
Abstract
Schizophrenia is a complex and unique disorder with processes of neurodevelopment and neurodegeneration. This is one of the most severe and chronic forms of mental disorders, and it affects roughly 1 % of the world’s population. Despite the recent advances, the molecular mechanisms underlying schizophrenia remain unclear. There is accumulating evidence for oxidative stress mechanisms as common pathophysiological pathways in this disorder. Specific markers of oxidative stress as a sensor of oxidation associated with schizophrenia might provide a key link connecting mechanisms underlying numerous processes described in schizophrenia. Mitochondrial dysfunction triggers extensive generation of free radicals leading to oxidative/nitrative stress, abnormalities, and pathologic consequences. Patients with schizophrenia present an increase in oxidative damage to lipids, proteins, and DNA, in both central and peripheral tissues with deficits of antioxidant defense, especially deficit of glutathione. The chapter presents biomarkers of oxidative damage and the role of oxidative stress in numerous abnormalities in biochemical pathways including apoptosis in the pathophysiology of schizophrenia. At present, no single theory provides all aspects of abnormalities in the disease. The development of new investigative techniques, especially neuroimaging, and studies of apoptotic pathway seem to prove neurodegenerative and neurodevelopmental theories; oxidative/nitrative damage may integrate and support basic mechanisms of neurodevelopment and neurodegeneration processes in schizophrenia.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Schizophrenia is one of the most severe and chronic forms of mental disorders with to date unknown pathophysiology and aetiology, and it affects roughly 1 % of the world’s population (Saha et al. 2007). At present, it is fully recognized as a multidimensional illness, with a profound impact on behavior, perception, thinking, emotions, neurocognition, and social function (O’Leary et al. 2000). A complex interaction between genetic and environmental factors appears to be critical to the pathogenesis of schizophrenia (van Os and Murray 2008), and data from many studies progressively contribute to a characterization of its mechanisms. The disorder is characterized by chronic, often recurrent course, and serious deterioration in cognitive and psychosocial functioning occurs in most patients, as early as during the first 5 years (Tamminga and Holcomb 2005).
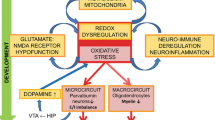
The etiopathogenesis of this illness due to heterogeneity of patient population, different symptoms, difficulties in diagnosis particularly in the early stage of the disease, long-term treatment, and various side effects is difficult to study. Neurodevelopmental (Murray and Lewis 1987; Lewis and Levitt 2002; Weinberger 1986, 1987), neurodegenerative (Lieberman 1999; Rund 2009), immunological (Kinney et al. 2010; Kliushnik et al. 2009; Strous et al. 2009; O’Donnell 2012), inflammatory (Covelli et al. 2003; Hanson and Gottesman 2005), infectious (Babulas et al. 2006; Brown et al. 2009; Yolken and Torrey 1995; Yolken et al. 2000), and metabolic (De Hert et al. 2009; Fan et al. 2010) hypotheses and various pathophysiological mechanisms have been proposed to explain the etiopathogenesis of schizophrenia. Molecular mechanisms, leading to oxidative stress, involved in the pathophysiology of schizophrenia are intricate and not yet fully explained. This diversity reflects the considerable neurobiological heterogeneity and complexity of clinical syndromes of schizophrenia. The implication of oxidative stress in inflammatory process, neurodegeneration, and neurodevelopment is emerging as an important mechanism underlying numerous pathological processes. This disorder is increasingly regarded as the result of pathological alterations in architecture and function of the brain circuits supporting perceptual, cognitive, and emotional processes (Insel 2009; Lewis and Sweet 2009; Palop et al. 2006) leading to disturbances of neural coordination within these networks. The current psychiatric classification systems, the International Statistical Classification of Diseases and Related Health Problems, 10th Revision (ICD-10; WHO 2008), and the Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV; APA 2000), are based solely on the diagnosis of schizophrenia dependent on its clinical symptomatology since there is a lack of specific biological markers. The identification of biological, clinical, or biochemical markers for schizophrenia could facilitate the understanding of its etiopathogenesis as well as its diagnosis. It is important to determine clear biological markers, which could accurately help to predict clinical outcome of schizophrenia and identify at-risk individuals in preclinical stages of the disorder (Schwarz and Bahn 2008). Thus, the search for effective biological markers for schizophrenia seems to be a main challenge for neuroscientific and psychiatric researches (Lakhan et al. 2010). Recently, markers of oxidative stress have been taken into account. Biochemical alterations in the brain, especially the dopamine system with free radical production and oxidative damage to the brain structure, might be partly responsible for the pathogenesis of this heterogeneous disease. Oxidative stress is involved in different intricate mechanisms of the disease and might contribute to the explanation of various pathological processes integrating some etiopathogenetic hypotheses about schizophrenia.
2 ROS Generation in Schizophrenic Patients
Imbalance between the pro- and anti-oxidative processes is a physiological phenomenon used in many important functions of the body, and this imbalance with a predominance of oxidation, defined as oxidative stress, may be involved in the pathogenesis of many diseases including schizophrenia. Oxidative stress causes changes in the function of several cellular components of the central nervous system (CNS) and is involved in inflammatory and neurodegenerative processes (Ng et al. 2008; Pedrini et al. 2012; Lin and Beal 2006; Halliwel 2006). The brain with its high metabolic rate is particularly vulnerable to oxidative damage (Halliwell 1992). The impairment of redox mechanisms in the brain manifested as an imbalance in the generation and scavenging of reactive oxygen species (ROS) and nitrogen species (RNS) and altered regulation of these fundamental redox mechanisms leading to oxidative stress are involved in the brain pathology in schizophrenia and can contribute to the pathogenesis of the disorder. Moreover, cognitive dysfunction in schizophrenia is accompanied by the changed redox mechanisms (Bitanihirwe and Woo 2011; Zhang et al. 2012). The brain, rich in essential polyunsaturated fatty acids (EPUFAs), is particularly susceptible to damage caused by free radicals, has a high demand for oxygen (20 % of the total oxygen consumed by the body), and produces large amounts of ROS (Halliwell and Gutteridge 1996). ROS are produced in mitochondria as the leak of electrons from the electron transport chain; oxidative phosphorylation in the mitochondrial electron transport chain is highly effective. Incomplete reduction in oxygen during the respiration produces superoxide anion (O2.−) that is enzymatically dismutated by superoxide dismutase (SOD) to hydrogen peroxide (H2O2). Hydroxyl radicals, in turn, are formed via Fenton reaction (Halliwell 1992). Superoxide anion (O2.−) of these three generated ROS is one of the most reactive and controlled via multiple enzyme systems: SOD, CAT, GSH transferase, and thioredoxin. NADPH oxidase (NOX2) and xanthine oxidase are also involved in the production of free radicals (Halliwell 1992; Halliwel et al. 2006).
In mitochondria (outer membranes), enzymatic oxidation of biogenic amines induced by monoamine oxidase (MAO) generates H2O2; dopamine as a substrate for MAO is also a source of ROS. Dopamine is a modulator of oxidative status; it can undergo autoxidation to generate ROS (Dalla Libera et al. 1996; Masserano et al. 2000; Grima et al. 2003; Bošković et al. 2011). During the mitochondrial respiration, the increased Ca ion influx also activates neuronal NO synthase (nNOS) to generate NO, which has been reported to regulate mitochondrial function. NMDA is also engaged in ROS production (O2 −). The activation of NMDA receptors in neurons and subsequent Ca ion influx can induce NO generation by nNOS (Nakamura and Lipton 2011). NO, in turn, inhibits the activity of NMDA receptor via S-nitrosylation (Lipton et al. 1993). The malfunction of NMDA receptors may have effect on the redox status in patients with schizophrenia. Damage to mitochondria in brain cells of schizophrenic individuals is connected with the impairment of oxidative phosphorylation and increased ROS generation (Ben-Shachar et al. 1999; Ben-Shachar 2002; Prabakaran et al. 2004; Clay et al. 2011; Martins-de-Souza et al. 2011). Approximately up to 50 % of ROS in the CNS derive from mitochondria. Especially neurons are sensitive to mitochondrial defects because they require high levels of energy (ATP) for maintenance of synapses, survival, and their specialized functions (Nakanishi and Wu 2009).
The impaired redox regulation caused by genetic and environmental factors, with the alterations of antioxidant defense system and oxidative stress in the brains of schizophrenic patients, is associated with various pathophysiological processes including mitochondrial dysfunction, inflammation, epigenetic changes, impairment of cell signaling, hypoactive NMDA receptors, or impairment of GABA interneurons (Benes and Berretta 2001; Reynolds et al. 2004; Picchioni and Murray 2007; Ben Sachar et al. 2002; Nakazawa et al. 2012). Schizophrenia is characterized by mitochondrial dysfunction (Ben-Shachar and Karry 2008) with oxidative damage to the mitochondrial membrane (Yao et al. 2001) and the mitochondrial electron transport chain dysfunction (Ben-Shachar 2002; Prabakaran et al. 2004). Dopamine may also impair mitochondrial membrane potential (Elkashef et al. 2002). The differences between schizophrenic and normal brains seem to be related partly to mitochondrial dysfunction and oxidative changes (Dror et al. 2002). In mitochondria, the toxic hydrogen peroxide is generated from the enzymatically dismutated O2 − by SOD, and glutathione is oxidized to glutathione disulfide (GSSG) (Dror et al. 2002). Mitochondria do not synthetize GSH and the low GSH level contributes to mitochondrial function impairment and oxidative damage (Griffith and Meister 1985). Recently, Seybolt (2010) has suggested that adjunctive use of alpha lipoic acid (its reduced form dihydrolipoic acid) and niacinamide as components of coenzymes NAD and NADP in the treatment of schizophrenia due to their properties to reduce ROS/RNS and improve the GSH/GSSG ratio could help alleviate mitochondrial dysfunction, reduce oxidative stress, and improve psychiatric symptoms in schizophrenia. S-nitrosylation and subsequent further oxidation of critical cysteine residues can also lead to mitochondrial dysfunction (Nakamura and Lipton 2011). Reduced level of GSH was observed in plasma of patients with schizophrenia (Dietrich-Muszalska et al. 2009c). GSH is the brain’s dominant antioxidant implicated in the pathophysiology of schizophrenia. An increased risk of schizophrenia is linked with polymorphisms of genes associated with GSH synthesis (Saadat et al. 2007; Tosic et al. 2006). In some brain structures, the presence of significant quantities of transition metal ions – iron (Fe), copper (Cu), and manganese (Mn) – can also contribute to the formation of ROS (Halliwell 1992). Free radical-induced membrane lipid/phospholipid peroxidation may cause damage to the cell membrane, due to changes in its biophysical properties, such as fluidity, and inactivation of receptors/enzymes associated with the membrane (Perluigi et al. 2012). These processes, in turn, may lead to disturbances in the physiological function of cells, especially neurons. Neurons are particularly sensitive to oxidative stress, and therefore, the efficient antioxidant defense system plays an important role in their normal functioning. The low antioxidant defense system observed in schizophrenia seems to be responsible in part for oxidative stress in this disorder (Yao et al. 1998a; Yao and Keshavan 2011a).
3 Markers of Oxidative and Nitrative Stress in Schizophrenia: Antioxidants
Multidimensional data support the fact that redox status in schizophrenia can be determined by the level of specific biomarkers of oxidative stress and the level of antioxidants (Lakhan and Kramer 2009). Data for the brain redox status are limited and contradictory. The majority of evidence for oxidative stress in schizophrenia is mostly received peripherally by the assessment of markers in plasma/serum or blood cells. Direct evidence of oxidative stress, especially when tardive dyskinesia occurred, has been obtained in animal models (Harrison 1999). The functional and biochemical consequences of oxidative stress were studied using animal models and postmortem brain analysis, but most measurements of oxidative stress in schizophrenia are in peripheral cells and fluids. They reflect redox processes occurring in the brain. Common markers used to assess the extent of oxidative/nitrative stress in schizophrenic patients are the products of oxidized and changed biomolecules – lipids, proteins, and nucleic acids – and also the activities of antioxidant defense system. The increased level of lipid peroxidation products measured commonly as TBARS or MDA, 4-hydroxynonenal, and isoprostanes (see Table 1), altered proteins and amino acids (estimated as the level of generated carbonyl groups or protein peroxides, 3-nitrotyrosine) (Dietrich-Muszalska and Olas 2009a, b), and the presence of DNA damage products (8-hydroxyguanosine, telomere shortening) (Miller et al. 2011; Jorgensen et al. 2013; Malaspina et al. 2001, 2014) as well as reduced antioxidant defense systems (Yao and Reddy 2011) are observed as the multiple pathophysiological consequences of increased toxicity of oxidative stress. Carbonyl groups formed by the oxidation of proteins may be readily estimated by an immunoassay technique (ELISA), but they seem not to be specific markers, contrary to 3-nitrotyrosine which is a specific and sensitive marker of protein alteration caused by nitrative stress.
The antioxidant defense system consists of antioxidant enzymes (superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), thioredoxin (Trx), and numerous nonenzymatic antioxidants). Changes in the activity of antioxidant enzymes and in total antioxidant status are presented in Tables 2 and 3. The individual antioxidants may act cooperatively and protect the biomolecules against oxidative damage.
To evaluate the antioxidant defense system in plasma reflecting the total activity of all antioxidants present in plasma, the total antioxidant status (TAS) should be measured together with the estimation of individual antioxidants. TAS depends mainly on a high concentration of albumin in plasma and the presence of ascorbic and uric acids, bilirubin, tocopherol, melatonin, and various exogenous antioxidants derived from diet, especially numerous polyphenols (see Chapter “Antioxidant Plant Polyphenols and Cognitive Disorders”).
The level of uric acid, a product of purine metabolism present in plasma, is relatively high and reaches approximately over half of the free radical scavenging activity in human blood (Korte et al. 1998; Murr et al. 2002; Chittiprol et al. 2010). Properties of uric acid include quenching of superoxide and singlet oxygen and protecting against oxidation of ascorbic acid through the chelation of iron. The ability of ascorbic acid to repair oxidized uric acid leads to synergy for maintaining antioxidant capacity by these two antioxidants. Due to its properties, uric acid is an important CNS antioxidant, since neurons contain high concentrations of ascorbic acid (Suboticanec et al. 1990; Rose and Bote 1993). Moreover, a lower serum level of this antioxidant may indicate that there is a significant loss of protection against NO and toxic peroxynitrite derived from NO because uric acid is a scavenger of peroxynitrite (Szabó et al. 2007). The level of uric acid in the CSF is about ten times lower than in serum (Bowman et al. 2010). This suggests that this purine metabolite is generated peripherally, and its migration to the CNS is limited by the blood-brain barrier (BBB). In schizophrenic patients, the lower level of uric acid has been reported (Yao et al. 1998b; Reddy et al. 2003).
Oxidative DNA damage may cause single- and double-stranded DNA breaks and modification of DNA components such as bases and sugar. The estimation of guanosine derivative is the most common method used (Cooke et al. 2003). Oxidative/nitrative changes induced by oxidative stress in schizophrenic patients are not restricted only to the brain, but may be observed in blood and peripheral cells: plasma, erythrocytes, and blood platelets. These elements are used to estimate the level of oxidative damage markers in schizophrenia. These markers may give clues about the relation of oxidative damage to the onset and progression of schizophrenia. Data from animal models and from postmortem studies in humans show an increase in the level of lipids, proteins, and DNA oxidation products implicated in oxidative stress in the brains of schizophrenic individuals. However, the results of the estimation of oxidative markers are often contradictory (see Tables 1 and 2). The discrepancies may depend on several reasons, such as differences in techniques of estimation, tested biological material (plasma, cell), the use of a single marker that may not accurately reflect the totality of oxidative damage, the choice of markers that can lack specificity and precision to detect oxidative stress in vivo, or measurement methods that may not be able to detect low levels of oxidative damage. Moreover, sample preparation methods may introduce artifactual oxidation during storage of samples. Improper matching of cases to the controls may also lead to the discrepant results, when lifestyle, diet, and ethnic origin are not taken into account. It is extremely important to study and compare the samples from schizophrenic patients in different stages of disease progression (naïve, first, chronic patients) and also after prescribed treatment, especially after different antipsychotics (both first and second generation). Altered gene expression may be attributed to the pathogenesis of schizophrenia (Konat 2003; Tsankova et al. 2007; Jiang et al. 2008). Genetic analysis has shown that the antioxidant pathway genes especially for GSH are involved in schizophrenia. Oxidative damage results in gene silencing expression of an affected genomic region. The mechanisms responsible for gene silencing are likely epigenetic and may be mediated through the alteration of DNA methylation (Lu et al. 2004). High level of homocysteine in patients with schizophrenia seems to be associated with DNA hypomethylation and changes in gene expression (James et al. 2002). Polymorphism in the glutathione S-transferase gene has been reported to be responsible partly for vulnerability to develop psychosis (methamphetamine abuse) leading to schizophrenia (Hashimoto et al. 2005). Polymorphism in the glutamate cysteine ligase gene has been also presented (Gysin et al. 2007). Moreover, in patients with schizophrenia, the capacity for the synthesis of GSH is lower than in controls and it may reflect a dysregulation of redox with an increased susceptibility to oxidative stress that may be of a genetic origin (Do et al. 2010; Gysin et al. 2007, 2009, 2011).
4 Lipid Peroxidation in Schizophrenia
Lipid peroxidation through free radical oxidation of unsaturated fatty acids (arachidonic acid, docosahexaenoic acid) is presumed to be involved in the oxidative injury in schizophrenia. Several methods have been developed for the measurement of the products of free radical-induced lipid peroxidation, such as lipid peroxides, 4-hydroxynonenal, malondialdehyde (MDA), and thiobarbituric acid reactive substances (TBARS), to assess oxidative injury in vivo in schizophrenia. The common colorimetric method used for the estimation of lipid peroxidation products is based on the reaction of unsaturated fatty acid products, mainly aldehydes with thiobarbituric acid, and the expression of the results as TBARS or MDA. The level of lipid peroxidation in peripheral cells and body fluid of schizophrenic patients is presented in Table 1. The assessment of MDA and TBARS, in spite of the lack of specificity leading to the overestimation of MDA, is still the most widely used marker of oxidative stress in clinical studies. Most of the published data concerning the oxidative stress and oxidative damage to lipids in schizophrenia are based on this assessment. There are other markers of free radical-induced lipid peroxidation: 4-hydroxynonenal, ethane (Puri et al. 2008; Ross et al. 2011), and isoprostanes (Dietrich-Muszalska and Olas 2009b). Grignon and Chianetta (2007) performed a meta-analysis of studies on MDA levels in schizophrenic patients published up to July 2006 and showed very large heterogeneity of the results. The analysis confirmed the value of these markers in the assessment of oxidative stress in schizophrenia.
The contribution of oxidative injury to the pathophysiology of schizophrenia has been indicated by the increase in lipid peroxidation products in the plasma and CSF of patients, and the altered levels of both enzymatic and nonenzymatic antioxidants in chronic, naïve, and first-episode patients (Mahadik and Scheffer 1996; Khan et al 2002; Dietrich-Muszalska et al. 2005; Zhang et al. 2010; Tsai et al 2013; see Tables 1 and 2). Recently, the increased level of breath ethane in patients with schizophrenia has been reported. The preliminary evidence suggests that oxidative stress can be assessed noninvasively by estimating breath ethane levels – the omega-3 PUFA oxidation product (Ross et al. 2011) and breath hydrocarbon analysis may represent a simple, noninvasive means to monitor the metabolic processes occurring in the course and treatment of schizophrenia.
According to Kartalci et al. (2011), the effects of electroconvulsive treatment (ECT) used in schizophrenic patients indicate that ECT, contrary to the effects of many antipsychotic drugs, may be a safe treatment alternative in terms of oxidative stress, inducing significant clinical improvement in severity of the disease associated with a significant decrease in the MDA serum level.
4.1 Isoprostanes
Isoprostanes (a group of prostaglandin isomers) belong to a family of prostaglandin derivatives synthesized from arachidonic acid in vivo through the nonenzymatic free radical-catalyzed mechanism. They circulate in peripheral blood and are excreted into the urine. Isoprostanes are generated in situ directly as an esterified form in membrane phospholipids and released as a free form, mainly by phospholipases. Phospholipase A2 has a dominant role in the release of arachidonic acid from phospholipids, removing the acyl chain of arachidonic acid from β-position of phospholipids. This nonenzymatic peroxidation of arachidonic acid associated with the creation of prostaglandin isomers (including the rearrangement) is important for the escalation of oxidative processes and damage to cellular biomolecules (Morrow and Roberts 1997; Chen et al. 1999; Brame et al. 1999). Morrow’s discovery of F2-isoprostanes (a group of isomers of PGF2α) in 1990 and the development of sensitive detection methods (immunoenzymatic assay) made their measurement a recommended, reliable method for assessing the status of oxidative stress in vivo (Morrow et al. 1990; Montuschi et al. 2004; Cracowski et al. 2002). F2-isoprostanes are important and recommended biomarkers of oxidative stress, and the method of measuring them is specific, highly sensitive, and noninvasive. The formation of isoprostanes indicates that the free radical oxidation of arachidonic acid occurs in patients with schizophrenia. The measurement of F2-isoprostanes is more sensitive than TBARS/MDA, lipid hydroperoxide, and conjugated diene level estimation, which is in vivo hampered by various methodological limitations (Morrow 2006; Morrow and Roberts 1997; Roberts et al. 1996; Roberts and Morrow 2000). Therefore, the method of F2-isoprostane evaluation can be used to monitor oxidative stress in patients with schizophrenia, according to clinical status of the disease (type of schizophrenia, predominance of positive or negative symptoms, first episode or recurrence, an acute episode or remission). Isoprostanes (prostaglandin isomer PGF2α such as 8-iso-PGF2α) are the primary end-products of lipid peroxidation in vivo. The measurement of isoprostanes can be useful for monitoring the effectiveness of schizophrenia treatment. The study (Dietrich-Muszalska and Olas 2009b) on the assessment of the increased F2-isoprostane concentration (8-iso-PGF2α) in patients with schizophrenia was the first one showing that the oxidation of arachidonic acid occurs through the nonenzymatic pathway (not associated with cyclooxygenase), as a result of free radical attack on membrane structures (Dietrich-Muszalska and Olas 2009b).
5 Cell Membrane Phospholipids and Polyunsaturated Fatty Acids (PUFAs) in Schizophrenia
Abnormal antioxidant enzyme activities and total antioxidant status, as well as greater abundance of oxidation products with the reduced level of PUFA, have been reported in schizophrenia (Yao et al. 2002b, 2003c; Ross et al. 2011). Metabolism of phospholipids is the most extensively studied in schizophrenia (Gattaz et al. 1995; Keshavan et al. 1993; Glen et al. 1994, 1996; Mahadik et al. 1994; Yao et al. 2002; Ripova et al. 1997, 1999; Strunecká and Rípová 1999; Rybakowski and Lehmann 1997). Phospholipids in the brain are extremely rich in polyunsaturated fatty acids (PUFAs), which in the neuronal membrane constitute as high as 65 % compared with other cells (15–35 %) (Horrocks et al. 1992). Membrane phospholipids such as phosphatidylserine (PS), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) are highly rich in arachidonic (AA) and docosahexaenoic (DHA) acids, which are predominantly released by phospholipase A2 (PLA2) after the stimulation of cell receptors (Skosnik and Yao 2003).
Polyunsaturated fatty acids (AA and DHA acids) as components of membrane phospholipids play an important role in cell membrane dynamics (Rana and Hokin 1990; Du Bois et al. 2005); their defects caused partly by oxidative stress have been observed in schizophrenic patients during the medication and in the course of the illness, suggesting PUFA dysregulation in schizophrenia (Skosnik and Yao 2003). The correlation between peripheral erythrocyte polyunsaturated fatty acids and cerebral phospholipid metabolism estimated by 31-phosphorus magnetic resonance spectroscopy (31P MRS) has also been documented for first-episode neuroleptic-naïve schizophrenic patients (Pettegrew et al. 1991; Yao et al. 2002; Fukuzako et al. 1999) and was selective for bilateral prefrontal cortex regions (Do et al. 2000 ). The postmortem cortical tissue study also implicates abnormal fatty acid composition in the frontal cortex in schizophrenic patients (McNamara et al. 2007; Yao and Keshavan 2011a). The altered composition of membrane phospholipids may be one of the most significant factors in the etiopathogenesis of schizophrenia, since it leads to disturbances in signal transduction dependent on receptors of numerous neurotransmitters (e.g., dopamine, serotonin, glutamate, acetylcholine, gamma-aminobutyric acid, and noradrenaline) and nerve growth factor (NGF) (Mahadik et al. 2001). Arachidonic acid, docosahexaenoic acid, and their metabolic products such as diacylglycerol (DAG), phosphatidylinositol, and prostaglandins act as second messengers and physiological mediators. The abnormalities in signal transduction caused by free radicals may be associated with altered membrane fluidity depending on phospholipid components or with changes in second messengers (arachidonic acid, docosahexaenoic acid, diacylglycerol, phosphatidylinositol, prostaglandins, cytokines, endocannabinoids), derived from membrane phospholipids and generated by neurotransmitters and growth factors (Du Bois et al. 2005; Yao and van Kammen 2003c).
It seems that the reduced level of second messengers derived from membrane arachidonic acid caused by oxidative stress may be a key factor in modified signal transduction and neuronal deficits in schizophrenia (Horrobin 1998; Skosnik and Yao 2003). The impairment of membrane phospholipids induced by free radicals and their dysfunction leading to disturbance of signal transduction can be also linked with function of neurotransmitters in schizophrenia, especially glutamatergic and serotoninergic systems (Du Bois et al. 2005; Skosnik and Yao 2003). Biochemical studies of blood platelets obtained from patients with schizophrenia have shown abnormalities in cellular phosphatidylinositol system, which is a second messenger for serotonin (5HT2) receptors (DeClerk 1990; Strunecká and Rípová 1999; Yao et al. 2004). The dysregulation of glutamatergic mechanisms, particularly hyperactive in exacerbation of psychosis and the related glutamate excitotoxicity, may be associated with oxidative stress and changes in the composition of membrane phospholipids (Tai et al. 1998). Membrane arachidonic acid is a precursor of second messengers in signal transduction of several neurotransmitters (dopamine, serotonin, acetylcholine, norepinephrine) (Axelrod 1990), and its oxidation may cause changes in signal transduction. Moreover, membrane phospholipid composition altered by oxidative stress may affect the activities of ion channels and enzymes, such as Na+/K+-ATPase and adenylate cyclase, leading to the production of cyclic AMP (Bourre et al. 1992). The omega-6/omega-3 PUFA ratio in membrane phospholipids has an effect not only on fluidity of membrane but also can affect the ligand-receptor interaction, possibly by increasing the availability of surface protein receptors and/or by increasing the concentration of receptors in the membrane (Farkas et al. 2002).
6 Nitrative Stress in Schizophrenia in Chapter 15
6.1 Glutathione
Glutathione (GSH), a tripeptide composed of glutamate, glycine, and cysteine, is involved in a number of diverse functions that include disulfide bond formation, detoxification, and antioxidant defense (Dringen 2000). The antioxidant function of GSH is due to the redox-active thiol group that becomes oxidized when GSH reduces target molecules. GSH may be oxidized to form glutathione disulfide (GSSG). The GSH/GSSG ratio is a marker of the redox status, which is low in schizophrenia (Dietrich-Muszalska et al. 2009c). GSH deficiency seems to be a major cause of oxidative stress in this disease. An impairment of the synthesis of glutathione seems to be one of the central causes of increased oxidative stress in schizophrenia (Do et al. 2009). GSH as a major nonprotein antioxidant in the brain plays a very important role in protecting the neurons against damage caused by ROS (which in the brain are produced additionally as a result of dopamine metabolism). The decrease in low-molecular-weight thiols in schizophrenic patients shows a significant reduction in the antioxidant defense system (Dietrich-Muszalska et al. 2009c), where GSH and thiols are important. The deficit of GSH can lead to the peroxidation of membrane lipids and microdamage in dopaminergic terminals, causing the loss of synaptic connectivity (Grima et al 2003). The reduced GSH levels in cerebrospinal fluid and prefrontal cortex in patients with schizophrenia have been described (Do et al 2000; Woo et al. 2008), and the deficit of GSH and its metabolite, γ-glutamylglutamine, in the cerebrospinal fluid of patients with schizophrenia, who were drug-naïve or drug-free at that time, was also found (see Table 4). In magnetic resonance spectroscopy studies in vivo, the significant decrease in GSH levels in the prefrontal cortex in patients with schizophrenia was proved (Do et al. 2000; Gawryluk et al. 2011a, b).
Glutathione may exert its antioxidant effects through several mechanisms. GSH as redox state-regulating antioxidant is involved in the detoxification of drugs and storage of cysteine and may affect gene expression and development of neurons (Dringen 2000). Moreover, glutathione also enhances the action of glutamate at the N-methyl-D-aspartate receptor (NMDA) (Köhr et al. 1994; Papadia et al. 2008); thus, the reduction in GSH concentration could also contribute to the hypofunction of NMDA receptor in the brain (Steullet et al. 2006). Metabolism of dopamine also plays a pathological role in schizophrenia. It was shown that dopamine in cultured cortical neurons decreased the GSH level by 40 % due to conjugation (Grima et al. 2003). Postmortem studies have revealed reduction in GSH level (about 40 %) in the caudate nucleus of schizophrenic patients (Yao et al. 2006) and in the prefrontal cortex (Gawryluk et al. 2011). The detoxication of ROS and harmful xenobiotics by GSH occurs either by nucleophilic scavenging or as a result of the reaction catalyzed by glutathione peroxidase (GPx). GSH acts as a cofactor for antioxidant enzymes such as glutathione peroxidase and glutathione transferase (Lu 2009); regenerates other crucial antioxidants, vitamins C and E; and may directly eliminate ROS (Do et al. 2009). In our work (Dietrich-Muszalska et al. 2009c), the significant reduction in the amount of low-molecular-weight thiols such as glutathione and its precursors, cysteine (CSH), and cysteinylglycine (CGSH) was described, while a significant increase in the amount of homocysteine (HCSH) in plasma of schizophrenic patients occurred. Oxidative stress in schizophrenia seems to be partly associated with the impairment of the synthesis of GSH, since a number of polymorphisms in the gene coding the key enzyme for GSH synthesis (glutamate cysteine ligase) have been demonstrated (Tosic et al. 2006; Saadat et al. 2007). The decreased level of GSH in blood cells (erythrocytes, platelets) from schizophrenic patients is presented in Table 5. Recently, Steullet et al. (2010) have demonstrated in the mouse model that redox dysregulation affected the ventral but not the dorsal hippocampus. GSH deficit caused impairment of parvalbumin neurons with concomitant reduction in gamma oscillations in the hippocampus (Steulett 2010). A novel treatment target in schizophrenia and other psychiatric disorders seems to be glutathione that possesses a dominance as the important cellular antioxidant. Recently, the results of several studies indicate the efficacy of N-acetylcysteine (NAC), a glutathione precursor, which is a useful agent in the treatment of various psychiatric disorders including schizophrenia (Matsuzawa and Hashimoto 2011; Berk et al. 2008a,b). The mechanism of NAC action is not clearly understood. NAC, which is capable of restoring thiol stores by shifting the redox balance in favor of GSH, may act as precursor of GSH and may be involved in the modulation of glutamatergic, neurotrophic, and inflammatory pathways.
7 Oxidative Stress and Pathological Mechanisms in Schizophrenia
Schizophrenia is a progressive disorder, as suggested by the clinical evidence, and is generally considered as a neurodevelopmental disorder. Many factors play an important role in its pathophysiological mechanisms. Despite the enormous advances that have been achieved, the pathogenesis of schizophrenia is still not clear. The brain pathology in schizophrenia is a neurodevelopmental, genetic, and environmental origin.
Biochemical alternations, especially in dopamine system in the brain with free radical production and ROS/RNS generation, leading to oxidative/nitrative damage to numerous biomolecules, might be partly responsible for the pathogenesis of this heterogeneous disorder. Oxidative stress with lower antioxidant defense and oxidative damage, reported by numerous studies, supports pathophysiological progression of schizophrenia and is consistent with the neurodegeneration hypothesis (Lieberman 1999). Inflammation has been postulated to be a factor in the pathophysiology of this disorder leading to the overproduction of prostaglandins from arachidonic acid, especially PGE2, and production of proinflammatory cytokines (IL-6) (Pedrini et al. 2012). The activity of cyclooxygenase 2 (COX-2) responsible for the synthesis of prostaglandins is also elevated.
Infection-induced developmental neuroinflammation may be pathologically relevant beyond the neonatal periods and may contribute to disease progression associated with the gradual development of the disease. Elevated risk of schizophrenia following prenatal exposure to infection is connected with cytokine-associated inflammatory events (prenatal cytokine hypothesis). The existence of the chronic inflammatory syndrome in schizophrenia has been described (Körschenhausen et al. 1996; Müller and Schwarz 2010). There is also increasing evidence that chronic inflammation, mediated by cytokines, contributes to the pathophysiology of this disorder (Potvin et al. 2008; Kim et al. 2000, 2009). Cytokines are an essential element of cell-to-cell communication in the immune system but also in the interaction between the immune system and the brain. Cytokines affect the differentiation and survival of neurons (Marx et al. 2001). Disturbances of brain development caused by prenatal maternal infections seem to be the result of cytokine-related inflammatory events (Meyer et al. 2008). The origins of increased cytokine levels in schizophrenia are not known yet. Schizophrenic patients had higher level of cytokines IL-2 and IL-6 than healthy controls (Lin et al. 1998; van Kammen et al. 1999; Behrens et al. 2008; Meyer et al. 2011). The elevated superoxide anion level in schizophrenia (with lower SOD activity) acts as a second messenger to activate NF-kB which initiates the transcription of many genes involved in the synthesis of cytokines such as IL-6, IL-1, and TNFα (Zhang et al. 2002).
Developmental neuroinflammation may affect processes that are pivotal for normal brain maturation, including myelination, synaptic pruning, and neuronal connectivity, all of which occur to a great extent during the postnatal brain maturation (Kasper and Papadimitriou 2009). Microglia that are resident macrophage of the brain are involved directly in the neuronal degeneration by producing various proinflammatory cytokines and free radicals (Yao et al. 2003; Bernstein et al. 2009). The neuropathology of schizophrenia is closely associated with microglial activation (Stefano et al. 2004). Changes in immune-inflammatory pathways with the activation of microglia increase proinflammatory cytokine generation and oxidative stress, autoimmune responses and activation of the tryptophan metabolite (TRYCAT) pathway and consequent modulation of NMDA receptors, and glutamate production (Yao et al. 2010b). These factors may account for the higher neurodevelopmental pathology in schizophrenia.
Various lines of evidence suggest immune dysfunction in schizophrenia (Yolken and Torrey 1995) and the association of schizophrenia with autoimmune disorders and increased levels of cytokines IL-1, IL-6, and IFNγ (Zhang et al. 2009b). Neopterin that is the catabolic product of GTP serves as a marker for immune system activation and might elucidate the interaction between immune pathogenesis and oxidative stress in schizophrenia (Yao et al. 2010a). It is produced in monocytes/macrophages upon stimulation with cytokine interferon γ. High level of neopterin is associated with increased production of ROS. Conflicting findings were presented by Chittiprol et al. (2010). They have reported that antipsychotic-naïve patients with schizophrenia had significantly higher levels of neopterin nitrates and lower levels of antioxidants; after treatment with antipsychotics, there were a significant decrease in the neopterin levels and an increase in antioxidants. These studies support the view that oxidative stress in schizophrenia might be linked with immune pathogenesis (Zhang et al. 2009b).
Tryptophan/kynurenine metabolism with kynurenic acid (KA) and neurotoxic TRYCAT production is regulated by cytokines (see Chapter “The Kynurenine Pathway at the Interface Between Neuroinflammation, Oxidative Stress and Neurochemical Disturbances: Emphasis in Schizophrenia”). Increased KA and TRYCAT pathway has the effect on NMDA receptor dysfunction and neuroprogression (Anderson and Maes 2013; Najjar et al. 2013). Maternal infection and subsequent immune-inflammatory responses are also associated with oxidative stress and lower level of an important antioxidant – glutathione. This process contributes to alterations in neurodegeneration and myelination. Oxidative stress and TRYCAT pathway could modulate the CNS glial-neuronal interactions that determine synaptic plasticity as well as myelin generation and maintenance (Anderson and Maes 2013). Schizophrenia is a brain disease with extensive abnormalities found in the cognitive function and brain structures. The use of magnetic resonance imaging (MRI) allowed to measure the amount of gray and white matter. In schizophrenic patients (first episode), the loss of gray matter is accelerated, particularly in the frontotemporal cortical regions and sulcal and ventricular expansion (Kasper and Papadimitriou 2009). However, there is considerable inhomogeneity of brain abnormalities in this disorder, and the alterations in mean volume of neuron and glial cell densities in different brain regions and changes in neurotransmitter/receptor systems, growth factors, hormones, regulatory proteins, and brain energy metabolism with dysfunction of mitochondria and ROS production have been reported (Rezin et al. 2009). There is some evidence that oxidative stress supports pathophysiological progression in schizophrenia. It is consistent with neurodegeneration hypothesis.
Based on the role of oxidative stress and lipid peroxidation, Horrobin in 1998 presented two hypotheses that:
-
1.
Alteration in membrane phospholipid composition and the lipid metabolism results in neuronal dysfunction leading to disruption of the membrane and cell damage.
-
2.
Deficiency in antioxidant defense in the cortex of maturational development and stressful physiological activity lead to increased concentrations of ROS, which cause cellular injury dysfunction and potentially cell death.
These hypotheses highly theoretical are supported by described oxidative stress in schizophrenia and integrate basic neurobiological mechanisms with the clinical dimensions of the illness. Clinical deterioration is manifested by the development and increasing severity and persistence of psychotic and negative symptoms and cognitive impairment.
Although a number of hypotheses have been proposed in an attempt to explain the pathophysiology of schizophrenia, no single theory seems to account for all aspects of the disease.
Each hypothesis explains only some of the phenomena associated with schizophrenia; however, many variables described in these hypotheses interact to produce a disorder characterized by heterogeneous symptomatology and its progression. Converging lines of evidence including reduced neuropil suggest that disrupted cortical synaptic circuitry is a central deficit in schizophrenia (Lewis and Lieberman 2000) and apoptotic mechanisms may also be involved in this process and the pathophysiology of schizophrenia (Jarskog et al. 2005). Cortical pyramidal neurons from individuals with schizophrenia exhibit smaller somal volume, decreased spine density, decreased dendritic length, and decreased terminals compared to healthy control. These postmortem findings contribute to the hypothesis that schizophrenia stems from altered synaptic circuitry. The cortical neuropathology of schizophrenia includes neuronal atrophy, decreased neuropil, and alterations in neuronal density with suggestion of altered synaptic circuitry (Kasper and Papadimitriou 2009). Moreover, neuroimaging studies also indicate that a progressive loss of cortical gray matter occurs in the early course of schizophrenia (Lewis and Sweet 2009; Lewis 2012). The underlying mechanisms of the defects and synaptic dysfunction suggest that the dysregulation of neuronal apoptosis may contribute to the pathophysiology of the disorder. Usually, the activation of complex apoptotic pathway may lead to rapid neuronal death. The dysregulation of neuronal apoptotic cascade of pro- and antiapoptotic proteins could lead to a limited form of apoptotic pathway in terminal neuritis and individual synapses to cause synaptic elimination without the cell death and with synaptic deficit and cortical dysfunction in schizophrenia. Apoptotic mechanism seems to be responsible for progressive gray matter volume loss (first onset of psychosis) when antioxidant activity is low (Glantz et al. 2006, 2010).
Apoptotic mechanisms that can influence synaptic connectivity, and neuronal complexity seem to support the apoptotic hypothesis of schizophrenia connected also with oxidative stress. Apoptotic hypothesis proposing that limited apoptotic activity with apoptotic dysregulation can contribute to a gradual reduction in neuronal viability and to synaptic deficits without causing neuronal death might be taken into account. Oxidative stress could contribute to complex apoptotic mechanisms.
Abbreviations
- (O2.−):
-
Superoxide anion
- 31P MRS:
-
31-Phosphorus magnetic resonance spectroscopy
- 4-HNE:
-
4-Hydroxynonenal
- 8-OH-Gua:
-
8-Hydroxyguanosine
- AA:
-
Arachidonic acid
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood-brain barrier
- cAMP:
-
3′-5′-Cyclic adenosine monophosphate (or cyclic AMP)
- CAT:
-
Catalase
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase 2
- CSF:
-
Cerebrospinal fluid
- CSH:
-
Cysteine CGSH – cysteinylglycine
- Cu:
-
Copper
- DAG:
-
Diacylglycerol
- DHA:
-
Docosahexaenoic acid
- ECT:
-
Electroconvulsive therapy
- ELISA:
-
Enzyme-linked immunosorbent assay
- EPUFAs:
-
Essential polyunsaturated fatty acids
- Fe:
-
Iron
- GABA:
-
Gamma-aminobutyric acid
- GCL:
-
Glutamate cysteine ligase
- GPx:
-
Glutathione peroxidase
- GR:
-
Glutathione reductase
- GSH:
-
Reduced glutathione
- GSH/GSSG:
-
Reduced glutathione/glutathione disulfide
- GSSG:
-
Glutathione disulfide
- GST:
-
Glutathione S-transferase
- GTP:
-
Guanosine triphosphate
- H2O2 :
-
Hydrogen peroxide
- HCSH:
-
Homocysteine
- IL-1:
-
Interleukin-1
- IL-2:
-
Interleukin-2
- IL-6:
-
Interleukin-6
- KA:
-
Kynurenic acid
- MAO:
-
Monoamine oxidase
- MDA:
-
Malondialdehyde
- Mn:
-
Manganese
- MRI:
-
Magnetic resonance imaging
- N2O:
-
Nitrous oxide
- NAC:
-
N-Acetylcysteine
- NAD:
-
Nicotinamide adenine dinucleotide
- NADP:
-
Nicotinamide adenine dinucleotide phosphate
- NF-kB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NGF:
-
Nerve growth factor
- NMDA:
-
N-methyl-D-aspartate receptor
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- NOX:
-
NADPH oxidase
- PE:
-
Phosphatidylethanolamine
- PGE2:
-
Prostaglandin E2
- PGF2α:
-
Prostaglandin F2 alpha
- PI:
-
Phosphatidylinositol
- PLA2:
-
Phospholipase A2
- PS:
-
Phosphatidylserine
- PUFAs:
-
Polyunsaturated fatty acids
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SOD:
-
Superoxide dismutase
- TAS:
-
Total antioxidant status
- TBARS:
-
Thiobarbituric acid reactive substances
- TNFα:
-
Tumor necrosis factor alpha
- Trx:
-
Thioredoxin
- TRYCAT:
-
Tryptophan catabolite
References
Abdalla DS, Monteiro HP, Oliveira JA, Bechara EJ (1986) Activities of superoxide dismutase and glutathione peroxidase in schizophrenic and manic-depressive patients. Clin Chem 32(5):805–807
Akyol O, Herken H, Uz E, Fadillioğlu E, Unal S, Söğüt S, Ozyurt H, Savaş HA (2002) The indices of endogenous oxidative and antioxidative processes in plasma from schizophrenic patients. The possible role of oxidant/antioxidant imbalance. Prog Neuropsychopharmacol Biol Psychiatry 6(5):995–1005
Altuntas I, Aksoy H, Coskun I, Cayköylü A, Akçay F (2000) Erythrocyte superoxide dismutase and glutathione peroxidase activities, and malondialdehyde and reduced glutathione levels in schizophrenic patients. Clin Chem Lab Med 38(12):1277–1281
American Psychiatric Association. Task Force on DSM-IV (2000) The diagnostic and statistical manual of mental disorders, 4th edn. (DSM-IV) American Psychiatric Association 2000, Washington, DC
Anderson G, Maes M (2013) Schizophrenia: linking prenatal infection to cytokines, the tryptophan catabolite (TRYCAT) pathway, NMDA receptor hypofunction, neurodevelopment and neuroprogression. Prog Neuropsychopharmacol Biol Psychiatry 5(42):5–19
Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP (2003a) Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr Res 62(3):195–204
Arvindakshan M, Sitasawad S, Debsikdar V, Ghate M, Evans D, Horrobin DF, Bennett C, Ranjekar PK, Mahadik SP (2003b) Essential polyunsaturated fatty acid and lipid peroxide levels in never-medicated and medicated schizophrenia patients. Biol Psychiatry 53(1):56–64
Axelrod J (1990) Receptor mediated activation of phospholipase A2 and arachidonic acid release in signal transduction. Biochem Soc Trans 18(4):503–507
Babulas V, Factor-Litvak P, Goetz R, Schaefer CA, Brown AS (2006) Prenatal exposure to maternal genital and reproductive infections and adult schizophrenia. Am J Psychiatry 163(5):927–929
Behrens MM, Ali SS, Dugan LL (2008) Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci 28(51):13957–13966
Ben Othmen L, Mechri A, Fendri C, Bost M, Chazot G, Gaha L, Kerkeni A (2008) Altered antioxidant defense system in clinically stable patients with schizophrenia and their unaffected siblings. Prog Neuropsychopharmacol Biol Psychiatry 32(1):155–159
Benes FM, Berretta S (2001) GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25:1–27
Ben-Shachar D (2002) Mitochondrial dysfunction in schizophrenia: a possible linkage to dopamine. J Neurochem 83(6):1241–1251
Ben-Shachar D, Karry R (2008) Neuroanatomical pattern of mitochondrial complex I pathology varies between schizophrenia, bipolar disorder and major depression. PLoS One 3(11):e3676
Ben-Shachar D, Zuk R, Gazawi H, Reshef A, Sheinkman A, Klein E (1999) Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int J Neuropsychopharmacol 2(4):245–253
Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, Anderson-Hunt M, Judd F, Katz F, Katz P, Ording-Jespersen S, Little J, Conus P, Cuenod M, Do KQ, Bush AI (2008a) N-acetyl cysteine as a glutathione precursor for schizophrenia – a double-blind, randomized, placebo-controlled trial. Biol Psychiatry 64(5):361–368
Berk M, Ng F, Dean O, Dodd S, Bush AI (2008b) Glutathione: a novel treatment target in psychiatry. Trends Pharmacol Sci 29(7):346–351
Bernstein HG, Steiner J, Bogerts B (2009) Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Rev Neurother 9:1059–1071
Bitanihirwe BK, Woo TU (2011) Oxidative stress in schizophrenia: an integrated approach. Neurosci Biobehav Rev 35:878–893
Bošković M, Vovk T, Kores Plesničar KB, Grabnar I (2011) Oxidative stress in schizophrenia. Curr Neuropharmacol 9(2):301–312
Bourre JM, Bonneil M, Chaudiere J, Clement M, Dumont O, Durand G, Lafont H, Nalbone G, Pascal G, Piciotti M (1992) Structural and functional importance of dietary polyunsaturated fatty acids in the nervous system. Adv Exp Med Biol 318:211–229
Bowman GL, Shannon J, Frei B, Kaye JA, Quinn JF (2010) Uric acid as a CNS antioxidant. J Alzheimer’s Dis 19(4):1331–1336
Brame CJ, Salomon RG, Morrow JD, Roberts LJ (1999) Identification of extremely reactive γ-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J Biol Chem 274(19):13139–13146
Brown K, Reid A, White T, Henderson T, Hukin S, Johnstone C, Glen A (1998) Vitamin E, lipids, and lipid peroxidation products in tardive dyskinesia. Biol Psychiatry 43(12):863–867
Brown AS, Deicken RF, Vinogradov S, Kremen WS, Poole JH, Penner JD, Kochetkova A, Kern D, Schaefer CA (2009) Prenatal infection and cavum septum pellucidum in adult schizophrenia. Schizophr Res 108(1–3):285–287
Chen Y, Morrow JD, Roberts LJ (1999) Formation of reactive cyclopentenone compounds in vivo as products of the isoprostane pathway. J Biol Chem 274(16):10863–10868
Chittiprol S, Venkatasubramanian G, Neelakantachar N, Babu SV, Reddy NA, Shetty KT, Gangadhar BN (2010) Oxidative stress and neopterin abnormalities in schizophrenia: a longitudinal study. J Psychiatr Res 44(5):310–313
Clay HB, Sillivan S, Konradi C (2011) Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int J Dev Neurosci 29(3):311–324
Cooke M, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17(10):1195–1214
Covelli V, Pellegrino NM, Jirillo E (2003) A point of view: the need to identify an antigen in psychoneuroimmunological disorders. Curr Pharm Des 9(24):1951–1955
Cracowski JL, Durand T, Bessard G (2002) Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol Sci 23(8):1–4
Dadheech G, Mishra S, Gautam S, Sharma P (2008) Evaluation of antioxidant deficit in schizophrenia. Indian J Psychiatry 50(1):16–20
Dakhale G, Mishra S, Gautam S, Sharma P (2008) Evaluation of antioxidant deficit in schizophrenia. Indian J Psychiatry 50(1):16–20
Dalla Libera A, Rigobello MP, Bindoli A (1996) Inhibitory action of neuroleptic drugs and serotonin on dopamine autoxidation and lipid peroxidation. Prog Neuropsychopharmacol Biol Psychiatry 19(2):291–298
Das I, Khan NS (1998) Increased arachidonic acid induced platelet chemiluminescence indicates cyclooxygenase overactivity in schizophrenic subjects. Prostaglandins Leukot Essent Fatty Acids 58(3):165–168
DeClerk F (1990) The role of serotonin in thrombogenesis. Clin Physiol Biochem, 8Supl 3:40–49
De Hert M, Schreurs V, Vancampfort D, van Winkel R (2009) Metabolic syndrome in people with schizophrenia: a review. World Psychiatry 8(1):15–22
Dietrich-Muszalska A, Kontek B (2010) Lipid peroxidation in patients with schizophrenia. Psychiatry Clin Neurosci 64:469–475
Dietrich-Muszalska A, Kwiatkowska A (2014) Generation of superoxide anion radicals and platelet glutathione peroxidase activity in patients with schizophrenia. Neuropsychiatr Dis Treat 10:703–709
Dietrich-Muszalska A, Olas B (2009a) Modifications of blood platelet proteins of patients with schizophrenia. Platelets 20(2):90–96
Dietrich-Muszalska A, Olas B (2009b) Isoprostanes as indicators of oxidative stress in schizophrenia. World J Biol Psychiatry 10(1):27–33
Dietrich-Muszalska A, Olas B, Rabe-Jablonska J (2005) Oxidative stress in blood platelets from schizophrenic patients. Platelets 16(7):386–391
Dietrich-Muszalska A, Olas B, Głowacki R, Bald E (2009) Oxidative/nitrative modifications of plasma proteins and thiols from patients with schizophrenia. Neuropsychobiology 59(1):1–7
Do KQ, Trabesinger AH, Kirsten-Krüger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuénod M (2000) Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci 12(10):3721–3728
Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M (2009) Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol 19(2):220–230
Do KQ, Conus P, Cuenod M (2010) Redox dysregulation and oxidative stress in schizophrenia: nutrigenetics as a challenge in psychiatric disease prevention. World Rev Nutr Diet 101:131–153
Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62(6):649–671
Dror N, Klein E, Karry R, Sheinkman A, Kirsh Z, Mazor M, Tzukerman M, Ben-Shachar D (2002) State-dependent alterations in mitochondrial complex I activity in platelets: a potential peripheral marker for schizophrenia. Mol Psychiatry 7(9):995–1001
du Bois TM, Deng C, Huang XF (2005) Membrane phospholipid composition, alterations in neurotransmitter systems and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 29(6):878–888
Elkashef AM, Al-Barazi H, Venable D, Baker I, Hill J, Apud J, Wyatt RJ (2002) Dopamine effect on the mitochondria potential in B lymphocytes of schizophrenic patients and normal controls. Prog Neuropsychopharmacol Biol Psychiatry 26(1):145–148
Evans DR, Parkih VV, Khan MM, Coussons C, Buckley PF, Mahadik SP (2003) Red blood cell membrane essential fatty acid metabolism in early psychotic patients following antipsychotic drug treatment. Prostaglandins Leukot Essent Fatty Acids 69(6):393–399
Fan X, Liu EY, Freudenreich O, Park JH, Liu D, Wang J, Yi Z, Goff D, Henderson DC (2010) Higher white blood cell counts are associated with an increased risk for metabolic syndrome and more severe psychopathology in non-diabetic patients with schizophrenia. Schizophr Res 118:211–217
Farkas E, de Willde MC, Kiliaan AJ, Meijer J, Keijser JN, Luiten PG (2002) Dietary long chain PUFAs differentially affect hippocampal muscarinic 1 and serotonergic 1A receptors in experimental cerebral hypoperfusion. Brain Res 954(1):32–41
Fukuzako H, Fukuzako T, Hashiguchi T, Kodama S, Takigawa M, Fujimoto T (1999) Changes in the level of phosphorus metabolites in temporal lobes of drug-naïve schizophrenic patients. Am J Psychiatry 156(8):1205–1208
Gama CS, Salvador M, Andreazza AC, Lobato MI, Berk M, Kapczinski F, Belmonte-de-Abreu PS (2008) Elevated serum thiobarbituric acid reactive substances in clinically symptomatic schizophrenic males. Neurosci Lett 433(3): 270–3.
Gama CS, Salvador M, Andreazza AC, Kapczinski F, Silva Belmonte-de-Abreu P (2006) Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in schizophrenia: a study of patients treated with haloperidol or clozapine. Prog Neuropsychopharmacol Biol Psychiatry 30(3):512–515
Gattaz WF, Schmitt A, Maras A (1995) Increased platelet phospholipase A2 activity in schizophrenia. Schizophr Res 16(1):1–6
Gawryluk JW, Wang JF, Andreazza AC, Shao L, Young LT (2011a) Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int J Neuropsychopharmacol 14(1):123–130
Gawryluk JW, Wang JF, Andreazza AC, Shao L, Yatham LN, Young LT (2011b) Prefrontal cortex glutathione S-transferase levels in patients with bipolar disorder, major depression and schizophrenia. Int J Neuropsychopharmacol 14(8):1069–1074
Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF (2006) Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr Res 8(1):47–63
Glantz LA, Gilmore JH, Overstreet DH, Salimi K, Lieberman JA, Jarskog LF (2010) Pro-apoptotic Par-4 and dopamine D2 receptor in temporal cortex in schizophrenia, bipolar disorder and major depression. Schizophr Res 118(1–3):292–299
Glen AI, Glen EM, Horrobin DF, Vaddadi KS, Spellman M, Morse-Fisher N, Ellis K, Skinner FS (1994) A red cell membrane abnormality in a subgroup of schizophrenic patients: evidence for two diseases. Schizophr Res 12(1):53–61
Glen AI, Cooper SJ, Rybakowski J, Vaddadi K, Brayshaw N, Horrobin DF (1996) Membrane fatty acids, niacin flushing and clinical parameters. Prostaglandins Leukot Essent Fatty Acids 55(1–2):9–15
Griffith OW, Meister A (1985) Origin and turnover of mitochondrial glutathione. Proc Natl Acad Sci U S A 82(14):4668–4672
Grignon S, Chianetta JM (2007) Assessment of malondialdehyde levels in schizophrenia: a meta-analysis and some methodological considerations. Prog Neuropsychopharmacol Biol Psychiatry 31(2):365–369
Grima G, Benz B, Parpura V, Cuenod M, Do KQ (2003) Dopamine-induced oxidative stress in neurons with glutathione deficit: implication for schizophrenia. Schizophr Res 62(3):213–224
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, Deppen P, Preisig M, Ruiz V, Steullet P, Tosic M, Werge T, Cuénod M, Do KQ (2007) Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci U S A 104(42):16621–16626
Gysin R, Riederer IM, Cuénod M, Do KQ, Riederer BM (2009) Skin fibroblast model to study an impaired glutathione synthesis: consequences of a genetic polymorphism on the proteome. Brain Res Bull 79(1):46–52
Gysin R, Kraftsik R, Boulat O, Bovet P, Conus P, Comte-Krieger E, Polari A, Steullet P, Preisig M, Teichmann T, Cuénod M, Do KQ (2011) Genetic dysregulation of glutathione synthesis predicts alteration of plasma thiol redox status in schizophrenia. Antioxid Redox Signal 15(7):2003–2010
Halliwel B (2006) Oxidative stress and neurodegeneration: where are we now? J Neurochem 97:1634–1658
Halliwell B (1992) Reactive oxygen species and the central nervous system. J Neurochem 59(5):1609–1623, Erratum in: J Neurochem, 2012, 120(5): 850
Halliwell B, Gutteridge JM (1996) Free radicals in biology and medicine. Clarendon Press, Oxford, UK
Hanson DR, Gottesman II (2005) Theories of schizophrenia: a genetic-inflammatory-vascular synthesis. BMC Med Genet 6:7
Harrison PJ (1999) The neuropathological effects of antipsychotic drugs. Schizophr Res 40(2):87–99
Hashimoto T, Hashimoto K, Matsuzawa D, Shimizu E, Sekine Y, Inada T, Ozaki N, Iwata N, Harano M, Komiyama T, Yamada M, Sora I, Ujike H, Iyo M (2005) A functional glutathione S-transferase P1 gene polymorphism is associated with methamphetamine-induced psychosis in Japanese population. Am J Med Genet B Neuropsychiatr Genet 135B(1):5–9
Herken H, Uz E, Ozyurt H, Akyol O (2001a) Red blood cell nitric oxide levels in patients with schizophrenia. Schizophr Res 52(3):289–290
Herken H, Uz E, Ozyurt H, Söğüt S, Virit O, Akyol O (2001b) Evidence that the activities of erythrocyte free radical scavenging enzymes and the products of lipid peroxidation are increased in different forms of schizophrenia. Mol Psychiatry 6(1):66–73
Horrobin DF (1998) The membrane phospholipid hypothesis as a biochemical basis for the neurodevelopmental concept of schizophrenia. Schizophr Res 30(3):193–208
Horrocks LA, Ansell GB, and Porcellati G (1992) Phospholipids in the Nervous System Vol. 1: Metabolism. New York, Raven
Huang TL, Liou CW, Lin TK (2010) Serum thiobarbituric acid-reactive substances and free thiol levels in schizophrenia patients: effects of antipsychotic drugs. Psychiatry Res 177(1–2):18–21
Insel TR (2009) Disruptive insights in psychiatry: transforming a clinical discipline. J Clin Invest 119(4):700–705
James SJ, Melnyk S, Pogribna M, Pogribny IP, Caudill MA (2002) Elevation in S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr 132(8 Suppl):2361S–2366S
Jarskog LF, Glantz LA, Gilmore JH, Lieberman JA (2005) Apoptotic mechanisms in the pathophysiology of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 29(5):846–858
Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, Kumar A, Nestler EJ, Akbarian S, Beckel-Mitchener AC (2008) Epigenetics in the nervous system. J Neurosci 28(46):11753–11759
Jorgensen A, Broedbaek K, Fink-Jensen A, Knorr U, Greisen Soendergaard M, Henriksen T, Weimann A, Jepsen P, Lykkesfeldt J, Poulsen HE, Balslev Jorgensen M (2013) Increased systemic oxidatively generated DNA and RNA damage in schizophrenia. Psychiatry Res 209(3):417–423
Kartalci S, Karabulut AB, Ozcan AC, Porgali E, Unal S (2011) Acute and chronic effects of electroconvulsive treatment on oxidative parameters in schizophrenia patients. Prog Neuropsychopharmacol Biol Psychiatry 35(7):1689–1694
Kasper S, Papadimitriou GN (2009) Schizophrenia, 2nd edn. Informa Health Care, New York
Keshavan MS, Mallinger AG, Pettegrew JW, Dippold C (1993) Erythrocyte membrane phospholipids in psychotic patients. Psychiatry Res 49(1):89–95
Khan MM, Evans DR, Gunna V, Scheffer RE, Parkih VV, Mahadik SP (2002) Reduced erythrocyte membrane essential fatty acids and increased lipid peroxides in schizophrenia at the never-medicated first-episode of psychosis and after years of treatment with antipsychotics. Schizophr Res 58(1):1–10
Kim YK, Kim L, Lee MS (2000) Relationships between interleukins, neurotransmitters and psychopathology in drug-free male schizophrenics. Schizophr Res 44:165–175
Kim YK, Myint AM, Verkerk R, Scharpe S, Steinbusch H, Leonard B (2009) Cytokine changes and tryptophan metabolites in medication-naïve and medication-free schizophrenic patients. Neuropsychobiology 59:123–129
Kinney DK, Hintz K, Shearer EM, Barch DH, Riffin C, Whitley K, Butler R (2010) A unifying hypothesis of schizophrenia: abnormal immune system development may help explain roles of prenatal hazards, post-pubertal onset, stress, genes, climate, infections, and brain dysfunction. Med Hypotheses 74(3):555–563
Kliushnik TP, Kalinina MA, Sarmanova ZV, Otman IN, Kozlovskaia GV (2009) Dynamics of immunological and clinical parameters in the treatment of childhood schizophrenia. Zh Nevrol Psikhiatr Im S S Korsakova 109(6):46–49
Köhr G, Eckardt S, Lüddens H, Monyer H, Seeburg PH (1994) NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron 12(5):1031–1040
Konat GW (2003) H2O2-induced higher order chromatin degradation: a novel mechanism of oxidative genotoxicity. J Biosci 28(1):57–60
Körschenhausen DA, Hampel HJ, Ackenheil M, Penning R, Müller N (1996) Fibrin degradation products in post mortem brain tissue of schizophrenics: a possible marker for underlying inflammatory processes. Schizophr Res 19(2–3):103–109
Korte S, Arolt V, Peters M, Weitzsch C, Rothermundt M, Kirchner H (1998) Increased serum neopterin levels in acutely ill and recovered schizophrenic patients. Schizophr Res 32(1):63–67
Kuloglu M, Ustundag B, Atmaca M, Canatan H, Tezcan AE, Cinkilinc N (2002) Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochem Funct 20(2):171–175
Kunz M, Gama CS, Andreazza AC, Salvador M, Ceresér KM, Gomes FA, Belmonte-de-Abreu PS, Berk M, Kapczinski F (2008) Elevated serum superoxide dismutase and thiobarbituric acid reactive substances in different phases of bipolar disorder and in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 32(7):1677–1681
Lakhan SE, Kramer A (2009) Schizophrenia genomics and proteomics: are we any closer to biomarker discovery? Behav Brain Funct 7(5):2. doi:10.1186/1744-9081-5-2
Lakhan SE, Vieira K, Hamlat E (2010) Biomarkers in psychiatry: drawbacks and potential for misuse. Int Arch Med 3:1
Lewis DA (2012) Cortical circuit dysfunction and cognitive deficits in schizophrenia: implications for preemptive interventions. Eur J Neurosci 35(12):1871–1878
Lewis DA, Levitt P (2002) Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 25:409–432
Lewis DA, Lieberman JA (2000) Catching up on schizophrenia: natural history and neurobiology. Neuron 28(2):325–334
Lewis DA, Sweet RA (2009) Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest 119(4):706–716
Li XF, Zheng YL, Xiu MH, da Chen C, Kosten TR, Zhang XY (2011) Reduced plasma total antioxidant status in first-episode drug-naive patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 35(4):1064–1067
Lieberman JA (1999) Is schizophrenia a neurodegenerative disorder? A clinical and neurobiological perspective. Biol Psychiatry 46(6):729–739
Lin MT, and Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443(7113):787–795
Lin A, Kenis G, Bignotti S, Tura GJ, DeJong R, Bosmans E, Pioli R, Altamura C, Scharpe S, Maes M (1998) The inflammatory response system in treatment-resistant schizophrenia: increased serum interleukin-6. Schizophr Res 32(1):9–15
Lipton SA, Choi YB, Pan Z-H, Lei SZ, Vincent C, Sucher NJ, Loscalzo J, Singiel DJ, Stamer JS (1993) A redox-based mechanism for the neuroprotective effects of nitric oxide and related nitroso-compounds. Nature 364(6438):626–632
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30(1–2):42–59
Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429(6994):883–891
Mahadik SP, Scheffer RE (1996) Oxidative injury and potential use of antioxidants in schizophrenia. Prostaglandins Leukot Essent Fatty Acids 55(1–2):45–54
Mahadik SP, Mukherjee S, Correnti EE, Kelkar HS, Wakade CG, Costa RM, Scheffer R (1994) Plasma membrane phospholipid and cholesterol distribution of skin fibroblasts from drug-naive patients at the onset of psychosis. Schizophr Res 13(3):239–247
Mahadik SP, Mukherjee S, Scheffer R, Correnti EE, Mahadik JS (1998) Elevated plasma lipid peroxides at the onset of nonaffective psychosis. Biol Psychiatry 43(9):674–679
Mahadik SP, Evans D, Lal H (2001) Oxidative stress and role of antioxidant and omega-3 essential fatty acid supplementation in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 25(3):463–493
Malaspina D, Harlap S, Fennig S, Heiman D, Nahon D, Feldman D, Susser ES (2001) Advancing paternal age and the risk of schizophrenia. Arch Gen Psychiatry 58(4):361–367
Malaspina D, Dracxler R, Walsh-Messinger J, Harlap S, Goetz RR, Keefe D, Perrin MC (2014) Telomere length, family history, and paternal age in schizophrenia. Mol Genet Genomic Med 2(4):326–331
Martins-de-Souza D, Harris LW, Guest PC, Bahn S (2011) The role of energy metabolism dysfunction and oxidative stress in schizophrenia revealed by proteomics. Antioxid Redox Signal 15(7):2067–2079
Marx CE, Jarskog LF, Lauder JM, Lieberman JA, Gilmore JH (2001) Cytokine effects on cortical neuron MAP-2 immunoreactivity: implications for schizophrenia. Biol Psychiatry 50(10):743–749
Masserano JM, Baker I, Venable D, Gong L, Zullo SJ, Merril CR, Wyatt RJ (2000) Dopamine induces cell death, lipid peroxidation and DNA base damage in a catecholaminergic cell line derived from the central nervous system. Neurotox Res 1(3):171–179
Matsuzawa D, Hashimoto K (2011) Magnetic resonance spectroscopy study of the antioxidant defense system in schizophrenia. Antioxid Redox Signal 15(7):2057–2065
Matsuzawa D, Obata T, Shirayama Y, Nonaka H, Kanazawa Y, Yoshitome E, Takanashi J, Matsuda T, Shimizu E, Ikehira H, Iyo M, and Hashimoto K (2008) Negative correlation between brain glutathione level and negative symptoms in schizophrenia: a 3T 1H-MRS study. PLoS One, 3(4):e1944
McCreadie RG, MacDonald E, Wiles D, Campbell G, Paterson JR (1995) The Nithsdale Schizophrenia Surveys. XIV: plasma lipid peroxide and serum vitamin E levels in patients with and without tardive dyskinesia, and in normal subjects. Br J Psychiatry 167:610–617
McNamara RK, Jandacek R, Rider T, Tso P, Hahn CG, Richtand NM, Stanford KE (2007) Abnormalities in the fatty acid composition of the postmortem orbitofrontal cortex of schizophrenic patients: gender differences and partial normalization with antipsychotic medications. Schizophr Res 91(1–3):37–50
Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J (2006) The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci 26(18):4752–4762
Meyer U, Engler A, Weber L, Schedlowski M, Feldon J (2008) Preliminary evidence for a modulation of fetal dopaminergic development by maternal immune activation during pregnancy. Neuroscience 154(2):701–709
Meyer U, Schwarz MJ, Müller N (2011) Inflammatory processes in schizophrenia: a promising neuroimmunological target for the treatment of negative/cognitive symptoms and beyond. Pharmacol Ther 132(1):96–110
Micó JA, Rojas-Corrales MO, Gibert-Rahola J, Parellada M, Moreno D, Fraguas D, Graell M, Gil J, Irazusta J, Castro-Fornieles J, Soutullo C, Arango C, Otero S, Navarro A, Baeza I, Martinez-Cengotitabengoa M, Gonzalez-Pinto A (2011) Reduced antioxidant defense in early onset first-episode psychosis: a case–control study. BMC Psychiatry 11:26
Miller B, Suvisaari J, Miettunen J, Jarvelin MR, Haukka J, Tanskanen A, Lönnqvist J, Isohanni M, Kirkpatrick B (2011) Advanced paternal age and parental history of schizophrenia. Schizophr Res 133(1–3):125–132
Montuschi P, Barnes PJ, Roberts LJ (2004) Isoprostanes: markers and mediators of oxidative stress. FASEB J 18(15):1791–1800
Morrow JD (2006) The isoprostanes – unique products of arachidonate peroxidation: their role as mediators of oxidant stress. Curr Pharm Des 12(8):895–902
Morrow JD, Roberts LJ (1997) The isoprostanes: unique bioactive products of lipid peroxidation. Prog Lipid Res 36(1):1–21
Morrow JD, Hill KE, Burk RF, Nammour TM, Badr KF, Roberts LJ (1990) A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc Natl Acad Sci U S A 97(23):9383–9387
Mukerjee S, Mahadik SP, Scheffer R, Correnti EE, Kelkar H (1996) Impaired antioxidant defense at the onset of psychosis. Schizophr Res 19(1):19–26
Müller N, Schwarz MJ (2010) Immune system and schizophrenia. Curr Immunol Rev 3:213–220
Murr C, Widner B, Wirleitner B, Fuchs D (2002) Neopterin as a marker for immune system activation. Curr Drug Metab 3(2):175–187
Murray R, Lewis S (1987) Is schizophrenia a neurodevelopmental disorder? Br Med J (Clin Res Ed) 295:681–682
Najjar S, Pearlman DM, Alper K, Najjar N, Devinsky O (2013) Neuroinflammation and psychiatric illness. J Neuroinflammation 10:43
Nakamura T, Lipton SA (2011) Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ 18(9):1478–1486
Nakanishi H, Wu Z (2009) Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav Brain Res 201(1):1–7
Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, Belforte JE (2012) GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62(3):1574–1583
Ng F, Berk M, Dean O, Bush AI (2008) Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol 11(6):851–876
O’Donnell P (2012) Cortical interneurons, immune factors and oxidative stress as early targets for schizophrenia. Eur J Neurosci 35(12):1866–1870
O’Leary DS, Flaum M, Kesler ML, Laura A, Flashman LA, Arndt S, Andreasen NC (2000) Cognitive correlates of the negative, disorganized, and psychotic symptom dimensions of schizophrenia. J Neuropsychiatry Clin Neurosci 12:4–15
Owe-Larsson B, Ekdahl K, Edbom T, Osby U, Karlsson H, Lundberg C, Lundberg M (2011) Increased plasma levels of thioredoxin-1 in patients with first episode psychosis and long-term schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 35(4):1117–1121
Padurariu M, Ciobica A, Dobrin I, Stefanescu C (2010) Evaluation of antioxidant enzymes activities and lipid peroxidation in schizophrenic patients treated with typical and atypical antipsychotics. Neurosci Lett 479(3):317–320
Palop JJ, Chin J, Mucke L (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443:768–773
Papadia S, Soriano FX, Leville F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, Craigon M, McKenzie G, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11(4):476–487
Pedrini M, Massuda R, Fries GR, de Bittencourt Pasquali MA, Schnorr CE, Moreira JC, Teixeira AL, Lobato MI, Walz JC, Belmonte-de-Abreu PS, Kauer-Sant’Anna M, Kapczinski F, Gama CS (2012) Similarities in serum oxidative stress markers and inflammatory cytokines in patients with overt schizophrenia at early and late stages of chronicity. J Psychiatr Res 46(6):819–824
Perluigi M, Coccia R, Butterfield DA (2012) 4-hydroxy-2-nonenal, a reactive product of lipid peroxidation, and neurodegenerative diseases: a toxic combination illuminated by redox proteomics studies. Antioxid Redox Signal 17(11):1590–1609
Pettegrew JW, Keshavan MS, Panchalingam K, Strychor S, Kaplan DB, Tretta MG, Allen M (1991) Alterations in brain high-energy phosphate and membrane phospholipid metabolism in first-episode, drug-naive schizophrenics. A pilot study of the dorsal prefrontal cortex by in vivo phosphorus 31 nuclear magnetic resonance spectroscopy. Arch Gen Psychiatry 48(6):563–568
Picchioni MM, Murray RM (2007) Schizophrenia. BMJ 335(7610):91–95
Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E (2008) Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 63(8):801–808
Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, Wayland M, Freeman T, Dudbridge F, Lilley KS, Karp NA, Hester S, Tkachev D, Mimmack ML, Yolken RH, Webster MJ, Torrey EF, Bahn S (2004) Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry 9(7):684–697, 643
Puri BK, Counsell SJ, Ross BM, Hamilton G, Bustos MG, Treasaden IH (2008) Evidence from in vivo 31-phosphorus magnetic resonance spectroscopy phosphodiesters that exhaled ethane is a biomarker of cerebral n-3 polyunsaturated fatty acid peroxidation in humans. BMC Psychiatry 8(Suppl 1):S2
Raffa M, Mechri A, Othman LB, Fendri C, Gaha L, Kerkeni A (2009) Decreased glutathione levels and antioxidant enzyme activities in untreated and treated schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry 33(7):1178–1183
Raffa M, Atig F, Mhalla A, Kerkeni A, Mechri A (2011) Decreased glutathione levels and impaired antioxidant enzyme activities in drug-naive first-episode schizophrenic patients. BMC Psychiatry 11:124
Raffa M, Barhoumi S, Atig F, Fendri C, Kerkeni A, Mechri A (2012a) Reduced antioxidant defense systems in schizophrenia and bipolar I disorder. Prog Neuropsychopharmacol Biol Psychiatry 39(2):371–375
Raffa M, Fendri C, Ben Othmen L, Slama H, Amri M, Kerkeni A, Mechri A (2012b) The reduction of superoxide dismutase activity is associated with the severity of neurological soft signs in patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 39(1):52–56
Rana RS, Hokin LE (1990) Role of phosphoinositols in transmembrane signaling. Physiol Rev 70(1):115–164
Ranjekar PK, Hinge A, Hegde MV, Ghate M, Kale A, Sitasawad S, Wagh UV, Debsikdar VB, Mahadik SP (2003) Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenic and bipolar mood disorder patients. Psychiatry Res 121(2):109–122
Reddy R, Sahebarao MP, Mukherjee S, Murthy JN (1991) Enzymes of the antioxidant defense system in chronic schizophrenic patients. Biol Psychiatry 30(4):409–412
Reddy R, Keshavan M, Yao JK (2003) Reduced plasma antioxidants in first-episode patients with schizophrenia. Schizophr Res 62(3A):205–212
Reynolds GP, Abdul-Monim Z, Neill JC, Zhang ZJ (2004) Calcium binding protein markers of GABA deficits in schizophrenia–postmortem studies and animal models. Neurotox Res 6(1):57–61
Rezin GT, Amboni G, Zugno AI, Quevedo J, Streck EL (2009) Mitochondrial dysfunction and psychiatric disorders. Neurochem Res 34(6):1021–1029
Ripova D, Strunecka A, Nemcova V, Farska I (1997) Phospholipids and calcium alterations in platelets of schizophrenic patients. Physiol Res 46(1):59–68
Ripova D, Strunecka A, Platilova V, Hoschl C (1999) Phosphoinositide signalling system in platelets of schizophrenic patients and the effect of neuroleptic therapy. Prostaglandins Leukot Essent Fatty Acids 61(2):125–129
Roberts LJ, Morrow JD (2000) Measurement of F2-isoprostanes as an index of oxidative stress in vivo. Free Radic Biol Med 28(4):505–513
Roberts LJ, Moore KP, Zackert WE, Oates JA, Morrow JD (1996) Identification of the major urinary metabolite of the F2-isoprostane 8-iso-prostaglandin F2α in humans. J Biol Chem 271(34):20617–20620
Rose RC, Bote AM (1993) Biology of free radical scavengers: an evaluation of ascorbate. FASEB J 7(12):1135–1142
Ross BM, Maxwell R, Glen I (2011) Increased breath ethane levels in medicated patients with schizophrenia and bipolar disorder are unrelated to erythrocyte omega-3 fatty acid abundance. Prog Neuropsychopharmacol Biol Psychiatry 35(2):446–453
Rund BR (2009) Is schizophrenia a neurodegenerative disorder? Nord J Psychiatry 63(3):196–201
Rybakowski J, Lehmann W (1997) Increased erythrocyte inositol monophosphatase activity in schizophrenia. Eur Psychiatry 12:44–45
Saadat M, Mobayen F, Farrashbandi H (2007) Genetic polymorphism of glutathione S-transferase T1: a candidate genetic modifier of individual susceptibility to schizophrenia. Psychiatry Res 153(1):87–91
Saha S, Chant D, McGrath J (2007) A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry 64(10):1123–1131
Schwarz E, Bahn S (2008) Biomarker discovery in psychiatric disorders. Electrophoresis 29:2884–2890
Scottish Schizophrenia Research Group (2000) Smoking habits and plasma lipid peroxide and vitamin E levels in never-treated first-episode patients with schizophrenia. Br J Psychiatry 176:290–293
Seybolt SE (2010) Is it time to reassess alpha lipoic acid and niacinamide therapy in schizophrenia? Med Hypotheses 75(6):572–575
Skinner AO, Mahadik SP, Garver DL (2005) Thiobarbituric acid reactive substances in the cerebrospinal fluid in schizophrenia. Schizophr Res 76(1):83–87
Skosnik PD, Yao JK (2003) From membrane phospholipid defects to altered neurotransmission: is arachidonic acid a nexus in the pathophysiology of schizophrenia? Prostaglandins Leukot Essent Fatty Acids 69(6):367–384
Srivastava N, Barthwal MK, Dalal PK, Agarwal AK, Nag D, Srimal RC, Seth PK, Dikshit M (2001) Nitrite content and antioxidant enzyme levels in the blood of schizophrenia patients. Psychopharmacology (Berl) 158(2):140–145
Stefano GB, Kim E, Liu Y, Zhu W, Casares F, Mantione KJ, Jones D, Cadet P (2004) Nitric oxide modulates microglial activation. Med Sci Monit 10(2):17–22
Steullet P, Neijt HC, Cuenod M, Do KQ (2006) Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience 137(3):807–819
Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP, Cuenod M, Do KQ (2010) Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci 30(7):2547–2558
Strous RD, Koppel M, Fine J, Nachliel S, Shaked G, Zivotofsky AZ (2009) Automated characterization and identification of schizophrenia in writing. J Nerv Ment Dis 197:585–588
Strunecká A, Rípová D (1999) What can the investigation of phosphoinositide signaling system in platelets of schizophrenic patients tell us? Prostaglandins Leukot Essent Fatty Acids 61(1):1–5
Suboticanec K, Folnegović-Smalc V, Korbar M, Mestrović B, Buzina R (1990) Vitamin C status in chronic schizophrenia. Biol Psychiatry 28(11):959–966
Szabó C, Ischiropoulos H, Radi R (2007) Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6(8):662–680
Tai G, Goff DC, Chang RW, Flood J, Baer L, Coyle JT (1998) Markers of glutamatergic neurotransmission and oxidative stress asssociated with tardive dyskinesia. Am. J. Psychiatry. 155(9):1207–1213
Tamminga CA, Holcomb HH (2005) Phenotype of schizophrenia: a review and formulation. Mol Psychiatry 10:27–39
Terpstra M, Vaughan TJ, Ugurbil K, Lim KO, Schulz SC, Gruetter R (2005) Validation of glutathione quantitation from STEAM spectra against edited 1H NMR spectroscopy at 4T: application to schizophrenia. MAGMA 18(5):276–282
Tosic M, Ott J, Barral S, Bovet P, Deppen P, Gheorghita F, Matthey ML, Parnas J, Preisig M, Saraga M, Solida A, Timm S, Wang AG, Werge T, Cuénod M, Do KQ (2006) Schizophrenia and oxidative stress: glutamate cysteine ligase modifier as a susceptibility gene. Am J Hum Genet 79(3):586–592
Tsai MC, Liou CW, Lin TK, Lin IM, Huang TL (2013) Changes in oxidative stress markers in patients with schizophrenia: the effect of antipsychotic drugs. Psychiatry Res 209(3):284–290
Tsankova N, Renthal W, Kumar A, Nestler EJ (2007) Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8(5):355–367
van Kammen DP, McAllister-Sistilli CG, Kelley ME, Gurklis JA, Yao JK (1999) Elevated interleukin-6 in schizophrenia. Psychiatry Res 87(2–3):129–136
van Os J, Murray R (2008) Gene-environment interactions in schizophrenia. Introduction. Schizophr Bull 34(6):1064–1065
Virit O, Altindag A, Yumru M, Dalkilic A, Savas HA, Selek S, Erel O, Herken H (2009) A defect in the antioxidant defense system in schizophrenia. Neuropsychobiology 60(2):87–93
Wang JF, Shao L, Sun X, Young LT (2009) Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord 11(5):523–529
Weinberger DR (1986) The pathogenesis of schizophrenia: a neurodevelopmental theory. In: Nasrallah HAW, Weinberger DR (eds) The neurobiology of schizophrenia. Elsevier, Amsterdam, pp 397–406
Weinberger DR (1987) Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44:660–669
World Health Organization (2008) International statistical classification of diseases and related health problems. 10th Revision (ICD-10). World Health Organization
Woo TU, Kim AM, Viscidi E (2008) Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res 1218:267–277
Wood SJ, Yücel M, Pantelis C, Berk M (2009) Neurobiology of schizophrenia spectrum disorders: the role of oxidative stress. Ann Acad Med Singapore 38(5):396–406
Wu Z, Zhang XY, Wang H, Tang W, Xia Y, Zhang F, Liu J, Fu Y, Hu J, Chen Y, Liu L, Chen DC, Xiu MH, Kosten TR, He J (2012) Elevated plasma superoxide dismutase in first-episode and drug naive patients with schizophrenia: inverse association with positive symptoms. Prog Neuropsychopharmacol Biol Psychiatry 36:34–38
Yao JK, Reddy RD (2003) Membrane pathology in schizophrenia: Implication for arachidonic acid signaling. Current Medicinal Chemistry-Central Nervous System Agents 3:57–65
Yao JK, Keshavan MS (2011) Antioxidants, redox signaling, and pathophysiology in schizophrenia: an integrative view. Antioxid Redox Signal 15(7):2011–2035
Yao JK, Reddy R (2011) Oxidative stress in schizophrenia: pathogenetic and therapeutic implications. Antioxid Redox Signal 15(7):1999–2002
Yao JK, van Kammen DP (1994) Red blood cell membrane dynamics in schizophrenia, I: membrane fluidity. Schizopr. Res. 209–216
Yao JK, van Kammen DP, Moss HB, Sokulski DE (1996c) Decreased serotonergic responsivity in platelets of drug-free patients with schizophrenia. Psychiatry Res. 63(2–3):123
Yao JK, Reddy R, McElhinny LG, van Kammen DP (1998a) Reduced status of plasma total antioxidant capacity in schizophrenia. Schizophr Res 32(1):1–8
Yao JK, Reddy R, van Kammen DP (1998b) Reduced level of plasma antioxidant uric acid in schizophrenia. Psychiatry Res 80(1):29–39
Yao JK, Reddy R, McElhinny LG, van Kammen DP (1998c) Effects of haloperidol on antioxidant defense system enzymes in schizophrenia. J Psychiatr Res 32(6):385–391
Yao JK, Reddy RD, van Kammen DP (2001) Oxidative damage and schizophrenia: an overview of the evidence and its therapeutic implications. CNS Drugs 15(4):287–310
Yao JK, Stanley JA, Reddy RD, Keshavan MS, Pettegrew JW (2002) Correlations between peripheral polyunsaturated fatty acid content and in vivo membrane phospholipid metabolites. Biol Psychiatry 52(8):823–830
Yao JK, Sistilli CG, van Kammen DP (2003) Membrane polyunsaturated fatty acids and CSF cytokines in patients with schizophrenia. Prostaglandins Leukot Essent Fatty Acids 69(6):429–436
Yao JK, Leonard S, Reddy RD (2004) Increased nitric oxide radicals in postmortem brain from patients with schizophrenia. Schizophr. Bull. 30(4):923–934
Yao JK, Leonard S, Reddy R (2006) Altered glutathione redox state in schizophrenia. Dis Markers 22(1–2):83–93
Yao JK, Dougherty GG Jr, Reddy RD, Keshavan MS, Montrose DM, Matson WR, McEvoy J, Kaddurah-Daouk R (2010a) Homeostatic imbalance of purine catabolism in first-episode neuroleptic-naïve patients with schizophrenia. PLoS One 5(3):e9508
Yao JK, Dougherty GG Jr, Reddy RD, Keshavan MS, Montrose DM, Matson WR, Rozen S, Krishnan RR, McEvoy J, Kaddurah-Daouk R (2010b) Altered interactions of tryptophan metabolites in first-episode neuroleptic-naive patients with schizophrenia. Mol Psychiatry 15(9):938–953
Yolken RH, Torrey EF (1995) Viruses, schizophrenia, and bipolar disorder. Clin Microbiol Rev 8:131–145
Yolken RH, Karlsson H, Yee F, Johnston-Wilson NL, Torrey EF (2000) Endogenous retroviruses and schizophrenia. Brain Res Brain Res Rev 31(2–3):193–199
Zhang XY, Zhou DF, Zhang PY, Wu GY, Cao LY, Shen YC (2002) Elevated interleukin-2, interleukin-6 and interleukin-8 serum levels in neuroleptic-free schizophrenia: association with psychopathology. Schizophr Res 57(2–3):247–258
Zhang XY, Zhou DF, Cao LY, Chen DC, Zhu FY, Wu GY (2003a) Blood superoxide dismutase level in schizophrenic patients with tardive dyskinesia: association with dyskinetic movements. Schizophr Res 62(3):245–250
Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY (2003b) Elevated blood superoxide dismutase in neuroleptic-free schizophrenia: association with positive symptoms. Psychiatry Res 117(1):85–88
Zhang XY, Zhou DF, Cao LY, Zhang PY, Wu GY, Shen YC (2003c) The effect of risperidone treatment on superoxide dismutase in schizophrenia. J Clin Psychopharmacol 23(2):128–131
Zhang XY, Tan YL, Cao LY, Wu GY, Xu Q, Shen Y, Zhou DF (2006) Antioxidant enzymes and lipid peroxidation in different forms of schizophrenia treated with typical and atypical antipsychotics. Schizophr Res 81(2–3):291–300
Zhang XY, da Chen C, Xiu MH, Wang F, Qi LY, Sun HQ, Chen HSC, Wu GY, Haile CN, Kosten TA, Lu L, Kosten TR (2009a) The novel oxidative stress marker thioredoxin is increased in first-episode schizophrenic patients. Schizophr Res 113(2–3):151–157
Zhang XY, Zhou DF, Qi LY, Chen S, Cao LY, da Chen C, Xiu MH, Wang F, Wu GY, Lu L, Kosten TA, Kosten TR (2009b) Superoxide dismutase and cytokines in chronic patients with schizophrenia: association with psychopathology and response to antipsychotics. Psychopharmacology (Berl) 204(1):177–184
Zhang M, Zhao Z, He L, Wan C (2010) A meta-analysis of oxidative stress markers in schizophrenia. Sci China Life Sci 53(1):112–124
Zhang XY, da Chen C, Xiu MH, Tang W, Zhang F, Liu L, Chen Y, Liu J, Yao JK, Kosten TA, Kosten TR (2012) Plasma total antioxidant status and cognitive impairments in schizophrenia. Schizophr Res 39(1–3):66–72
Zhang XY, da Chen C, Xiu MH, Yang FD, Tan YL, He S, Kosten TA, Kosten TR (2013) Thioredoxin, a novel oxidative stress marker and cognitive performance in chronic and medicated schizophrenia versus healthy controls. Schizophr Res 143(2–3):301–306
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Dietrich-Muszalska, A. (2015). Oxidative Stress in Schizophrenia. In: Dietrich-Muszalska, A., Chauhan, V., Grignon, S. (eds) Studies on Psychiatric Disorders. Oxidative Stress in Applied Basic Research and Clinical Practice. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-0440-2_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0440-2_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-0439-6
Online ISBN: 978-1-4939-0440-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)