot toxin of the V. cholerae and has been show to play a pivotal role in TJ regulation in different autoimmune disorders such as type 1 diabetes and celiac disease [233]. Food allergies are adverse reactions against food antigens that are IgE and mast cell mediated. Altered intestinal permeability has also been involved in the pathophysiology of food allergy, as these patients display an enhancement of intestinal permeability even in the absence of food allergens [234]. Moreover, patients under tacrolimus treatment have been shown to develop new-onset food allergies that could be related to tacrolimus-induced increase in intestinal permeability [235].
In contrast with these observations, several diet products such as glutamine or butyrate have been shown to exert a protective effect on the intestinal barrier. Butyrate, a short chain fatty acid produced by intestinal microbial fermentation of dietary fibres, maintains intestinal barrier function through an increase in mucus production [236] and an enhancement in TJ protein expression [237]. Glutamine has also been shown to protect intestinal barrier function through the regulation of TJ proteins such as claudin-1, and occludin [238].
Drugs and Toxins
Ethanol has been shown to promote separation of ZO-1 proteins in Caco-2 monolayers and disassembly and displacement of perijunctional actin and myosin filaments from the perijunctional areas and MLCK activation [239]. Recent findings point to one of its metabolites, acetaldehyde, as the main toxic product for intestinal barrier because it raises tyrosine phosphorylation of ZO-1, e-cadherin, and β-catenin [240]. Further investigations revealed that the deleterious effects of ethanol require the presence of resident microflora, to oxidize ethanol into acetaldehyde in situ, and downstream mast cell activation [241], and that the ethanol-mediated increase in intestinal permeability is modulated through iNOS-mediated activation of RhoA [242] and IL-22 [243].
NSAIDs can increase intestinal permeability. Several factors play a role in NSAIDs-induced intestinal barrier dysfunction. In vitro experiments with gastric epithelial monolayers showed that barrier dysfunction was associated with decreased expression of claudin 7 and involved phosphorylation of p38 MAPK [244]. NSAIDs also affect intestinal barrier through inhibition of intestinal epithelial restitution by decreasing calpain activity and membrane-associated expression of calpain-2 [245], and also through the increase of intestinal NO synthase [246].
Other drugs causing intestinal barrier dysfunction appear in Table 4.2. It is of particular interest the development of new drugs, such as larazotide, that may decrease intestinal permeability in celiac disease by acting on TJs.
Other Disorders Associated with Barrier Dysfunction
Many other conditions such as chronic kidney disease [247], type 1 diabetes [248], primary biliary cirrhosis and primary sclerosing cholangitis [249], liver cirrhosis [250], alcoholic liver disease [251], autoimmune thyroiditis [252] and IgA nephropathy [253] have been associated with TJ dysfunction. In addition, some life threatening conditions have been related to intestinal barrier dysfunction and translocation of bacteria or/and endotoxin from gastrointestinal tract. In this line, hemorrhagic shock has been associated with increased intestinal permeability and bacterial translocation [254] through mucus damage and the generation of free radical species [255]. Estrogens exert a protective role against hemorrhagic shock-induced gut and lung injury by the activation of estrogen receptor-α, β or both [256] receptors. Similarly, gut inflammation and loss of gut barrier function has been related to splachnic ischemia-reperfusion through HIF-1 activation [257]. Multiple injured patients also show an increased intestinal permeability that correlates with IL-6 levels [258]. Severe burn injury also results in the loss of intestinal barrier function involving MLCK-dependent MLC phosphorylation signalling pathway [259] and p38 MAPK activation [260] in a TLR-4-dependent process [261].
Intestinal Barrier and Disorders of the Brain-Gut Axis
The pathophysiology of several gastrointestinal disorders involves intestinal barrier dysfunction and dysregulation of brain-gut interactions, particularly functional gastrointestinal disorders including IBS and functional dyspepsia. In recent years, due to new imaging techniques, such as positron emission tomography, it has been possible to characterize the role of the CNS in modulating gut motility and visceral pain in patients with functional gastrointestinal diseases. There is significant overlap between the brain regions responsible for modulating visceral sensitivity and regions involved in emotion processing in these patients. IBS patients display a higher activation of the anterior cingulate cortex in response to rectal distension [262] that correlates with the presence of psychosocial disorders when compared to healthy subjects [263].
At the peripheral level, mucosal inflammation, increased intestinal permeability and visceral hypersensitivity are findings associated with clinical manifestations of IBS. Mast cells play a key role in IBS pathophysiology because they modulate intestinal permeability, and target visceral afferents involved in abdominal pain [155]. Stress has been associated with the development, exacerbation and perpetuation of IBS through the brain-gut-axis. Early life stress plays a major role in the vulnerability of individuals to develop IBS in adult life [264–266]. Post-traumatic stress syndrome or sexual abuse are also important risk factors in the development of IBS and functional gastrointestinal disorders [267] and both acute psychological and physical stress have been associated with enhancement of visceral sensitivity [268] and small-intestine motility in IBS [269].
Functional dyspepsia is characterized by postprandial fullness and early satiation or by epigastric pain or burning in the absence of an organic cause Functional dyspepsia has been show to share some of the pathophysiological features of IBS. Particularly, patients with functional dyspepsia display low-grade inflammation in the duodenal mucosa, characterized by an increased infiltration of mucosal mast cells and eosinophils, and increased duodenal permeability [270]. Acute gastroenteritis has been shown to be a risk factor for functional dyspepsia development [271], as well as the presence of psychosocial comorbidities such as anxiety and depression [272], and life stress [273].
Stress, acting through the brain-gut axis, also modulates intestinal inflammatory conditions such as IBD. Social Gibbon monkeys submitted to social upheaval develop spontaneous colon inflammation [274]. Intracolonic infusion of TNBS induced a significantly higher inflammatory reaction in maternally deprived rats than in control animals [275]. Collins et al. [276] found that rats recovering from TNBS-induced colitis and submitted to mild restraint stress displayed a significant increase in myeloperoxidase activity. Moreover, overt inflammation was induced when animals were exposed to stress in combination with a small dose of TNBS, suggesting an additive effect [277]. In keeping with these findings, a significant association between stress and relapse in IBD has been reported, especially in patients with ulcerative colitis [278, 279]. Although the mechanism underlying the association between stress and IBD remains unclear, disturbances of brain-gut axis, peripheral neuroendocrine-immune interactions and altered intestinal barrier function [280–284] have been demonstrated in IBD patients [285].
Finally, a heterogeneous group of conditions associated with chronic manifestations affecting the CNS and the gut may possibly reflect the existence of primary or secondary alterations of brain-gut axis, intestinal microbiota and barrier function. This is the case of diabetes and the metabolic syndrome [286], liver encephalopathy [287], neuropsychiatric disorders [288], autism [289], chronic fatigue [290] or fibromyalgia [291], although the ultimate pathophysiological mechanisms are not well known.
Abbreviations
- ACTH:
-
Corticotropin
- CNS:
-
Central nervous system
- CRF:
-
Corticotropin-releasing-factor
- DSS:
-
Dextran sulphate sodium
- ENS:
-
Enteric nervous system
- GALT:
-
Gut-associated lymphoid tissue
- GCs:
-
Goblet cells
- HNPs:
-
Human neutrophil peptides
- HPA:
-
Hypothalamic pituitary-adrenal axis
- IBD:
-
Inflammatory bowel disease
- IBS:
-
Irritable bowel syndrome
- JAMs:
-
Junctional adhesion molecules
- MAPKs:
-
Mitogen-activated protein kinases
- MARVEL:
-
MAL and related proteins for vesicle trafficking and membrane link
- MLC:
-
Myosin light chain
- MLCK:
-
Myosin light chain kinase
- MUC:
-
Mucins
- NGF:
-
Nerve growth factor
- NLRs:
-
Nod-like receptors
- NOD:
-
Nucleotide-binding oligomerization domain
- PAMP:
-
Pathogen-associated molecular patterns
- POFUT1:
-
Protein O-fucosyltransferase 1
- PRR:
-
Pattern recognition receptors
- RELM:
-
Resistin-like molecule
- TJs:
-
Tight junctions
- TNBS:
-
Trinitrobenzene sulphonic acid
- ZO:
-
Zonula occludens
References
Turner JR (2009) Intestinal mucosal barrier function in health and disease. Nat Rev Immunol 9(11):799–809
Kayama H, Nishimura J, Takeda K (2013) Regulation of intestinal homeostasis by innate immune cells. Immune Netw 13(6):227–234
Knoop KA, Miller MJ, Newberry RD (2013) Transepithelial antigen delivery in the small intestine: different paths, different outcomes. Curr Opin Gastroenterol 29(2):112–118
Wood JD (2007) Neuropathophysiology of functional gastrointestinal disorders. World J Gastroenterol 13(9):1313–1332
Sansonetti PJ (2004) War and peace at mucosal surfaces. Nat Rev Immunol 4(12):953–964
Santos J, Bienenstock J, Perdue MH (2002) Innervation of lymphoid tissue and functional consequences of neurotransmitter and neuropeptide release. In: Brostoff J, Challacombe SJ (eds) Food allergy and intolerance. W.B. Saunders, London, pp 51–67
Hall JE (2011) General principles of gastrointestinal function. Guyton and Hal textbook of medical physiology, 12th edn. Saunders Elsevier, p 755. ISBN: 978-1416045748
Shao L, Serrano D, Mayer L (2001) The role of epithelial cells in immune regulation in the gut. Semin Immunol 13(3):163–176
Yamada T, Sartor RB, Marshall S, Specian RD, Grisham MB (1993) Mucosal injury and inflammation in a model of chronic granulomatous colitis in rats 2. Gastroenterology 104(3):759–771
Gibson P, Rosella O, Nov R, Young G (1995) Colonic epithelium is diffusely abnormal in ulcerative colitis and colorectal cancer. Gut 36:857–863
Farhadi A, Banan A, Fields J, Keshavarzian A (2003) Intestinal barrier: an interface between health and disease. J Gastroenterol Hepatol 18(5):479–497
Liévin-Le Moal V, Servin AL (2006) The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev 19(2):315–337
Corfield AP, Carroll D, Myerscough N, Probert CS (2001) Mucins in the gastrointestinal tract in health and disease. Front Biosci 6:D1321–D1357
Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, He B, Cassis L, Bigas A, Cols M, Comerma L, Huang B, Blander JM, Xiong H, Mayer L, Berin C, Augenlicht LH, Velcich A, Cerutti A (2013) Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science 342(6157):447–453
Brandtzaeg P (1995) Molecular and cellular aspects of the secretory immunoglobulin system. APMIS 103:1–19
Bevins CL, Salzman NH (2011) Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9(5):356–368
Qin X, Caputo FJ, Xu DZ, Deitch EA (2008) Hydrophobicity of mucosal surface and its relationship to gut barrier function. Shock 29(3):372–376
Shirazi T, Longman RJ, Corfield AP, Probert CS (2000) Mucins and inflammatory bowel disease. Postgrad Med J 76(898):473–478
Allen A, Pearson JP (1993) Mucus glycoproteins of the normal gastrointestinal tract. Eur J Gastroenterol Hepatol 5:193–199
Rhodes JM (1997) Mucins and inflammatory bowel disease. Q J Med 90:79–82
Kim YS, Ho SB (2010) Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 12(5):319–330
Pigny P, Guyonnet-Duperat V, Hill AS, Pratt WS, Galiegue-Zouitina S, d’Hooge MC, Laine A, Van-Seuningen I, Degand P, Gum JR, Kim YS, Swallow DM, Aubert JP, Porchet N (1996) Human mucin genes assigned to 11p15.5: identification and organization of a cluster of genes. Genomics 38(3):340–352
Gum JR Jr, Hicks JW, Toribara NW, Siddiki B, Kim YS (1994) Molecular cloning of human intestinal mucin (MUC2) cDNA. Identification of the amino terminus and overall sequence similarity to prepro-von Willebrand factor. J Biol Chem 269(4):2440–2446
Tytgat KM, Büller HA, Opdam FJ, Kim YS, Einerhand AW, Dekker J (1994) Biosynthesis of human colonic mucin: Muc2 is the prominent secretory mucin. Gastroenterology 107(5):1352–1363
Johansson ME, Larsson JM, Hansson GC (2011) The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci U S A 108(Suppl 1):4659–4665
Atuma C, Strugala V, Allen A, Holm L (2001) The adherent gastrointestinal mucus gel layer: thickness and physical state in vivo. Am J Physiol Gastrointest Liver Physiol 280(5):G922–G929
Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW (2006) Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131(1):117–129
Hasnain SZ, Wang H, Ghia JE, Haq N, Deng Y, Velcich A, Grencis RK, Thornton DJ, Khan WI (2010) Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology 138(5):1763–1771
Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, Xia L, Vallance BA (2013) The mucin Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella enterica serovar Typhimurium colitis. Infect Immun 81(10):3672–3683
Einerhand AW, Renes IB, Makkink MK, van der Sluis M, Buller HA, Dekker J (2002) Role of mucins in inflammatory bowel disease: important lessons from experimental models. Eur J Gastroenterol Hepatol 14:757–765
Larsson JM, Karlsson H, Crespo JG, Johansson ME, Eklund L, Sjövall H, Hansson GC (2011) Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis 17(11):2299–2307
Kyo K, Parkes M, Takei Y, Nishimori H, Vyas P, Satsangi J, Simmons J, Nagawa H, Baba S, Jewell D, Muto T, Lathrop GM, Nakamura Y (1999) Association of ulcerative colitis with rare VNTR alleles of the human intestinal mucin gene, MUC3. Hum Mol Genet 8(2):307–311
Kyo K, Muto T, Nagawa H, Lathrop GM, Nakamura Y (2001) Associations of distinct variants of the intestinal mucin gene MUC3A with ulcerative colitis and Crohn’s disease. J Hum Genet 46:5–20
Guilmeau S, Flandez M, Bancroft L, Sellers RS, Tear B, Stanley P, Augenlicht LH (2008) Intestinal deletion of Pofut1 in the mouse inactivates notch signaling and causes enterocolitis. Gastroenterology 135(3):849–860
Krimi RB, Kotelevets L, Dubuquoy L, Plaisancié P, Walker F, Lehy T, Desreumaux P, Van Seuningen I, Chastre E, Forgue-Lafitte ME, Marie JC (2008) Resistin-like molecule beta regulates intestinal mucous secretion and curtails TNBS-induced colitis in mice. Inflamm Bowel Dis 14(7):931–941
Hogan SP, Seidu L, Blanchard C, Groschwitz K, Mishra A, Karow ML, Ahrens R, Artis D, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rothenberg ME (2006) Resistin-like molecule beta regulates innate colonic function: barrier integrity and inflammation susceptibility. J Allergy Clin Immunol 118(1):257–268
Taupin D, Podolsky DK (2003) Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol 4:721–732
Kindon H, Pothoulakis C, Thim L, Lynch-Devaney K, Podolsky DK (1995) Trefoil peptide protection of intestinal epithelial barrier function: cooperative interaction with mucin glycoprotein. Gastroenterology 109(2):516–523
Mashimo H, Wu DC, Podolsky DK, Fishman MC (1996) Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274:262–265
Beck PL, Wong JF, Li Y, Swaminathan S, Xavier RJ, Devaney KL, Podolsky DK (2004) Chemotherapy- and radiotherapy-induced intestinal damage is regulated by intestinal trefoil factor. Gastroenterology 126:796–808
Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP (2001) Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med 193:1027–1034
Menard S, Cerf-Bensussan N, Heyman M (2010) Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol 3(3):247–259
Booth C, Potten CS (2000) Gut instincts: thoughts on intestinal epithelial stem cells 1. J Clin Invest 105(11):1493–1499
Jeon MK, Klaus C, Kaemmerer E, Gassler N (2013) Intestinal barrier: molecular pathways and modifiers. World J Gastrointest Pathophysiol 4(4):94–99
Pott J, Hornef M (2012) Innate immune signalling at the intestinal epithelium in homeostasis and disease. EMBO Rep 13(8):684–698
Muehlhoefer A, Saubermann LJ, Gu X, Luedtke-Heckenkamp K, Xavier R, Blumberg RS, Podolsky DK, MacDermott RP, Reinecker HC (2000) Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol 164(6):3368–3376
Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt Rde W, Omori M, Zhou B, Ziegler SF (2007) TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol 25:193–219
Saitou M, Fujimoto K, Doi Y, Itoh M, Fujimoto T, Furuse M, Takano H, Noda T, Tsukita S (1998) Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. J Cell Biol 141(2):397–408
Rao R (2009) Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci 1165:62–68
Dörfel MJ, Huber O (2012) Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J Biomed Biotechnol 2012:807356
Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S (2002) Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 156(6):1099–1111
Cereijido M, Contreras RG, Shoshani L, Flores-Benitez D, Larre I (2008) Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim Biophys Acta 1778:770–793
Fihn BM, Sjoqvist A, Jodal M (2000) Permeability of the rat small intestinal epithelium along the villus-crypt axis: effects of glucose transport. Gastroenterology 119:1029–1036
Schumann M, Günzel D, Buergel N, Richter JF, Troeger H, May C, Fromm A, Sorgenfrei D, Daum S, Bojarski C, Heyman M, Zeitz M, Fromm M, Schulzke JD (2012) Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut 61(2):220–228
Zeissig S, Bürgel N, Günzel D, Richter J, Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M, Schulzke JD (2007) Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 56:61–72
Escaffit F, Boudreau F, Beaulieu JF (2005) Differential expression of claudin-2 along the human intestine: implication of GATA-4 in the maintenance of claudin-2 in differentiating cells. J Cell Physiol 203(1):15–26
Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA (2000) Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 113(Pt 13):2363–2374
Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, Dermody TS, Nusrat A, Parkos CA (2007) JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med 204(13):3067–3076
Vetrano S, Rescigno M, Cera MR, Correale C, Rumio C, Doni A, Fantini M, Sturm A, Borroni E, Repici A, Locati M, Malesci A, Dejana E, Danese S (2008) Unique role of junctional adhesion molecule-a in maintaining mucosal homeostasis in inflammatory bowel disease. Gastroenterology 135(1):173–184
Wilcz-Villega EM, McClean S, O’Sullivan MA (2013) Mast cell tryptase reduces junctional adhesion olecule-A (JAM-A) expression in intestinal epithelial cells: implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am J Gastroenterol 108(7):1140–1151
Ebnet K, Suzuki A, Ohno S, Vestweber D (2004) Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci 117(Pt 1):19–29
Tsukita S, Katsuno T, Yamazaki T, Umeda K, Tamura A, Tsukita S (2009) Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann N Y Acad Sci 1165:44–52
Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M, Tsukita S (2006) ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell 126:741–754
Shen L, Turner JR (2006) Role of epithelial cells in initiation and propagation of intestinal inflammation. Eliminating the static: tight junction dynamics exposed. Am J Physiol Gastrointest Liver Physiol 290(4):G577–G582
Utech M, Mennigen R, Bruewer M (2010) Endocytosis and recycling of tight junction proteins in inflammation. J Biomed Biotechnol 2010:484987
Schulzke JD, Bojarski C, Zeissig S, Heller F, Gitter AH, Fromm M (2006) Disrupted barrier function through epithelial cell apoptosis. Ann N Y Acad Sci 1072:288–299
Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A (2003) Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci 116(Pt 4):725–742
Fasano A, Not T, Wang W, Uzzau S, Berti I, Tommasini A, Goldblum SE (2000) Zonulin, a newly discovered modulator of intestinal permeability, its expression in coeliac disease. Lancet 358:1518–1519
Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, Fasano A (2005) Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U S A 102:2916–2921
Kandori H, Hirayama K, Takeda M, Doi K (1996) Histochemical, lectin-histochemical and morphometrical characteristics of intestinal goblet cells of germfree and conventional mice. Exp Anim 45:155–160
Lin PW, Simon PO Jr, Gewirtz AT, Neish AS, Ouellette AJ, Madara JL, Lencer WI (2004) Paneth cell cryptdins act in vitro as apical paracrine regulators of the innate inflammatory response. J Biol Chem 279(19):19902–19907
Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV (2008) Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A 105:20858–20863
Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nuñez G, Keshav S (2003) Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology 125(1):47–57
Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, Greenson JK, Keshav S, Nuñez G (2003) Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 52(11):1591–1597
McElroy SJ, Underwood MA, Sherman MP (2013) Paneth cells and necrotizing enterocolitis: a novel hypothesis for disease pathogenesis. Neonatology 103(1):10–20
Underwood MA (2012) Paneth cells and necrotizing enterocolitis. Gut Microbes 3(6):562–565
Sternini C, Anselmi L, Rozengurt E (2008) Enteroendocrine cells: a site of ‘taste’ in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes 15(1):73–78
Flemström G, Sjöblom M (2005) Epithelial cells and their neighbors. II. New perspectives on efferent signaling between brain, neuroendocrine cells, and gut epithelial cells. Am J Physiol Gastrointest Liver Physiol 289(3):G377–G380
Bienenstock J, McDermott M, Befus D, O’Neill M (1978) A common mucosal immunologic system involving the bronchus, breast and bowel 4. Adv Exp Med Biol 107:53–59
Campbell DJ, Kim CH, Butcher EC (2003) Chemokines in the systemic organization of immunity. Immunol Rev 195:58–71
Gill N, Wlodarska M, Finlay BB (2011) Roadblocks in the gut: barriers to enteric infection. Cell Microbiol 13(5):660–669
Nürnberger T, Brunner F, Kemmerling B, Piater L (2004) Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev 198:249–266
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4(7):499–511
Abreu MT, Vora P, Faure E, Thomas LS, Arnold ET, Arditi M (2001) Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J Immunol 167(3):1609–1616
Cario E, Podolsky DK (2000) Differential alteration in intestinal epithelial cell expression of Toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 68(12):7010–7017
Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL (2001) Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 167:1882–1885
Lee J, Mo JH, Katakura K, Alkalay I, Rucker AN, Liu YT, Lee HK, Shen C, Cojocaru G, Shenouda S, Kagnoff M, Eckmann L, Ben-Neriah Y, Raz E (2006) Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat Cell Biol 8(12):1327–1336
Franchi L, McDonald C, Kanneganti TD, Amer A, Núñez G (2006) Nucleotide-binding oligomerization domain-like receptors: intracellular pattern recognition molecules for pathogen detection and host defense. J Immunol 177(6):3507–3513
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jéhanno M, Viala J, Tedin K, Taha MK, Labigne A, Zähringer U, Coyle AJ, DiStefano PS, Bertin J, Sansonetti PJ, Philpott DJ (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300(5625):1584–1587
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278(11):8869–8872
Tschopp J, Martinon F, Burns K (2003) NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4:95–104
Kim JG, Lee SJ, Kagnoff MF (2004) Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by Toll-like receptors. Infect Immun 72:1487–1495
Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L, Selvanantham T, Girardin SE, Philpott DJ (2010) Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun 78:5107–5115
Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS (2009) Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A 106:15813–15818
Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, Schröder JM, Bevins CL, Fellermann K, Stange EF (2004) NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 53(11):1658–1664
Ho S, Pothoulakis C, Koon HW (2013) Antimicrobial peptides and colitis. Curr Pharm Des 19(1):40–47
Papagianni M (2003) Ribosomally synthesized peptides with antimicrobial properties: biosynthesis, structure, function, and applications. Biotechnol Adv 21(6):465–499
Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415(6870):389–395
Yang D, Biragyn A, Hoover DM, Lubkowski J, Oppenheim JJ (2004) Multiple roles of antimicrobial defensins, cathelicidins, and eosinophil-derived neurotoxin in host defense. Annu Rev Immunol 22:181–215
Cunliffe RN, Kamal M, Rose FR, James PD, Mahida YR (2002) Expression of antimicrobial neutrophil defensins in epithelial cells of active inflammatory bowel disease mucosa. J Clin Pathol 55(4):298–304
Cunliffe RN (2003) Alpha-defensins in the gastrointestinal tract. Mol Immunol 40:463–467
Ishikawa C, Tanabe H, Maemoto A, Ito T, Watari J, Kono T, Fujiya M, Ashida T, Ayabe T, Kohgo Y (2010) Precursor processing of human defensin-5 is essential to the multiple functions in vitro and in vivo. J Innate Immun 2(1):66–76
Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL (2003) Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422:522–526
Chu H, Pazgier M, Jung G, Nuccio SP, Castillo PA, de Jong MF, Winter MG, Winter SE, Wehkamp J, Shen B, Salzman NH, Underwood MA, Tsolis RM, Young GM, Lu W, Lehrer RI, Bäumler AJ, Bevins CL (2012) Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets. Science 337(6093):477–481
Harder J, Bartels J, Christophers E, Schroder JM (2001) Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J Biol Chem 276(8):5707–5713
Wehkamp J, Fellermann K, Herrlinger KR, Baxmann S, Schmidt K, Schwind B, Duchrow M, Wohlschlager C, Feller AC, Stange EF (2002) Human beta-defensin 2 but not beta-defensin 1 is expressed preferentially in colonic mucosa of inflammatory bowel disease. Eur J Gastroenterol Hepatol 14:745–752
Fahlgren A, Hammarstrom S, Danielsson A, Hammarstrom ML (2004) beta-defensin-3 and -4 in intestinal epithelial cells display increased mRNA expression in ulcerative colitis. Clin Exp Immunol 137:379–385
O’Neil DA (2003) Regulation of expression of beta-defensins: endogenous enteric peptide antibiotics. Mol Immunol 40(7):445–450
Cowland JB, Johnsen AH, Borregaard N (1995) hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett 368(1):173–176
Frohm NM, Sandstedt B, Sorensen O, Weber G, Borregaard N, Stahle-Backdahl M (1999) The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and colocalizes with interleukin-6. Infect Immun 67(5):2561–2566
Murakami M, Dorschner RA, Stern LJ, Lin KH, Gallo RL (2005) Expression and secretion of cathelicidin antimicrobial peptides in murine mammary glands and human milk. Pediatr Res 57:10–15
Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF (2002) Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun 70(2):953–963
Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, Gudmundsson G (2001) Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med 7(2):180–185
Kida Y, Shimizu T, Kuwano K (2006) Sodium butyrate up-regulates cathelicidin gene expression via activator protein-1 and histone acetylation at the promoter region in a human lung epithelial cell line, EBC-1. Mol Immunol 43(12):1972–1981
Koon HW, Shih DQ, Chen J, Bakirtzi K, Hing TC, Law I, Ho S, Ichikawa R, Zhao D, Xu H, Gallo R, Dempsey P, Cheng G, Targan SR, Pothoulakis C (2011) Cathelicidin signaling via the Toll-like receptor protects against colitis in mice. Gastroenterology 141(5):1852–1863
He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, Knowles DM, Rescigno M, Cerutti A (2007) Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity 26:812–826
Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, Cahenzli J, Velykoredko Y, Balmer ML, Endt K, Geuking MB, Curtiss R 3rd, McCoy KD, Macpherson AJ (2010) Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328:1705–1709
Cicchetti D, Cohen DJ (2006) Developmental psychopathology: developmental neuroscience. Wiley, New York
de Kloet ER, Joels M, Holsboer F (2005) Stress and the brain: from adaptation to disease. Nat Rev Neurosci 6(6):463–475
Gunnar M, Quevedo K (2007) The neurobiology of stress and development. Annu Rev Psychol 58:145–173
Saunders PR, Kosecka U, McKay DM, Perdue MH (1994) Acute stressors stimulate ion secretion and increase epithelial permeability in rat intestine. Am J Physiol 267(5 Pt 1):G794–G799
Santos J, Saunders PR, Hanssen NP, Yang PC, Yates D, Groot JA, Perdue MH (1999) Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. Am J Physiol 277(2 Pt 1):G391–G399
Santos J, Benjamin M, Yang PC, Prior T, Perdue MH (2000) Chronic stress impairs rat growth and jejunal epithelial barrier function: role of mast cells. Am J Physiol Gastrointest Liver Physiol 278(6):G847–G854
Vicario M, Guilarte M, Alonso C, Yang P, Martínez C, Ramos L, Lobo B, González A, Guilà M, Pigrau M, Saperas E, Azpiroz F, Santos J (2010) Chronological assessment of mast cell-mediated gut dysfunction and mucosal inflammation in a rat model of chronic psychosocial stress. Brain Behav Immun 24(7):1166–1175
Barclay GR, Turnberg LA (1987) Effect of psychological stress on salt and water transport in the human jejunum. Gastroenterology 93(1):91–97
Barclay GR, Turnberg LA (1988) Effect of cold-induced pain on salt and water transport in the human jejunum. Gastroenterology 94(4):994–998
Santos J, Saperas E, Nogueiras C, Mourelle M, Antolin M, Cadahia A, Malagelada JR (1998) Release of mast cell mediators into the jejunum by cold pain stress in humans. Gastroenterology 114(4):640–648
Alonso C, Guilarte M, Vicario M, Ramos L, Ramadan Z, Antolin M, Martinez C, Rezzi S, Saperas E, Kochhar S, Santos J, Malagelada JR (2008) Maladaptive intestinal epithelial responses to life stress may predispose healthy women to gut mucosal inflammation. Gastroenterology 135(1):163–172
Meddings JB, Swain MG (2000) Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology 119(4):1019–1028
Soderholm JD, Perdue MH (2001) Stress and gastrointestinal tract. II. Stress and intestinal barrier function. Am J Physiol Gastrointest Liver Physiol 280(1):G7–G13
Kiliaan AJ, Saunders PR, Bijlsma PB, Berin MC, Taminiau JA, Groot JA, Perdue MH (1998) Stress stimulates transepithelial macromolecular uptake in rat jejunum. Am J Physiol 275(5 Pt 1):G1037–G1044
Saunders PR, Santos J, Hanssen NP, Yates D, Groot JA, Perdue MH (2002) Physical and psychological stress in rats enhances colonic epithelial permeability via peripheral CRH. Dig Dis Sci 47(1):208–215
Santos J, Perdue MH (2000) Stress and neuroimmune regulation of gut mucosalfunction. Gut 47(Suppl 4):iv49–51; discussion iv52
Ferrier L, Mazelin L, Cenac N, Desreumaux P, Janin A, Emilie D, Colombel JF, Garcia-Villar R, Fioramonti J, Bueno L (2003) Stress-induced disruption of colonic epithelial barrier: role of interferon-gamma and myosin light chain kinase in mice. Gastroenterology 125(3):795–804
Dunlop SP, Hebden J, Campbell E, Naesdal J, Olbe L, Perkins AC, Spiller RC (2006) Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am J Gastroenterol 101(6):1288–1294
Wallon C, Yang PC, Keita AV, Ericson AC, McKay DM, Sherman PM, Perdue MH, Soderholm JD (2008) Corticotropin-releasing hormone (CRH) regulates macromolecular permeability via mast cells in normal human colonic biopsies in vitro. Gut 57(1):50–58
Guilarte M, Santos J, Alonso C, Vicario M, Antolin M, Saperas E, Malagelada JR (2004) Corticotropin-releasing hormone (CRH) triggers jejunal mast cell and eosinophil activation in IBS patients. Gastroenterology 126(Suppl 2):A38 (ref type: abstract)
Vanuytsel T, van Wanrooy S, Vanheel H, Vanormelingen C, Verschueren S, Houben E, Salim Rasoel S, Tóth J, Holvoet L, Farré R, Van Oudenhove L, Boeckxstaens G, Verbeke K, Tack J (2013) Psychological stress and corticotropin-releasing hormone increase intestinal permeability in humans by a mast cell-dependent mechanism. Gut. doi:10.1136/gutjnl-2013-305690 (Epub ahead of print)
Zhou Q, Souba WW, Croce CM, Verne GN (2010) MicroRNA-29a regulates intestinal membrane permeability in patients with irritable bowel syndrome. Gut 59(6):775–784
Castagliuolo I, LaMont JT, Qiu B, Fleming SM, Bhaskar KR, Nikulasson ST, Kornetsky C, Pothoulakis C (1996) Acute stress causes mucin release from rat colon: role of corticotropin releasing factor and mast cells. Am J Physiol 271(5 Pt 1):G884–G892
Castagliuolo I, Wershil BK, Karalis K, Pasha A, Nikulasson ST, Pothoulakis C (1998) Colonic mucin release in response to immobilization stress is mast cell dependent. Am J Physiol 274(6 Pt 1):G1094–G1100
Söderholm JD, Yang PC, Ceponis P, Vohra A, Riddell R, Sherman PM, Perdue MH (2002) Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology 123(4):1099–1108
Bailey MT, Coe CL (1999) Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol 35(2):146–155
Bailey MT, Lubach GR, Coe CL (2004) Prenatal stress alters bacterial colonization of the gut in infant monkeys. J Pediatr Gastroenterol Nutr 38(4):414–421
Eutamene H, Bueno L (2007) Role of probiotics in correcting abnormalities of colonic flora induced by stress. Gut 56(11):1495–1497
Gareau MG, Jury J, MacQueen G, Sherman PM, Perdue MH (2007) Probiotic treatment of rat pups normalises corticosterone release and ameliorates colonic dysfunction induced by maternal separation. Gut 56(11):1522–1528
Spitz JC, Ghandi S, Taveras M, Aoys E, Alverdy JC (1996) Characteristics of the intestinal epithelial barrier during dietary manipulation and glucocorticoid stress. Crit Care Med 24(4):635–641
Velin AK, Ericson AC, Braaf Y, Wallon C, Soderholm JD (2004) Increased antigen and bacterial uptake in follicle associated epithelium induced by chronic psychological stress in rats. Gut 53(4):494–500
Lizko NN, Silov VM, Syrych GD (1984) Events in he development of dysbacteriosis of the intestines in man under extreme conditions. Nahrung 28(6–7):599–605
Lyte M, Ernst S (1992) Catecholamine induced growth of gram negative bacteria. Life Sci 50(3):203–212
Santos J, Guilarte M, Alonso C, Malagelada JR (2005) Pathogenesis of irritable bowel syndrome: the mast cell connection. Scand J Gastroenterol 40(2):129–140
Eutamene H, Theodorou V, Fioramonti J, Bueno L (2003) Acute stress modulates the histamine content of mast cells in the gastrointestinal tract through interleukin-1 and corticotropin-releasing factor release in rats. J Physiol 553(Pt 3):959–966
Stead RH, Dixon MF, Bramwell NH, Riddell RH, Bienenstock J (1989) Mast cells are closely apposed to nerves in the human gastrointestinal mucosa. Gastroenterology 97(3):575–585
Barbara G, Stanghellini V, De GR, Cremon C, Cottrell GS, Santini D, Pasquinelli G, Morselli-Labate AM, Grady EF, Bunnett NW, Collins SM, Corinaldesi R (2004) Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology 126(3):693–702
Jiang W, Kreis ME, Eastwood C, Kirkup AJ, Humphrey PP, Grundy D (2000) 5-HT(3) and histamine H(1) receptors mediate afferent nerve sensitivity to intestinal anaphylaxis in rats. Gastroenterology 119(5):1267–1275
Barbara G, Wang B, Stanghellini V, De GR, Cremon C, Di NG, Trevisani M, Campi B, Geppetti P, Tonini M, Bunnett NW, Grundy D, Corinaldesi R (2007) Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 132(1):26–37
Vergnolle N (2005) Clinical relevance of proteinase activated receptors (pars) in the gut10. Gut 54(6):867–874
Sander LE, Lorentz A, Sellge G, Coeffier M, Neipp M, Veres T, Frieling T, Meier PN, Manns MP, Bischoff SC (2006) Selective expression of histamine receptors H1R, H2R, and H4R, but not H3R, in the human intestinal tract. Gut 55(4):498–504
Hauger RL, Grigoriadis DE, Dallman MF, Plotsky PM, Vale WW, Dautzenberg FM (2003) International Union of Pharmacology. XXXVI. Current status of the nomenclature for receptors for corticotropin-releasing factor and their ligands. Pharmacol Rev 55(1):21–26
Kiank C, Tache Y, Larauche M (2010) Stress-related modulation of inflammation in experimental models of bowel disease and post-infectious irritable bowel syndrome: role of corticotropin-releasing factor receptors. Brain Behav Immun 4(1):41–48
la Fleur SE, Wick EC, Idumalla PS, Grady EF, Bhargava A (2005) Role of peripheral corticotropin-releasing factor and urocortin II in intestinal inflammation and motility in terminal ileum. Proc Natl Acad Sci U S A 102(21):7647–7652
Kokkotou E, Torres D, Moss AC, O’Brien M, Grigoriadis DE, Karalis K, Pothoulakis C (2006) Corticotropin-releasing hormone receptor 2-deficient mice have reduced intestinal inflammatory responses. J Immunol 177(5):3355–3361
Moss AC, Anton P, Savidge T, Newman P, Cheifetz AS, Gay J, Paraschos S, Winter MW, Moyer MP, Karalis K, Kokkotou E, Pothoulakis C (2007) Urocortin II mediates pro-inflammatory effects in human colonocytes via corticotropin-releasing hormone receptor 2alpha 1. Gut 56(9):1210–1217
Saruta M, Takahashi K, Suzuki T, Torii A, Kawakami M, Sasano H (2004) Urocortin 1 in colonic mucosa in patients with ulcerative colitis. J Clin Endocrinol Metab 89(11):5352–5361
Delgado M, Pozo D, Ganea D (2004) The significance of vasoactive intestinal peptide in immunomodulation. Pharmacol Rev 56(2):249–290
Toumi F, Neunlist M, Denis MG, Oreshkova T, Laboisse CL, Galmiche JP, Jarry A (2004) Vasoactive intestinal peptide induces IL-8 production in human colonic epithelial cells via MAP kinase-dependent and PKA-independent pathways. Biochem Biophys Res Commun 317(1):187–191
Cao SG, Wu WC, Han Z, Wang MY (2005) Effects of psychological stress on small intestinal motility and expression of cholecystokinin and vasoactive intestinal polypeptide in plasma and small intestine in mice. World J Gastroenterol 11(5):737–740
Neunlist M, Toumi F, Oreschkova T, Denis M, Leborgne J, Laboisse CL, Galmiche JP, Jarry A (2003) Human ENS regulates the intestinal epithelial barrier permeability and a tight junction-associated protein ZO-1 via VIPergic pathways. Am J Physiol Gastrointest Liver Physiol 285(5):G1028–G1036
Koon HW, Pothoulakis C (2006) Immunomodulatory properties of substance P: the gastrointestinal system as a model. Ann N Y Acad Sci 1088:23–40
Wang L, Stanisz AM, Wershil BK, Galli SJ, Perdue MH (1995) Substance P induces ion secretion in mouse small intestine through effects on enteric nerves and mast cells. Am J Physiol 269(1 Pt 1):G85–G92
Zheng PY, Feng BS, Oluwole C, Struiksma S, Chen X, Li P, Tang SG, Yang PC (2009) Psychological stress induces eosinophils to produce corticotrophin releasing hormone in the intestine. Gut 58(11):1473–1479
Barreau F, Cartier C, Ferrier L, Fioramonti J, Bueno L (2004) Nerve growth factor mediates alterations of colonic sensitivity and mucosal barrier induced by neonatal stress in rats. Gastroenterology 127(2):524–534
Kobayashi H, Yamataka A, Fujimoto T, Lane GJ, Miyano T (1999) Mast cells and gut nerve development: implications for Hirschsprung’s disease and intestinal neuronal dysplasia. J Pediatr Surg 34(4):543–548
Theodosiou M, Rush RA, Zhou XF, Hu D, Walker JS, Tracey DJ (1999) Hyperalgesia due to nerve damage: role of nerve growth factor. Pain 81(3):245–255
Pearce FL, Thompson HL (1986) Some characteristics of histamine secretion from rat peritoneal mast cells stimulated with nerve growth factor. J Physiol 372:379–393
Marshall JS, Stead RH, McSharry C, Nielsen L, Bienenstock J (1990) The role of mast cell degranulation products in mast cell hyperplasia. I. Mechanism of action of nerve growth factor. J Immunol 144(5):1886–1892
Barreau F, Cartier C, Leveque M, Ferrier L, Moriez R, Laroute V, Rosztoczy A, Fioramonti J, Bueno L (2007) Pathways involved in gut mucosal barrier dysfunction induced in adult rats by maternal deprivation: corticotrophin-releasing factor and nerve growth factor interplay. J Physiol 580(Pt 1):347–356
Winston JH, Xu GY, Sarna SK (2010) Adrenergic stimulation mediates visceral hypersensitivity to colorectal distension following heterotypic chronic stress. Gastroenterology 138(1):294–304
Dahlman-Wright K, Cavailles V, Fuqua SA, Jordan VC, Katzenellenbogen JA, Korach KS, Maggi A, Muramatsu M, Parker MG, Gustafsson JA (2006) International Union of Pharmacology. LXIV. Estrogen receptors. Pharmacol Rev 58:773–781
Konstantinopoulos PA, Kominea A, Vandoros G, Sykiotis GP, Andricopoulos P, Varakis I, Sotiropoulou-Bonikou G, Papavassiliou AG (2003) Oestrogen receptor-β (ERβ) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour’s dedifferentiation. Eur J Cancer 39:1251–1258
Condliffe SB, Doolan CM, Harvey BJ (2001) 17beta-oestradiol acutely regulates Cl- secretion in rat distal colonic epithelium. J Physiol 530(Pt 1):47–54
Mayol JM, Arbeo-Escolar A, Alarma-Estrany P, Adame-Navarrete Y, Fernandez-Represa JA (2002) Progesterone inhibits chloride transport in human intestinal epithelial cells. World J Surg 26(6):652–656
Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, Warner M, Gustafsson JA (2006) Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci U S A 103:2959–2964
Braniste V, Leveque M, Buisson-Brenac C, Bueno L, Fioramonti J, Houdeau E (2009) Oestradiol decreases colonic permeability through oestrogen receptor beta-mediated up-regulation of occludin and junctional adhesion molecule-A in epithelial cells. J Physiol 587:3317–3328
Berkes J, Viswanathan VK, Savkovic SD, Hecht G (2003) Intestinal epithelial responses to enteric pathogens: effects on the tight junction barrier, ion transport, and inflammation. Gut 52(3):439–451
Wu Z, Nybom P, Magnusson KE (2000) Distinct effects of Vibrio cholera haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Microbiol 2(1):11–17
Fullner KJ, Lencer WI, Mekalanos JJ (2001) Vibrio cholerae-induced cellular responses of polarized T84 intestinal epithelial cells are dependent on production of cholera toxin and the RTX toxin. Infect Immun 69:6310–6317
Fasano A, Baudry B, Pumplin DW, Wasserman SS, Tall BD, Ketley JM, Kaper JB (1991) Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc Natl Acad Sci U S A 88(12):5242–5246
Fasano A, Fiorentini C, Donelli G, Uzzau S, Kaper JB, Margaretten K, Ding X, Guandalini S, Comstock L, Goldblum SE (1995) Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest 96(2):710–720
Hecht G, Pothoulakis C, LaMont JT, Madara JL (1988) Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 82(5):1516–1524
Hecht G, Koutsouris A, Pothoulakis C, LaMont JT, Madara JL (1992) Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 102(2):416–423
Chen M, Pothoulakis C, LaMont T (2002) Protein kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile toxin A. J Biol Chem 277:4247–4254
Katahira J, Sugiyama H, Inoue N, Horiguchi Y, Matsuda M, Sugimoto N (1997) Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem 272(42):26652–26658
McClane B (1994) Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 87:43–67
Simonovic I, Rosenberg J, Koutsouris A, Hecht G (2000) Enteropathogenic Escherichia coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol 2(4):305–315
Yuhan R, Koutsouris A, Savkovic SD, Hecht G (1997) Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 113(6):1873–1882
Lyte M (2013) Microbial endocrinology in the microbiome-gut-brain axis: how bacterial production and utilization of neurochemicals influence behavior. PLoS Pathog 9(11):e1003726
Cryan JF, O’Mahony SM (2011) The microbiome-gut-brain axis: from bowel to behavior. Neurogastroenterol Motil 23(3):187–192
Sekirov I, Russell SL, Antunes LC, Finlay BB (2010) Gut microbiota in health and disease. Physiol Rev 90(3):859–904
Qin HL, Shen TY, Gao ZG, Fan XB, Hang XM, Jiang YQ, Zhang HZ (2005) Effect of Lactobacillus on the gut microflora and barrier function of the rats with abdominal infection. World J Gastroenterol 11(17):2591–2596
Zyrek AA, Cichon C, Helms S, Enders C, Sonnenborn U, Schmidt MA (2007) Molecular mechanisms underlying the probiotic effects of Escherichia coli Nissle 1917 involve ZO-2 and PKCzeta redistribution resulting in tight junction and epithelial barrier repair. Cell Microbiol 9:804–816
Miyauchi E, O’Callaghan J, Buttó LF, Hurley G, Melgar S, Tanabe S, Shanahan F, Nally K, O’Toole PW (2012) Mechanism of protection of transepithelial barrier function by Lactobacillus salivarius: strain dependence and attenuation by bacteriocin production. Am J Physiol Gastrointest Liver Physiol 303(9):G1029–G1041
Patel RM, Myers LS, Kurundkar AR, Maheshwari A, Nusrat A, Lin PW (2012) Probiotic bacteria induce maturation of intestinal claudin 3 expression and barrier function. Am J Pathol 180(2):626–635
Lin PW, Nasr TR, Berardinelli AJ, Kumar A, Neish AS (2008) The probiotic Lactobacillus GG may augment intestinal host defense by regulating apoptosis and promoting cytoprotective responses in the developing murine gut. Pediatr Res 64(5):511–516
Ishii Y, Sugimoto S, Izawa N, Sone T, Chiba K, Miyazaki K. Oral administration of Bifidobacterium breve attenuates UV-induced barrier perturbation and oxidative stress in hairless mice skin. Arch Dermatol Res. 2014 Jan 11
Fujita R, Iimuro S, Shinozaki T, Sakamaki K, Uemura Y, Takeuchi A, Matsuyama Y, Ohashi Y (2013) Decreased duration of acute upper respiratory tract infections with daily intake of fermented milk: a multicenter, double-blinded, randomized comparative study in users of day care facilities for the elderly population. Am J Infect Control 41(12):1231–1235
Luoto R, Ruuskanen O, Waris M, Kalliomäki M, Salminen S, Isolauri E (2013) Prebiotic and probiotic supplementation prevents rhinovirus infections in preterm infants: a randomized, placebo-controlled trial. J Allergy Clin Immunol S0091–6749(13):01307–01309
Dicksved J, Schreiber O, Willing B, Petersson J, Rang S, Phillipson M, Holm L, Roos S (2012) Lactobacillus reuteri maintains a functional mucosal barrier during DSS treatment despite mucus layer dysfunction. PLoS One 7(9):e46399
Ukena SN, Singh A, Dringenberg U, Engelhardt R, Seidler U, Hansen W, Bleich A, Bruder D, Franzke A, Rogler G, Suerbaum S, Buer J, Gunzer F, Westendorf AM (2007) Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS One 2(12):e1308
Toumi R, Abdelouhab K, Rafa H, Soufli I, Raissi-Kerboua D, Djeraba Z, Touil-Boukoffa C (2013) Beneficial role of the probiotic mixture Ultrabiotique on maintaining the integrity of intestinal mucosal barrier in DSS-induced experimental colitis. Immunopharmacol Immunotoxicol 35(3):403–409
Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N, Bruewer M (2009) Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol 296(5):G1140–G1149
Persborn M, Gerritsen J, Wallon C, Carlsson A, Akkermans LM, Söderholm JD (2013) The effects of probiotics on barrier function and mucosal pouch microbiota during maintenance treatment for severe pouchitis in patients with ulcerative colitis. Aliment Pharmacol Ther 38(7):772–783
Ait-Belgnaoui A, Durand H, Cartier C, Chaumaz G, Eutamene H, Ferrier L, Houdeau E, Fioramonti J, Bueno L, Theodorou V (2012) Prevention of gut leakiness by a probiotic treatment leads to attenuated HPA response to an acute psychological stress in rats. Psychoneuroendocrinology 37(11):1885–1895
Palomar MM, Maldonado Galdeano C, Perdigón G (2014) Influence of a probiotic Lactobacillus strain on the intestinal ecosystem in a stress model mouse. Brain Behav Immun 35:77–85
Agostini S, Goubern M, Tondereau V, Salvador-Cartier C, Bezirard V, Lévèque M, Keränen H, Theodorou V, Bourdu-Naturel S, Goupil-Feuillerat N, Legrain-Raspaud S, Eutamene H (2012) A marketed fermented dairy product containing Bifidobacterium lactis CNCM I-2494 suppresses gut hypersensitivity and colonic barrier disruption induced by acute stress in rats. Neurogastroenterol Motil 24(4):376–e172
Whelan K, Quigley EM (2013) Probiotics in the management of irritable bowel syndrome and inflammatory bowel disease. Curr Opin Gastroenterol 29(2):184–189
Cryan JF, Dinan TG (2012) Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13(10):701–712
Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, Patterson PH, Mazmanian SK (2013) Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155(7):1451–1463
Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, Mrsny RJ, Turner JR (2002) A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 123(1):163–172
Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR (2005) Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol 166(2):409–419
Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, Schulzke JD (2000) Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci 113(Pt 11):2085–2090
Barmeyer C, Amasheh S, Tavalali S, Mankertz J, Zeitz M, Fromm M, Schulzke JD (2004) IL-1beta and TNFalpha regulate sodium absorption in rat distal colon. Biochem Biophys Res Commun 317(2):500–507
Clayburgh DR, Musch MW, Leitges M, Fu YX, Turner JR (2006) Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest 116(10):2682–2694
Zünd G, Madara JL, Dzus AL, Awtrey CS, Colgan SP (1996) Interleukin-4 and interleukin-13 differentially regulate epithelial chloride secretion. J Biol Chem 271(13):7460–7464
Ceponis PJ, Botelho F, Richards CD, McKay DM (2000) Interleukins 4 and 13 increase intestinal epithelial permeability by a phosphatidylinositol 3-kinase pathway. Lack of evidence for STAT 6 involvement. J Biol Chem 275(37):29132–29137
Madsen KL, Lewis SA, Tavernini MM, Hibbard J, Fedorak RN (1997) Interleukin 10 prevents cytokine-induced disruption of T84 monolayer barrier integrity and limits chloride secretion. Gastroenterology 113(1):151–159
Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN (1999) Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis 5(4):262–270
Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W (1993) Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75(2):263–274
Blaschitz C, Raffatellu M (2010) Th17 cytokines and the gut mucosal barrier. J Clin Immunol 30(2):196–203
Al-Sadi R, Boivin M, Ma T (2009) Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 14:2765–2778
Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, Rallabhandi P, Shea-Donohue T, Tamiz A, Alkan S, Netzel-Arnett S, Antalis T, Vogel SN, Fasano A (2008) Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology 135(1):194–204
Wang W, Uzzau S, Goldblum SE, Fasano A (2000) Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci 113(Pt 24):4435–4440
Ventura MT, Polimeno L, Amoruso AC, Gatti F, Annoscia E, Marinaro M, Di Leo E, Matino MG, Buquicchio R, Bonini S, Tursi A, Francavilla A (2006) Intestinal permeability in patients with adverse reactions to food. Dig Liver Dis 38(10):732–736
Ozdemir O, Arrey-Mensah A, Sorensen RU (2006) Development of multiple food allergies in children taking tacrolimus after heart and liver transplantation. Pediatr Transplant 10(3):380–383
Finnie IA, Dwarakanath AD, Taylor BA, Rhodes JM (1995) Colonic mucin synthesis is increased by sodium butyrate. Gut 36:93–99
Ma X, Fan PX, Li LS, Qiao SY, Zhang GL, Li DF (2012) Butyrate promotes the recovering of intestinal wound healing through its positive effect on the tight junctions. J Anim Sci 90(Suppl 4):266–268
Li N, Lewis P, Samuelson D, Liboni K, Neu J (2004) Glutamine regulates Caco-2 cell tight junction proteins. Am J Physiol Gastrointest Liver Physiol 287(3):G726–G733
Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N (1999) Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol 276:G965–G974
Atkinson KJ, Rao RK (2001) Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am J Physiol 280:G1280–G1288
Ferrier L, Bérard F, Debrauwer L, Chabo C, Langella P, Buéno L, Fioramonti J (2006) Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol 168(4):1148–1154
Tong J, Wang Y, Chang B, Zhang D, Liu P, Wang B (2013) Activation of RhoA in alcohol-induced intestinal barrier dysfunction. Inflammation 36(3):750–758
Rendon JL, Li X, Akhtar S, Choudhry MA (2013) Interleukin-22 modulates gut epithelial and immune barrier functions following acute alcohol exposure and burn injury. Shock 39(1):11–18
Oshima T, Miwa H, Joh T (2008) Aspirin induces gastric epithelial barrier dysfunction by activating p38 MAPK via claudin-7. Am J Physiol Cell Physiol 295(3):C800–C806
Silver K, Leloup L, Freeman LC, Wells A, Lillich JD (2010) Non-steroidal anti-inflammatory drugs inhibit calpain activity and membrane localization of calpain 2 protease. Int J Biochem Cell Biol 42(12):2030–2036
Tanaka A, Kunikata T, Mizoguchi H, Kato S, Takeuchi K (1999) Dual action of nitric oxide in pathogenesis of indomethacin-induced small intestinal ulceration in rats. J Physiol Pharmacol 50(3):405–417
Vaziri ND, Yuan J, Norris K (2013) Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am J Nephrol 37(1):1–6
Watts T, Berti I, Sapone A, Gerarduzzi T, Not T, Zielke R, Fasano A (2005) Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U S A 102(8):2916–2921
Sakisaka S, Kawaguchi T, Taniguchi E, Hanada S, Sasatomi K, Koga H, Harada M, Kimura R, Sata M, Sawada N, Mori M, Todo S, Kurohiji T (2001) Alterations in tight junctions differ between primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 33(6):1460–1468
Pijls KE, Jonkers DM, Elamin EE, Masclee AA, Koek GH (2013) Intestinal epithelial barrier function in liver cirrhosis: an extensive review of the literature. Liver Int 33(10):1457–1469
Rao RK, Seth A, Sheth P (2004) Recent advances in alcoholic liver disease I. Role of intestinal permeability and endotoxemia in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 286(6):G881–G884
Rebuffat SA, Kammoun-Krichen M, Charfeddine I, Ayadi H, Bougacha-Elleuch N, Peraldi-Roux S (2013) IL-1β and TSH disturb thyroid epithelium integrity in autoimmune thyroid diseases. Immunobiology 218(3):285–291
Peng SN, Zeng HH, Fu AX, Chen XW, Zhu QX (2013) Effects of rhein on intestinal epithelial tight junction in IgA nephropathy. World J Gastroenterol 19(26):4137–4145
Li Y, Guo M, Shen J, Zheng L, Wang J, Wang P, Li J (2014) Limited Fluid Resuscitation Attenuates lung and intestine injury caused by hemorrhagic shock in rats. J Invest Surg 27:81–87 (Epub 2013 Oct 2)
Fishman JE, Levy G, Alli V, Sheth S, Lu Q, Deitch EA (2013) Oxidative modification of the intestinal mucus layer is a critical but unrecognized component of trauma hemorrhagic shock-induced gut barrier failure. Am J Physiol Gastrointest Liver Physiol 304(1):G57–G63
Doucet D, Badami C, Palange D, Bonitz RP, Lu Q, Xu DZ, Kannan KB, Colorado I, Feinman R, Deitch EA (2010) Estrogen receptor hormone agonists limit trauma hemorrhage shock-induced gut and lung injury in rats. PLoS One 5(2):e9421
Feinman R, Deitch EA, Watkins AC, Abungu B, Colorado I, Kannan KB, Sheth SU, Caputo FJ, Lu Q, Ramanathan M, Attan S, Badami CD, Doucet D, Barlos D, Bosch-Marce M, Semenza GL, Xu DZ (2010) HIF-1 mediates pathogenic inflammatory responses to intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 299(4):G833–G843
Spindler-Vesel A, Wraber B, Vovk I, Kompan L (2006) Intestinal permeability and cytokine inflammatory response in multiply injured patients. J Interferon Cytokine Res 26(10):771–776
Chen C, Wang P, Su Q, Wang S, Wang F (2012) Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS One 7(4):e34946
Costantini TW, Peterson CY, Kroll L, Loomis WH, Eliceiri BP, Baird A, Bansal V, Coimbra R (2009) Role of p38 MAPK in burn-induced intestinal barrier breakdown. J Surg Res 156(1):64–69
Peterson CY, Costantini TW, Loomis WH, Putnam JG, Wolf P, Bansal V, Eliceiri BP, Baird A, Coimbra R (2010) Toll-like receptor-4 mediates intestinal barrier breakdown after thermal injury. Surg Infect (Larchmt) 11(2):137–144
Mertz H, Morgan V, Tanner G, Pickens D, Price R, Shyr Y, Kessler R (2000) Regional cerebral activation in irritable bowel syndrome and control subjects with painful and nonpainful rectal distention. Gastroenterology 118(5):842–848
Drossman DA, Ringel Y, Vogt BA, Leserman J, Lin W, Smith JK, Whitehead W (2003) Alterations of brain activity associated with resolution of emotional distress and pain in a case of severe irritable bowel syndrome. Gastroenterology 124(3):754–761
Barreau F, Ferrier L, Fioramonti J, Bueno L (2007) New insights in the etiology and pathophysiology of irritable bowel syndrome: contribution of neonatal stress models. Pediatr Res 62(3):240–245
O’Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho AM, Quigley EM, Cryan JF, Dinan TG (2009) Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry 65(3):263–267
Klooker TK, Braak B, Painter RC, de Rooij SR, van Elburg RM, van den Wijngaard RM, Roseboom TJ, Boeckxstaens GE (2009) Exposure to severe wartime conditions in early life is associated with an increased risk of irritable bowel syndrome: a population-based cohort study. Am J Gastroenterol 104(9):2250–2256
Koloski NA, Talley NJ, Boyce PM (2005) A history of abuse in community subjects with irritable bowel syndrome and functional dyspepsia: the role of other psychosocial variables. Digestion 72(2–3):86–96
Murray CD, Flynn J, Ratcliffe L, Jacyna MR, Kamm MA, Emmanuel AV (2004) Effect of acute physical and psychological stress on gut autonomic innervation in irritable bowel syndrome. Gastroenterology 127(6):1695–1703
Kellow JE, Langeluddecke PM, Eckersley GM, Jones MP, Tennant CC (1992) Effects of acute psychologic stress on small-intestinal motility in health and the irritable bowel syndrome. Scand J Gastroenterol 27(1):53–58
Vanheel H, Vicario M, Vanuytsel T, Van Oudenhove L, Martinez C, Keita AV, Pardon N, Santos J, Söderholm JD, Tack J, Farré R (2014) Impaired duodenal mucosal integrity and low-grade inflammation in functional dyspepsia. Gut 63:262–271 (Epub 2013 Mar 8)
Pike BL, Porter CK, Sorrell TJ, Riddle MS (2013) Acute gastroenteritis and the risk of functional dyspepsia: a systematic review and meta-analysis. Am J Gastroenterol 108(10):1558–1563
Van Oudenhove L, Aziz Q (2013) The role of psychosocial factors and psychiatric disorders in functional dyspepsia. Nat Rev Gastroenterol Hepatol 10(3):158–167
Van Oudenhove L, Vandenberghe J, Vos R, Fischler B, Demyttenaere K, Tack J (2011) Abuse history, depression, and somatization are associated with gastric sensitivity and gastric emptying in functional dyspepsia. Psychosom Med 73(8):648–655
Stout C, Snyder RL (1969) Ulcerative colitis-like lesion in Siamang gibbons. Gastroenterology 57(3):256–261
Barreau F, Ferrier L, Fioramonti J, Bueno L (2004) Neonatal maternal deprivation triggers long term alterations in colonic epithelial barrier and mucosal immunity in rats. Gut 53(4):501–506
Collins SM, McHugh K, Jacobson K, Khan I, Riddell R, Murase K, Weingarten HP (1996) Previous inflammation alters the response of the rat colon to stress. Gastroenterology 111(6):1509–1515
Qiu BS, Vallance BA, Blennerhassett PA, Collins SM (1999) The role of CD4+ lymphocytes in the susceptibility of mice to stress-induced reactivation of experimental colitis. Nat Med 5(10):1178–1182
Bitton A, Sewitch MJ, Peppercorn MA, deB Edwardes MD, Shah S, Ransil B, Locke SE (2003) Psychosocial determinants of relapse in ulcerative colitis: a longitudinal study. Am J Gastroenterol 98(10):2203–2208
Levenstein S, Prantera C, Varvo V, Scribano ML, Andreoli A, Luzi C, Arca M, Berto E, Milite G, Marcheggiano A (2000) Stress and exacerbation in ulcerative colitis: a prospective study of patients enrolled in remission. Am J Gastroenterol 95(5):1213–1220
Buhner S, Buning C, Genschel J, Kling K, Herrmann D, Dignass A, Kuechler I, Krueger S, Schmidt HH, Lochs H (2006) Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut 55(3):342–347
D’Inca R, Annese V, Di Leo V, Latiano A, Quaino V, Abazia C, Vettorato MG, Sturniolo GC (2006) Increased intestinal permeability and NOD2 variants in familial and sporadic Crohn’s disease. Aliment Pharmacol Ther 23(10):1455–1461
Peeters M, Geypens B, Claus D, Nevens H, Ghoos Y, Verbeke G, Baert F, Vermeire S, Vlietinck R, Rutgeerts P (1997) Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology 113(3):802–807
D’Inca R, Di Leo V, Corrao G, Martines D, D’Odorico A, Mestriner C, Venturi C, Longo G, Sturniolo GC (1999) Intestinal permeability test as a predictor of clinical course in Crohn’s disease. Am J Gastroenterol 94(10):2956–2960
Jorgensen J, Ranlov PJ, Bjerrum PJ, Diemer H, Bisgaard K, Elsborg L (2001) Is an increased intestinal permeability a valid predictor of relapse in Crohn disease? Scand J Gastroenterol 36(5):521–527
Langhorst J, Cobelens PM, Kavelaars A, Heijnen CJ, Benson S, Rifaie N, Dobos GJ, Schedlowski M, Elsenbruch S (2007) Stress-related peripheral neuroendocrine-immune interactions in women with ulcerative colitis. Psychoneuroendocrinology 32(8–10):1086–1096
Bekkering P, Jafri I, van Overveld FJ, Rijkers GT (2013) The intricate association between gut microbiota and development of Type 1, Type 2 and Type 3 diabetes. Expert Rev Clin Immunol 9(11):1031–1041
Summa KC, Voigt RM, Forsyth CB, Shaikh M, Cavanaugh K, Tang Y, Vitaterna MH, Song S, Turek FW, Keshavarzian A (2013) Disruption of the circadian clock in mice increases intestinal permeability and promotes alcohol-induced hepatic pathology and inflammation. PLoS One 8(6):e67102
Fetissov SO, Déchelotte P (2011) The new link between gut-brain axis and neuropsychiatric disorders. Curr Opin Clin Nutr Metab Care 14(5):477–482
Heberling CA, Dhurjati PS, Sasser M (2013) Hypothesis for a systems connectivity model of autism spectrum disorder pathogenesis: links to gut bacteria, oxidative stress, and intestinal permeability. Med Hypotheses 80(3):264–270
Maes M, Coucke F, Leunis JC (2007) Normalization of the increased translocation of endotoxin from gram negative enterobacteria (leaky gut) is accompanied by a remission of chronic fatigue syndrome. Neuro Endocrinol Lett 28(6):739–744
Goebel A, Buhner S, Schedel R, Lochs H, Sprotte G (2008) Altered intestinal permeability in patients with primary fibromyalgia and in patients with complex regional pain syndrome. Rheumatology (Oxford) 47(8):1223–1227
Hadjiyanni I, Li KK, Drucker DJ (2009) Glucagon-like peptide-2 reduces intestinal permeability but does not modify the onset of type 1 diabetes in the nonobese diabetic mouse. Endocrinology 150(2):592–599
Moran GW, O’Neill C, McLaughlin JT (2012) GLP-2 enhances barrier formation and attenuates TNFα-induced changes in a Caco-2 cell model of the intestinal barrier. Regul Pept 178(1–3):95–101
Tang ZF, Ling YB, Lin N, Hao Z, Xu RY (2007) Glutamine and recombinant human growth hormone protect intestinal barrier function following portal hypertension surgery. World J Gastroenterol 13(15):2223–2228
Yi C, Cao Y, Wang SR, Xu YZ, Huang H, Cui YX, Huang Y (2007) Beneficial effect of recombinant human growth hormone on the intestinal mucosa barrier of septic rats. Braz J Med Biol Res 40(1):41–48
Lorenzo-Zúñiga V, Rodríguez-Ortigosa CM, Bartolí R, Martínez-Chantar ML, Martínez-Peralta L, Pardo A, Ojanguren I, Quiroga J, Planas R, Prieto J (2006) Insulin-like growth factor I improves intestinal barrier function in cirrhotic rats. Gut 55(9):1306–1312
Dong CX, Zhao W, Solomon C, Rowland KJ, Ackerley C, Robine S, Holzenberger M, Gonska T, Brubaker PL (2014) The intestinal epithelial insulin-like growth factor-1 receptor links glucagon-like peptide-2 action to gut barrier function. Endocrinology 155:370–379 (Epub 2013 Nov 21)
Bansal V, Ryu SY, Blow C, Costantini T, Loomis W, Eliceiri B, Baird A, Wolf P, Coimbra R (2010) The hormone ghrelin prevents traumatic brain injury induced intestinal dysfunction. J Neurotrauma 27(12):2255–2260
Bettenworth D, Buyse M, Böhm M, Mennigen R, Czorniak I, Kannengiesser K, Brzoska T, Luger TA, Kucharzik T, Domschke W, Maaser C, Lügering A (2011) The tripeptide KdPT protects from intestinal inflammation and maintains intestinal barrier function. Am J Pathol 179(3):1230–1242
Hamada K, Shitara Y, Sekine S, Horie T (2010) Zonula Occludens-1 alterations and enhanced intestinal permeability in methotrexate-treated rats. Cancer Chemother Pharmacol 66(6):1031–1038
Jung S, Fehr S, Harder-d’Heureuse J, Wiedenmann B, Dignass AU (2001) Corticosteroids impair intestinal epithelial wound repair mechanisms in vitro. Scand J Gastroenterol 36(9):963–970
Hopkins AM, McDonnell C, Breslin NP, O’Morain CA, Baird AW (2002) Omeprazole increases permeability across isolated rat gastric mucosa pre-treated with an acid secretagogue. J Pharm Pharmacol 54(3):341–347
Yang J, Liu KX, Qu JM, Wang XD (2013) The changes induced by cyclophosphamide in intestinal barrier and microflora in mice. Eur J Pharmacol 714(1–3):120–124
Lee SE, Choi Y, Kim SE, Noh EB, Kim SC (2013) Differential effects of topical corticosteroid and calcineurin inhibitor on the epidermal tight junction. Exp Dermatol 22(1):59–61
Gabe SM, Bjarnason I, Tolou-Ghamari Z, Tredger JM, Johnson PG, Barclay GR, Williams R, Silk DB (1998) The effect of tacrolimus (FK506) on intestinal barrier function and cellular energy production in humans. Gastroenterology 115(1):67–74
Kong J, Zhang Z, Musch MW, Ning G, Sun J, Hart J et al (2008) Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol 294:G208–G216
Paterson BM, Lammers KM, Arrieta MC, Fasano A, Meddings JB (2007) The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther 26:757–766
Kelly CP, Green PH, Murray JA, Dimarino A, Colatrella A, Leffler DA, Alexander T, Arsenescu R, Leon F, Jiang JG, Arterburn LA, Paterson BM, Fedorak RN, Larazotide Acetate Celiac Disease Study Group (2013) Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther 37(2):252–262
Gopalakrishnan S, Tripathi A, Tamiz AP, Alkan SS, Pandey NB (2012) Larazotide acetate promotes tight junction assembly in epithelial cells. Peptides 35(1):95–101
Gopalakrishnan S, Durai M, Kitchens K, Tamiz AP, Somerville R, Ginski M, Paterson BM, Murray JA, Verdu EF, Alkan SS, Pandey NB (2012) Larazotide acetate regulates epithelial tight junctions in vitro and in vivo. Peptides 35(1):86–94
Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G et al (2002) Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol 97:2000–2004
Bode L, Salvestrini C, Park PW, Li JP, Esko JD, Yamaguchi Y et al (2008) Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest 118:229–238
Acknowledgements
Supported in part by the Fondo de Investigación Sanitaria and Ciberehd, Instituto Carlos III, Subdirección General de Investigación Sanitaria, Ministerio de Ciencia e Innovación (PI12/00314, Alonso, C.; CM08/00229, Lobo, B; CM10/00155, Pigrau, M; CP10/00502, PI13/00935, Vicario, M; PI11/00716 & CB06/04/0021, Santos, J.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer New York
About this chapter
Cite this chapter
Alonso, C., Vicario, M., Pigrau, M., Lobo, B., Santos, J. (2014). Intestinal Barrier Function and the Brain-Gut Axis. In: Lyte, M., Cryan, J. (eds) Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. Advances in Experimental Medicine and Biology(), vol 817. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0897-4_4
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0897-4_4
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0896-7
Online ISBN: 978-1-4939-0897-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)