
Abstract
The synthesis and characterization of new lithium salts has been a core component of electrolyte research for the past three decades. Upon the commercialization of Li-ion batteries with a graphite anode, LiPF6 became the dominant salt for lithium battery electrolytes. But the advent of new electrodes/cell chemistries (e.g., Si alloy anodes, high-voltage cathodes, Li-air, Li-S), as well as the need for exceptional battery safety, higher/lower temperature operation, improved durability/longer lifetimes, etc., has resulted in the pressing need for new electrolyte formulations. Lithium salts, either as a substitute for LiPF6 or as an additive, are one central focus for this electrolyte transformation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1.1 Introduction
Early lithium battery electrolyte research in the 1970s used available lithium salts—i.e., principally LiClO4, LiAlCl4, LiBF4, LiPF6, and LiAsF6. Efforts at the time were devoted to stabilizing the stripping/plating of Li metal [1–3], as well as the use of intercalation electrodes [4]. LiPF6 was reported to not provide the best Li metal stripping/plating efficiency amongst the salts studied [5, 6] and LiPF6 was also found initially to be problematic for the cycling of carbon electrodes [7, 8]. LiPF6 did not become the paramount salt for lithium battery electrolytes until carbon coke replaced Li metal (later to be replaced by graphite) and the solvents were optimized for the electrolyte utilized in the Li-ion batteries initially commercialized by Sony in 1991 [9–19].
Many/most of the anions used for electrolyte lithium salts were initially developed in efforts to generate stronger superacids. The term superacid generally refers to acids which are more acidic than mineral Brønsted acids [20–27]. In particular, a widely used definition for superacids was given by Gillespie who defined this term to be applicable to acids which are more acidic than sulfuric acid [28–30]. The stronger the acidity, the weaker the coordination of the anions is with the associated protons (H+ cations). The gas-phase acidity is given by:
and
In practice, however, the determination of the acidity of superacid anions in solution is not a trivial matter as the acidity of a given acid (HA) is a function of the solvent (S) used:
which is influenced by factors such as the steric bulk of the anions and solvent, ion pairing, etc. Three major substitution effects contribute to increasing the acidity of a neutral acid functional group (C–H, O–H, N–H, S–H, B–H, etc.) [20, 26]:
-
1.
field/inductive effects
-
2.
π-electron acceptor (resonance) and negative hyperconjugation effects
-
3.
substituent polarization effects
The influence of these factors is evident from the acidity variations noted for different substituents (Figs. 1.1 and 1.2) [20, 26]. No simple patterns in acidity behavior are found for these effects, however, due to the interplay between them (i.e., these effects do not operate independently of one another) [20, 26]. For =Z(X) n substituents (replacing an =O: M=O → M=Z(X) n , where M=O might be CH3C(=O)H, HC(=O)NH2, HC(=O)OH, etc.), the acidification for the same X increases with increasing n [22]:

Experimental gas-phase acidity values (kcal mol−1) for various monosubstituents [20]

In addition, the acidifying effect of fluorosulfonyl groups is greater than that of the corresponding cyano groups (Fig. 1.2) [22].
Classical strong mineral acids include:
(experimental gas-phase acidity (ΔG acid) values in kcal mol−1) [31]. Interestingly, the gas-phase acidity may be increased somewhat by the replacement of fluorine atoms with chlorine [32, 33]. The values noted above for HF and HCl are one example. The corresponding values for HCO2CH2F, HCO2CH2Cl, HCO2CHF2, and HCO2CHCl2 are 337.6, 335.4, 330.0, and 328.4 kcal mol−1, respectively [32]. This effect, which is opposite to that expected from electronegativity alone, is attributed to the greater delocalization of the charge on the chlorine atom relative to fluorine. Thus, HSO3Cl is expected to be a stronger acid than HSO3F [26, 32, 33].
Replacement of the acid protons with Li+ cations results in the corresponding lithium salts. Due to differences between the gas and condensed liquid-phase interactions, as well as the varying factors which determine proton (H+) and Li+ cation coordination/solvation, the exact trends noted for anion acidity do not always hold for the relative strength of lithium salt interactions. But, overall, the ionic association tendency of anions to coordinate Li+ cations is governed by the same effects noted above for acidity, as exemplified by DMSO electrolytes with lithium acetate salts with increasing anion fluorination which have the following increasing ionic association trend [34]:
Efforts to develop new lithium salts began in the late 1970s and 1980s with the use of perfluoroalkylsulfate (e.g., SO3CF3 −) and bis(perfluoroalkanesulfonyl)imide (e.g., N(SO2CF3)2 −) anions. New salt development efforts gained momentum throughout the next three decades, but none of the developed salts have offered significant advantages over—while retaining the benefits of—the LiPF6 salt, which serves as the standard for industry. Thus, LiPF6 continues to be the predominant salt used for most commercial Li-ion batteries.
Some of the new lithium salts, however, have been found to be very effective as electrolyte additives for the modification of electrolyte–electrode interfaces. Other lithium salts hold promise as future primary salts for lithium batteries (i.e., replacements for some or all of the LiPF6), especially for demanding battery electrolyte requirements such as low/high-temperature operation and superb safety characteristics, which cannot be met by the sole use of LiPF6 [35–38]. Thus, there is a continued need for rapid advances in lithium salts for the diverse range of lithium battery chemistries which are now the focus of worldwide efforts to greatly improve vehicular and stationary energy storage technologies.
1.2 Electrolyte Salt Properties
Electrolyte salts must meet a broad and demanding range of properties—some of these include [12, 37, 39, 40] the following:
-
1.
Ionic Conductivity: A high Li+ cation transport rate is necessary to achieve high power (i.e., a high rate for the overall battery reaction) as the Li+ cation mobility within the bulk electrolyte is often one of the main sources of impedance for the battery [41]. The choice of a lithium salt’s anion dramatically influences the electrolyte’s conductivity due to the variations in the Li+ cation solvation and ionic association interactions resulting from the differences in anion structure and coordination strength. Although electrolyte conductivity is the parameter most frequently considered, the Li+ cation mobility is actually obtained from the product of the conductivity and Li+ cation transference (or transport) number (i.e., t Li+) (fraction of the current carried by the Li+ cations). This latter parameter is frequently not reported in the literature for a given electrolyte composition. The most common t Li+ measurement method is that reported by Bruce and Vincent [42–44]. Caution should be exercised in interpreting results from such measurements, however, as the data can be skewed by the reaction of the electrolyte with Li metal (active electrodes) resulting in erroneous tLi+ values, especially for liquid electrolytes.
-
2.
Salt Solubility/Crystalline Solvates: Achieving a reasonably high lithium salt solubility in the electrolyte solvent(s) is necessary to provide sufficient charge carriers for rapid ionic conduction, as well as to prevent salting out of the salt (i.e., precipitation). It is important to distinguish between salt solubility and crystalline solvates. A salt may be highly soluble, but also may readily form a crystalline solvate phase with a high melting point (T m) resulting in the formation of solids in the electrolyte which effectively extracts the salt from the electrolyte solution (causing the conductivity to plummet). This is a particular concern for low-temperature cell operation.
-
3.
Stability: Electrolytes must, in general, be robust (nonreactive with other cell components) within the electrochemical potential window utilized for the battery charge/discharge reactions [45], as well as at elevated temperature, to achieve thousands of charge/discharge cycles with low capacity loss (fade). The temperature and voltage(s) at which oxidation and/or reduction of the electrolyte components occur are not independent of one another. As for chemical stability, the electrochemical stability of electrolytes (i.e., potential stability window) is strongly temperature dependent with a modest increase in temperature, in some cases, resulting in a substantial decrease in stability [46]. The potential window is also a strong function of the materials in contact with the electrolyte [46], as well as the presence of impurities. Thus, the potential window, as measured on an inert glassy carbon electrode, is not a clear indicator of the stability of an electrolyte in contact with active electrode materials (although a poor stability on this electrode likely also indicates a poor stability with more active electrodes). Stability is a complicated factor, however, as it may also be necessary for the salt’s anions to selectively degrade—e.g., to form a solid-electrolyte interface (SEI) layer on the anode and/or cathode [12] and to stabilize the Al current collector [47].
-
4.
SEI Formation: The SEI is a layer formed between the electrode surface and the electrolyte through the degradation/reaction of both electrolyte components and electrode material(s) [12]. Ideally, only a limited amount of materials react, with the resulting SEI layer preventing further electrode–electrolyte reactions and enabling the facile transport of only Li+ cations between the electrode and electrolyte (resulting in a low impedance). The lithium salt(s) present in the electrolyte, whether as a bulk salt or an additive, can dramatically influence the SEI’s composition, properties, and stability [48].
-
5.
Al Corrosion: The use of Al as a cathode current collector in commercial Li-ion batteries is nearly ubiquitous [47]. A given electrolyte must passivate the electrolyte–Al interface to prevent corrosive pitting of the current collector during cell cycling to high potential (>3.6 V vs. Li/Li+).
-
6.
Hydrolysis Stability: Many anions hydrolyze when exposed to water, especially at elevated temperature—often resulting in the formation of HF. This results in additional costs associated with the salt’s preparation, storage, and handling. This also strongly influences the cycling behavior and lifetime of batteries, especially when cycled to high temperature and/or high potential (>4.8 V vs. Li/Li+) [49–54]. HF formation may also result from the reaction of the anions with solvent molecules (abstraction of a hydrogen) [55].
Other desirable features include low cost and low toxicity. Failure to meet one or more of these criterions prevents the practical use of a salt in lithium and Li-ion batteries. It is important to note, however, that many of these properties are strongly dependent upon the electrolyte formulation (e.g., solvents used, salt concentration, additives). Thus, electrolyte compositions need to be tailored to specific battery applications/demands.
1.3 Established Salts
A number of lithium salts are well established as salts which are used or have been previously used for lithium battery electrolytes, although many of these do not meet the necessary criterion for commercial battery electrolytes. Frequently studied lithium salts include (Fig. 1.3) [12, 40] the following:

Widely used lithium salt anions: (a) AsF6 −, (b) PF6 −, (c) ClO4 −, (d) BF4 −, (e) SO3CF3 −, and (f) N(SO2CF3)2 − (TFSI−) (C 1 (cis) and C 2 (trans) conformations) (B—tan, C—gray, N—blue, O—red, F—light green, P—orange, S—yellow, Cl—dark green, As—purple)
-
1.
LiClO 4 : Lithium perchlorate was widely used for battery electrolyte research in the 1970s and 1980s due to its high ionic conductivity, high solubility in aprotic solvents, high thermal/electrochemical stability, and favorable SEI-forming properties [12, 56]. Electrolytes with the LiClO4 salt, however, typically do not passivate the Al current collector as well as those with LiPF6 [38, 57–60]. The high oxidation state of the ClVII atom also makes the anion a strong oxidant and thus the salt a potential explosive [61–63]. This has largely precluded the use of LiClO4 for commercial batteries.
-
2.
LiAsF 6 : Like LiClO4, lithium hexafluoroarsenate was widely used for electrolyte research in the 1970s and 1980s. In particular, it was found to improve the efficiency of Li metal plating/stripping relative to electrolytes with LiClO4 [12]. LiAsF6 has many properties in common with LiPF6 [12, 40, 64], but the potential hazards associated with the salt have largely prevented its commercial usage. Although the AsV oxidation state is not toxic, the AsIII and As0 states, which might be formed from electrochemical reduction, are highly toxic.
-
3.
LiPF 6 : Lithium hexafluorophosphate is used almost exclusively in commercial Li-ion batteries. This salt has thus far demonstrated the best balance of essential properties necessary for a primary Li-ion electrolyte salt [12, 40]. In aprotic solvents, the resulting electrolytes have some of the highest conductivity values measured. LiPF6-based electrolytes react to form a stable interface with the Al current collector at high potential [58–60, 65–68] and a stable SEI with graphite electrodes when used with carbonate solvents [12, 40]. The P–F bond is labile, however, and the salt thus readily undergoes hydrolysis [69–73] and has a relatively low thermal stability [56, 71, 74–78]. The presence of HF in LiPF6-based electrolytes, and its impact on cell performance, is one of the principal concerns associated with this salt’s usage [49–54].
-
4.
LiBF 4 : Electrolytes with lithium tetrafluoroborate tend to have a significantly lower conductivity, relative to those with LiPF6 [79–81], which has been a major impediment to its use in commercial Li-ion cells. The B–F bond is less labile than the P–F bond. Thus, the LiBF4 salt is less susceptible to hydrolysis and more thermally stable than LiPF6 [56, 82–85], but electrolytes with this salt do passivate Al well at high potential [59, 67]. Despite the lower conductivity, electrolytes with LiBF4 have been shown to have improved cell cycling performance at low/high temperature, relative to cells with LiPF6-based electrolytes, due to the formation of a less resistive SEI layer and improved thermal stability [81, 82, 86–89]. LiBF4 may also serve as a useful additive to electrolytes with LiPF6 [90] and it enables the use of γ-butyrolactone (GBL) as an electrolyte solvent (which is unstable with LiPF6) [91–96].
-
5.
LiSO 3 CF 3 : Lithium trifluoromethanesulfonate (triflate—most commonly abbreviated as “CF3SO3 −” in the scientific literature) was at one time widely used for electrolytes, especially for polymer electrolytes [97–107]. This salt has a high thermal stability [56] and is not susceptible to hydrolysis due to the stability of the C–F bond. Electrolytes with this salt, however, are found to be notably less conductive than those with LiPF6 [5, 108–110], and LiSO3CF3-based electrolytes corrode the Al current collector at high potential [59, 65–67]. Thus, while this salt has been extensively used for research purposes, it is not used in commercial Li-ion batteries.
-
6.
LiTFSI (i.e., LiN(SO 2 CF 3 ) 2 ): Many acronyms are used for the lithium bis(trifluoromethanesulfonyl)imide salt: LiTFSI, LiTFSA, LiNTf2, LiTf2N, etc. The terms “imide” and “amide” are frequently interchanged. Much of the initial interest in the LiTFSI salt was due to its tendency to form amorphous mixtures with poly(ethylene oxide), rather than crystalline phases, when mixed to form polymer electrolytes [97, 111–113]. The bis(perfluoroalkanesulfonyl)imide anions, such as TFSI−, are highly flexible with two low-energy conformations in which the –CF3 groups are either cis (C 1) or trans (C 2) to one another (Fig. 1.3f) [114–117]. The combination of the strongly electron-withdrawing fluorine atoms and resonance structures due to the sulfonyl groups results in extensive negative-charge delocalization across the –SO2–N–SO2– backbone of the anion [108, 118]. Thus, TFSI−…Li+ cation coordination occurs predominantly through oxygen atom coordination to the Li+ cations (rather than N…Li+ or F…Li+ cation coordination) [119–124]. Electrolytes with this salt generally are somewhat less conductive than the corresponding LiPF6 electrolytes [5, 11, 125]. The TFSI− anion has a high thermal stability and is not susceptible to hydrolysis due to the very stable C–F bonds [56, 126], but dilute aprotic solvent-base electrolytes with LiTFSI are known to strongly corrode the Al current collector at high potential [59, 60, 65–68]. This observation should be qualified with the fact that electrolytes with ionic liquids with the TFSI− anion (with or without LiTFSI) do not corrode the Al current collector [127–132], nor do electrolytes with very high concentrations of LiTFSI [133]. Thus, simple generalizations about electrolyte salt properties (e.g., “the LiTFSI salt corrodes Al”) may be flawed.
1.4 Electrolyte Characterization Tools
Rigorous electrolyte characterization requires a thorough understanding of not only the properties of the electrolyte, but also the solution structure. Anion solvation in protic solvents (e.g., water, methanol, ammonia) occurs through hydrogen-bonding interactions. Protic solvents, however, have poor electrochemical stability due to the acidic protons (i.e., O–H, N–H, S–H). The aprotic solvents useful for electrolyte applications do not have acidic protons. Thus, in general, the anions remain unsolvated (naked) in electrolytes with such solvents. Dissolution of a lithium salt therefore occurs through the solvation of the Li+ cations by the formation of coordination bonds between the Li+ cations and electron lone-pairs of the solvent donor atoms. Anions also form coordination bonds to the Li+ cations using the electron lone-pairs on donor atoms (F, O, N, etc.). The competition between the solvent and anions for Li+ cation coordination—the occupancy of the Li+ cation’s coordination shell—determines the solvate species which are present in the electrolyte. Thus, the ion solvation (solvent–Li+ cation interactions) and ionic association tendency of the anions (anion–Li+ cation interactions) are important features of electrolytes which are governed by the solvent/anion structure: steric factors which influence the coordination bond formation and packing around the cation, polarizability, charge delocalization, etc. The following discussion provides a short overview of a methodology which may be used to identify the electrolyte interactions and how these are determinants for electrolyte properties [134–136]:
-
1.
Phase Diagrams and Solvate Crystal Structures: Solvent-lithium salt phase diagrams are an underutilized, but highly informative tool for examining electrolyte interactions. Acetonitrile (AN) is a particularly useful model solvent for comparing a salt’s phase behavior as this solvent has only a single electron lone-pair and thus is either uncoordinated or coordinated to a single Li+ cation. Figure 1.4 compares the phase diagrams of (AN) n –LiX mixtures with LiPF6, LiTFSI, LiClO4, LiBF4, and LiCO2CF3 [134, 135]. Figure 1.4 also shows the solvent/ion coordination in the solvate crystal structures determined for some of the indicated phases in the phase diagrams: (AN)6:LiPF6 [137], (AN)5:LiPF6 [138], (AN)1:LiTFSI [139], (AN)4:LiClO4 [140], (AN)2:LiBF4 [141], and (AN)1:LiBF4 [142]. Notable features include the tendency of the (AN) n –LiPF6 and (AN) n –LiClO4 mixtures to form crystalline solvates with a high T m. This is attributed to the small, symmetric, and relatively weakly coordinating anions which readily pack well within the solvate crystalline lattices. In contrast, dilute (AN) n –LiTFSI mixtures form low T m solvates and a crystallinity gap exists for more concentrated mixtures for which it is difficult or impossible to crystallize the electrolytes—these features may be attributable to the bulky anion with lower symmetry and its influence on solvate formation. The ClO4 − and BF4 − anions are both tetrahedral and nearly of the same size (Fig. 1.3c, d). Interestingly, although the (AN) n –LiClO4 and (AN) n –LiBF4 mixtures form the same solvate crystalline phases (i.e., 4/1, 2/1, and 1/1 AN/LiX compositions), significant differences exist in the T m of the 4/1 phases in which the Li+ cations are fully solvated by four AN molecules and the anions are uncoordinated (Fig. 1.4d). This can be explained by the difference in the solvation/ionic association tendency of the two salts, as the BF4 − anions have a greater tendency to displace the solvent molecules in the Li+ cation coordination shells (and thus a greater tendency to disrupt the solvate structure). The 6/1 and 5/1 solvates with LiPF6 also form solvates with four-fold Li+ cation coordination (Fig. 1.4a, b), but also include uncoordinated solvent molecules to facilitate the packing of the solvated Li+ cations and PF6 − anions together. Finally, the (AN) n –LiCO2CF3 mixtures do not form crystalline solvates. Only some of the excess AN is able to crystallize as a pure solvent phase.
Fig. 1.4 
Phase diagrams of (AN) n –LiX mixtures with LiPF6, LiTFSI, LiClO4, LiBF4, and LiCO2CF3 [134, 135] and ion/solvent coordination within the solvate crystal structures: (a) (AN)6:LiPF6 [137], (b) (AN)5:LiPF6 [138], (c) (AN)1:LiTFSI [139], (d) (AN)4:LiClO4 [140], (e) (AN)2:LiBF4 [141], and (f) (AN)1:LiBF4 [142] (the sample T g values are indicated by an “x” for fully amorphous samples and by a triangle for partially crystalline samples)
-
2.
Li + Cation Solvation: Electrolyte (average) solvation numbers are most commonly determined using vibrational spectroscopy by examining the solvent’s vibrational band(s). Upon coordination to a Li+ cation through an electron lone-pair, the electron density of the solvent molecule (and thus bond lengths/angles) changes resulting in variations in the solvent band positions [134, 135]. Integration of the peak area of the bands associated with the uncoordinated and coordinated solvent enables the calculation of the fraction of coordination solvent molecules—this number multiplied times the total number of solvent molecules in the electrolytes gives the average solvation number (Fig. 1.5). This analysis can be confounded, however, by overlapping vibrational bands, as well as variations in the relative intensity (scaling) of the peaks. Failure to account for these factors can result in a highly misleading interpretation of the experimental data. The scientific literature related to the determination of solvation numbers is rife with these problems. From an analysis of the AN Raman C–C and C≡N stretching vibrations [134, 135], ion solvation for (AN) n –LiX mixtures increases in the order:
Fig. 1.5 
Calculated Li+ cation average solvation numbers (AN/Li) for (AN) n –LiX electrolytes. The dark solid line corresponds to the average of the data obtained from the analysis of the C–C and C≡N Raman vibrational stretching bands (note that values for approximately n > 9 are unreliable due to a compilation of experimental errors associated with the Raman solvent band deconvolution) [134, 135]
$$ {\mathrm{LiPF}}_6>\mathrm{LiTFSI}\ge {\mathrm{LiClO}}_4>{\mathrm{LiBF}}_4>>{\mathrm{LiCO}}_2C{F}_3 $$The maximum ion solvation number (average number of solvent molecules coordinated to the Li+ cations) for the AN mixtures is found to be about 4 [134, 135]. Wide differences are noted for the solvation numbers for the varying anions. For example, for highly concentrated (>3 M) (AN) n –LiX (n = 4) liquid mixtures (i.e., four AN molecules present per Li+ cation) at 60 °C, the solvation numbers are approximately 3.2 for LiPF6, 2.8 for LiTFSI, 2.7 for LiClO4, 2.1 for LiBF4, and 1.0 for LiCO2CF3 [134, 135]. Note that these numbers are not the coordination numbers for the Li+ cations. Rather, they represent the average number of coordinated solvent molecules with the anions making up the difference in the Li+ cation coordination shells. Thus, the Li+ cations in the LiPF6 electrolytes are expected to be well solvated, whereas those for the LiCO2CF3 electrolytes are instead expected to be highly associated to the anions over the entire concentration range. Despite this, the solubility of LiCO2CF3 in AN is exceptionally high (>5 M). Thus, salt solubility and ionic association are not necessarily directly correlated with one another.
-
3.
Ionic Association: The ionic association present in electrolytes is most commonly also determined using vibrational spectroscopy by examining one or more anion vibrational bands which shift upon coordination to the Li+ cations [103–105, 121, 134, 135, 143, 144]. Often, the assignment of the bands to specific modes of coordination is somewhat ambiguous and the analysis of the data is simply based upon guesswork. This can be very misleading. To resolve this problem, the study of the vibrational bands for model crystalline (solid) solvates with known structure is particularly useful for assigning the vibrational band positions to particular anion…Li+ cation coordination modes. This evaluation has been done for the salts LiClO4 [145, 146], LiBF4 [141], and LiDFOB [147]. Work is currently in progress to provide similar analyses for LiPF6, LiTFSI, and LiSO3CF3. An example of the modes of coordination for the BF4 − anion and the corresponding anion band positions obtained from crystalline solvates (such as AGG-I (AN)2:LiBF4 and AGG-II (AN)1:LiBF4—Fig. 1.4e, f) is shown in Fig. 1.6 [141]. This characterization “tool” is quite useful for the evaluation (deconvolution) of the Raman band data for the liquid electrolytes shown in Fig. 1.7b. This form of analysis is greatly complemented by molecular dynamics (MD) simulations (validated by the experimental data) which provide a visual representation of the solvates present in solution (Fig. 1.8) [134–136]. As an example of the utility of this marriage of methods, the solvates shown in Fig. 1.8c, d represent contact ion pairs (CIP-I and CIP-II, respectively). But the solvates in Fig. 1.8b, e, g–j also contain BF4 − anions coordinated to a single-Li+ cation. Thus the spectral signature of these anions would be that of CIP-I anion coordination (Fig. 1.6b). The experimental spectroscopic data, therefore, does not provide direct information about the solvates present. Instead, it indicates the fraction of the anions present with different modes of anion…Li+ cation coordination. This is an important distinction which is generally not made throughout the published scientific literature on electrolyte characterization. The ionic association tendency of lithium salts is found to increase in the order [134–136, 148]:
Fig. 1.6 
Varying modes of BF4 −…Li+ cation coordination: (a) SSIP, (b) CIP-I, (c) CIP-II, (d) AGG-I, (e) AGG-II, and (f) AGG-III and Raman band peak positions for the BF4 − anion v 1 vibrational band for different crystalline solvates (each line corresponds to a different crystalline solvate) (no crystalline solvates with CIP-II coordination were available for analysis) [141]
Fig. 1.7 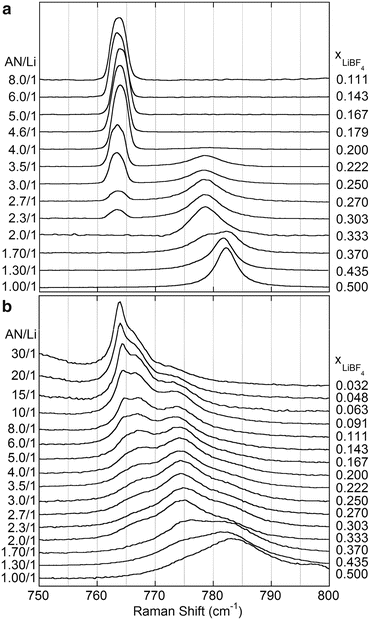
BF4 − anion band variation with concentration for the (AN) n –LiBF4 mixtures at (a) −80 °C and (b) 60 °C [134]
Fig. 1.8 
Representative Li+ cation solvate species (i.e., coordination shells) extracted from the MD simulations for the (AN) n –LiBF4 mixtures (n = 30, 20, and 10) at 60 °C with BF4 − coordination: (a) SSIP; (b) CIP-I, CIP-II; (c) CIP-I; (d) CIP-II; (e) CIP-I, AGG-I; (f) AGG-I; (g) CIP-I, AGG-I (×3); (h) CIP-I, AGG-I (×3), AGG-III; (i) CIP-I, AGG-I (×2); (j) CIP-I (×2), AGG-I; and (k) AGG-I (×3). Only solvent and BF4 − anions within 3.33 Å of a Li+ cation are shown (Li—purple, B—tan, C—gray, N—blue, F—light green) [134]
$$ {\mathrm{LiAsF}}_6,{\mathrm{LiPF}}_6<\mathrm{LiTFSI}\le {\mathrm{LiClO}}_4<{\mathrm{LiBF}}_4<{\mathrm{LiSO}}_3C{F}_3<{\mathrm{LiCO}}_2C{F}_3 $$A comparison of this with the Li+ cation solvation data (Fig. 1.5) indicates that this increasing ionic association tendency is opposite to that noted for the Li+ cation solvation. This is due to the competitive coordination of the solvent molecules and anions to the Li+ cations. Quantum chemical (QC) calculations and MD simulations find that (AN) n –LiX mixtures consist predominantly of Li+ cations with four-fold coordination to anions and/or AN solvent molecules (very little five-fold Li+ cation coordination is found for AN-based electrolytes) [134–136]. As noted above, aprotic solvent molecules, such as AN, have only weak interactions with anions. Thus, the Li+ cation coordination shell in solution consists of anions and/or solvent molecules. The competitive coordination between these determines the solvate distribution present in solution (Fig. 1.8) [134–136, 141].
-
4.
Transport Properties: The transport properties (e.g., viscosity and ionic conductivity) of electrolytes are one of the key metrics used to gauge electrolytes. The viscosity may be correlated with the wettability of an electrolyte with the porous separator and electrodes. Figure 1.9 shows the variation in the viscosity of (AN) n –LiX mixtures at 60 °C with different lithium salts [136]. Perhaps contrary to expectations, the most dissociated salts result in the highest viscosity for the more dilute mixtures. The differences in the viscosity for the different salts and concentrations can be explained by the differences in solution structure (amount of uncoordinated solvent and number/distribution of solvates) (Figs. 1.8 and 1.10) [136]. Figure 1.11 shows the variation in the conductivity of the (AN) n –LiX mixtures at 60 °C with the different lithium salts [136]. The differences noted in terms of both salt concentration and anion structure can once again be explained by the differences in electrolyte solution structure (types and distribution of solvates and uncoordinated anions, as well as the solvate formation/evolution dynamics) [136]. It is important to note that the AN electrolytes with LiPF6 have the highest conductivity, but also the highest viscosity (Fig. 1.9). Similarly, the AN electrolytes with LiCO2CF3 have the lowest conductivity and the lowest viscosity. This clearly shows that the conventional wisdom that conductivity is directly linked with viscosity—“a high conductivity is achieved for electrolytes with a low viscosity”—is inaccurate. Instead, both properties originate from the molecular-level interactions between the ions and solvent (i.e., solution structure). These properties are thus only indirectly correlated with one another. Frequently, it is found that a maximum in electrolyte conductivity is obtained near a 1 M salt concentration (Fig. 1.12) (for the AN electrolytes shown in Fig. 1.11, this corresponds to an AN/Li ratio of about 14 to 17—depending upon the salt’s formula weight [136]). In addition, the choice of aprotic solvent or solvent mixtures used greatly impacts the conductivity of an electrolyte (Figs. 1.12 and 1.13) [5, 149–153], but importantly the trend in the conductivity for different salts remains largely the same irrespective of the solvent(s) used. For example, the conductivity with different lithium salts in an EC:DMC equimolar binary mixture with the solvents 3-methylsydnone (3-MSD) or 3-ethylsydnone (3-ESD) added is found to increase in the order [154]:
Fig. 1.9 
(a) Viscosity of (AN) n –LiX mixtures at 60 °C (AN/LiX (n) noted in plots) and (b) the same data for the dilute mixtures alone. Data for concentrated mixtures with LiPF6 and LiClO4 were not gathered as these samples crystallize during the measurements [136]
Fig. 1.10 
Snapshot of the molecular simulations of (AN) n –LiX mixtures (n = 20) with (a) LiPF6 and (b) LiBF4 (Li—purple, B—tan, C—gray, N—blue, F—light green, P—orange). Uncoordinated AN solvent molecules have been removed to aid in discerning the solvates present [136]
Fig. 1.11 
Ionic conductivity of (AN) n –LiX mixtures at 60 °C (solvent/LiX ratio (n) noted at the top of the plot) (LiSO3CF3 data not shown due to crystalline solvate formation) [136]
Fig. 1.12 
Ionic conductivity of EC:PC (50:50 v:v) and 2-MeTHF:EC:PC (75:12.5:12.5 v:v:v) mixtures with LiAsF6 for different temperatures and salt concentrations [5]
Fig. 1.13 
Ionic conductivity of 1 M LiAsF6 electrolytes with the indicated solvents (either a single solvent or a 50:50 v:v binary mixture) [5]
$$ {\mathrm{LiPF}}_6>{\mathrm{LiClO}}_4>\mathrm{LiN}{\left({\mathrm{SO}}_2C{F}_3\right)}_2(LiTFSI)>\mathrm{LiN}{\left(S{O}_2{C}_2{F}_5\right)}_2>{\mathrm{LiBF}}_4>{\mathrm{LiSO}}_3C{F}_3 $$which is similar to the trend in Fig. 1.11 (if LiClO4 and LiTFSI are interchanged). A similar order is also noted for the electrolytes in Table 1.1 and Fig. 1.14 [5, 108–110, 125, 150–153, 155, 156]. This latter figure indicates that the EC:PC electrolyte with LiPF6 is slightly more conductive than the corresponding electrolyte with LiAsF6, whereas the opposite is true for the 2-MeTHF:EC:PC electrolytes. Also, there is a crossover in the conductivity for the 2-MeTHF:EC:PC electrolytes with LiBF4 or LiSO3CF3 with varying temperature. These points demonstrate that the ion solvation and ionic association interactions within the electrolytes are a function of numerous factors and generalizations about salt properties/behavior should be used with caution, as noted above.
Table 1.1 Conductivity (mS cm−1) of electrolytes with various lithium salts (1 M) at 25 °C (av:v; bw:w) [108–110, 125, 155, 156] Fig. 1.14 
Ionic conductivity of EC:PC (50:50 v:v) and 2-MeTHF:EC:PC (75:12.5:12.5 v:v:v) mixtures with the indicated lithium salts (1 M) [5]











1.5 Advanced Salts—Fluoroborates and -Phosphates
Lithium salts with tetraalkylborate anions are highly soluble in dioxolane (up to 3 M) [157]. This is due to the poorly coordinating tetraalkylborate anions which lack donor atoms with electron lone-pairs for Li+ cation coordination [158]. Thus, solvent molecules readily displace the anions in the Li+ cation coordination shells. This also accounts for the poor chemical stability of the salts [159]: lithium tetramethylborate (i.e., LiB(CH3)4) (Figs. 1.15a and 1.16a) [158, 160–162] is stable in air [157], but lithium tetrabutylborate (i.e., LiB(C4H9)4) (Fig. 1.15c) is pyrophoric [157]. In the former salt, the Li+ cations are coordinated by the methyl hydrogens [158], but less favorable coordination may occur for anions with longer alkyl chains making the anions more reactive. Lithium tetraphenylborate (LiBPh 4 ) (i.e., LiB(C6H5)4) (Figs. 1.15h and 1.16c) is stable only when the Li+ cations are fully solvated—this salt is typically sold commercially as the (DME)3:LiBPh4 solvate, while crystal structures have been reported for the (H2O)4:LiBPh4 [163], (H2O)2(DME)1:LiBPh4 [164], (H2O)2(THF)2:LiBPh4 [165], (THF)1(12C4)1:LiBPh4 [166], (triglyme)1:LiBPh4 [167], and P(EO)5:LiBPh4 [168] solvates—all with fully solvated Li+ cations and uncoordinated BPh4 − anions. The unsolvated LiBPh4 salt slowly reacts with dry air. PC-based electrolytes with LiBPh4 (and some DME) have a conductivity which is similar (slightly lower) to that of electrolytes with LiBF4, but the conductivity of THF- and DME-based electrolytes with LiBPh4 is higher than for those with LiBF4 [169]. This may be attributable to variations in the ionic association interactions for the LiBF4 salt with the different solvents, whereas the LiBPh4 salt remains fully dissociated for all of the electrolyte formulations.


Anion structures: (a) B(CH3)4 −, (b) B(C2H5)4 −, (c) B(C6H5)4 − (BPh4 −), (d) BF3(CF3)− (e) BF3(C2F5)− (FAB−), (f) BF2(CF3)2 −, (g) BF(CF3)3 −, (h) B(CF3)4 −, (i) BF3Cl−, (j) BF3(C6H5)−, (k) BF2(C6F5)2 −, (l) B(C6F5)4 − (BArF−), and (m) PF3(C2F5)3 − (FAP−) (B—tan, C—gray, F—light green, P—orange)
The anions from conjugate Brønsted–Lewis superacids represent the core lithium salts used for commercial lithium batteries (i.e., LiPF6 and LiBF4). The acidity order determined from QC calculations is as follows: HBF4 (287.7) < HPF6 (276.6) < HTaF6 (268.3) < HAlCl4 (257.4) < HSbF6 (255.5) (DFT-calculated ΔG acid values in kcal mol−1) [21]. LiBF4 and LiPF6 have both been extensively used for lithium battery research, and the latter is used in commercial Li-ion cells. The synthesis of LiTaF6 and LiNbF6 has been reported [170–175]. Although LiTaF6 is highly soluble in THF, after the salt dissolution the electrolyte subsequently polymerized [175]. The conductivity of a 0.33 M LiTaF6 electrolyte with sulfolane is 3.8 × 10−3 S cm−1 at 75 °C [175] (which is somewhat lower than a 0.33 M LiPF6 electrolyte with sulfolane at this temperature [5]). The conductivity of PC-based electrolytes with either LiTaF6 or LiNbF6 was also found to be lower than the corresponding electrolytes with LiPF6 [174]. Electrolytes with LiTaF6 result in very poor Li metal cycling efficiency suggesting that the TaF6 − anions may also have poor reductive stability [6]. The reductive stability of the MF6 − anions is reported to follow the order SbF6 − < AsF6 − < PF6 − [176]. Electrolytes with LiSbF6 have a similar conductivity to those with LiPF6, but the LiSbF6 salt may be corrosive to metals [6]. The Li2SiF6 and Li3AlF6 salts tend to be poorly soluble in aprotic solvents, and the resulting electrolytes have a low conductivity [6]. In contrast to these salts, LiAsF6 is highly soluble and its use results in electrolytes with comparable and, in some cases, superior conductivity and properties to those with LiPF6 (Table 1.1 and Fig. 1.14) [5]. But the potential to reduce the anion AsV oxidation state to the highly toxic AsIII or As0 oxidation states, as noted above, has largely limited the commercial use of LiAsF6 [177].
LiAlCl4 has been widely used for Li/SO2Cl2 batteries [178] (as has LiGaCl4 [179–186]), but LiAlCl4 has also been studied for use with intercalation cathodes. For example, an electrolyte composed of LiAlCl4⋅3SO2 was used for a Li/LiCoO2 cell [187, 188]. This electrolyte has a very high conductivity (70–80 mS cm−1 at 0–20 °C), but the salt undergoes a degradation side reaction to produce Cl2 which then reacts with Li metal to form LiCl [187, 188]:
A diverse range of analogues of BF4 − anions (Fig. 1.17) has been reported with the replacement of fluorine atoms with perfluoroalkyl chains. The conductivity of 1 M salt in EC:EMC electrolytes with lithium salts with these anions is given in Table 1.2. Perhaps contrary to expectations, it is interesting to note that the electrolyte conductivity increases with increasing size/mass of the anions. One might instead expect that bulkier anions would be less mobile, thus decreasing the conductivity. This increase in the conductivity may be due to decreased ionic association interactions with increasing perfluoroalkyl chain lengths and numbers (Figs. 1.1 and 1.2). A separate publication, however, indicated that an electrolyte with LiBF3C2F5 (LiFAB) (Figs. 1.16e and 1.17a) has a lower conductivity than the corresponding LiBF4 electrolyte (1 M salt in EC:EMC 30:70 v:v) above 0 °C, but a higher conductivity at < −20 °C [190]. Yet another publication indicates that LiFAB electrolytes (EC:EMC 30:70 v:v) are more conductive than LiBF4 electrolytes (but less conductive than LiPF6-based electrolytes) from −10 to 25 °C [192]. The LiFAB salt has good compatibility with Al at high potential, as well as a high stability with a graphite anode and nickel oxide-based cathode. The cell cycling behavior is comparable to an electrolyte with LiPF6 and far better than one with LiBF4. Cells with LiFAB also had improved capacity retention relative to cells with LiPF6 after storage at 60 °C [190]. For cells with graphite anodes and LiCoO2 cathodes, however, comparable performance to LiPF6 electrolytes was found at room temperature, but inferior performance was found at elevated temperature reportedly due to a degradation reaction of the LiFAB electrolyte with the cathode [192]. Electrolytes with variants of BF4 − anions with perfluoroalkylsulfonyl groups (–SO2C n F2n+1) (Fig. 1.18a–d) have conductivity values somewhat higher than those with anions with the corresponding perfluoroalkyl groups (Table 1.2) [189]. Analogous anions with the fluorine atoms of BF4 − replaced with –PO2F2 groups have also been reported (Fig. 1.18e–g) [210].

Examples of analogues of BF4 − and PF6 − anions: (a) BF3(C n F2n+1)− [189–193], (b) BF2(C n F2n+1)2 − [189, 191], (c) BF(C n F2n+1)3 − [189, 191], (d) B(C n F2n+1)4 − [189, 194, 195], (e) BF3Cl− [196], (f) BF3(C6H n F5−n )− [160, 197, 198], (g) BF2(C6H n F5−n )2 − [160, 197, 198], (h) BF(C6H n F5−n )3 − [160, 197, 198], (i) B(C6H n F5−n )4 − [160, 198–201], (j) BF3O(C2H4O) n CH3 − [202], (k) PF5(C n F2n+1)− [203, 204], (l) PF4(C n F2n+1)2 − [203, 204], (m) PF3(C n F2n+1)3 − [70, 203–206], (n) PF2(C n F2n+1)4 − [63, 70, 203–209], and (o) PF(C n F2n+1)5 − [204]

Examples of BF4 − and PF6 − analogue anions with perfluoroalkylsulfonyl and phosphorodifluoridato groups: (a) BF3(SO2C n F2n+1)− [189], (b) BF2(SO2C n F2n+1)2 − [189], (c) BF(SO2C n F2n+1)3 − [189], (d) B(SO2C n F2n+1)4 − [189], (e) BF3(PO2F2)− [210], (f) BF2(PO2F2)2 − [210], (g) BF(PO2F2)3 − [210], (h) PF5(SO2C n F2n+1)− [204], (i) PF4(SO2C n F2n+1)2 − [204], (j) PF3(SO2C n F2n+1)3 − [204], (k) PF2(SO2C n F2n+1)4 − [204], (l) PF(SO2C n F2n+1)5 − [204], (m) PF5(PO2F2)− [210], (n) PF4(PO2F2)2 − [210], and (o) PF3(PO2F2)3 − [210]
The LiBF3Cl salt (Figs. 1.16i and 1.17e) has a higher solubility than LiBF4 in aprotic solvents and hinders crystallization of electrolytes to a greater extent at low temperature [196]. In common with LiBF4, the LiBF3Cl salt passivates Al well at high potential. The salt also makes a more favorable SEI with graphite than comparable electrolytes with LiBF4 [196].
Lithium salts with fluorinated phenylfluoroborate anions have been reported (Fig. 1.17f–i) [198]. The conductivity values for 0.5 M salt in PC:DMC (v:v) electrolytes at 30 °C are reported to be LiBF4 (3.7 mS cm−1), LiBF3(C6F5) (Fig. 1.17f) (4.0 mS cm−1), and LiBF3C3F7 (Fig. 1.17a) (6.1 mS cm−1), but the t Li+ values are 0.31, 0.71, and 0.43, respectively [198]. The lithium tetrakis(pentafluorophenyl)borate salt (LiBArF) (i.e., LiB(C6F5)4) (Figs. 1.16l and 1.17i) [199, 200] has a very weakly coordinating anion with 20 fluorine atoms per Li+ cation. Although this salt does form crystalline solvates with fully solvated Li+ cations (i.e., (AN)4:LiB(C6F5)4 [211] and (Et2O)4:LiB(C6F5)4 [212]), it also crystallizes as solvates in which the Li+ cations are partially coordinated by the anion fluorine atoms (i.e., (Et2O)1:LiB(C6F5)4⋅CH2Cl2 [212], (toluene)1:LiB(C6F5)4⋅toluene [212], and (benzene)1:LiB(C6F5)4 [213]). Thus, the extensive fluorination of the anion actually facilitates the coordination of the anion to the Li+ cations (relative to LiBPh4). The properties of a wide variety of different LiBR4 salts with varying alkyl and/or aryl groups and substituents (–CH3, –OCH3, –F, –CF3) have been extensively explored as electrolytes by researchers at Exxon Research and Engineering Company in the early 1980s [161, 214]. The anodic oxidative stability of the salts varies by more than 1.6 V depending upon the R group, with aryl groups resulting in higher stability than alkyl groups and the addition of electron-withdrawing substituents further increasing the stability [157, 160]. Related salts have been reported, such as LiB(OC6F5)4 [215] and LiB(SC6F5)4 [215, 216], but these salts tend to result in PEO-based electrolytes with a very low conductivity.
Lithium trifluoroalkoxyborate salts (Fig. 1.17j) are liquid at room temperature with a neat salt conductivity on the order of 10−4 S cm−1 [202]. Carbonate-based electrolytes (EC:PC:DMC 1:1:3 v:v:v) with these salts have a conductivity >3 mS cm−1 at 20 °C (higher than comparable electrolytes with LiBF4) [202].
Analogues of PF6 − anions (Fig. 1.17k–o) have also been reported [63, 70, 203–209]. Electrolytes (1 M salt in EC:DMC 50:50 w:w) with the LiPF3(C2F5)3 (LiFAP) salt (Figs. 1.16m and 1.17m) have a conductivity which is only slightly lower than those with LiPF6 [70]. Half cells with Li metal and LiMn2O4 cycled better with the electrolyte with LiFAP (relative to those with LiPF6) [70, 208]. LiFAP was found to not undergo hydrolysis (in sharp contrast to LiPF6) and to have improved thermal stability relative to LiPF6 [63, 70, 207, 209]. In addition, carbonate solvent-based electrolytes with a mixture of LiFAP and LiPF6 were reported to have superior cycling performance (relative to comparable electrolytes with the individual LiFAP and LiPF6 salts), especially at 80 °C [206]. Anions in which the PF6 − fluorine atoms have been replaced with –SO2C n F2n+1 or –PO2F2 groups have also been reported (Fig. 1.18h–o) [204, 210].
1.6 Advanced Salts—Perfluoroalkylacetates, -Sulfonates, and -Phosphates
The experimental gas-phase acidity values (ΔG acid in kcal mol−1) for alkyl- and perfluoroalkylacetates and sulfonates follow the order HCO2CH3 (341.1) < HCO2CF3 (316.3) ≤ HSO3CH3 (315.0) < HSO3F (299.8) < HSO3CF3 (299.5) (Fig. 1.1) [21, 26]. The fluorocarbonate anion (i.e., CO2F−) is highly reactive and may be considered to be a fluorine anion, F−, solvated by CO2 [217]. Other perfluoroalkylacetate anions (i.e., CO2C n F2n+1 −) (Figs. 1.19a and 1.20a) have not been used to any significant extent for lithium battery electrolytes. The lithium salts with these anions tend to be highly aggregated (extensive anion…Li+ cation coordination interactions) in PC electrolytes [218]. This is in agreement with the results noted above for the (AN) n –LiCO2CF3 mixtures which have a very low solvation number over the entire concentration range (Fig. 1.5) and very low conductivity relative to other (AN) n –LiX mixtures (Fig. 1.11).

Examples of fluoroalkylacetate, -sulfonate, and -phosphate anions: (a) CO2C n F2n+1 − [218], (b) SO3C n F2n+1 − [219–222], (c) SO4(C2H4O) n CH3 − [223, 224], (d) SO3(C6F5)− [175, 221], (e) SO3(CF2) n SO3 2− [175, 222, 225], (f) SO3(C6F4)SO3 2− [226], (g) PO3(C n F2n+1)2− [72, 227–232], and (h) PO2(C n F2n+1)2 − [72, 227, 230, 233–235]

Anion structures: (a) CO2CF3 −, (b) SO3C4F9 −, (c) SO3C8F17 −, (d) SO3(C6F5)−, (e) SO3C3F6SO3 2−, (f) PO3C2F5 2−, (g) PO3F2−, and (h) PO2F2 − (C—gray, O—red, F—light green, P—orange, S—yellow)
Fluorosulfonic acid (i.e., HSO3F) (Fig. 1.19b) was first reported in 1918 [236]. LiSO3F-based electrolytes with several aprotic solvents were found to have a similar oxidative stability to electrolytes with other lithium salts such as LiSO3CF3 [237]. The conductivity of 1 M LiSO3F electrolytes with PC and GBL is 1.4 and 3.6 mS cm−1, respectively [237]. For a mixed-solvent electrolyte, 1 M LiSO3F in GBL:DME (1:1 mol:mol), however, the conductivity is 7.4 mS cm−1 at 25 °C [237] (to be compared with 1 M GBL:DME (1:1 mol:mol) electrolytes with LiBF4, LiPF6, and LiAsF6 which have conductivity values of 7.7, 11.2, and 11.8 mS cm−1 [238]). The crystal structure for LiSO3F has been reported [239].
Trifluoromethanesulfonic acid (i.e., HSO3CF3) (Fig. 1.19b) and the corresponding sodium salt were first reported in 1954 [240, 241]. A 3M patent in 1956 then reported the preparation of a variety of perfluoroalkylsulfonic acids and the corresponding sodium and potassium salts [242]. The use of LiSO3CF3 in battery electrolytes began in the 1970s and early 1980s [243, 244]. Lithium salts with perfluoroalkylsulfonate anions (Figs. 1.19b and 1.20b, c) tend to be more dissociated than those with perfluoroalkylacetate anions due to the larger size of the sulfur atoms relative to carbon (making the anion “softer”) and more extensive resonance (and thus charge delocalization) due to the additional oxygen atom. This results in the sulfonate salts having a higher conductivity than the corresponding acetate salts (Table 1.3) [219, 221]. The conductivity of electrolytes with the perfluoroalkylsulfonate anions is not correlated with the mass of the anions (Table 1.3). All of the electrolytes with these salts, however, have a conductivity which is significantly lower than for the comparable electrolyte with LiPF6 (Tables 1.1 and 1.3, Fig. 1.14). The crystal structure for LiSO3CF3 is known for both low- and high-temperature phases [245–247]. The conductivity of amorphous PEO-based polymer electrolytes with the perfluoroalkylsulfonate salts follows the order [220]:
This ordering is likely due to a reduction in the partial negative charge on the anion oxygen atoms upon increasing chain length from –CF3 to –C4F9 (which reduces the ionic association tendency of the anions). An additional increase in the chain length (i.e., –C8F17 and –C10F21), however, does not then significantly decrease the ionic association interactions further and the more bulky anions decrease the anion mobility (and perhaps the Li+ cation mobility due to the correlated interactions of the cations with the anions), thus lowering the conductivity (relative to LiSO3C4F9).
Note that lithium salts with nonfluorinated alkylsulfonate (e.g., LiSO3CH3) or benzenesulfonate (e.g., LiSO3(C6H5)) anions have a very low solubility in aprotic solvents and a correspondingly low conductivity (Table 1.3) [219]. Lithium salts with oligoethersulfate anions (Fig. 1.19c) are soluble in EC:DMC mixtures, but these have a much lower conductivity (<10−3 S cm−1 at 30 °C) than comparable electrolytes with LiPF6 [223, 224]. The lithium salt with fluorinated benzenesulfonate anions (i.e., LiSO3(C6F5)) (Figs. 1.19d and 1.20d) is somewhat more soluble in aprotic solvents, but the conductivity of such electrolytes is relatively low (lower than for electrolytes with LiSO3CF3) (Table 1.3) [219, 248].
Monofluorophosphoric acid (i.e., H2PO3F) (Figs. 1.19g and 1.20g) and difluorophosphoric acid (i.e., HPO2F2) (Figs. 1.19h and 1.20h) were first reported in 1929 and 1927, respectively [227–229, 235]. Lithium salts with these anions have been prepared [230, 231, 233, 234]. The related HPHO2F acid and corresponding LiPHO2F salt, however, were much more challenging to prepare [249]. Trifluoromethanephosphonic acid (i.e., H2PO3CF3) (Fig. 1.19g) and bis(trifluoromethane)phosphonic acid (i.e., HPO2(CF3)2) (Fig. 1.19h) were synthesized in 1954–1955 [250, 251]. The relative acidity of these anions was reported to follow the order [251]:
The lithium trifluoromethanephosphate salt (i.e., Li2PO3CF3) (Fig. 1.19g) has also been synthesized [252]. This salt was reportedly soluble, forming a 1 M electrolyte with a PC:DME mixture which was utilized for battery cycling [252]. Perfluorodiphenylphosphinic acid (i.e., HPO2(C6F5)2) and the crystal structure of the corresponding potassium salt have also been reported [253].
1.7 Advanced Salts—Imides, Methides, and Phosphorylimides
The bis(fluorocarbonyl)imide acid (i.e., H[N(COF)2]) (Fig. 1.21a) was first reported in 1973 [291], but anions with the X–CO–N–CO–X (X = C or F) backbone have not received much attention for battery electrolytes. In contrast, a diverse range of lithium salts with imide (sometimes called amide) anions with the X–SO2–N–SO2–X (X = C or F) backbone have been prepared. This difference in focus is due to the difference, for example, in the acidifying capability of the –COCF3 and –SO2CF3 groups (Fig. 1.2)—experimental gas-phase acidity values (ΔG acid in kcal mol−1) are HN(COCF3)2 (307.5) and HN(SO2CF3)2 (291.8) (ref: H2SO4 (302.2)) (Fig. 1.2) [20, 26]. The X–SO2–N–SO2–X backbone is able to adopt two low-energy conformations (Fig. 1.3f) [114–117]. This flexibility, combined with the extensive charge delocalization due to resonance and the electron-withdrawing fluorine atoms [20, 26], tends to make these lithium salts highly soluble. In addition, solvent–salt mixtures with such salts often form crystalline solvates with a low T m (in contrast with LiPF6) or crystallinity gaps (concentration ranges in which it is difficult or impossible to crystallize some or all of the electrolyte) (Fig. 1.4).

Examples of imide anions: (a) N(COC n F2n+1)2 − [254–257], (b) N(SO2C n H2n+1)2 − [258], (c) N(SO2C n F2n+1)2 − [259–270], (d) N(COC n F2n+1)(SO2C n F2n+1)− [256, 257, 271–273], (e) N(SO2(C6H5))(SO2CF3)− [274–276], (f) N(SO2(C6F5))2 − [277], (g) N(SO2C2F4SO2)− [278, 279], (h) N(SO2C3F6SO2)− [269, 278, 279], (i) N(SO2C4F8SO2)− [278, 279], (j) CO(NSO2F)2 2− [280], (k) SO2 (NSO2C n F2n+1)2 2− [281–283], (l) (CF2) n (SO2NSO2CF3)2 2− [284, 285], (m) N(SO2C2F4O(C2H4O)CH3)2 − [277], (n) N(SO2C4H8SO3)2 3− [286], (o) N(SO2CH2CO2)2 3− [286], (p) N(SO2CH2CO(C6H4)SO3)2 3− [286], (q) (C6H4)(SO2NSO2CF3)2 2− [287], (r) O((C6H5)SO2NSO2CF3)2 2− [287], (s) (C6H2)(OC2H2O)(SO2NSO2CF3)2 2− [287], (t) N(SO2NSOF2)2 − [288], (u) N(CONSNSO2)− [289], and (v) N(SO2NSNSO2)− [290]
Lithium bis(methanesulfonyl)imide (or dimesylamide) (i.e., LiN(SO2CH3)2) (Figs. 1.21b and 1.22a) [258, 292–295] and lithium bis(butanesulfonyl)imide (i.e., LiN(SO2C4H9)2) [258] have been reported. In sharp contrast to the lithium salts with tetraalkylborate anions which are highly soluble in aprotic solvents, lithium salts with nonfluorinated bis(alkanesulfonyl)imide anions have poor solubility in cyclic/acyclic carbonate and ether solvents [258]. This is likely due to the poor Li+ cation-coordinating ability of the tetraalkylborate anions—thus favoring solvent coordination to the Li+ cations, whereas the nonfluorinated bis(alkanesulfonyl)imide anions instead readily coordinate the Li+ cations with the anion oxygen atoms and the lack of electron-withdrawing fluorine atoms results in high electron density on the oxygen atoms (i.e., strong ionic association tendency)—thus restricting the solvent coordination to the Li+ cations. LiN(SO2CH3)2 is insoluble in an EC:DMC mixture and has poor solubility in DMSO (<0.1 M), while LiN(SO2C4H9)2 has low solubility in EC:DMC (<0.1 M) and fair solubility in DMSO (~0.3 M) [258]. The conductivity at 25 °C of DMSO electrolytes with these salts (3.2 mS cm−1) was considerably lower than for a 0.5 M LiPF6 electrolyte with DMSO (9.8 mS cm−1). In addition, ionic liquid salts with the N(SO2CH3)2 − anion have a higher viscosity, lower conductivity, lower thermal stability, and lower electrochemical stability than the corresponding salts with the TFSI− anion [296].

Anion structures: (a) N(SO2CH3)2 − (trans conformation), (b) N(SO2(C6H5))2 − (trans conformation), (c) N(SO2(C6H5))2 − (cis conformation), (d) N(SO2F)2 − (FSI−), (e) N(SO2C2F5)2 − (BETI−) (trans conformation), (f) N(SO2C4F9)2 − (cis conformation), (g) N(SO2C4F9)2 − (trans conformation), (h) N(SO2C2F4SO2)−, (i) SO2(NSO2CF3)22 −, ( j) C(SO2CF3)3 − (TriTFSM−), (k) C(SO2CF3)2(C6F5)−, (l) CH(SO2CF3)2 − (TFSM−) (trans conformation), and (m) CH(SO2CF3)2 − (TFSM−) (cis conformation) (C—gray, N—blue, O—red, F—light green, S—yellow)
The =NSO2CF3 group has a very strong acidifying effect when replacing an =O [26]. The (trifluorosulfonyl)(sulfonate)imide anion [297] can be viewed as the SO4 2− anion with a =NSO2CF3 group replacing an =O:

Similarly, the TFSI− anion can be viewed as the SO3CF3 − anion with a =NSO2CF3 group replacing an =O:

Additional =NSO2CF3 groups give [298, 299]

Fluorination of the imide anions results in lithium salts with exceptionally high solubility in common aprotic solvents. Bis(trifluoromethanesulfonyl)imide acid or HTFSI (i.e., HN(SO2CF3)2) (Figs. 1.3f and 1.21c) was first reported in 1982 by Foropoulos and DesMarteau [300, 301], while the longer chain anions—N(SO2RF)2 −—were reported a decade earlier by Meussdorffer and Niederprum [302]. In 1990, Armand patented the synthesis of LiTFSI and related fluorinated sulfonyl imide salts [303]. LiTFSI is now the most widely studied salt for this class of anions, but lithium bis(perfluoroethanesulfonyl)imide (LiBETI) (i.e., LiN(SO2C2F5)2) (Figs. 1.21c and 1.22e) has also been widely examined [49, 69, 207, 262, 304–310] with more than 200 publications reported for this latter salt. Electrolytes with LiBETI have a lower conductivity than those with LiPF6 or LiTFSI (Table 1.4), but this salt, like LiTFSI, has an exceptionally high thermal stability and does not undergo hydrolysis due to the high stability of the C–F bonds. In addition, unlike LiTFSI, electrolytes with aprotic solvents and LiBETI are reported to not strongly corrode Al at high potential [66, 68, 311–313]. Numerous other perfluoroalkanesulfonyl imide anions have also been prepared [259, 314], including cyclic anions such as lithium cyclic-1,3-perfluoroethanedisulfonylimide (i.e., LiN(SO2C2F4SO2)) (Figs. 1.21g and 1.22h) and lithium cyclic-1,3-perfluoropropanedisulfonylimide (i.e., LiN(SO2C3F6SO2)) (Fig. 1.21h)—PC:DME electrolytes with the latter have the highest conductivity of the imide salts noted in Table 1.4 [314]. The properties of most of these salts have not been extensively explored by the battery research community.
Anions with fluorosulfonyl groups (–SO2F) have garnered tremendous interest in recent years. Chief among these is lithium bis(fluorosulfonyl)imide (LiFSI) (i.e., LiN(SO2F)2) (Figs. 1.21c and 1.22d) [156, 260, 263, 315–333]. Bis(fluorosulfonyl)imide acid or HFSI (i.e., HN(SO2F)2) was first reported in 1962 [334], and, even though the synthesis of LiFSI was reported in 1995 [335], the limited availability and high cost of this salt have restricted its use in research studies until quite recently. Electrolytes with LiFSI typically have a conductivity equivalent to comparable electrolytes with LiPF6 (making this one of the most conductive salts known) [156]. The thermal and hydrolytic stability of the FSI− anion is lower than for the TFSI− anion due to the lower stability of the S–F bond (relative to a C–F bond), but the LiFSI salt has improved thermal/hydrolysis stability relative to LiPF6 [156]. It was reported that use of the LiFSI salt in electrolytes results in severe Al corrosion at high potential [320], but it has recently been shown that this is likely due to chloride impurities in the salt rather than the LiFSI salt itself [156]. An additional favorable property (relative to LiPF6) is the wide liquidus range of electrolytes with LiFSI. As for LiTFSI-based electrolytes, LiFSI-based electrolytes tend to form solvates with a low T m or crystallinity gaps for specific electrolyte compositions.
Lithium salts with asymmetric anions may also be of interest as these tend to be more soluble and form solvates with a lower T m than for salts with symmetric anions. Examples include lithium (fluorosulfonyl)(trifluoromethanesulfonyl)imide (LiFTI or LiFTA) (i.e., LiN(SO2F)(SO2CF3)) (Fig. 1.21c) [263–265, 336] and lithium (fluorosulfonyl)(nonafluorobutanesulfonyl)imide (LiFNFSI) (i.e., LiN(SO2F)(SO2C4F9)) (Fig. 1.21c) [264, 265, 337]. The LiFNFSI salt has a high thermal stability (>200 °C), forms electrolytes with a high conductivity (comparable to those with LiClO4), and does not significantly corrode the Al current collector at high potential. For battery testing, graphite/LiCoO2 cells with LiFNFSI had a much improved cycling performance over cells with LiPF6 when cycled at elevated temperature (60 °C). Asymmetric imide anions with carbonyl groups such as TSAC − (i.e., N(COCF3)(SO2CF3)−) (Fig. 1.21d) have also been reported [256, 266]. The experimental gas-phase acidity value (ΔG acid in kcal mol−1) for HTSAC (298.2) is somewhat higher than for HTFSI (291.8), but lower than that for HFSI (301.2) [26]. The TSAC− anion, however, has been found to have a poor electrochemical stability relative to other imide anions. The anodic oxidative stability was slightly lower relative to anions such as FTI− and TFSI−, but the cathodic reductive stability of the TSAC− anion was notably poorer (almost 1 V less stable) [273, 338].
Many other variants of imide anions have been reported (Fig. 1.21). In general, these do not have improved properties or other advantages over more widely used anions (i.e., TFSI− and BETI−). One possible exception to this may be the bis(trifluoromethanesulfonamido) sulfone anion (i.e., LiSO2(NSO2CF3)2) (Figs. 1.21k and 1.22i) [281]. If this dilithium salt has a high solubility in aprotic solvents, then it may offer an advantage in terms of having a greater Li+/anion mass ratio with less fluorine atoms per Li+ cation than for LiPF6 and LiTFSI. The properties of the dilithium salt with this anion, however, are not yet available. The related acid H2[CO(NSO2F)2] (Fig. 1.21j) and the corresponding alkali metal salts (with Na+ or K+) have been reported [280].
Nonfluorinated lithium tris(alkanesulfonyl)methide salts have been examined for their suitability for battery electrolytes (Fig. 1.23a) [258]. The LiC(SO2CH3)3 salt has poor solubility in EC:DMC (<0.1 M), but good solubility in DMSO (~0.5 M). Increasing the alkyl chain length from methyl to ethyl (i.e., LiC(SO2C2H5)3) increases the solubility, EC:DMC (~0.3 M) and DMSO (>4 M), while using asymmetric alkyl chain lengths (i.e., LiC(SO2CH3)(SO2C2H5)2) further increases the salt solubility, EC:DMC (~0.5 M) and DMSO (>4 M). These salts have a considerably lower conductivity (4.3 mS cm−1), however, in 0.5 M DMSO electrolytes at 25 °C than that for the corresponding LiPF6 electrolyte (9.8 mS cm−1) [258].

Lithium tris(perfluoroalkanesulfonyl)methide salts have also received some attention for battery electrolytes, especially the salt with the C(SO2CF3)3 − anion (TriTFSM −) (Figs. 1.22j and 1.23b). The experimental gas-phase acidity values (ΔG acid in kcal mol−1) are HC(COCF3)3 (300.6) and HC(SO2CF3)3 (289.0) (ref: HN(SO2CF3)2 (291.8)) (Fig. 1.2) [20, 26]. Despite the higher acidity of the TriTFSM− anion relative to TFSI− and calculations which indicate that the former anion will have a weaker Li+ cation affinity (i.e., lower ionic association tendency), the conductivity of polyether-based electrolytes with LiTriTFSM is lower than for similar electrolytes with LiTFSI [111, 341]. This may perhaps be related to the greater size/mass of the TriTFSM− anion. For liquid 1 M electrolytes with EC:DMC (50:50 v:v), the conductivity (mS cm−1) at 25 °C is LiAsF6 (11.0), LiTFSI (9.0), and LiTriTFSM (7.1) [343]. Despite the recent attention devoted to LiFSI (i.e., LiN(SO2F)2), no published work is yet available regarding the properties of electrolytes with lithium tris(fluorosulfonyl)methide salt, LiC(SO2F)3 [340, 350], and the bis(fluoromethanesulfonyl)methane (i.e., CH(SO2F)2 −) anion has not yet been reported. Some limited studies have, however, been reported for the related bis(trifluoromethanesulfonyl)methide anion (TFSM −) (i.e., CH(SO2CF3)2 −) (Figs. 1.22l, m and 1.23c). The acid, bis(trifluoromethanesulfonyl)methane (i.e., HCH(SO2CF3)2), was first prepared by Gramstad and Haszeldine in 1956 [355]. Poly(ethylene oxide) (PEO) electrolytes with the lithium salt (LiTFSM) (i.e., LiCH(SO2CF3)2) were found in one report to have a lower oxidative and reductive electrochemical stability than for similar electrolytes with LiTFSI [341], whereas another study indicated that such electrolytes with LiTFSM were stable with a voltage stability window of approximately 4.5 V [359]. As for LiTFSI and LiTriTFSM, the LiTFSM salt tends to plasticize poly(ethylene oxide) resulting in amorphous electrolytes with a relatively high conductivity, although somewhat lower than for comparable LiTFSI electrolytes [341, 359].
Lithium perfluoroalkanephosphorylimide salts (Fig. 1.24a) have not yet been reported, but such anions have been explored using QC calculations [361] and the sodium and potassium salts with the N(PO(C2F5)2)2 − anion (Fig. 1.25) have been prepared [365]. The lithium bis(difluorophosphoryl)imide salt (i.e., LiN(POF2)2) (Fig. 1.24a), however, has been synthesized [362, 363], as has the acid HN(PSF2)2 (Fig. 1.24b) [367]. Nonfluorinated alkyl or phenylphosphorylimide salts are also known, such as LiN(PS(C6H5)2)2(Fig. 1.24c) [368–370] and LiN(PO(C6H5)2)(PS(CH3)2) [371]. No data is yet available, however, regarding the electrolyte characteristics of lithium salts with such anions.


Anion structure: N(PO(C2F5)2)2 − (C—gray, N—blue, O—red, F—light green, P—orange)
1.8 Advanced Salts—Organoborates, -Phosphates, and -Aluminates
In 1995, Barthel and Gores reported a new class of inexpensive and chemically, electrochemically, and thermally stable salts based upon boron chelate complex anions with aromatic or aliphatic diols or carboxylic acids. The first such salt reported was lithium bis(1,2-benzenediolato(2-)-O,O′)borate (LiBBB) (Fig. 1.26a) [372]. The acid with this anion was originally reported in 1949 by Schafer [388]. The LiBBB salt has a high solubility in aprotic solvents (>1 M), but the oxidative stability is relatively low. A number of other nonfluorinated lithium salts with benzenediol nonfluorinated chelates were subsequently prepared: bis(2,3-naphthalenediolato(2-)-O,O′)borate (LiBNB) (Figs. 1.26d and 1.27e), bis(salicylato(2-))borate (LiBSB) (Fig. 1.26e and 1.27g), and bis(2,2′-biphenyldiolato(2-)-O,O′)borate (LiBBPB) (Figs. 1.26f and 1.27h) [373]. The conductivity of electrolytes with these salts was found to increase in the order LiBBPB < LiBSB < LiBNB ~ LiBBB (Table 1.5) [169, 374, 375]. All of these electrolytes have a significantly lower conductivity than comparable electrolytes with LiPF6, LiTFSI, and LiBETI (Table 1.5) [375]. The LiBSB-based electrolyte was found to have a relatively high Li cycling efficiency, however, in contrast to the other salts [375]. A number of crystalline solvates have been reported for the LiBBB and LiBSB salts: (H2O)2(THF)1:LiBBB, (H2O)1(AN)1:LiBBB, (AN)2:LiBSB, and (THF)2:LiBSB [389]. For the former solvate, the Li+ cations are coordinated to the diol oxygen atoms, but for the latter solvate these oxygens do not participate in the cation coordination. Instead, the carbonyl oxygens are coordinated to the Li+ cations.

Examples of chelated organoborate anions: (a) B(O(C6H4)O)2 − (BBB−) [169, 372–378], (b) B(O(C6H3F)O)2 − (FLBBB−) [373, 379, 380], (c) B(O(C6F4)O)2 − (4FLBBB−) [373, 381], (d) B(O(C10H6)O)2 − (BNB−) [56, 169, 373–375, 378], (e) B(O(C6H4)CO2)2 − (BSB−) [56, 373, 375, 382–385], (f) B(O(C6H4)(C6H4)O)2 − (BBPB−) [56, 169, 373, 375], (g) B(O(C6H3F)SO2)2 − (FSB−) [386], and (h) B(O(C5NH3)O)2 − (BPB−) [387]

Anion structures: (a) B(CO2CO2)2 − (BOB−), (b) BF2(CO2CO2)2 − (DFOB−), (c) B(OCH3)4 −, (d) B(CO2CH(CH3)O)2 −, (e) B(O(C10H6)O)2 − (BNB−), (f) B(OC(CH3)2C(CH3)2O)2 −, (g) B(O(C6H4)CO2)2 − (BSB−), (h) B(O(C6H4)(C6H4)O)2 − (BBPB−), (i) B(O(C6H2F2)(C6H2F2)O)2 −, (j) B(CO2CH2CO2)2 −(BMB−), and (k) B(CO2CH3)4 − (B—tan, C—gray, O—red, F—light green)
The preparation of fluorinated chelated organoborate salts (i.e., LiB(C6H4−x F x O2) demonstrated that with increasing fluorination (x = 0, 1, or 4) (Fig. 1.26a, b, c) both the conductivity and oxidative stability increased [379, 381]. The oxidative stability limit (vs. Li/Li+) was found to vary considerably for these salts: 3.6 V for BBB−, 3.8 V for FLBBB−/BNB−, 4.1 V for 4FLBBB−/BBPB−, and 4.5 V for BSB− [373]. It was suggested that these anions anodically degrade on the cathode surface to form thin, electrically insulated, but Li+ cation-conducting polymeric films which passify the electrode surface from further degradation of the salts or the solvents. A similar high salt solubility and oxidative stability limit was noted for the lithium bis(5-fluoro-2-olato-benzenesulfonato(2-)-O,O′)borate salt (LiFSB) (Fig. 1.26g) which has a stability limit of 4.6 V vs. Li/Li+ [386]. The current density decreased upon repeated cycling on a Pt electrode due to electrode passivation. The salt also passivated an Al electrode at high potential. The introduction of a nitrogen to the benzenediol chelate—lithium bis(2,3-pyridinediolato(2-)-O,O′)borate (LiBPB) (Fig. 1.26h), however, resulted in a salt with low solubility in DMC and DEC (in contrast to the other salts prepared), but the solubility was higher in EC or PC [387]. The conductivity of electrolytes with LiBPB was lower than for the other borate salts, but the salt did passivate Pt and Al electrodes, as for the other salts studied [387].
The first organoborate salt to attract significant interest from the broader battery research community was lithium bis(oxalato)borate (LiBOB) (i.e., LiB(CO2CO2)2) (Figs. 1.27a and 1.28a) [48, 79, 376, 380, 385, 390–484]. The first publication with LiBOB was from Xu and Angell in 2001 [393], but a German patent application was filed for this salt by Metallgesellschaft AG in 1999 [548]. The acid and tetraalkylammonium salts with BOB− (called borodicatecholate) and related anions had been reported in 1994 by Ue [549], as well as earlier by others [550]. LiBOB electrolytes have a conductivity which is comparable to or lower than that for electrolytes with LiBF4 [79, 414], moderate stability to hydrolysis, high electrochemical stability (>4.5 V vs. Li/Li+), and high thermal stability [393, 394, 414, 448]. The salt has a relatively low solubility in acyclic carbonate solvents (i.e., the solubility limit is 0.8 M in EC:DMC 3:7) but is more soluble (>1 M) in nitrile, ester, and cyclic carbonate solvents [447]. The salt has been used as both a primary salt (replacement for LiPF6) and an additive (addition of small amounts to LiPF6 electrolytes). Notably, the electrode surface layers formed by this salt on both the anode and cathode enable cells with LiBOB to have excellent capacity retention when cycled at elevated temperature (≥60 °C) and when cycling cathodes up to 5 V (vs. Li+/Li).

Examples of chelated organoborate anions: (a) B(CO2CO2)2 − (BOB−) [48, 79, 376, 380, 385, 390–484], (b) B(O(C6H4)O)(CO2CO2)− (BDOB−) [376, 377, 485, 486], (c) B(CO2CH2O)2 − [384, 487], (d) B(CO2CH(CH3)O)2 − [384, 487, 488], (e) B(CO2C(CH3)2O)2 − (BMLB−) [385], (f) B(OC(CH3)2C(CH3)2O)2 − [489, 490], (g) BF2(CO2CO2)− (DFOB−) [390–392, 491–535], (h) BF2(O(C6H4)O)− (DFBDB−) [485, 536, 537], (i) BF2(O(C6H3F)O)− (FLDFBDB−) [485, 537],(j) B(O(C6H3F)O)(CO2CO2)− (FLBDOB−) [377, 380, 485, 486, 536], (k) BF2(O(C6F4)O)− (4FLDFBDB−) [486, 537], (l) B(O(C6H4)O)(C5O5)− (BDCB−) [538], (m) B(CO2CO2)(C5O5)− (OCB−) [538], (n) B(C5O5)2 − (BCB−) [382, 383, 538], (o) B(O(C6H4)CO2)(C5O5)− (CSB−) [382, 383], (p) BF2(CO2CH2CO2)− [539], (q) BF2(CO2C(CH3)2CO2)− [539], (r) B(CO2CO2)(CO2CH2CO2)− (MOB−) [540], (s) B(CO2CH2CO2)2 − (BMB−) [385, 484, 540, 541], (t) B(CO2C(CH3)2CO2)2 − [539], (u) B(CO2CHFCO2)2 − [540, 542], (v) B(CO2CF2CO2)2 − [392, 543], (w) BF2(CO2CH2CO2)− [392, 543], (x) BF2(CO2CF2CO2)− [392, 543], (y) BF2(CO2C2F4CO2)− [392, 543], (z) B(CO2CO2)(CO2CF3)2 − [544], (aa) B(CO2C(CF3)2O)(CO2CF3)2 − [392], (bb) B(CO2CO2)(OCH(CF3)2)2 − [392], (cc) B(CO2C(CF3)2O)(OCH(CF3)2)2 − [392, 543, 545, 546], and (dd) B(OC(CF3)2C(CF3)2O)2 − (BPFPB−) [484, 541, 547]
Lithium difluoro(oxalato)borate (LiDFOB) (also called lithium oxalyldifluoroborate (LiODFB)) (i.e., LiBF2(CO2CO2)) (Figs. 1.27b and 1.28g), like LiBOB, has also received a great deal of attention from the lithium battery research community. This salt was first reported in Central Glass Company patents filed in 2000 [390–392] and then later in a US Army Research Laboratory (ARL) publication in 2006 [491]. Many studies have now demonstrated that LiDFOB is quite useful both as a primary salt (replacement for LiPF6) and as an additive to LiPF6 electrolytes [491–535]. LiDFOB has a higher solubility than LiBOB in linear carbonate solvents, but it is still lower than for other salts such as LiBF4, LiTFSI, and LiPF6. Electrolytes with the LiDFOB salt are better than those with LiBOB at passivating the Al current collector and also tend to have a higher conductivity (i.e., the conductivity of a 1 M LiDFOB electrolyte in EC:DMC (1:1 v:v) at 25 °C is 8.6 mS cm−1 [506], which is somewhat lower than the conductivity of comparable electrolytes with LiPF6 or LiClO4 (Table 1.1)). As for LiBOB, enhanced battery performance is noted upon addition of LiDFOB to electrolytes including the cyclability/stabilization of electrode materials (such as LiFePO4 and Li4Ti5O12) at elevated temperature (60 °C) and of high-voltage cathode materials when cycled to 5 V due to favorable surface layers formed on the anode and cathode [40].
In addition to LiBOB and LiDFOB, a wide variety of additional organoborate anions have been synthesized (Figs. 1.28 and 1.29). Only limited information is available about the properties of the corresponding lithium salts. The LiB(CO2C(CF3)2O)2 salt (Fig. 1.29a), however, reportedly does not undergo hydrolysis and is thermally stable at 100 °C for 1 month, and electrolytes with this salt do not corrode Al at high potential [545]. The conductivity of electrolytes with 1 M salt in EC:DMC 1:1 at 25 °C is 7.0, 8.3, 6.3, and 1.9 mS cm−1, respectively, for the LiB(CO2C(CF3)2O)2, LiBF2(CO2C(CF3)2O), LiB(CO2CH(CF3)O)2, and LiB(CO2CH2C(CF3)2O)2 salts (Fig. 1.29a–d) [545]. Note that the concentration for the latter salt is only 0.8 M (instead of 1 M) due to its limited solubility in EC:DMC. For the fluorinated salts in Fig. 1.29h–p, the maximum conductivity (mS cm−1) (differing concentrations) for DME-based electrolytes at 25 °C is 5.88 (Fig. 1.29h), 5.39 (Fig. 1.29i), 6.57 (Fig. 1.29j), 6.89 (Fig. 1.29k), 6.39 (Fig. 1.29l), 7.89 (Fig. 1.29m), 7.55 (Fig. 1.29n), 7.79 (Fig. 1.29o), and 8.39 (Fig. 1.29p) [551, 552]. The conductivity of the electrolytes thus increases with increasing fluorination of the anions and is dependent upon the positioning of the fluorine atoms (with the para position less favorable for increasing the conductivity). Increased fluorination of the anions also increased the oxidative stability of DME- or EC:DMC-based electrolytes with the salts [551, 552]. With regard to Al passivation at high potential for these electrolytes (as well as those with EC:DMC), only the electrolytes with Fig. 1.29h, j, and n anions passivated the Al electrode—the electrolytes with the other six anions did not [551, 552]. Seemingly, the anions with fluorine atoms in the ortho or the para position on the benzene ring do not decompose to leave a passivating film on the Al surface.

Additional examples of chelated organoborate anions: (a) B(CO2C(CF3)2O)2 − [392, 545, 546], (b) BF2(CO2C(CF3)2O)− [392, 545, 546], (c) B(CO2CH(CF3)O)2 − [392, 545, 546], (d) B(CO2CH2C(CF3)2O)2 − [392, 545, 546], (e) B(CO2(C6H3(CH3))O)2 − (3-MLBSB−) [56], (f) B(CO2(C6H2Cl2)O)2 − (DCLBSB−) [56], (g) B(CO2(C6HCl3)O)2 − (TCLBSB−) [56], (h) B(O(C6H4)C(CF3)2O)2 − [551, 552], (i) B(O(C6H3F)C(CF3)2O)2 − [551, 552], (j) B(O(C6H3F)C(CF3)2O)2 − [551, 552], (k) B(O(C6H3F)C(CF3)2O)2 − [551, 552], (l) B(O(C6H2F2)C(CF3)2O)2 − [551, 552], (m) B(O(C6H2F2)C(CF3)2O)2 − [551, 552], (n) B(O(C6H2F2)C(CF3)2O)2 − [551, 552], (o) B(O(C6HF3)C(CF3)2O)2 − [551, 552], and (p) B(O(C6HF3)C(CF3)2O)2 − [551, 552]
The lithium bis(perfluoropinacolato)borate (LiBPFPB) salt (Fig. 1.28dd) is also reported to have a high oxidative stability [547]. No information is available about the Al corrosion behavior of electrolytes with this salt, but a 0.6 M LiBPFPB electrolyte with DME at 25 °C has a conductivity of 11.1 mS cm−1 [547]. The conductivity of a 1 M electrolyte with the salt in PC at 25 °C, however, is 2.1 mS cm−1 [547]—for comparison, the conductivity for a 1 M LiAsF6 electrolyte with PC at 20 °C is 5.28 mS cm−1 [5].
A number of lithium salts with tetrakis(haloacyloxy)borate anions (i.e., LiB(CO2R)4) have also been synthesized (Fig. 1.30a–d) [553, 554]. These are fluorinated and/or chlorinated versions of the tetra(acetato)borate anion (i.e., B(CO2CH3)4 −) (Figs. 1.27k and 1.30e). The acid and cesium salt with the B(CO2CF3)4 − anion were first reported in 1971 [561]. The lithium salt (i.e., LiB(CO2CF3)4) was subsequently reported in 1972 [562]. Electrolytes with these non-chelate salts have a relatively high conductivity (although lower than for LiPF6), high oxidative stability, and high cycle efficiency with a graphite electrode. The most conductive salt is LiB(CO2CF3)4 (comparable to the conductivity of electrolytes with LiTFSI). Lengthening the perfluoralkyl chains from –CF3 to –C2F5 decreases the conductivity, as does replacing the fluorine atoms with chlorine atoms [553]. A lithium salt with the tetrakis(chlorosulfato)borate anion (i.e., B(SO3Cl)4 −) has been reported (Fig. 1.30f) [556], as has the acid with the tetrakis(trifluoromethanesulfonato)borate anion (i.e., B(SO3CF3)4 −) (Fig. 1.30g) [557]. Note that this anion differs from the B(SO2CF3)4 − anion noted in Fig. 1.18d.

Examples of nonchelated organoborate anions: (a) B(CO2CF3)4 − [553, 554], (b) B(CO2C2F5)4 − [553, 554], (c) B(CO2CF2Cl)4 − [553, 554], (d) B(CO2CCl3)4 − [553, 554], (e) B(CO2CH3)4 − [554], (f) B(SO3Cl)4 − [555, 556], (g) B(SO3CF3)4 − [557], (h) B(O(CH2CH2O) n CH3)3(C4H9)− [558], (i) B(C6F5)2(O(CH2CH2O) n CH3)2 − [559], (j) B(CO2CF3)2(O(CH2CH2O) n CH3)2 − [559], and (k) B(C6F5)(OCH3)3 − [560]
The reaction of trialkoxyborates with butyllithium produces salts which are liquid (for n ≥ 2) at ambient temperature without solvents (i.e., ionic liquids) (Fig. 1.30h) [558]. The ambient temperature conductivity for the n = 3 salt is 2 × 10−5 S cm−1, while 1 M electrolytes of the salt in EC:PC have a conductivity <10−3 S cm−1.
The nonfluorinated lithium tris(1,2-benzenediolato(2)-O,O′)phosphate (LiTBP) salt (Figs. 1.31a and 1.32a), reported by Sasaki and co-workers, has a relatively low thermal (<200 °C) and electrochemical (about 3.7 V (vs. Li/Li+)) stability [378, 564, 565]. Adding a methyl group—lithium tris(4-methyl-1,2-benzenediolato(2)-O,O′)phosphate (Li4-MLTBP) (Fig. 1.31b)—improves the thermal stability somewhat, but decreases the electrolyte conductivity (relative to LiTBP) [566]. Adding a fluorine atom—lithium tris(3-fluoro-1,2-benzenediolato(2)-O,O′)phosphate (Li3-FLTBP ) (Fig. 1.31c)—improves the electrolyte conductivity and thermal/electrochemical stability (relative to LiTBP and Li4-MLTBP) [564, 565]. The conductivity of 0.5 M electrolytes with EC:DMC at 25 °C is about 2.62, 2.25, and 3.16 mS cm−1, respectively, for LiTBP, Li4-MLTBP, and Li3-FLTBP (as compared to 9.66 mS cm−1 for LiPF6) [378, 564, 566]. Fully fluorinating the anion—lithium tris(3,4,5,6-tetrafluoro-1,2-benzenediolato(2)-O,O′)phosphate (Fig. 1.31d)—further increases the electrochemical stability, but the conductivity of a 0.6 mol kg−1 EC:DEC (2:1) electrolyte at 25 °C is relatively low (2.1 mS cm−1) [567]. The high mass, high fluorination (12 F/Li+), and lack of improved properties (relative to LiPF6) have limited interest in this salt.

Examples of chelated organophosphate anions: (a) P(O(C6H4)O)3 − (TBP−) [378, 563–567], (b) P(O(C6H3(CH3))O)3 − (4-MLTBP−) [564, 566], (c) P(O(C6H3F)O)3 − (3-FLTBP−) [564, 567], (d) P(O(C6F4)O)3 − [567], (e) P(CO2CH2O)3 − [568], (f) PF2(CO2CH2O)2 − [568], (g) PF4(CO2CH2O)− [568], (h) P(CO2CO2)3 − (TOP−) [568, 569], (i) PF2(CO2CO2)2 − [568], (j) PF4(CO2CO2)− (FOP−) [392, 543, 568, 570–576], (k) PF2(OC(CF3)2C(CF3)2O)2 − [577], (l) PF4(OC(CF3)2C(CF3)2O)− [577], (m) PF4(CO2C(CF3)2O)2 − [545, 546], (n) P(O(C6H4)(C6H4)O)3 − (TBPP−) [578], and (o) P(CO2(C6H4)O)3 − (TSP−) [579]

Anion structures: (a) P(O(C6H4)O)3 −, (b) P(CO2CO2)3 − (TOP−), (c) P(CO2(C6H4))3 − (not TSP−), and (d) P((C6H4)(C6H4))3 − (not TBPP−) (C—gray, O—red, P—orange)
The nonfluorinated lithium tri(oxalato)phosphate (LiTOP) (i.e., LiP(CO2CO2)3) salt (Figs. 1.31h and 1.32b) has a high solubility and high (oxidative) electrochemical stability [569]. The related lithium tetrafluoro(oxalato)phosphate salt (LiFOP) (i.e., LiPF4(CO2CO2)) (Fig. 1.31j) was first formed in electrolytes with mixtures of LiPF6 and LiBOB [576]. The properties of this salt have been extensively characterized in battery electrolytes [570–576]. In many respects, LiFOP has similar properties to LiPF6. Both salts have a similar thermal stability and result in electrolytes with a high conductivity [574, 575], but a carbonate solvent-based electrolyte (i.e., 1 M in EC:DEC:DMC 1:1:1) with LiFOP may be stored at 85 °C for weeks with no evident degradation, whereas extensive salt degradation occurs for a similar electrolyte with LiPF6 [574]. As for the LiBOB and LiDFOB salts, the addition of small amounts of LiFOP is found to improve the capacity retention of MCMB/NMC cells, as well as the stability of the lithiated negative electrode [573]. This is attributed to the oxalate group reacting to form surface layers on both the cathode and carbon anode [574]. The first cycle irreversible capacity losses due to SEI formation on the anode, however, are strongly dependent upon the type of carbon used for the anode [571, 572].
Other organophosphate anions have also been reported, such as those with perfluoropinacol (Fig. 1.31k, l), biphenylene (i.e., tris(2,2′-biphenylylene)phosphate (TBPP −)) (Fig. 1.31n), and salicylate (i.e., tris(salicylato(2-))phosphate (TSP −)) (Fig. 1.31o). No information is available about the use of these anions as lithium salts for battery electrolytes.
Fluorinated organoaluminate anions have also been examined for battery electrolytes (Figs. 1.33 and 1.34) [552, 580]. For smaller bidentate ligands (e.g., oxalate), the AlIII is typically coordinated by three ligands in solid-state salts, instead of two, due to its larger size relative to boron (Fig. 1.34a) [581]. This results in a six-coordinate Al(CO2CO2)3 3− trianion instead of a four-coordinate Al(CO2CO2)2 − anion, although there is spectroscopic evidence for the latter in aqueous solutions as the additional coordination sites are occupied by water molecules (i.e., Al(CO2CO2)2 −⋅2H2O) [582–584]. For more bulky ligands, however, four-coordinate anions are formed. The conductivity values (mS cm−1) (0.2 M salt in DME at 25 °C) for the lithium salts with the anions shown in Fig. 1.33a–f are reported to be 6.4 (Fig. 1.33a), 6.2 (Fig. 1.33b), 1.6 (Fig. 1.33c), 6.2 (Fig. 1.33d), 3.5 (Fig. 1.33e), and 1.3 (Fig. 1.33f) [552, 580]. PC-based electrolytes with the LiAl(HFIP)4 (Figs. 1.33b and 1.34c) and LiAl(HFPP)4 (Figs. 1.33e and 1.34e) salts (0.3 M in PC) both passivated an Al electrode at high potential in a similar manner to a LiPF6 electrolyte [552, 580].

Examples of organoaluminate anions: (a) Al(OC(CF3)3)4 − (Al(PFTB)4 −) [552, 580], (b) Al(OCH(CF3)2)4 − (Al(HFIP)4 −) [552, 580], (c) Al(OCH2(CF3))4 − (Al(TFE)4 −) [552, 580], (d) Al(OC(CH3)(CF3)2)4 − (Al(HFTB)4 −) [552, 580], (e) Al(OC(C6H5)(CF3)2)4 − (Al(HFPP)4 −) [552, 580], and (f) AlF(OC(C6H5)(CF3)2)3 − (AlF(HFPP)3 −) [552, 580]

Anion structures: (a) Al(CO2CO2)3 3−, (b) Al(OC(CF3)3)4 − (Al(PFTB)4 −), (c) Al(OCH(CF3)2)4 − (Al(HFIP)4 −), (d) Al(OC(CH3)(CF3)2)4 − (Al(HFTB)4 −), and (e) Al(OC(C6H5)(CF3)2)4 − (Al(HFPP)4 −) (C—gray, O—red, F—light green, Al—tan)
1.9 Advanced Salts—Other Anions
HNF2 (Fig. 1.35a) is a gas with a T b of −24 °C [590, 591]. This gas loses hydrogen when contacted with various materials to form tetrafluorohydrazine (N2F4). At low temperature (crystalline solid), this acid tends to detonate spontaneously [591]. The LiNF2 salt has not been reported, but it is predicted to be a dimeric complex [592, 593]. HN(CF3)2 (Fig. 1.35a) is a gas with a T b of −6 °C [586–588]. The experimental gas-phase acidity value for HN(CF3)2 is 324.3 kcal mol−1, which is significantly higher (less acidic) than the corresponding values for HN(COCF3)2 (307.5) and HN(SO2CF3)2 (291.8) (Fig. 1.2) [26]. A German patent for Merck has been issued for the preparation of N(CF3)2 − salts (Fig. 1.35a), but this does not include the lithium salt [589]. Another report indicates that the alkali metal bis(trifluoromethyl)amides, -phosphides, and -arsenides (i.e., MN(CF3)2, MP(CF3)2, and MAs(CF3)2) all have a high nucleophilic reactivity [594]. For the related methides, the experimental gas-phase acidity values for HCH(CF3)2 and HC(CF3)3 (Fig. 1.35b) are 343.9 and 326.6 kcal mol−1, respectively (Fig. 1.2) [26]. The value for the latter is comparable to the gas-phase acidity of HCl (328.1 kcal mol−1) [595, 596].

Trifluoromethanol (i.e., HOCF3) (Fig. 1.35c) is unstable at room temperature due to the elimination of HF [618, 619]:
MOCF3 salts (M = K, Rb, and Cs) (Fig. 1.35c) have been prepared, however, by passing O=CF2 through an acetonitrile solution of the fluoride MF [620, 621]:
and the crystal structures determined [597]. No reaction occurred when LiF or NaF was used [597], and some decomposition occurred for KF, suggesting that the lithium trifluoromethoxide (or trifluoroorthocarbonate) salt is unstable (i.e., harder cations are more reactive). Additional salts with perfluoralkyl groups have also been prepared [598–600]:
with M = Rb or Cs and RF = –CF3, –C2F5, or –C3F7. It was found that the cesium salts were more thermally stable than the corresponding rubidium salts. The LiOCH(CF3)2 (Figs. 1.35d and 1.36a) and LiOC(CF3)3 (Figs. 1.35d and 1.36b) salts are both volatile with the former subliming at 50 °C under vacuum (0.05 mmHg) and the latter having a T b of 218 °C [601].

Anion structures: (a) OCH(CF3)2 −, (b) OC(CF3)3 −, and (c) N=C(CF3)2 − (C—gray, N—blue, O—red, F—light green)
The difluoromethanimine acid (i.e., HN=CF2) (Fig. 1.35e) has been reported [604, 605]. This is a colorless gas which is stable at ambient temperature for hours in the gas phase at pressures lower than 5 mm Hg, but which disproportionates at higher pressure:
The lithium hexafluoroisopropylidenimine salt (i.e., LiN=C(CF3)2) (Figs. 1.35e and 1.36c) has been reported [606–611]. The strong electron-withdrawing –CF3 groups make the double bond susceptible to nucleophilic attack. The difluorosulfur oxyimine (i.e., HN=S(O)F2) and bis(trifluoromethyl)sulfur oxyimine (i.e., HN=S(O)(CF3)2) acids (Fig. 1.32f) are also known [612–617], and the lithium bis(trifluoromethyl)sulfur oxyimine salt (i.e., LiN=S(O)F2) (Fig. 1.32f) has been prepared [615]. No information is available, however, about the properties of this latter salt. Note that none of the anions shown in Fig. 1.35 have resonance to stabilize the negative charge, except for the N=S(O)(C n F2n+1)2 − anions.
A family of lithium salts based upon monoanionic species with one or more Lewis acid groups (i.e., BF3) complexed to a Lewis base have also been reported (Fig. 1.37) [622–626]. The most promising of these for electrolyte applications is lithium bis(trifluoroborane)imidazolide (i.e., LiC3N2H3(BF3)2) (Fig. 1.37a). This salt has a high solubility—up to 2 M solutions in EC:EMC (1:3 v:v)—and a conductivity of 5.1 mS cm−1 at 20 °C for a 1 M electrolyte (as compared to equivalent LiBF4 and LiPF6 electrolytes with conductivity values of 1.78 and 7.71 mS cm−1, respectively) [622]. The salt also has a reasonably high (>4.5 V vs. Li/Li+) oxidative stability in DMC. Li/LiNi0.2Co0.8O2 cells with this electrolyte have a comparable performance to cells with LiPF6-based electrolytes [622].

The addition of Lewis acids such as B(OCH(CF3)2)3 and B(OC6F5)3—the so-called anion receptors—to electrolyte solutions containing highly associated salts, such as LiF and LiCO2CF3, has been demonstrated to result in much more conductive electrolytes (than the same electrolytes without the anion receptors) [627–636]. An earlier series of publications by Brownstein explored in detail the formation, or lack of formation, in solution of complex fluoroanions when Lewis acids were added to solutions with a wide variety of fluoroanion Lewis bases [637–642]. Two of the strongest superacids known—HSO3F-SbF5 (“magic acid”) and HF-SbF5 (fluoroantimonic acid) [21, 557]—are also examples of complex formation between a Lewis base and Lewis acid.
The calculated (rather than experimental) gas-phase acidity values for methanides with –NO, –NO2, and –CN substituents are as follows (QC-calculated ΔG acid values in kcal mol−1) relative to CH4 (407.1): HCH2NO (353.5), HCH(NO)2 (318.5) (Fig. 1.38b), HC(NO)3 (303.9) (Fig. 1.38c), HCH2NO2 (348.5), HCH(NO2)2 (311.5) (Fig. 1.38e), HC(NO2)3 (298.0) (Fig. 1.38f), HCH2CN (363.9), HCH(CN)2 (322.9), and HC(CN)3 (288.8) (Fig. 1.38k) [693]. The addition of a single –NO2 group is therefore expected to be more effective at increasing the acidity than a single –CN group. But, while double and triple substitution significantly increases the acid strength further, the gains are much smaller than for the initial substituent and vary for the different functional groups. Thus, trisubstitution with the –CN groups is more effective (as determined from the calculations) at increasing the acidity than for the –NO and –NO2 groups [693]. Therefore, while resonance effects strongly influence the acidity of these methanides, the magnitude of the effect differs markedly depending upon both the degree of substitution and the identity of the functional groups.

Examples of nitroso-, nitro-, and cyano-substituted anions: (a) N(NO)2 − [643], (b) CH(NO)2 − [644], (c) C(NO)3 −, (d) N(NO2)2 − [645], (e) CH(NO2)2 − [646], (f) C(NO2)3 − [646], (g) CH(NO2)(CN)− [646], (h) C(NO)(NO2)(CN)− [646], (i) C(NO2)(CN)2 − [646], (j) N(CN)2 − (DCA−) [647, 648], (k) C(CN)3 − (TCM−) [648, 649], (l) B(CN)4 − (TCB−) [648, 650–654], (m) C(CN)2=C(CN)C(CN)2 − [655–659], (n) C5(CN)5 − [660–662], (o) C5B(CN)6 − [663], (p) C2N3(CN)2 − (DCTA−) [661, 664–668], (q) C3N2(CN)3 − [661, 669–671], (r) C4N(CN)4 − [661, 672–674], (s) C3N2(CN)2(C n F2n+1)− (TDI−) (n = 1), (PDI−) (n = 2) and HDI− (n = 3) [675–686], (t) C7N2(CN)4(C n F2n+1)− [680], (u) C3N2(C2H5)(CF3)2 − [687], (v) C4N(CF3)4 − [688, 689], (w) C5(CF3)5 − [688, 690–692], and (x) C5B(CF3)6 − [663]
Lithium salts with anions having nitroso or nitro functional groups have not been used for lithium battery electrolytes, despite the strong electron-withdrawing properties of these groups. This is due to the energetic characteristics of such anions [694–697]. For example, lithium dinitrosomethanide (i.e., LiCH(NO)2) (Fig. 1.38b), while stable at room temperature, is reported to be heat/shock sensitive and highly explosive, as well as highly toxic [644]. Anions with mixtures of the –NO, –NO2, and –CN substituents have also been prepared (Fig. 1.38g–i) [646], but salts with the nitrosodicyanomethanide (i.e., C(NO)(CN)2 −), nitrodicyanomethanide (i.e., C(NO2)(CN)2 −) (Fig. 1.38i), and other related anions are also predicted to be highly energetic and thus favorable for propellant applications [646]. Further, the trinitrogen dioxide or dinitrosamide anion (i.e., N(NO)2 −) is unstable [643]. This may also be the case for the C(NO)3 − anion as there are no reports for salts with this anion in the scientific literature, although salts with the CH(NO)2 − anion are known [447]. Lithium salts with the dicyanamide anion (LiDCA) (i.e., LiN(CN)2) (Fig. 1.38j), tricyanomethanide (LiTCM) (i.e., LiC(CN)3) (Fig. 1.38k), and tetracyanoborate (LiTCB) (i.e., LiB(CN)4) (Fig. 1.38l) have been little utilized for lithium battery applications [647–653]. The most likely explanation for this is the limited electrochemical stability of these anions to oxidation, relative to other lithium salt anions.
Other cyanocarbon acids are thought to be some of the strongest acids known. For example, the QC-calculated gas-phase acidity (ΔG acid in kcal mol−1) of 1,1,2,3,3-pentacyanopropene (i.e., C(CN)2=C(CN)CH(CN)2) (267.2) (Fig. 1.38m) and pentacyano-cyclo-pentadienide (i.e., HC5(CN)5) (250.1) (Figs. 1.38n and 1.39j) (as compared to a similarly calculated value for HC(CN)3 (287.6)) are some of the lowest values known [21]. Sodium salts with the C(CN)2=C(CN)C(CN)2 − and C5(CN)5 − anions have been prepared [655, 656, 698–700].

Anion structures: (a) N(NO2)2 −, (b) C(NO2)3 −, (c) N(CN)2 −, (d) C(CN)3 −, (e) B(CN)4 −, (f) C(NO2)(CN)2 −, (g) C2N3(CN)2 − (DCTA−), (h) C3N2(CN)2(CF3)− (TDI−), (i) C3N2(CN)2(C2F5)− (PDI−), (j) C5(CN)5 −, and (k) C5(CF3)5 − (B—tan, C—gray, N—blue, O—red, F—light green)
The lithium salt with the 4,5-dicyano-1,2,3-triazolate anion (DCTA −) (also known as 1,2,3-triazole-4,5-dicarbonitrile (TADC −)) (Fig. 1.38p) was first studied by Michot in 1995 [701] and reported in two publications in 2003 [664, 665]. Little has been reported regarding the properties of the lithium salt. Alkali and alkali earth salts with the DCTA− anion have also been prepared [666, 667]. The thermal stability of these salts was found to be excellent (>350 °C) [668]. In general, triazole salts are known to be energetic. The alkali metal salts, however, were found to have low sensitivity towards impact, friction, electrostatic discharge, and fast heating [668]. The acid with the related pyrazole-3,4,5-tricarbonitrile anion (PATC −) (Fig. 1.38q) was first reported in 1962 [669], but the lithium salt has not been investigated extensively as an electrolyte salt [661, 670, 671]. Similarly, the acid with the tetracyanopyrrolide anion (TCP −) (Fig. 1.38r) was also reported in 1962 [672] and the sodium salt with this anion has been prepared [674]. Trifluoromethane-substituted versions of the C5(CN)5 − and TCP− anions have also been reported (Fig. 1.38v, w) [688–692], but the lithium salt has only been reported for LiC5(CF3)5 (Fig. 1.39k) [692].
Lithium salts with the 4,5-dicyano-2-(trifluoromethyl)imidazole (TDI −) (Fig. 1.39h) and 4,5-dicyano-2-(pentafluoroethyl)imidazole (PDI −) (Fig. 1.39i) anions (Fig. 1.38s) were reported by Niedzicki in 2009 [677]. The acid with TDI− (i.e., HTDI) was first reported by Begland in 1974 [675]. A number of publications related to the use of these lithium salts for battery applications have been published [675–686], including a salt with the 4,5-dicyano-2-(n-heptafluoropropyl)imidazolide (HDI−) anion (Fig. 1.38s). The LiTDI, LiPDI, and LiHDI salts have been found to have a high thermal stability (>250 °C), negligible hydrolysis, high oxidative stability on Pt electrodes (4.8 V vs. Li/Li+), and passivate Al at high potential [679, 681]. Half cells with LiMn2O4 electrodes cycled (to 4.3 V vs. Li/Li+) with electrolytes (1 M in EC:DMC 50:50 w:w) containing LiTDI or LiPDI have a similar capacity and capacity retention to the same electrode cycled with an electrolyte with LiPF6 [681]. The rate performance of such cells is only slightly diminished relative to the cell with the LiPF6 electrolyte due to the somewhat lower conductivity of the LiTDI and LiPDI electrolytes (Table 1.6) [681]. Although PC-based electrolytes with these salts have been tested to determine the change in impedance with time upon storage in contact with Li metal electrodes [682], it is not yet clear if these anions are stable when charged to low potential [679] as nitriles are known to have poor reductively stability [702]. A computational study has suggested that the related 4,5,6,7-tetracyano-2-fluoroalkyl benzimidazole anions (Fig. 1.38t) may also have favorable electrolyte properties [680].
A number of other anions have also been reported which do not fit readily into the classifications noted above (Figs. 1.40 and 1.41). Little information is available about the properties of these anions (Table 1.7). The relative ionic association tendency (which influences electrolyte conductivity) for some of these may be estimated by considering the impact of different substituents on an anion’s acidity (Figs. 1.1 and 1.2) and the additional information reported above. For example, the experimental gas-phase acidity values (ΔG acid in kcal mol−1) for HC(C6F5)(SO2CF3)2 (301.3) (Fig. 1.22k) and HC(C6F5)(CN)2 (303.6) are higher than those for HN(SO2CF3)2 (291.8), HC(SO2CF3)3 (289.0), and HC(CN)3 (~294) due to the weaker electron-withdrawing effect of the –C6F5 group relative to –SO2CF3 and –CN for trisubstitution (in contrast to monosubstitution) (Figs. 1.1 and 1.2) [26].

Examples of additional anions: (a) SO2(NCN)2 − [703], (b) N(SO2C n F2n+1)(CN)− [704], (c) C(SO2C n F2n+1)(CN)2 − [704–706], (d) C(SO2C n F2n+1)2(CN)− [705, 706], (e) SO2(C(CN)2)2 − [706], (f) N(COC n F2n+1)(C n F2n+1)− [254], (g) N(SO2C n F2n+1)(C n F2n+1)− [707], (h) N(SO2C n F2n+1)(C6F5)− [708], (i) N(SO2(CN))2 − [361, 709], (j) N(PO(CN)2)2 − [361], (k) C(SO2C2N2S(CF3))(CN)2 − [706], (l) N(SO2C n F2n+1)(SO2CN)− [361], (m) N(PO(C n F2n+1)2)(PO(CN)2)− [361], (n) S(OC2H5)(NSO2CF3)2 − [289], (o) N(C(CF3)NSO2NC(CF3))− [255], (p) Al(N(SO2CF3)2)2(O(CH2CH2O) n CH3)2 − [559], and (q) PO(NSO2CF3)3 3− [710]

Anion structure: C(SO2CF3)2(CN)− (C—gray, N—blue, O—red, F—light green, S—yellow)
Finally, a relatively unique but diverse class of anions for battery electrolytes are the carboranes and boranes [711–727]. These anions are often chemically inert and superweakly coordinating [728]. Carboranes are composed of clusters of carbon and boron atoms, whereas boranes only have boron. These carbon and boron atoms are typically bonded to H, F, Cl, Br, and/or I atoms or other groups [718]. A variety of classifications are used for carboranes and boranes including closo- for a complete polyhedron (e.g., B n X n 2− with X = H, F, Br, Cl, I, and 6 ≤ n ≤ 12) and for polyhedron missing one, two, or more vertices: nido- (e.g., B5X9, B6X10), arachno-, etc. The basicity of the dodecaborate [B12X12]2− anions increases in the order X = F < Cl < Br < I [721]. It is noteworthy that carborane synthesis is time consuming and costly, whereas closo-dodecaborates have similar stability, but are easier and cheaper to prepare [713]. Lithium closo-borane salts (i.e., Li2B10Cl10 and Li2B12Cl12) dissolved in SOCl2 were used in electrolytes for Li/SOCl2 liquid cathode cells in 1979 [729]. Johnson and Whittingham then used these salts for electrolytes for the early Exxon work with Li/TiS2 cells in 1980 [712–714]. These salts were found to be poorly soluble in individual ether solvents, but did have a higher solubility in dioxolane: DME mixtures [712]. More recently, Li2B12F n H12−n (n = 9 and 12) salts have been developed by Air Products (as the Stabilife fluorinated electrolyte salts) with a number of intriguing electrolyte properties. A TGA analysis of the Li2B12F12 salt (Li 2 DFB) (Figs. 1.42 and 1.43) indicates that no mass loss occurs up to 450 °C. The salt is also inert in 98 % sulfuric and 70 % nitric acid, as well as 3 M KOH (in contrast with the B12H12 2− anion which reacts with sulfuric acid) [718]. The conductivity of Li2DFB electrolytes is lower than for comparable electrolytes with LiPF6 [717, 718], but the electrolyte solvent/salt formulation can be tuned to obtain promising cell cycling performance, especially at elevated temperature (60 °C) [718]. In addition, the B12F12 2− anion is not reduced at 0 V (vs. Li/Li+) and undergoes a quasi-reversible oxidation reaction at 4.7 V (vs. Li/Li+) to form the stable radical cation B12F12 − [717]. The potential at which this oxidation reaction occurs can be tuned by changing the anion structure. The Li2B12H3F9 salt, for example, has a reversible oxidation reaction at 4.5 V (vs. Li/Li+) [716, 717]. These anions are thus able to provide overcharge protection for 4+ V Li-ion batteries, as has been demonstrated for MCMB/spinel cells [716, 724]. Note that many organic molecules designed for overcharge protection have poor solubility in carbonate solvents [724]. This is not the case for the Li2B12F9H3 salt which has a reasonably high solubility (≥0.5 M) while also serving as a divalent Li+ cation salt [717, 723].


Anion structure: B12F12 2− (B—tan, F—light green)
1.10 Adoption Criterion for New Salts
Lithium salts have not been prepared for many of the anions noted above. For other anions, the corresponding lithium salts have been reported, but only in a single report with or without some limited electrolyte data provided. Such salts are often not widely available to the greater battery research community…and many of the practitioners in this community do not have the time, resources, or expertise necessary to prepare and purify such salts. Thus, the useful properties and/or limitations of the salts often remain unknown and unutilized.
Ultimately, when developing new lithium salts, it is important to ask what advantages are sought over the widely utilized salt LiPF6. This salt has been a cornerstone of Li-ion battery R&D for well over two decades. But it is clear that LiPF6 has limitations. Properties or features of salts which would make them attractive candidates for commercial battery electrolytes include the following:
-
(a)
Simplified synthesis—If low-cost and/or nontoxic reagents which are easily handled are available for salt synthesis, this may reduce the overall expense of the salt production. Waste from the synthesis/purification methods is another consideration, as is the ease of salt purification to an electrochemical-grade material. The ownership of the intellectual property (IP) rights may be another important factor.
-
(b)
Reduced hazards—In some cases, the reagents, reaction intermediates, and/or final salts are highly toxic or have other undesirable properties (e.g., energetic materials, corrosive) which requires specialized handling of the materials and makes them undesirable for commercial batteries. The degradation products (if battery failure occurs) may also be highly toxic, as has been noted for the fluoro-organic products obtained from the reaction of LiPF6 and carbonate solvents at high temperature when in contact with cathode materials [730]. Thus, the elimination of fluorine from the anions is a worthy goal, but one which is difficult to achieve based upon the information noted in this review.
-
(c)
Hydrolysis—LiPF6 readily hydrolyzes when contacted with water, especially at elevated temperature. This makes the synthesis, storage, and handling of this salt more onerous, thus adding cost to its use. A salt which is not susceptible to hydrolysis would greatly simplify the handing/transportation of the salt for large-scale battery production.
-
(d)
Divalent anions—Divalent anions may offer a means of increasing the Li+ cation content (for a fixed number of anions) or decreasing the amount of salt needed to retain the same number of Li+ cations in the electrolyte. This is dependent upon both of the Li+ cations having weak interactions with the anions, which is difficult to achieve, and the salt having a moderate–high solubility in aprotic solvents, which is also difficult to achieve.
-
(e)
Redox shuttle mechanism—The use of a salt which has other functionality, such as a redox shuttle mechanism (e.g., B12F12 2−) (Figs. 1.42 and 1.43) which protects the cell against overcharging, would be desirable.
-
(f)
Thermal stability—The use of salts, either as primary salts or as additives, which result in electrolytes with high thermal stability (alone and in contact with electrodes)—enabling cell cycling at >60 °C—is a long sought after trait.
-
(g)
New solvents—LiPF6 was optimized for use with the cyclic/acyclic carbonate solvents used for Li-ion batteries. Often the use of LiPF6 for electrolytes with other solvents does result in the most conductive electrolytes (relative to other salts), but there are exceptions. For example, LiPF6 reacts with GBL (forming brown or black solutions), but this solvent has many promising features for electrolytes [91–96]. Thus, LiPF6 has generally been replaced with LiBF4, LiTFSI, LiBOB, LiDFOB, etc. when GBL is used as an electrolyte solvent.
-
(h)
Low-temperature operation—The state-of-the-art carbonate-based electrolytes with LiPF6 do not perform well at low temperature. Typically, approaches to optimize electrolytes for low temperature involve the addition of other solvents (e.g., methyl butyrate) to the carbonate solvents [478]. The difference in conductivity for electrolytes with different salts, however, becomes less significant at low temperature (Fig. 1.14). LiPF6 tends to make high-melting crystalline solvates (Fig. 1.4) with a wide variety of solvents. This may be a factor for long running-time operation of batteries at low temperature. Thus, other salts which form solvates with a low T m or do not crystallize (for specific compositions) (Fig. 1.4)—such as LiFSI, LiTFSI, or LiBETI—may be useful for such applications.
-
(i)
SEI formation—Salts which preferentially decompose to form protective layers on the anode, cathode, Al current collector, etc. (i.e., LiBOB, LiDFOB) are likely to be required for electrodes such as Si alloys, sulfur, and high-voltage cathodes [54, 731, 732]. In particular, the formation of HF in electrolytes with LiPF6 (perhaps due to the reaction of the anions with solvent molecules, instead of water), especially at high potentials and high temperature, is deemed to be particularly problematic [49–54]. Replacing the LiPF6, the use of HF scavengers and the formation of protective layers are all ways to mitigate this problem.
Ultimately, the electrolyte formulations must meet the demanding criterion necessary for long-term battery operation (high stability with selective reactions, high safety, etc.). Conductivity remains an important consideration for high-power applications. Increasingly, salt mixtures (rather than just solvent mixtures) are being used to tailor electrolyte formulations. This trend is likely to increase in the future due to the necessity of simultaneously optimizing so many electrolyte properties which influence the battery’s usable energy and power, lifetime, safety, cost, etc. New salts therefore remain one of the key variables available for the development of advanced electrolyte formulations.
1.11 Summary
A diverse range of anions have been prepared over the past three decades for lithium salts intended for lithium battery electrolyte applications. Many of these anions were originally prepared in efforts to generate stronger superacids. In general, it is found that selectively fluorinating the anions decreases the anion… Li+ cation (ionic association) interactions, thereby increasing the conductivity of electrolyte solutions with the corresponding lithium salts. Anion fluorination also tends to increase the anodic stability of the anions to oxidation at high potential—an important consideration for electrolyte formulations intended for use with high-voltage cathodes. In recent years, nitrile groups have also been used in lieu of fluorination. In some cases, however, salt stability is undesireable. For example, a common approach to improving electrolytes is the use of salt additives—sacrificial anions which degrade to produce interfacial layers with the anode, cathode and/or Al current collector—which stablize the interfaces with the electrolyte resulting in dramatic improvements in battery performance. New salts therefore remain one of the key variables available for the development of advanced electrolyte formulations with tailored properties for demanding energy storage applications.
Acknowledgement The author wishes to express his gratitute to the U.S. Department of Energy (DOE) Office of Basic Energy Science-Division of Materials Sciences and Engineering which supported the preparations of this review under Award DE-SC0002169.
References
R. Selim, P. Bro, J. Electrochem. Soc. 1974, 121, 1457-1459. Some observations on rechargeable lithium electrodes in a propylene carbonate electrolyte.
F. W. Dampier, S. B. Brummer, Electrochim. Acta 1977, 22, 1339-1345. The cycling behavior of the lithium electrode in LiAsF6/methyl acetate solutions.
I. Epelboin, M. Froment, M. Garreau, J. Thevenin, D. Warin, J. Electrochem. Soc. 1980, 127, 2100-2104. Behavior of secondary lithium and aluminum-lithium electrodes in propylene carbonate.
K. Brandt, Solid State Ionics 1994, 69, 173-183. Development of secondary lithium batteries.
J. T. Dudley, D. F. Wilkinson, G. Thomas, R. LeVae, S. Woo, H. Blom, C. Horvath, M. W. Juzkow, B. Denis, P. Juric, P. Aghakian, J. R. Dahn, J. Power Sources 1991, 35, 59-82. Conductivity of electrolytes for rechargeable lithium batteries.
S.-I. Tobishima, K. Hayashi, K.-I. Saito, J.-I. Yamaki, Electrochim. Acta 1995, 40, 537-544. Ethylene carbonate-based ternary mixed solvent electrolytes for rechargeable lithium batteries.
M. Ishikawa, M. Morita, M. Asao, Y. Matsuda, J. Electrochem. Soc. 1994, 141, 1105-1108. Charge/discharge characteristics of carbon fiber with graphite structure in organic electrolytes containing lithium trifluoromethanesulfate and lithium hexafluorophosphate.
K. Zaghib, K. Tatsumi, H. Abe, T. Ohsaki, Y. Sawada, S. Higuchi, J. Power Sources 1995, 54, 435-439. Electrochemical behavior of an advanced graphite whisker anodic electrode for lithium-ion rechargeable batteries.
C.-K. Huang, S. Surampudi, D. H. Shen, G. Halpert, Proc. Electrochem. Soc. 1994, 94-4, 105-110. Effect of electrolyte composition on carbon electrode performance.
K. Sekai, H. Azuma, A. Omaru, S. Fujita, H. Imoto, T. Endo, K. Yamaura, Y. Nishi, S. Mashiko, M. Yokogawa, J. Power Sources 1993, 43–44, 241-244. Lithium-ion rechargeable cells with LiCoO2 and carbon electrodes.
J. M. Tarascon, D. Guyomard, Solid State Ionics 1994, 69, 293-305. New electrolyte compositions stable over the 0 to 5 V voltage range and compatible with the Li1+xMn2O4/carbon Li-ion cells.
K. Xu, Chem. Rev. 2004, 104, 4303-4417. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries.
A. Yoshino, Angew. Chem. Int. Ed. 2012, 51, 5798-5800. The birth of the lithium-ion battery.
M. S. Whittingham, Proc. IEEE 2012, 100, 1518-1536. History, evolution, and future status of energy storage.
B. Scrosati, J. Solid-State Electrochem. 2011, 15, 1623-1630. History of lithium batteries.
K. Brandt, Solid State Ionics 1994, 69, 173-183. Historical development of secondary lithium batteries.
Z.-I. Takehara, K. Kanamura, Electrochim. Acta 1993, 38, 1169-1177. Historical development of rechargeable lithium batteries in Japan.
B. Scrosati, J. Electrochem. Soc. 1992, 139, 2776-2781. Lithium rocking chair batteries: An old concept?
J. M. Tarascon, D. Guyomard, Electrochim. Acta 1993, 38, 1221-1231. The Li1-xMn2O4/C rocking-chair system: A review.
I. A. Koppel, R. W. Taft, F. Anvia, S.-Z. Zhu, L.-Q. Hu, K.-S. Sung, D. D. Desmarteau, L. M. Yagupolskii, Y. L. Yagupolskii, N. V. Igat’ev, N. V. Kondratenko, A. Y. Volkonskii, V. M. Vlasor, R. Notario, P.-C. Maria, J. Am. Chem. Soc. 1994, 116, 3047-3057. The gas-phase acidities of very strong neutral Brønsted acids.
I. A. Koppel, P. Burk, I. Koppel, I. Leito, T. Sonoda, M. Mishima, J. Am. Chem. Soc. 2000, 122, 5114-5124. Gas phase acidities of some neutral Bronsted superacids: A DFT and ab initio study.
I. A. Koppel, P. Burk, I. Koppel, I. Leito, J. Am. Chem. Soc. 2002, 124, 5594-5600. Generalized principle of designing neutral superstrong Brønsted acids.
E. S. Stoyanov, K.-C. Kim, C. A. Reed, J. Am. Chem. Soc. 2006, 128, 8500-8506. An infrared vNH scale for weakly basic anions. Implications for single-molecule acidity and superacidity.
I. Leito, E. Raamat, A. Kütt, J. Saame, K. Kipper, I. A. Koppel, I. Koppel, M. Zhang, M. Mishima, L. M. Yagupolskii, R. Y. Garlyauskayte, A. A. Filatov, J. Phys. Chem. A 2009, 113, 8421-8424. Revision of the gas-phase acidity scale below 300 kcal mol-1.
A. Kütt, T. Rodima, J. Saame, E. Raamat, V. Mäemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner, I. Leito, J. Org. Chem. 2011, 76, 391-395. Equilibrium acidities of superacids.
P. Burk, I. A. Koppel, I. Koppel, L. M. Yagupolskii, R. W. Taft, J. Comp. Chem. 1996, 17, 30-41. Superacidity of neutral Brönsted acids in gas phase.
G. A. Olah, G. K. Surya Prakash, J. Sommer, Science, 1979, 206, 13-20. Superacids.
G. A. Olah, G. K. S. Prakash, Á. Molnár, J. Sommer, Superacid Chemistry, Wiley, Hoboken, NJ, 2009.
R. J. Gillespie, T. E. Peel, Adv. Phys. Org. Chem. 1971, 9, 1-24. Superacid systems.
R. J. Gillespie, T. E. Peel, J. Am. Chem. Soc. 1973, 95, 5173-5178. The Hammett Acidity function for some superacid systems. 11. The systems H2SO4-HSO3F, KSO3F-HSO3F, HSO3F-SO2, HSO3F-AsF5, HSO3F-SbF5, and HSO3F-SbF5-SO2.
I. A. Koppel, V. Pihl, J. Koppel, F. Anvia, R. W. Taft, J. Am. Chem. Soc. 1994, 116, 8654-8657. Thermodynamic acidity of (CF3)3CH and 1H-undecafluorobicyclo[2.2.1]heptane: The concept of anionic (fluorine) hyperconjugation.
A. H. Otto, S. Schrader, T. Steiger, M. Schneider, J. Chem. Soc. Faraday Trans. 1997, 93, 3927-3930. Gas-phase deprotonation of sulfuric acid, perchloric acid, chlorosulfonic acid and fluorosulfonic acid.
R. Steudel, A. H. Otto, Eur. J. Inorg. Chem. 2000, 2379-2386. Geometries, acidities, and dissociation reactions of the gaseous superacids H2S2O3, H2SO5, HSO3F, and HSO3Cl.
J. Barthel, H.-J. Gores, L. Kraml, J. Phys. Chem. 1996, 100, 1283-1287. Effects of electronegative substituents of anions on ion-pair formation. 1. Temperature dependence of the conductivity of lithium fluoroacetate and alkali-metal acetate solutions in dimethyl sulfoxide.
D. Lisbona, T. Snee, Proc. Saf. Environ. 2011, 89, 434-442. A review of hazards associated with primary lithium and lithium-ion batteries.
P. G. Balakrishnan, R. Ramesh, T. Prem Kumar, J. Power Sources 2006, 155, 401-414. Safety mechanisms in lithium-ion batteries.
J. B. Goodenough, Y. Kim, Chem. Mater. 2010, 22, 587-603. Challenges for rechargeable Li batteries.
L. Hu, Z. Zhang, K. Amine, J. Power Sources 2013, 236, 175-180. Electrochemical investigation of carbonate-based electrolytes for high voltage lithium-ion cells.
D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich, E. Zinigrad, L. Asraf, J. S. Gnanaraj, H.-J. Kim, Electrochim. Acta 2004, 50, 247-254. Design of electrolyte solutions for Li and Li-ion batteries: a review.
V. Aravindan, J. Gnanaraj, S. Madhavi, H.-K. Liu, Chem. Eur. J. 2011, 17, 14326-14346. Lithium-ion conducting electrolyte salts for lithium batteries.
C.-K. Park, Z. Zhang, Z. Xu, A. Kakirde, K. Kang, C. Chai, G. Au, L. Cristo, J. Power Sources 2007, 165, 892-896. Variables study for the fast charging lithium ion batteries.
P. G. Bruce, C. A. Vincent, J. Electroanal. Chem. 1987, 255, 1-17. Steady state current flow in solid binary electrolyte cells.
P. G. Bruce, J. Evans, C. A. Vincent, Solid State Ionics 1988, 28–30, 918-922. Conductivity and transference number measurements on polymer electrolytes.
F. M. Gray, Solid Polymer Electrolytes Fundamentals and Technological Applications, VCH, New York, NY 1991.
M. Ue, M. Takeda, M. Takehara, S. Mori, J. Electrochem. Soc. 1997, 144, 2684-2688. Electrochemical properties of quaternary ammonium salts for electrochemical capacitors.
H. Zheng, J. Qin, Y. Zhao, T. Abem Z. Ogumi, Solid State Ionics 2005, 176, 2219-2226. Temperature dependence of the electrochemical behavior of LiCoO2 in quaternary ammonium-based ionic liquid electrolyte.
C. Iwakura, Y. Fukumoto, H. Inoue, S. Ohashi, S. Kobayashi, H. Tada, M. Abe, J. Power Sources 1997, 68, 301-303. Electrochemical characterization of various metal foils as a current collector of positive electrode for rechargeable lithium batteries.
M.-H. Ryou, J.-N. Lee, D. J. Lee, W.-K. Kim, Y. K. Jeong, J. W. Choi, J.-K. Park, Y. M. Lee, Electrochim. Acta 2012, 83, 259-263. Effects of lithium salts on thermal stabilities of lithium alkyl carbonates in SEI layer.
H. Lee, J.-J. Cho, J. Kim, H.-J. Kim, J. Electrochem. Soc. 2005, 152, A1193-A1198. Comparison of voltammetric responses over the cathodic region in LiPF6 and LiBETI with and without HF.
E. Markevich, R. Sharabi, H. Gottlieb, V. Borgel, K. Fridman, G. Salitra, D. Aurbach, G. Semrau, M. A. Schmidt, N. Schall, C. Bruenig, Electrochem. Commun. 2012, 15, 22-25. Reasons for capacity fading of LiCoPO4 cathodes in LiPF6 containing electrolyte solutions.
J. Wu, N. Membreno, W.-Y. Yu, J. D. Wiggins-Camacho, D. W. Flaherty, C. B. Mullins, K. J. Stevenson, J. Phys. Chem. C 2012, 116, 21208-21215. Influence of hydrofluoric acid formation on lithium ion insertion in nanostructured V2O5.
R. Sharabi, E. Markevich, V. Borgel, G. Salitra, G. Gershinsky, D. Aurbach, M. A. Schmidt, N. Schall, C. Stinner, J. Power Sources 2012, 203, 109-114. Raman study of structural stability of LiCoPO4 cathodes in LiPF6 containing electrolytes.
S. F. Lux, I. T. Lucas, E. Pollak, S. Passerini, M. Winter, R. Kostecki, Electrochem. Commun. 2012, 14, 47-50. The mechanism of HF formation in LiPF6 based organic carbonate electrolytes.
H. Zheng, Q. Sun, G. Liu, X. Song, V. S. Battaglia, J. Power Sources 2012, 207, 134-140. Correlation between dissolution behavior and electrochemical cycling performance for LiNi1/3Co1/3Mn1/3O2-based cells.
O. Borodin, W. Behl, T. R. Jow, J. Phys. Chem. C 2013, 117, 8661-8682. Oxidative stability and initial decomposition reactions of carbonate, sulfone, and alkyl phosphate-based electrolytes.
Y. Sasaki, M. Handu, K. Kurashima, T. Tonuma, K. Usami, J. Electrochem. Soc. 2001, 148, A999-A103. Application of lithium organoborate with salicyclic ligand to lithium battery electrolyte.
R. Marom, O. Haik, D. Aurbach, I. C. Halalay, J. Electrochem. Soc. 2010, 157, A972-A983. Revisiting LiClO4 as an electrolyte for rechargeable lithium-ion batteries.
B. Markovsky, F. Amalraj, H. E. Gottlieb, Y. Gofer, S. K. Martha, D. Aurbach, J. Electrochem. Soc. 2010, 157, A423-A429. On the electrochemical behavior of aluminum electrodes in nonaqueous electrolyte solutions of lithium salts.
S. S. Zhang, T. R. Jow, J. Power Sources 2002, 109, 458-464. Aluminum corrosion in electrolyte of Li-ion battery.
K. Kanamura, J. Power Sources 1999, 81–82, 123-129. Anodic oxidation of nonaqueous electrolytes on cathode materials and current collectors for rechargeable lithium batteries.
G. H. Newman, R. W. Francis, L. H. Gaines and B. M. Rao, J. Electrochem. Soc. 1980, 127, 2025-2027. Hazard investigations of LiClO4/dioxolane electrolyte.
R. Jasinski, S. Carroll, J. Electrochem. Soc. 1970, 117, 218-219. Thermal stability of a propylene carbonate electrolyte.
J. S. Gnanaraj, E. Zinigrad, L. Asraf, H. E. Gottieb, M. Sprecher, D. Aurbach, M. Schmidt, J. Power Sources 2003, 119–121, 794-798. The use of accelerating rate calorimetry (ARC) for the study of the thermal reactions of Li-ion battery electrolyte solutions.
Y. Ein-Eli, S. R. thomas, R. Chadha, T. J. Blakley, V. R. Koch, J. Electrochem. Soc. 1997, 144, 823-829. Li-ion battery electrolyte formulated for low-temperature applications.
K. Kanamura, T. Umegaki, S. Shiraishi, M. Ohashi, Z.-I. Takehara, J. Electrochem. Soc. 2002, 149, A185-A194. Electrochemical behavior of Al current collector of rechargeable lithium batteries in propylene carbonate with LiCF3SO3, Li(CF3SO2)2N, or Li(C4F9SO2)(CF3SO2)N.
L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch, R. Atanasocki, J. Power Sources 1997, 68, 320-325. Corrosion of aluminum at high voltages in non-aqueous electrolytes containing perfluoroalkylsulfonyl imides; new lithium salts for lithium-ion cells.
H. Yang, K. Kwon, T. M. Devine, J. W. Evans, J. Electrochem. Soc. 2000, 147, 4399-4407. Aluminum corrosion in lithium batteries. An investigation using the electrochemical quartz crystal microbalance.
M. Morita, T. Shibata, N. Yoshimoto, M. Ishikawa, Electrochim. Acta 2002, 47, 2787-2793. Anodic behavior of aluminum in organic solutions with different electrolytic salts for lithium batteries.
C. G. Barlow, Electrochem. Solid-State Lett. 1999, 2, 362-364. Reaction of water with hexafluorophosphates and with Li bis(perfluoroethylsulfonyl)imide salt.
M. Schmidt, U. Heider, A. Kuehner, R. Oesten, M. Jungnitz, N. Ignat’ev, P. Sartori, J. Power Sources 2001, 97–98, 557-560. Lithium fluorophosphates: A new class of conducting salts for electrolytes for high energy lithium-ion batteries.
T. Kawamura, S. Okada, J. Yamaki, J. Power Sources 2006, 156, 547-554. Decomposition reaction of LiPF6-based electrolytes for lithium ion cells.
A. V. Plakhotnyk, L. Ernst, R. Schmutzler, J. Fluor. Chem. 2005, 126, 27-31. Hydrolysis in the system LiPF6—propylene carbonate—dimethyl carbonate—H2O.
R. Yazami, A. Martinent, in Fluorinated Materials for Energy Conversion, Chap. 9 (T. Nakajima, H. Groult, Eds.), Elsevier, New York, 2005. Fluorinated anions and electrode/electrolyte stability in lithium batteries.
H. Yang, G. V. Zhuang, P. N. Ross Jr., J. Power Sources 2006, 161, 573-579. Thermal stability of LiPF6 salt and Li-ion battery electrolytes containing LiPF6.
B. Ravdel, K. M. Abraham, R. Gitzendanner, J. DiCarlo, B. Lucht, C. Campion, J. Power Sources 2003, 119–121, 805-810. Thermal stability of lithium-ion battery electrolytes.
N.-S. Choi, I. A. Profatilova, S.-S. Kim, E.-H. Song, Thermochim. Acta 2008, 480, 10-14. Thermal reactions of lithiated graphite anode in LiPF6-based electrolyte.
W. Li, B. L. Lucht, J. Electrochem. Soc. 2006, 153, A1617-A1625. Lithium-ion batteries: Thermal reactions of electrolyte with the surface of metal oxide cathode particles.
H. Yang, X.-D. Shen, J. Power Sources 2007, 167, 515-519. Dynamic TGA–FTIR studies on the thermal stability of lithium/graphite with electrolyte in lithium-ion cell.
M. S. Ding, T. R. Jow, J. Electrochem. Soc. 2004, 151, A2007-A2015. How conductivities and viscosities of PC-DEC and PC-EC solutions of LiBF4, LiPF6, LiBOB, Et4NBF4, and Et4NPF6 differ and why.
T. R. Jow, M. S. Ding, K. Xu, S. S. Zhang, J. L. Allen, K. Amine, G. L. Hendriksen, J. Power Sources 2003, 119–121, 343-348. Nonaqueous electrolytes for wide-temperature-range operation of Li-ion cells.
J. Li, C. F. Yuan, Z. H. Guo, Z. A. Zhang, Y. Q. Lai, J. Liu, Electrochim. Acta 2012, 59, 69-74. Limiting factors for low-temperature performance of electrolytes in LiFePO4/Li and graphite/Li half cells.
S. S. Zhang, K. Xu, T. R. Jow, J. Electrochem. Soc. 2002, 149, A586-A590. Study of LiBF4 as an electrolyte salt for a Li-ion battery.
E.-S. Hong, S. Okada, T. Sonoda, S. Gopukumar, J. Yamaki, J. Electrochem. Soc. 2004, 151, A1836-A1840. Thermal stability of electrolytes with mixtures of LiPF6 and LiBF4 used in lithium-ion cells.
N. Katayama, T. Kawamura, Y. Baba, J. Yamaki, J. Power Sources 2002, 109, 321-326. Thermal stability of propylene carbonate and ethylene carbonate-propylene carbonate-based electrolytes for use in Li cells.
P. Ping, Q. Wang, J. Sun, H. Xiang, C. Chen, J. Electrochem. Soc. 2010, 157, A1170-A1176. Thermal stabilities of some lithium salts and their electrolyte solutions with and without contact to a LiFePO4 electrode.
N. Takami, M. Sekino, T. Ohsaki, M. Kanda, M. Yamamoto, J. Power Sources 2001, 97–98, 677-680. New thin lithium-ion batteries using a liquid electrolyte with thermal stability.
N. Takami, T. Ohsaki, H. Hasebe, M. Yamamoto, J. Electrochem. Soc. 2002, 149, A9-A12. Laminated thin Li-ion batteries using a liquid electrolyte.
S. S. Zhang, K. Xu, T. R. Jow, Electrochem. Commun. 2002, 4, 928-932. A new approach toward improved low temperature performance of Li-ion battery.
S. S. Zhang, K. Xu, T. R. Jow, J. Solid State Electrochem. 2003, 7, 147-151. Low-temperature performance of Li-ion cells with a LiBF4-based electrolyte.
X. Zuo, C. Fan, J. Liu, X. Xiao, J. Wu, J. Nan, J. Electrochem. Soc. 2013, 160, A1199-A1204. Lithium tetrafluoroborate as an electrolyte additive to improve the high voltage performance of lithium-ion battery.
A. Chagnes, B. Carré, P. Willmann, R. Dedryvère, D. Gonbeau, D. Lemordant, J. Electrochem. Soc. 2003, 150, A1255-A1261. Cycling ability of γ-butyrolactone-ethylene carbonate based electrolytes.
J. Kasnatscheew, R. W. Schmitz, R. Wagner, M. Winter, R. Schmitz, J. Electrochem. Soc. 2013, 160, A1369-1374. Fluoroethylene carbonate as an additive for γ-butyrolactone based electrolytes.
D. Belov, D.-T. Shich, J. Solid State Electrochem. 2012, 16, 603-615. GBL-based electrolyte for Li-ion battery: thermal and electrochemical performance.
K. Zaghib, K. Striebel, A. Guerfi, J. Shim, M. Armand, M. Gauthier, Electrochim. Acta 2004, 50, 263-270. LiFePO4/polymer/natural graphite: Low cost Li-ion batteries.
T. Ohsaki, N. Takami, M. Kanda, M. Yamamoto, Stud. Surf. Sci. Catal. 2001, 132, 925-928. High performance thin lithium-ion battery using an aluminum-plastic laminated film bag.
K. Zaghib, M. Dontigny, A. Guerfi, J. Trottier, J. Hamel-Paquet, V. Gariepy, K. Galoutov, P. Hovington, A. Mauger, H. Groult, C.M. Julien, J. Power Sources 2012, 216, 192-200. An improved high-power battery with increased thermal operating range: C-LiFePO4//C-Li4Ti5O12.
A. Vallée, S. Besner, J. Prud’homme, Electrochim. Acta 1992, 37, 1579-1583. Comparative study of poly(ethylene oxide) electrolytes made with LiN(CF3SO2)2, LiCF3SO3 and LiClO4: Thermal properties and conductivity behavior.
M. Minier, C. Berthier, W. Gorecki, J. Phys. 1984, 45, 739-744. Thermal analysis and NMR study of a poly(ethylene oxide) complex electrolyte: PEO(LiCF3SO3)x.
D. Fauteux, Solid State Ionics 1985, 17, 133-138. Formation of a passivating film at the lithium poly(ethylene oxide) (PEO)-lithium trifluoromethanesulfonate (LiCF3SO3) interface.
R. Neat, M. Glasse, R. Linford, A. Hooper, Solid State Ionics 1986, 18–19, 1088-1092. Thermal history and polymer electrolyte structure: implications for solid-state battery design.
P. Lightfoot, M. A. Mehta, P. G. Bruce, Science 1993, 262, 883-885. Crystal structure of the polymer electrolyte poly(ethylene oxide)3:LiCF3SO3.
T. Shodai, B. B. Owens, H. Ohtsuka, J. Yamaki, J. Electrochem. Soc. 1994, 141, 2978-2981. Thermal stability of the polymer electrolyte (PEO)8LiCF3SO3.
S. Chintapalli, R. Frech, Electrochim. Acta 1998, 43, 1395-1400. Kinetic effects in the ionic association of poly(ethylene oxide)-lithium triflate complexes.
R. Frech, S. Chintapalli, P. G. Bruce, C. A. Vincent, Macromolecules 1999, 32, 808-813. Crystalline and amorphous phases in the poly(ethylene oxide)-LiCF3SO3 system.
C. P. Rhodes, R. Frech, Macromolecules 2001, 34, 2660-2666. Local structures in crystalline and amorphous phases of diglyme-LiCF3SO3 and poly(ethylene oxide)-LiCF3SO3 systems: Implications for the mechanism of ionic transport.
G. B. Appetecchi, F. Alessandrini, R. G. Duan, A. Arzu, J. Power Sources 2001, 101, 42-46. Electrochemical testing of industrially produced PEO-based polymer electrolytes.
W. A. Henderson, Solid State Ionics 2012, 217, 1-5. Influence of polymer chain length and chains ends on polyelectrolyte solvate structure and melting point.
A. Webber, J. Electrochem. Soc. 1991, 138, 2586-2590. Conductivity and viscosity of solutions of LiCF3SO3, Li(CF3SO2)N, and their mixtures.
S.-I. Tobishima, T. Okada, Electrochim. Acta 1985, 30, 1715-1722. Lithium cycling efficiency and conductivity in high dielectric solvent/low viscosity solvent mixed systems.
M. Morita, O. Yamada, M. Ishikawa, J. Power Sources 1999, 81–82, 425-429. Charge and discharge performances of lithiated metal oxide cathodes in organic electrolyte solutions with different compositions.
L. A. Dominey, V. R. Koch, T. J. Blakley, Electrochim. Acta 1992, 37, 1551-1554. Thermally stable lithium salts for polymer electrolytes.
S. Sylla, J.-Y. Sanchez, M. Armand, Electrochim. Acta 1992, 37, 1699-1701. Electrochemical study of linear and crosslinked PEO-based polymer electrolytes.
M. Armand, W. Gorecki, R. Andreani, Int. Symp. Polym. Electrolytes 1990, 91-97. Perfluorosulfonimide salts as solute for polymer electrolytes.
P. Johansson, S. P. Gejji, J. Tegenfeldt, J. Lindgren, Electrochim. Acta 1998, 43, 1375-1379. The imide ion: Potential energy surface and geometries.
I. Rey, P. Johansson, J. Lindgren, J. C. Lassègues, J. Grondin, L. Servant, J. Phys. Chem. A 1998, 102, 3249-3258. Spectroscopic and theoretical study of (CF3SO2)2N- (TFSI-) and (CF3SO2)2NH (HTFSI).
S. P. Gejji, C. H. Suresh, K. Babu, S. R. Gadre, J. Phys. Chem. A 1999, 103, 7474-7480. Ab initio structure and vibrational frequencies of (CF3SO2)2N–Li+ ion pairs.
L. Xue, C. W. Padgett, D. D. DesMarteau, W. T. Pennington, Solid State Sci. 2002, 4, 1535-1545. Synthesis and structures of alkali metal salts of bis[(trifluoromethyl)sulfonyl]imide.
D. Benrabah, R. Arnaud, J.-Y. Sanchez, Electrochim. Acta 1995, 40, 2437-2443. Comparative ab initio calculations on several salts.
W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young Jr., R. Frech, Chem. Mater. 2005, 17, 2284-2289. Glyme-lithium bis(trifluoromethanesulfonyl)imide and glyme-lithium bis(perfluoroethanesulfonyl)imide phase behavior and solvate structures.
M. G. Davidson, P. R. Raithby, A. L. Johnson, P. D. Bolton, Eur. J. Inorg. Chem. 2003, 18, 3445-3452. Structural diversity in Lewis-base complexes of lithium triflamide.
D. Brouillette, D. E. Irish, N. J. Taylor, G. Perron, M. Odziemkowski, J. E. Desnoyers, Phys. Chem. Chem. Phys. 2002, 4, 6063-6071. Stable solvates in solution of lithium bis(trifluoromethylsulfone)imide in glymes and other aprotic solvents: Phase diagrams, crystallography and Raman spectroscopy.
Q. Zhou, P. D. Boyle, L. Malpezzi, A. Mele, J.-H. Shin, S. Passerini, W. A. Henderson, Chem. Mater. 2011, 23, 4331-4337. Phase behavior of ionic liquid-LiX mixtures: Pyrrolidinium cations and TFSI– anions - Linking structure to transport properties.
J. L. Nowinski, P. Lightfoot, P. G. Bruce, J. Mater. Chem. 1994, 4, 1579–1580. Structure of LiN(CF3SO2)2, A novel salt for electrochemistry.
W. A. Henderson, D. M. Seo, Q. Zhou, P. D. Boyle, J.-H. Shin, H. C. De Long, P. C. Trulove, S. Passerini, Adv. Energy Mater. 2012, 2, 8014-8019. An alternative ionic conductivity mechanism for plastic crystalline salt–lithium salt electrolyte mixtures.
A. Chagnes, B. Carré, P. Willmann, D. Lemordant, J. Power Sources 2002, 109, 203-213. Modeling viscosity and conductivity of lithium salts in γ-butyrolactone.
S. Steudte, J. Neumann, U. Bottin-Weber, M. Diedenhofen, J. Arning, P. Stepnowski, S. Stolte, Green Chem. 2012, 14, 2474-2483. Hydrolysis study of fluoroorganic and cyano-based ionic liquid anions – consequences for operational safety and environmental stability.
J. L. Goldman, A. B. McEwen, Electrochem. Solid-State Lett. 1999, 2, 501-503. EMIIm and EMIBeti on aluminum. Anodic stability dependence on lithium salt and propylene carbonate.
B. Garcia, M. Armand, J. Power Sources 2004, 132, 206-208. Aluminium corrosion in room temperature molten salt.
R.-S. Kühnel, M. Lübke, M. Winter, S. Passerini, A. Balducci, J. Power Sources 2012, 214, 178-184. Suppression of aluminum current collector corrosion in ionic liquid containing electrolytes.
C. Peng, L. Yang, Z. Zhang, K. Tachibana, Y. Yang, J. Power Sources 2007, 173, 510-517. Anodic behavior of Al current collector in 1-alkyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl] amide ionic liquid electrolytes.
C. Peng, L. Yang, Z. Zhang, K. Tachibana, Y. Yang, S. Zhao, Electrochim. Acta 2008, 53, 4764-4772. Investigation of the anodic behavior of Al current collector in room temperature ionic liquid electrolytes.
J. L. Allen, D. W. McOwen, S. A. Delp, E. T. Fox, J. S. Dickmann, S.-D. Han, Z.-B. Zhou, T. R. Jow, W. A. Henderson, J. Power Sources 2013, 237, 104-111. N-Alkyl-N-methylpyrrolidinium difluoro(oxalato)borate ionic liquids: Physical/electrochemical properties and Al corrosion.
D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle, W. A. Henderson, Energy Environ. Sci. 2014, 7, 416-426. Concentrated electrolytes: Decrypting electrolyte properties and reassessing Al corrosion mechanisms.
D. M. Seo, O. Borodin, Sang-Don Han, Q. Ly, P. D. Boyle, W. A. Henderson, J. Electrochem. Soc. 2012, 159, A553-A565. Electrolyte solvation and ionic association. I. Acetonitrile-lithium salt mixtures: Intermediate and highly associated salts.
D. M. Seo, O. Borodin, S.-D. Han, P. D. Boyle, W. A. Henderson, J. Electrochem. Soc. 2012, 159, A1489-A1500. Electrolyte solvation and ionic association. II. Acetonitrile-lithium salt mixtures: Highly dissociated salts.
D. M. Seo, O. Borodin, D. Balogh, M. O'Connell, Q. Ly, S.-D. Han, S. Passerini, W. A. Henderson, J. Electrochem. Soc. 2013, 160, A1061-A1070. Electrolyte solvation and ionic association. III. Acetonitrile-lithium salt mixtures—Transport properties.
D. M. Seo, P. D. Boyle, O. Borodin, W. A. Henderson, RSC Adv. 2012, 2, 8014-8019. Li+ cation coordination by acetonitrile—Insights from crystallography.
D. M. Seo, P. D. Boyle, W. A. Henderson, Acta Crystallogr. 2011, E67, m1148. Tetrakis(acetonitrile-κN)lithium hexafluoridophosphate acetonitrile monosolvate.
D. M. Seo, P. D. Boyle, W. A. Henderson, Acta Crystallogr. 2011, E67, m534. Poly[bis(acetonitrile-κN) bis[μ3-bis(trifluoromethanesulfonyl)imido-κ4O, O':O'':O''']dilithium].
Y. Yokota, V. G. Young Jr., J. G. Verkade, Acta Crystallogr. 1999, C55, 196-198. The homoleptic lithium complex [Li(CH3CN)4]ClO4.
D. M. Seo, P. D. Boyle, J. L. Allen, S.-D. Han, E. Jónsson, P. Johansson, W. A. Henderson, J. Phys. Chem. C 2013, in-press. Solvate structures and computational/spectroscopic characterization of LiBF4 electrolytes.
D. M. Seo, P. D. Boyle, W. A. Henderson, Acta Crystallogr. 2011, E67, m547. Poly[[(acetonitrile)-lithium(I)]-μ3-tetrafluoridoborato].
W. A. Alves, Vib. Spectrosc. 2007, 44, 197-200. Vibrational spectroscopic and conductometric studies of lithium battery electrolyte solutions.
W. Huang, R. Frech, P. Johansson, J. Lindgren, Electrochim. Acta 1995, 40, 2147-2151. Cation-polymer interaction and ionic association in diglyme-LiCF3SO3 and diglyme-propylene carbonate-LiCF3SO3 complexes.
J. Grondin, D. Talaga, J.-C. Lassègues, W. A. Henderson, Phys. Chem. Chem. Phys. 2004, 6, 938-944. Raman study of crystalline solvates between glymes CH3(OCH2CH2)nOCH3 (n = 1, 2 and 3) and LiClO4.
J. Grondin, J.-C. Lassègues, M. Chami, L. Servant, D. Talaga, W. A. Henderson, Phys. Chem. Chem. Phys. 2004, 6, 4260-4267. Raman study of tetraglyme–LiClO4 solvate structures.
S.-D. Han, J. L. Allen, E. Jonsson, P. Johansson, D. W. McOwen, P. D. Boyle, W. A. Henderson, J. Phys. Chem. C 2013, 117, 5521-5531. Solvate structures and computational/spectroscopic characterization of lithium difluoro(oxalato)borate (LiDFOB) electrolytes.
W. A. Henderson, J. Phys. Chem. B 2006, 110, 13177-13183. Glyme-lithium salt phase behavior.
Y. Matsuda, M. Morita, F. Tachihara, Bull. Chem. Soc. Jpn. 1986, 59, 1967-1973. Conductivity of lithium salts in the mixed systems of high permittivity solvents and low viscosity solvents.
Y. Matsuda, M. Morita, K. Yamada, K. Hirai, J. Electrochem. Soc. 1985, 132, 2538-2543. Characteristics of sulfolane-based electrolytes for rechargeable lithium batteries.
M. Morita, F. Tachihara, Y. Matsuda, Electrochim. Acta 1987, 32, 299-305. Dimethyl sulfoxide-based electrolytes for rechargeable lithium batteries.
Y. Matsuda, M. Morita, J. Power Sources 1987, 20, 273-278. Organic electrolyte solutions for rechargeable lithium batteries.
M. Morita, Y. Matsuda, J. Power Sources 1987, 20, 299-304. Effect of solvent blending on cycling characteristics of lithium.
Y. Sasaki, H. Miura, K. Tominaga, N. Nanbu, Electrochemistry 2002, 70, 334-336. Electrolytic properties and application to a lithium battery of ternary solvent electrolytes with ethylene carbonate - dimethyl carbonate - 3-methylsydnone and 3-ethylsydnone.
G. Moumouzias, G. Ritzoulis, D. Siapkas, D. Terzidis, J. Power Sources 2003, 122, 57-66. Comparative study of LiBF4, LiAsF6, LiPF6, and LiClO4 as electrolytes in propylene carbonate-diethyl carbonate solutions for Li/LiMn2O4 cells.
H.-B. Han, S.-S. Zhou, D.-Z. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand, Z.-B. Zhou, J. Power Sources 2011, 196, 3623-3632. Lithium bis(fluorosulfonyl)imide (LiFSI) as conducting salt for nonaqueous liquid electrolytes for lithium-ion batteries: Physicochemical and electrochemical properties.
L. P. Klemann, G. H. Newman, J. Electrochem. Soc. 1981, 128, 13-18. LiB(CH3)4-dioxolane electrolyte in rechargeable Li/TiS cells.
W. E. Rhine, G. Stucky, S. W. Peterson, J. Am. Chem. Soc. 1975, 97, 6401-6406. Stereochemistry of polynuclear compounds of the main group elements. A neutron and x-ray diffraction investigation of lithium-hydrogen-carbon interactions in LiB(CH3)4.
A. Haag, G. Hesse, Liebigs Ann. Chem. 1971, 751, 95-108. Uber die umsetzung von lithium-tetraalkylboranaten mit alkylierungsmitteln.
H. H. Horowitz, J. I. Haberman, L. P. Klemann, G. H. Newman, E. L. Stogryn, T. A. Whitney, Proc. Electrochem. Soc. 1981, 81–4, 131-143. The anodic oxidation stability of lithium electrolytes.
L. P. Klemann, G. H. Newman, U.S. Patent 4,060,674, 1977. Alkali metal anode-containing cells having electrolytes of organometallic-alkali metal salts and organic solvents.
L. P. Klemann, G. H. Newman, E. L. Stogryn, U.S. Patent 4,139,681, 1978. Electrochemical cells having alkali metal anodes and electrolyte salt complex compositions including haloorganometallic alkali metal salt complexes.
P. K. Bakshi, S. V. Sereda, O. Knop, Can. J. Chem. 1994, 72, 2144-2152. Crystal chemistry of tetraradial species. Part 6. Embarras de richesses: Lithium cation coordinated by eight 0-H…phenyl bonds.
J. M. Butler, G. M. Gray, J. Chem. Crystallogr. 2002, 32, 171-175. Diaquo(1,2-dimethoxyethane)lithium tetraphenylborate.
H. Schödel, T. Vaupel, H. Bock, Acta Crystallogr. 1996, C52, 637-640. Lithium tetraphenylborate·2THF·2H2O.
B. Wilde, A. Jaenschke, F. Olbrich, Z. Anorg. Allg. Chem. 2008, 634, 166-172. Komplexe der alkalimetalltetraphenylborate mit makrocyclischen kronenethern.
W. A. Henderson, N. R. Brooks, W. W. Brennessel, V. G. Young Jr., Chem. Mater. 2003, 15, 4679-4684. Triglyme-Li+ cation solvate structures: Models for amorphous concentrated liquid and polymer electrolytes (I).
W. A. Henderson, N. R. Brooks, V. G. Young Jr., J. Am. Chem. Soc. 2003, 125, 12098-12099. Single-crystal structures of polymer electrolytes.
Y. Sasaki, S. Sekiya, M. Handa, K. Usami, J. Power Sources 1999, 79, 91-96. Chelate complexes with boron as lithium salts for lithium battery electrolytes.
V. Gutmann, H. J. Emeléus, J. Chem. Soc. 1950, 1046-1050. The preparation of hexafluoroniobates, hexafluorotantalates, and hexafluorobismuthates by means of bromine trifluoride.
B. Cox, J. Chem. Soc. 1956, 876-878. Complex fluorides. Part IV. The structural chemistry of complex fluorides of the general formula ABF6.
V. T. Kalinnikov, Y. I. Balabanov, L. A. Agulyanskaya, A. I. Agulyanskii, Y. A. Serebryakov, Zh. Obshch. Khim. 1984, 54, 1929-1932. Physicochemical study of the reaction of lithium metatantalate with ammonium hydrogen difluoride.
E. G. Rakov, M. V. Melkumyants, V. F. Sukhoverkhov, Zh. Neorg. Khim. 1990, 35, 1123-1126. Reaction of lithium niobate and tantalate with bromine trifluoride.
J.-H. Kim, S. Yonezawa, M. Takashima, J. Fluor. Chem. 2006, 127, 1054-1057. Reaction between LiF and MF5 (M = Ta, Nb) in a fluorine atmosphere.
V. R. Koch, L. A. Dominey, J. L. Goldman, M. E. Langmuir, J. Power Sources 1987, 20, 287-291. New anions as supporting electrolytes for rechargeable lithium batteries.
C. Nanjundiah, J. L. Goldman, L. A. Dominey, V. R. Koch, J. Electrochem. Soc. 1988, 135, 2914-2917. Electrochemical stability of lithium M hexafluoride (M = P, As, Sb) in tetrahydrofuran and sulfolane.
V. R. Koch, J. Electrochem. Soc. 1979, 126, 181-187. Reactions of tetrahydrofuran and lithium hexafluoroarsenate with lithium.
M. Alamgir, K. M. Abraham, J. Appl. Electrochem. 1994, 24, 288-297. Studies to improve the Li/SO2Cl2 cell.
W. L. Bowden, J. S. Miller, D. Cubbison, A. N. Dey, J. Electrochem. Soc. 1984, 131, 1768-1772. New electrolyte salts for Li/SOCl2 cells.
K. A. Klinedinst, R. A. gary, J. Electrochem. Soc. 1987, 134, 1884-1890. Performance and abuse resistance of a high-rate prismatic lithium/sulfuryl chloride cell.
T. J. Lee, P. C. Yao, G. T.-K. Fey, J. Power Sources 1991, 35, 143-151. Raman and 7Li NMR spectroscopic studies of Li/SO2 rechargeable cells.
W. P. Kilroy, C. Schlakikjer, P. Polsonetti, M. Jones, J. Power Sources 1993, 43–44, 715-723. Optimized lithium oxyhalide cells.
M. M. Morrison, N. Marincic, J. Power Sources 1993, 45, 343-352. Studies in lithium oxyhalide cells for downhole instrumentation. Use of lithium tetrachlorogallate electrolyte in Li/SOC12 cells.
M. Goledzinowski, R. J. Doré, I. R. Hill, J. Power Sources 1995, 54, 356-361. Cyclic voltammetric and Raman spectroscopic studies of the Li/LiAlCl4/SO2 and Li/LiGaCl4/SO2 rechargeable systems.
I. R. Hill, B. G. Anderson, M. Goledzinowski, R. J. Doré, J. Electrochem. Soc. 1995, 142, 3267-3273. Investigations of the chemistry of rechargeable Li/SO2 cells containing retrachlorometallate:SO2 electrolytes.
W. C. West, A. Shevade, J. Soler, J. Kulleck, M. C. Smart, B. V. Ratnakumar, M. Moran, R. Haiger, K. O. Christie, G. K. Surya Prakash, J. Electrochem. Soc. 2010, 157, A571-A577. Sulfuryl and thionyl halide-based ultralow temperature primary batteries.
J. Dreher, B. Haas, G. Hambitzer, J. Power Sources 1993, 43–44, 583-587. Rechargeable LiCoO2 in inorganic electrolyte solution.
C. P. Park, S. M. Oh, J. Power Sources 1997, 68, 338-343. Performances of Li/LixCoO2 cells in LiAlCl4·3SO2 electrolyte.
K. Sonoda, A. Ueda, K. Iwamoto, Eur. Patent Appl. EP 1174941 A2, 2002. Non-aqueous electrolyte and electrochemical device comprising the same.
Z.-B. Zhou, M. Takeda, T. Fujii, M. Ue, J. Electrochem. Soc. 2005, 152, A351-A356. Li[C2F5BF3] as an electrolyte salt for 4 V class lithium-ion cells.
G. Pawelke, H. Bürger, Coord. Chem. Rev. 2001, 215, 243-266. Perfluoroalkyl borates: Congeners of perfluoroalkanes.
M. Ue, T. Fujii, Z.-B. Zhou, M. Takeda, S. Kinoshita, Solid State Ionics 2006, 177, 323-331. Electrochemical properties of Li[CnF2n+1BF3] as electrolyte salts for lithium-ion cells.
Z.-B. Zhou, M. Takeda, M. Ue, J. Fluor. Chem. 2003, 123, 127-131. Novel electrolyte salts based on perfluoroalkyltrifluoroborate anions 1. Synthesis and characterization.
E. Bernhardt, G. Henkel, H. Willner, G. Pawelke, H. Bürger, Chem. Eur. J. 2001, 7, 4696-4705. Synthesis and [properties of the tetrakis(trifluoromethyl)borate anion, [B(CF3)4]-: Structure determination of Cs[B(CF3)4] by single-crystal x-ray diffraction.
T. Kubota, M. Adachi, S. Fujita, Eur. Patent Appl. EP 1465267 A2, 2004. Battery with specified electrolyte.
S. S. Zhang, J. Power Sources 2008, 180, 586-590. LiBF3Cl as an alternative salt for the electrolyte of Li-ion batteries.
H.-J. Frohn, H. Franke, P. Fritzen, V. V. Bardin, J. Organomet. Chem. 2000, 598, 127-135. (Fluoroorgano)fluoroboranes and -fluoroborates I: Synthesis and spectroscopic characterization of potassium fluoroaryltrifluoroborates and fluoroaryldifluoroboranes.
H. S. Lee, X.-Q. Yang, J. McBreen, Int. Patent WO 2011/031401 A2, 2011. Lithium non-fluorinated and fluorinated phenyl trifluoro borate salts for non-aqueous battery electrolytes.
A. G. Massey, A. J. Park, J. Org. Chem. 1964, 2, 245-250. Perfluorophenyl derivatives of the elements. I. Tris(pentafluorophenyl)boron.
B. Zhang, M. Köberl, A. Pöthig, M. Cokoja, W. A. Hermann, F. E. Kühn, Z. Naturforsch. 2012, 67b, 1030-1036. Synthesis and characterization of imidazolium salts with the weakly coordinating [B(C6F5)4]– anion.
M. Kuprat, M. Lehmann, A. Schulz, A. Villinger, Organometallics 2010, 29, 1421-1427. Synthesis of pentafluorophenyl silver by means of Lewis Acid catalysis: Structure of silver solvent complexes.
E. Zygadło-Monikowska, Z. Florjańczyk, J. Ostrowska, P. Bołtromiuk, J. Frydrych, W. Sadurski, N. Langwald, Electrochim. Acta 2011, 57, 66-73. Synthesis and characterization of new trifluoroalkoxyborates lithium salts of ionic liquid properties.
F. Kita, H. Sakata, A. Kawakami, H. Kamizori, T. Sonoda, H. Nagashima, N. V. Pavlenko, Y. L. Yagupolskii, J. Power Sources 2001, 97–98, 581-583. Electronic structures and electrochemical properties of LiPF6-n(CF3)n.
A. Omaru, T. Nirasawa, Eur. Patent Appl. EP1318562 A2, 2002. Secondary battery and electrolyte used therefore.
R. Oesten, U. Heider, M. Schmidt, Solid State Ionics 2002, 148, 391-397. Advanced electrolytes.
J. S. Gnanaraj, E. Zinigrad, L. Asraf, M. Spreche, H. E. Gottlieb, W. Geissler, M. Schmidt, D. Aurbach, Electrochem. Commun. 2003, 5, 946-951. On the use of LiPF3(CF2CF3)3 (LiFAP) solutions for Li-ion batteries. Electrochemical and thermal studies.
J. S. Gnanaraj, E. Zinigrad, M. D. Levi, D. Aurbach, M. Schmidt, J. Power Sources 2003, 119–121, 799-904. A comparison among LiPF6, LiPF3(CF2CF3)3 (LiFAP), and LiN(SO2CF2CF3)2 (LiBETI) solutions: Electrochemical and thermal studies.
J. S. Gnanaraj, M. D. Levi, Y. Gofer, D. Aurbach, M. Schmidt, J. Electrochem. Soc. 2003, 150, A445-A454. LiPF3(CF2CF3)3: A salt for rechargeable lithium ion batteries.
E. Zinigrad, L. Larush-Asraf, J. S. Gnanaraj, H. E. Gottieb, M. Sprecher, D. Aurbach, J. Power Sources 2005, 146, 176. Calorimetric studies of the thermal stability of electrolyte solutions based on alkyl carbonates and the effect of the contact with lithium.
S. Tsujioka, T. Mitsui, K. Kondo, A. Fujiwara, Int. Patent WO 2010122867 A1, 2010. Electrolyte for electrochemical device, electrolyte solution using same, and nonaqueous electrolyte battery.
B. Zhang, M. Köberl, A. Pöthig, M. Cokoja, W. A. Herrmann, F. E. Kühn, Z. Naturforsch. 2012, 67b, 1030-1036. Synthesis and characterization of imidazolium salts with the weakly coordinating [B(C6F5)4]– anion.
M. Kuprat, M. Lehmann, A. Schulz, A. Villinger, Organometallics 2010, 29, 1421-1427. Synthesis of pentafluorophenyl silver by means of Lewis Acid catalysis: Structure of silver solvent complexes.
M. Bolte, I. Ruderfer, T. Müller, Acta Crystallogr. 2005, E61, m1581-m1582. Lithium–tetrakis(pentafluorophenyl)borate–benzene (1/1/2).
L. P. Klemann, G. H. Newman, T. A. Whitney, E. L. Stogryn, D. Farcasiu, Proc. Electrochem. Soc. 1981, 81–4, 79-88. Investigations of anion structure in lithium tetraorganoborate salts - steps toward the design of an organic electrolyte.
T. Aoki, T. Fujinami, J. Electrochem. Soc. 2005, 152, A2352-A2356. Lithium ion conductivity of polymer electrolytes based on insoluble lithium tetrakis(pentafluorobenzenethiolato)borate and poly(ethylene oxide).
T. Aoki, A. Konno, T. Fujinami, J. Power Sources 2005, 146, 412-417. Polymer electrolytes composed of lithium tetrakis(pentafluorobenzenethiolato) borate and poly(fluoroalkylcarbon)s.
X. Zhang, U. Gross, K. Seppelt, Angew. Chem. Int. Ed. Engl. 1995, 34, 1858-1860. Fluorocarbonate, [FCO2]– : Preparation and structure.
M. Hojo, T. Ueda, H. Hamada, Z. Chen, S. Umetani, J. Mol. Liq. 2009, 145, 24-32. Conductometric studies on higher ion-aggregation from lithium fluoroalkanoates in propylene carbonate and N,N-dimethylformamide.
F. Kita, H. Sakata, S. Sinomoto, A. Kawakami, H. Kamizori, T. Sonoda, H. Nagashima, J. Nie, N. V. Pavlenko, Y. L. Yagupolskii, J. Power Sources 2000, 90, 27-32. Characteristics of the electrolyte with fluoro organic lithium salts.
G. Nagasubramanian, D. H. Shen, S. Surampudi, Q. Wang, G. K. Surya Prakash, Electrochim. Acta 1995, 40, 2277-2280. Lithium superacid salts for secondary lithium batteries.
F. Kita, A. Kawakami, J. Nie, T. Sonoda, H. Kobayashi, J. Power Sources 1997, 68, 307-310. On the characteristics of electrolytes with new lithium imide salts.
S. A. Ullrich, G. L. Gard, R. L. Nafshun, M. M. Lerner, J. Fluor. Chem. 1996, 79, 33-38. Lithium salts of bis (perfluoroalkyl) sulfonic acids: synthesis, characterization and conductivity studies.
C. Chauvin, F. Alloin, P. Judeinstein, C. P. del Valle, J.-Y. Sanchez, ChemPhysChem 2006, 7, 1921-1929. NMR and electrochemical study on lithium oligoether sulfate in polymeric and liquid electrolytes.
C. Chauvin, X. Ollivrin, F. Alloin, J.-F. LeNest, J.-Y. Sanchez, Electrochim. Acta 2005, 50, 3843-3852. Lithium salts based on oligoether sulfate esters.
W. M. Lamanna, A. Xiao, M. J. Triemert, P. T. Pham, Int. Patent Appl. WO 2012170240 A1, 2012. Lithium ion electrochemical cells including fluorocarbon electrolyte additives.
P. Sartori, G. Bauer, J. Fluor. Chem. 1978, 12, 203-210. 2,3,5,6-Tetrafluorbenzoldisulf-onsäure aus pentafluorbenzolsulfonsäure.
M. B. Herath, S. E. Creager, A. Kitaygorodskiy, D. D. DesMarteau, ChemPhysChem 2010, 11, 2871-2878. Perfluoroalkyl phosphonic and phosphinic acids as proton conductors for anhydrous proton-exchange membranes.
W. Lange, Ber. Dtsch. Chem. Ges. 1929, 62B, 793-801. Monofluorophosphoric acid and the similarity between its salts and the sulfates.
W. Lange, Ber. Dtsch. Chem. Ges. 1927, 60B, 962-970. The resemblance of the fluorosulfonates to the perchlorates in chemical and crystallographic relationships, and a fluorophosphate.
U. Schülke, R. Kayser, Z. Anorg. Allg. Chem. 1991, 600, 221-226. Herstellung von fluorophosphaten, difluorophosphaten, fluorophosphonaten und fluorophosphiten in fluoridhaltigen harnstoffschmelzen.
G. A. Bukhalova, I. V. Mardirosova, Izv. An. SSR Neorg. Mater. 1966, 2, 2015-2019. Complex formation in binary systems of alkali metal fluorides and metaphosphates.
F. W. Bennett, H. J. Emeléus, R. N. Haszeldine, J. Chem. Soc. 1954, 3598-3603. Organometallic and organometalloidal fluorine compounds. Part X. Trifluoromethyl-phosphonous and -phosphonic acids.
R. C. Thompson, W. Reed, Nucl. Chem. Lett. 1969, 5, 581-585. Preparation and infrared spectra of alkali metal difluorophosphates.
P. Vast, A. Semmoud, A. Addou, G. Palavit, J. Fluor. Chem. 1988, 38, 297-302. Etude methodologique de la synthese des difluorodioxophosphates metalliques a partir de l'oxyde du difluorure de phosphoryle.
W. Lange, Ber. Dtsch. Chem. Ges. 1929, 62B, 786-792. Difluophosphoric acid and formation from it of salts similar to those of perchloric acid.
W. Traube, J. Hoerenz, F. Wunderlich, Ber. Dtsch. Chem. Ges. 1918, 52B, 1272-1284. Fluorosulfonic acid, fluorosulfonates and sulfuryl fluoride.
V. I. Sirenko, V. D. Prisyazhnyi, T. A. Zmievskaya, A. A. Kramarenko, Russ. J. Electrochem. 1999, 35, 1133-1136. Aprotic electrolytes containing lithium fluorosulfonate.
A. Bałkowska, G. Szymański, L. Werblan, J. Electroanal. Chem. 1990, 287, 229-238. Conductivities of lithium salts in a mixed solvent: Comparison of conductance equations for their analysis.
Z. Žák, M. Kosiŭka, Acta Crystallogr. 1978, B34, 38-40. The crystal structure of lithium fluorosulphaste LiSO3F.
R. N. Haszeldine, J. M. Kidd, J. Chem. Soc. 1954, 4228-4232. Polyfluoroalkyl derivatives of sulfur. Part I. Trifluoromethanesulfphonic acid.
T. Gramstad, R. N. Haszeldine, J. Chem. Soc. 1956, 173-180. Perfluoroalkyl derivatives of sulphur. Part IV. Perfluoroalkanesulphonic acids.
T. J. Brice, P. W. Trott, U.S. Patent 2,732398, 1956. Fluorocarbon sulfonic acids and derivatives.
W. H. Tiedemann, D. N. Bennion, J. Chem. Engr. Data 1971, 16, 368-370. Density, viscosity, and conductivity of lithium trifloromethanesulfonate solutions in dimethylsulfite.
T. Fujinaga, I. Sakamoto, Pure Appl. Chem. 1980, 52, 1387-1396. Electrochemical characteristics of trifluoromethanesulfphonic acid and its salts in non-aqueous solvents.
M. Tremayne, P. Lightfoot, M. A. Mehta, P. G. Bruce, K. D. M. Harris, K. Shankland, C. J. Gilmore, G. Bricogne, J. Solid State Chem. 1992, 100, 191-196. Ab initio structure determination of LiCF3SO3 from X-ray powder diffraction data using entropy maximization and likelihood ranking.
M. Bolte, H.-W. Lerner, Acta Crystallogr. 2001, E57, m231-m232. Lithium trifluoromethanesulfonate.
R. Dinnebier, N. Sofina, M. Jansen, Z. Anorg. Allg. Chem. 2004, 630, 1613-1616. The structure of the high temperature modification of lithium triflate (γ-LiSO3CF3).
G. Hirankumar, C. Iojoiu, F. Alloin, J. Y. Sanchez, T. Pagnier, J. Raman. Spectrosc. 2007, 38, 1570-1576. Dissociation of C6F5SO3Li in two organic solvents with different dielectric constants: A Raman spectroscopic study.
H. Falius, Angew. Chem., Int. Ed. 1970, 9, 733-734. Simple synthesis of phosphorofluoridous acid, H[PHO2F].
R. N. Haszeldine, Angew. Chem. 1954, 693-728. Neuere chemie des fluors. Organometall- und organometalloid-verbindungen des fluors.
H. J. Emeléus, R. N. Haszeldine, R. C. Paul, J. Chem. Soc. 1955, 563-574. Organometallic and organometalloidal fluorine compounds. Part XII. Bistrifluoromethylphos-phinic acid and related phosphorus oxy-acids.
S. Furukawa, S. Yoshimura, M. Takahashi, Jap. Patent JP 03040372, 1991. Nonaqueous lithium batteries with new electrolytes.
K. W. Oliver, S. J. Rettig, R. C. Thompson, J. Trotter, S. Xia, J. Fluor. Chem. 1997, 83, 47-50. Synthesis of perfluorodiphenylphosphinic acid and its potassium and oxonium salts; Crystal structure of oxonium perfluorodiphenylphosphinate.
G. H. Sprenger, K. J. Wright, J. M. Shreeve, Inorg. Chem. 1973, 12, 2890-2893. Polar additions of chlorine monofluoride and chlorine fluorosulfate to fluorinated isocyanates.
H. W. Roesky, Angew. Chem. Int. Ed. 1969, 8, 510. 3,5-Bis(trifluorornethyl)-1,2,4,6-thiatriaza-2,5-cyclohexadiene-1,1-dione.
F. Ye, R. E. Noftle, J. Fluor. Chem. 1997, 81, 193-196. Preparation of fluorinated imides.
F. Ye, R. E. Noftle, D. D. DesMarteau, Syn. Met. 1993, 60, 141-144. Preparation and properties of conducting polypyrrole doped with fluorinated imides.
B. Mandal, T. Sooksimuang, B. Griffin, A. Padhi, R. Filler, Solid State Ionics 2004, 175, 267-272. New lithium salts for rechargeable battery electrolytes.
L. Conte, G. P. Gambaretto, G. Caporiccio, F. Alessandrini, S. Passerini, J. Fluor. Chem. 2004, 125, 243-252. Perfluoroalkanesulfonylimides and their lithium salts: Synthesis and characterisation of intermediates and target compounds.
A. Vij, R. L. Kirchmeier, J. M. Shreeve, R. D. Verma, Coord. Chem. Rev. 1997, 158, 413-432. Some fluorine-containing nitrogen acids and their derivatives.
M. Beran, J. Příhoda, J. Taraba, Polyhedron, 2010, 29, 991-994. A new route to the syntheses of N-(fluorosulfuryl)fulfonamide salts: Crystal structure of Ph4P+ [CF3SO2NSO2F]–.
W. Xianming, E. Yasukawa, S. Kasuya, Electrochim. Acta 2001, 46, 813-819. Electrochemical properties of tetrahydropyran-based ternary electrolytes for 4 V Lithium metal rechargeable batteries.
K. Kubota, J. Matsumoto, Chem. Lett. 2011, 40, 1105-1106. Melting and crystallization behaviors of alkali metal (fluorosulfonyl)(trifluoromethylsulfonyl)amides.
R. D. Howells, W. M. Lamanna, A. D. Fanta, J. E. Waddell, Int. Patent Appl. WO 97/23448, 1997. Preparation of bis(fluoroalkylenesulfonyl) imides and (fluoroalkylsulfonyl) (fluorosulfonyl) imides.
R. D. Howells, W. M. Lamanna, A. D. Fanta, J. E. Waddell, U.S. Patent WO 5,874,616, 1999. Preparation of bis(fluoroalkylenesulfonyl) imides and (fluoroalkylsulfonyl) (fluorosulfonyl) imides.
H. V. Venkatasetty, Annual Battery Conference on Applications and Advances, 16th, Long Beach, CA, United States, Jan. 9-12, 2001, 277-282. New and novel lithium imide electrolytes and copolymers: Synthesis and characterization for lithium rechargeable batteries.
H. W. Roesky, H. H. Giere, Inorg. Nucl. Chem. Lett. 1971, 7, 171-175. Sulfur-nitrogen compounds. 33. Preparation of N-(trifluoromethylsulfonyl)sulfamoyl fluoride and some reactions.
H. V. Venkatasetty, J. Power Sources 2001, 97–98, 671-673. Novel superacid-based lithium electrolytes for lithium ion and lithium polymer rechargeable batteries.
W. M. Lamana, L. J. Krause, J. W. Summerfield, Int. Patent WO 9711504 A1, 1997. Battery containing bis(perfluoroalkylsulfonyl)imide and cyclic perfluoroalkylenedisulfonylimide.
W. Gorecki, C. Roux, M. Clémancey, M. Armand, E. Belorizky, ChemPhysChem 2002, 620-625. NMR and conductivity study of polymer electrolytes in the imide family: P(EO)/Li[N(SO2CnF2n+1)(SO2CmF2m+1)].
J. A. Roderiguez, R. E. Noftle, Inorg. Chem. 1971, 10, 1874-1877. The reaction of fluorosulfuryl isocyanate with alkali metal fluorides.
R. E. Noftle, J. W. Green, S. K. Yarbro, J. Fluor. Chem. 1976, 7, 221-227. Reaction of fluorosulfuryl isocyanate with group IIb metal fluorides.
H. Matsumoto, H. Sakaebe, K. Tatsumi, J. Power Sources 2005, 146, 45-50. Preparation of room temperature ionic liquids based on aliphatic onium cations and asymmetric amide anions and their electrochemical properties as a lithium battery electrolyte.
M. B. Herath, S. E. Creager, R. V. Rajagopal, O. E. Geiculescu, D. D. DesMarteau, Electrochim. Acta 2009, 54, 5877-5883. Ionic conduction in polyether-based lithium arylfluorosulfonimide ionic melt electrolytes.
S. Hamrock, P. Pham, Int. Patent Appl. WO 98/50349, 1997. Fluorinated sulphonamide and sulphone derivatives.
A. Chakrabarti, R. Filler, B. Mandal, ECS Trans. 2007, 6, 77-88. Synthesis and characterization of a new class of mono-lithium salts for PEO-based solid polymer electrolytes.
B. K. Mandal, R. Filler, J. Fluor. Chem. 2005, 126, 845-848. New fluorine-containing plasticized low lattice energy lithium salt for plastic batteries.
R. J. Koshar, Eur. Patent EP 57327 A1, 1982. Cyclic perfluoroaliphatic disulfonimides.
L. J. Krause, J. W. Summerfield, U.S. Patent 5,691,081, 1997. Battery containing bis(perfluoroalkylsulfonyl)imide and cyclic perfluoroalkulene disulfonylimide salts.
H. W. Roesky, A. Hoff, Chem. Ber. 1968, 101, 162-173. Darstellung und untersuchung von fluorsulfurylverbindungen.
J. Taraba, Z. Žak, Acta Crystallogr. 2004, C60, o79-o81. The polysulfonylamines bis(methylsulfonamido) sulfone and bis(trifluoromethylsulfonamido) sulfone.
R. Appel, H. Rittersbacher, Chem. Ber. 1964, 97, 849-851. Reaction of sulfuryl diisocyanate with halosulfuric acids. A simple method for the preparation of fluorosulfonyl isocyanate and imidobis(sulfuryl fluoride).
Z.-B. Zhou, H. Han, J. Nie, W. Song, H. Zhang, Chin. Patent CN 102786443, 2012. Dibasic or tribasic fluoro-containing sulfonylimide alkaline metal salt and ionic liquid, and application thereof.
O. E. Geiculescu, Y. Xie, R. Rajagopal, S. E. Creager, D. D. DesMarteau, J. Fluor. Chem. 2004, 125, 1179-1185. Dilithium bis[(perfluoroalkyl)sulfonyl]diimide salts as electrolytes for rechargeable lithium batteries.
R. Jüschke, M. Koeckerling, P. Sartori, Z. Naturforsch. B 1998, 53, 135-144. Synthesis of novel lithium salts with doubly charged anions for secondary batteries.
K. Luo, R. Filler, B. K. Mandal, Solid State Ionics 2006, 177, 857-861. Synthesis and characterization of novel non-fluorinated tri-lithium imide salts for use in lithium-ion battery electrolytes.
A. Chakrabarti, R. Filler, B. K. Mandal, Solid State Ionics 2010, 180, 1640-1645. Synthesis and properties of a new class of fluorine-containing dilithium salts for lithium-ion batteries.
W. Buckendahl, O. Glemser, Chem. Ber. 1977, 110, 1154-1158. Some substitution reactions of nitrogen-sulfur-chlorine compounds.
H. W. Roesky, W. Schmieder, W. Isenberg, W. S. Sheldrick, G. M. Sheldrick, Chem. Ber. 1982, 115, 2714-2727. Anions of sulfur with the coordination number 3: Synthesis, structure, and range of existence.
H. W. Roesky, W. G. Böwing, J. Chem. Soc. Chem. Comm. 1975, 735-736. Preparation and x-ray structure of sulfur-nitrogen oxides.
C. Jäckh, W. Sundermeyer, Chem. Ber. 1973, 106, 1752-1757. Darstellung und reaktionen von carbonylhalogeniden und -pseudohalogeniden.
D. Schomburg, A. Blaschette, S. Kassomenakis, Z. Anorg. Allg. Chem. 1991, 604, 47-51. Polysulfonyl amines. XXVI. A polymeric lithium amide with alternating tetra- and hexacoordinate lithium atoms: Crystal structure of lithium dimesylamide monohydrate.
S. Ukawa, Y. Yamamoto, Jap. Patent JP 2005209523 A, 2005. Lithium batteries inhibiting self discharge under high-temperature environment, and their electrolytes.
O. Moers, A. Blaschette, J. G. Jones, Z. Anorg. Allg. Chem. 2002, 628, 2086-2090. Polysulfonylamines. Part 161. Crystal structures of metal di(methanesulfonyl)amides. Part 8. A surprising pair of isostructural compounds. Crystal structure of AgNa[(CH3SO2)2N]2·H2O and space-group revision for Li[(CH3SO2)2N]·H2O.
A. Blaschette, K. H. Nagel, P. G. Jones, Z. Naturforsch. B 1994, 49, 36-42. Polysulfonylamines. LIV. (12-Crown-4)lithium dimesylamide-acetonitrile (3/2): A crystal with two fundamentally different conformations of the same coronand.
J. M. Pringle, J. Golding, K. Baranyai, C. M. Forsyth, G. B. Deacon, J. L. Scott, D. R. MacFarlane, New J. Chem. 2003, 27, 1504-1510. The effect of anion fluorination in ionic liquids—physical properties of a range of bis(methanesulfonyl)amide salts.
A. Scozzafava, M. D. Banciu, A. Popescu, C. T. Supuran, J. Enzym. Inhib. 2000, 15, 443-453. Carbonic anhydrase inhibitors: Inhibition of isozymes I, II and IV by sulfamide and sulfamic acid derivatives: 85.
Z.-B. Zhou, H. Han, J. Nie, W. Song, H. Zhang, Chin. Patent CN 102786443, 2012. Dibasic or tribasic fluoro-containing sulfonylimide alkaline metal salt and ionic liquid, and application thereof.
R. Y. Garlyauskayte, A. N. Chernega, C. Michot, M. Armand, Y. L. Yagupolskii, L. M. Yagupolskii, Org. Biomol. Chem. 2005, 3, 2239-2243. Synthesis of new organic super acids—N-(trifluoromethylsulfonyl)imino derivatives of trifluoromethanesulfonic acid and bis(trifluoromethylsulfonyl)imide.
J. Foropoulos Jr., D. D. DesMarteau, J. Am. Chem. Soc. 1982, 104, 4260-4261. Bis[bis(trifluoromethanesulfonyl)imido]xenon: A new compound possessing xenon-nitrogen bonds.
J. Foropoulos Jr., D. D. DesMarteau, Inorg. Chem. 1984, 23, 3720-3723. Synthesis, properties, and reactions of bis((trifluoromethyl)sulfonyl) imide, (CF3SO2)2NH.
J. N. Meussdorffer, H. Niederprum, Chem. Ztg. 1972, 96, 582-583. Bis(perfluoralkanesulfonyl)imides (RfSO2)2NH.
M. Armand, Int. Patent WO 90/11999, 1990. Process for the synthesis of sulfonylimidides.
J. Grondin, D. Talaga, J. C. Lassègues, P. Johansson, W. A. Henderson, J. Raman Spectrosc. 2007, 38, 53-60. Spectroscopic and ab initio characterization of the conformational states of the bis(perfluoroethanesulfonyl) imide anion (BETI–).
W. A. Henderson, F. McKenna, M. A. Khan, N. R. Brooks, V. G. Young Jr., R. Frech, Chem. Mater. 2005, 17, 2284-2289. Glyme-lithium bis(trifluoromethanesulfonyl)imide and glyme-lithium bis(perfluoroethanesulfonyl)imide phase behavior and solvate structures.
P. Johansson, J. Tegenfeldt, J. Lindgren, J. Phys. Chem. A 2000, 104, 954-961. Vibrational spectroscopy and ab initio calculations on [N(C2F5SO2)2]– and the corresponding superacid HN(C2F5SO2)2.
K. Naoi, M. Mori, Y. Naruoka, W. M. Lamanna, R. Atanasoski, J. Electrochem. Soc. 1999, 146, 462-469. The surface film formed on a lithium metal electrode in a new imide electrolyte, lithium bis(perfluoroethylsulfonylimide) [LiN(C2F5SO2)2].
C. Capiglia, Y. Saito, H. Kageyama, P. Mustarelli, T. Iwamoto, T. Tabuchi, H. Tukamoto, J. Power Sources 1999, 81–82, 859-862. 7Li and 19F diffusion coefficients and thermal properties of non-aqueous electrolyte solutions for rechargeable lithium batteries.
X. Wang, E. Yasukawa, S. Mori, J. Electrochem. Soc. 1999, 146, 3992-3998. Electrochemical behavior of lithium imide/cyclic ether electrolytes for 4 V lithium metal rechargeable batteries.
X. Wang, E. Yasukawa, S. Kasuya, J. Electrochem. Soc. 2000, 147, 2421-2426. Lithium imide electrolytes with two-oxygen-atom-containing cycloalkane solvents for 4 V lithium metal rechargeable batteries.
L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch, R. Atanasoski, J. Power Sources 1997, 68, 320-325. Corrosion of aluminum at high voltages in non-aqueous electrolytes containing perfluoroalkylsulfonyl imides; New lithium salts for lithium-ion cells.
L. Péter, J. Arai, J. Appl. Electrochem. 1999, 29, 1053-1061. Anodic dissolution of aluminium in organic electrolytes containing perfluoroalkylsulfonyl imides.
X. Wang, E. Yasukawa, S. Mori, Electrochim. Acta 2000, 45, 2677-2684. Inhibition of anodic corrosion of aluminum cathode current collector on recharging in lithium imide electrolytes.
L. J. Krause, J. W. Summerfield, U.S. Patent 5,691,081, 1997. Battery containing bis(perfluoroalkylsulfonyl)imide and cyclic perfluoroalkylene disulfonylimide salts.
K. Zaghib, P. Charest, A. Guerfi, J. Shim, M. Perrier, K. Striebel, J. Power Sources 2004, 134, 124-129. Safe Li-ion polymer batteries for HEV applications.
K. Zaghib, P. Charest, A. Guerfi, J. Shim, M. Perrier, K. Striebel, J. Power Sources 2005, 146, 380-385. LiFePO4 safe Li-ion polymer batteries for clean environment.
M. Beran, J. Prihoda, Z. Zak, M. Cernik, Polyhedron 2006, 25, 1292-1298. A new route to the syntheses of alkali metal bis(fluorosulfuryl)imides: Crystal structure of LiN(SO2F)2.
A. Guerfi, S. Duchesne, Y. Kobayashi, A. Vijh, K. Zaghib, J. Power Sources 2008, 175, 866-873. LiFePO4 and graphite electrodes with ionic liquids based on bis(fluorosulfonyl)imide (FSI)- for Li-ion batteries.
K. Kuboto, T. Nohira, T. Goto, R. Hagiwara, Electrochem. Commun. 2008, 10, 1886-1888. Novel inorganic ionic liquids possessing low melting temperatures and wide electrochemical windows: Binary mixtures of alkali bis(fluorosulfonyl)amides.
A. Abouimrane, J. Ding, I. J. Davidson, J. Power Sources 2009, 189, 693-696. Liquid electrolyte based on lithium bis-fluorosulfonyl imide salt: Aluminum corrosion studies and lithium ion battery investigations.
A. Guerfi, M. Gontigny, Y. Kobayashi, A. Vijh, K. Zaghib, J. Solid State Electrochem. 2009, 13, 1003-1014. Investigations on some electrochemical aspects of lithium-ion ionic liquid/gel polymer battery systems.
K. Kuboto, T. Nohira, R. Hagiwara, J. Chem. Engr. Data 2010, 55, 3142-3146. Thermal properties of alkali bis(fluorosulfonyl)amides and their binary mixtures.
A. S. Best, A. I. Bhatt, A. F. Hollehkamp, J. Electrochem. Soc. 2010, 157, A903-A911. Ionic liquids with the bis(fluorosulfonyl)imide anion: Electrochemical properties and applications in battery technology.
J. Huang, A. F. Hollenkamp, J. Phys. Chem. C 2010, 114, 21840-21847. Thermal behavior of ionic liquids containing the FSI anion and the Li+ cation.
S. Tsuzuki, K. Hayamizu, S. Seki, J. Phys. Chem. B 2010, 114, 16329-16336. Origin of the low viscosity of [emim][(FSO2)2N] ionic liquid and its lithium salt mixture: Experimental and theoretical study of self-diffusion coefficients, conductivities, and intermolecular interactions.
K. Hiyamizu, S. Tsuzuki, S. Seki, K. Fujii, M. Suenaga, Y. Umebayashi, J. Chem. Phys. 2010, 133, 194505/1-194505/13. Studies on the translational and rotational motions of ionic liquids composed of N-methyl-N-propyl-pyrrolidinium (P13) cation and bis(trifluoromethanesulfonyl)amide and bis(fluorosulfonyl)amide anions and their binary systems including lithium salts.
L. Li, S. Zhou, H. Han, H. Li, J. Nie, M. Armand, Z. Zhou, X. Huang, J. Electrochem. Soc. 2011, 158, A74-A82. Transport and electrochemical properties and spectral features of non-aqueous electrolytes containing LiFSI in linear carbonate solvents.
S. Seki, K. Takei, H. Miyashiro, M. Watanabe, J. Electrochem. Soc. 2011, 158, A769-A774. Physicochemical and electrochemical properties of glyme-LiN(SO2F)2 complex for safe lithium-ion secondary battery electrolyte.
M. Nadherna, J. Reiter, J. Moskon, R. Dominko, J. Power Sources 2011, 196, 7700-7706. Lithium bis(fluorosulfonyl)imide-PYR14TFSI ionic liquid electrolyte compatible with graphite.
K. Hayamizu, S. Tsuzuki, S. Seki, Y. Umebayashi, J. Chem. Phys. 2011, 135, 084505/1-084505/11. Nuclear magnetic resonance studies on the rotational and translational motions of ionic liquids composed of 1-ethyl-3-methylimidazolium cation and bis (trifluoromethanesulfonyl)amide and bis(fluorosulfonyl)amide anions and their binary systems including lithium salts.
K. Kubota, T. Nohira, R. Hagiwara, Electrochim. Acta 2012, 66, 320-324. New inorganic ionic liquids possessing low melting temperatures and wide electrochemical windows: Ternary mixtures of alkali bis(fluorosulfonyl)amides.
J. Reiter, M. Nadherna, R. Dominko, J. Power Sources 2012, 205, 402-407. Graphite and LiCo1/3Mn1/3Ni1/3O2 electrodes with piperidinium ionic liquid and lithium bis(fluorosulfonyl)imide for Li-ion batteries.
G. G. Eshetu, S. Grugeon, G. Gachot, D. Mathiron, M. Armand, S. Laruelle, Electrochim. Acta 2013, 102, 133-141. LiFSI vs. LiPF6 electrolytes in contact with lithiated graphite: Comparing thermal stabilities and identification of specific SEI-reinforcing additives.
R. Appel, G. Eisenhauer, Chem. Ber. 1962, 95, 246-248. The synthesis of HN(SO2F)2.
C. Michot, M. Armand, J.-Y. Sanchez, Y. Choquette, M. Gauthier, Int. Patent WO 95/26056, 1995. Ionic conducting material having good anticorrosive properties.
K. Kubota, T. Nohira, R. Hagiwara, H. Matsumoto, Chem. Lett. 2010, 39, 1303-1304. Thermal properties of alkali (fluorosulfonyl)(trifluoromethylsulfonyl)amides.
H. Han, J. Guo, D. Zhang, S. Feng, W. Feng, J. Nie, Z. Zhou, Electrochem. Commun. 2011, 13, 265-168. Lithium (fluorosulfonyl)(nonafluorobutanesulfonyl)imide (LiFNFSI) as conducting salt to improve the high-temperature resilience of lithium-ion cells.
H. Matsumoto, N. Terasawa, T. Umecky, S. Tsuzuki, H. Sakaebe, K. Asaka, K. Tatsumi, Chem. Lett. 2008, 37, 1020-1021. Low melting and electrochemically stable ionic liquids based on asymmetric fluorosulfonyl(trifluoromethylsulfonyl)amide.
J. V. Silverton, D. T. Gibson, Acta Crystallogr. 1965, 19, 651-658. The disordered crystal and molecular structure of tris(methylsulfonyl)methane.
G. Klöter, H. Pritzkow, K. Seppelt, Angew. Chem. Int. Ed. Engl. 1980, 19, 942. Tris(fluorosulfonyl)methane, HC(SO2F)3.
F. Alloin, J.-Y. Sanchez, J. Power Sources 1999, 81–82, 795-803. Electrochemical investigation of organic salts in polymeric and liquid electrolytes.
P. Johansson, J. Phys. Chem. A 2001, 105, 9258-9264. Ab initio structures and vibrational spectra of Li[C(CF3SO2)3] and Li[CH(CF3SO2)2].
C. W. Walker Jr., J. D. Cox, M. Salomon, J. Electrochem. Soc. 1996, 143, L80-L82. Conductivity and electrochemical stability of electrolytes containing organic solvent mixtures with lithium tris(trifluoromethanesulfonyl)methide.
Y. L. Yagupolskii, T. I. Savina, Zh. Obshch. Khim. 1983, 19, 79-81. Methanetrisulfonic acid derivatives.
Y. L. Yagupolskii, T. I. Savina, Zh. Obshch. Khim. 1985, 21, 2048-2054. Aryl ethers of the aci-forms of methanetrisulfonyl trifluoride and their reactions.
Y. L. Yagupolskii, I. I. Gerus, T. I. Savina, Zh. Obshch. Khim. 1988, 24, 73-77. Addition of methanetris(sulfonyl fluoride) to unsaturated compounds.
Y. L. Yagupolskii, T. I. Savina, I. I. Gerus, R. K. Orlova, Zh. Obshch. Khim. 1990, 26, 2030-2031. New Friedel-Crafts reaction catalyst: tris(fluorosulfonyl)methane.
Y. L. Yagupolskii, T. I. Savina, N. O. Pavlenko, A. A. Pankov, S. V. Pazenok, Zh. Obshch. Khim. 1991, 61, 1512-1518. Tris(fluorosulfonyl)methanide -C(SO2F)3 - an effective stabilizing nonnucleophilic carbanion.
P. Burk, I. A. Koppel, J. Tapfer, F. Anvia, R. W. Taft, J. Mol. Struct. 1994, 121, 191-196. Theoretical study of prototropic tautomerism and acidity of tris(fluorosulfonyl)methane.
G. Klöter, H. Pritzkow, K. Seppelt, Angew. Chem. Int. Ed. 1980, 19, 942. Tris(fluorosulfonyl)methane, HC(SO2F).
Y. L. Yagupolskii, W. Tyrra, R. Gnann, N. Maggiarosa, D. Naumann, J. Fluor. Chem. 2002, 113, 143-146. 2,6-Difluorophenylxenon(II)bis(fluorsulfonyl)amid, -bis(trifluormethyl-sulfonyl)amid, -tris(fluorsulfonyl)methanid und -bis(trifluormethylsulfonyl)methanid-neue C-Xe-N- und C-Xe-C-verbindungen?
L. Turowsky, K. Seppelt, Inorg. Chem. 1988, 27, 2135-2137. Tris((trifluoromethyl)sulfonyl)methane, HC(SO2CF3)3.
D. Aurbach, O. Chusid, I. Weissman, P. Dan, Electrochim. Acta 1996, 41, 747-760. LiC(SO2CF3)3, a new salt for Li battery systems. A comparative study of Li and non-active metal electrodes in its ethereal solutions using in situ FTIR spectroscopy.
R. Arnaud, D. Benrabah, J.-Y. Sanchez, J. Phys. Chem. 1996, 100, 10882-10891. Theoretical study of CF3SO3Li, (CF3SO2)2NLi, and (CF3SO2)2CHLi ion pairs.
T. Gramstad, R. N. Haszeldine, J. Chem. Soc. 1957, 4069-4079. Perfluoroalkyl derivatives of sulphur. Part VII. Alkyl trifluoromethanesulphonates as alkylating agents, trifluoromethanesulphonic anhydride as a promoter for esterification, and some reactions of trifluoromethanesulphonic acid.
R. J. Koshar, R. A. Mitsch, J. Org. Chem. 1973, 38, 3358-3363. Bis(perfluoroalkylsulfonyl)methanes and related disulfones.
D. D. Desmarteau, W. T. Pennington, K. S. Sung, S. Z. Zhu, R. Scott, Eur. J. Solid State Inorg. Chem. 1991, 28, 905-917. Novel layered structures in metal salts of bis(sulfonyl)methanes.
D. Benrabah, D. Baril, J.-Y. Sanchez, M. Armand, G. G. Gard, J. Chem. Soc. Faraday Trans. 1993, 89, 355-359. Comparative electrochemical study of new poly(oxyethylene)-Li salt complexes.
N. R. Holcomb, P. G. Nixon, G. L. Gard, R. L. Nafshun, M. M. Lerner, J. Electrochem. Soc. 1996, 143, 1297-1300. Synthesis of LiCH(SO2CF3)2 and ionic conductivity of polyether-saIt complexes.
R. Jüschke, P. Häsel, P. Sartori, J. Fluor. Chem 1998, 91, 9-12. Comparative study of physical properties of bis(trifluoromethylsulfonyl) dihalogenomethanes and bis(fluorosulfonyl) dihalogenomethanes.
J. Scheers, E. Jonsson, P. Jacobsson, P. Johansson, Electrochemistry 2012, 80, 18-25. Novel lithium imides; Effects of -F, -CF3, and -C≡N substituents on lithium battery salt stability and dissociation.
E. Fluck, E. Beuerle, Z. Anorg. Allg. Chem. 1975, 412, 65-70. Bis(difluorophosphoryl)amine and some N-derivatives.
Z.-B. Zhou, H. Han, J. Nie, Chin. Patent CN 101654229, 2010. Method for preparation of fluorine-containing sulfonimide (phosphonimide) and its alkali metal salt.
E. Fluck, E. Beuerle, Z. Anorg. Allg. Chem. 1975, 412, 65-70. Bis(difluorophosphoryl)amine and some N-derivatives.
D. Bejan, H. Willner, N. Ignatiev, C. W. Lehmann, Inorg. Chem. 2008, 47, 9085-9089. Synthesis and characterization of bis[bis(pentafluoroethyl)phosphinyl]imides, M+N[(C2F5)2P(O)]2 –, M = H, Na, K, Cs, Ag, Me4N.
L. Kling III, C. B. Colburn, W. E. Hill, Inorg. Nucl. Chem. - Herbert H. Hyman Mem. Vol. 1976, 5-7. The reactions of phosphorus fluorides with lithium bis(trimethylsilyl)amide.
T. L. Charlton, R. G. Cavell, Inorg. Chem. 1970, 9, 379-387. Preparation and properties of some oxygen-, sulfur-, and nitrogen-bridged phosphoryl and thiophosphoryl difluorides.
A. Schmidpeter, H. Groeger, Z. Anorg. Allg. Chem. 1966. 345, 106-118. Über phosphazene. II. Darstellung und struktur der tetraphenyl-dithio-imidodiphosphinsäure.
M. Hernández-Arganis, S. Hernández-Ortega, R. A. Toscano, V. García-Montalvo, R. Cea-Olivares, Chem. Commun. 2004, 310-311. A powerful novel strategy for the preparation of discrete inorganic carbon-free rings containing alkaline cations.
F. Chen, M. Kapon, J. D. Woollins, M. S. Eisen, Organometallics 2009, 28, 2391-2400. Bis(imidodithiodiphosphinato) titanium and zirconium complexes: Synthesis, characterization, and their catalytic activity in the polymerization of α-olefins.
I. Ghesner, C. palotas, A. Silvestru, C. Silvestru, J. E. Drake, Polyhedron 2001, 20, 1101-1105. Lithium tetraorganodichalcogenoimidodiphosphinates. Crystal structure of [Li{(OPPh2)(SPMe2)N}·2H2O]2, a dimer formed through oxygen-bridging phosphorus ligands.
J. Barthel, M. Wühr, R. Buestrich, H. J. Gores, J. Electrochem. Soc. 1995, 142, 2527-2531. A new class of electrochemically and thermally stable lithium salts for lithium battery electrolytes I. Synthesis and properties of lithium bis[1,2-benzenediolato(2-)-O,O']borate.
J. Barthel, R. Buestrich, H. J. Gores, M. Schmidt, M. Wühr, J. Electrochem. Soc. 1997, 144, 3866-3870. A new class of electrochemically and thermally stable lithium salts for lithium battery electrolytes IV. Investigations of the electrochemical oxidation of lithium organoborates.
M. Handa, S. Fukuda, Y. Sasaki, K. Usami, J. Electrochem. Soc. 1997, 144, L235-L237. Use of a chelate complex with boron as a lithium salt for lithium battery electrolytes.
Y. Sasaki, M. Handa, S. Sekiya, K. Kurashima, K. Usami, J. Power Sources 2001, 97–98, 561-565. Application to lithium battery electrolyte of lithium chelate compound with boron.
Z.-M. Xue, J.-F. Zhao, J. Ding, C.-H. Chen, J. Power Sources 2010, 195, 853-856. LBDOB, a new lithium salt with benzenediolato and oxalato complexes of boron for lithium battery electrolytes.
Z.-M. Xue, W. Zhou, J. Ding, C.-H. Chen, Electrochim. Acta 2010, 55, 5342-5348. Electronic structures and molecular properties of FLBDOB and its derivatives: A combined experimental and theoretical study.
M. Handa, M. Suzuki, J. Suzuki, H. Kanematsu, Y. Sasaki, Electrochem. Solid-State Lett. 1999, 2, 60-62. A new lithium salt with a chelate complex of phosphorus for lithium battery electrolytes.
J. Barthel, R. Buestrich, E. Carl, H. J. Gores, J. Electrochem. Soc. 1996, 143, 3565-3571. A new class of electrochemically and thermally stable lithium salts for lithium battery electrolytes II. Conductivity of lithium organoborates in dimethoxyethane and propylene carbonate.
Z.-M. Xue, C.-Q. Ji, W. Zhou, C.-H. Chen, J. Power Sources 2010, 195, 3689-3692. A new lithium salt with 3-fluoro-1,2-benzenediolato and oxalato complexes of boron for lithium battery electrolytes.
J. Barthel, R. Buestrich, E. Carl, H. J. Gores, J. Electrochem. Soc. 1996, 143, 3572-3575. A new class of electrochemically and thermally stable lithium salts for lithium battery electrolytes III. Synthesis and properties of some lithium organoborates.
Z.-M. Xue, K.-N. Wu, B. Liu, C.-H. Chen, J. Power Sources 2007, 171, 944-947. New lithium salts with croconato-complexes of boron for lithium battery electrolytes.
Z.-M. Xue, Y.-Z. Ding, C.-H. Chen, Electrochim. Acta 2007, 53, 990-997. A DFT study of electronic structures, energies, and molecular properties of lithium bis[croconato]borate and its derivatives.
O. Hiruta, M. Mizutani, Y. Takeuchi, Y. Ukyo, K. Kaneko, S. Iida, K. Torii, Jap. Patent JP 2009266644 A, 2009. Nonaqueous electrolyte solutions and lithium ion secondary batteries.
W. Xu, L.-M. Wang, R. A. Nieman, C. A. Angell, J. Phys. Chem. B 2003, 107, 11749-11756. Ionic liquids of chelated orthoborates as model ionic glassformers.
J. Barthel, M. Schmidt, H. J. Gores, J. Electrochem. Soc. 1998, 145, L17-L20. Lithium bis[5-fluoro-2-olato-1-benzenesulfonato (2-)-O,O']borate(l-), a new anodically and cathodically stable salt for electrolytes of lithium-ion cells.
J. Barthel, A. Schmid, H. J. Gores, J. Electrochem. Soc. 2000, 147, 21-24. A new class of electrochemically and thermally stable lithium salts for lithium battery electrolytes V. Synthesis and properties of lithium bis[2,3-pyridinediolato(2-)-O,O´]borate.
H. Schafer, Z. Anorg. Chem. 1949, 259, 86-91. Boric acid and hydroxy compounds. IV. New salts of dipyrocatechol boric acid.
A. Downard, M. Nieuwenhuyzen, K. R. Seddon, J.-A. van den Berg, Cryst. Growth Des. 2002, 2, 111-119. Structural features of lithium organoborates.
S. Tsujioka, H. Takase, M. Takahashi, H. Sugimoto, Jap. Patent JP 2001247306 A, 2001. Synthesis and purification of ionic metal complexes.
S. Tsujioka, H. Takase, M. Takahashi, H. Sugimoto, Jap. Patent JP 2001302675 A, 2001. Preparation of lithium bis(oxalato)borate as ionic metal complex.
S. Tsujioka, H. Takase, M. Takahashi, Y. Isono, Eur. Patent EP 1308449 A2, 2003. Preparation of lithium (alkanoato)borates and (alkanoato)phosphates.
W. Xu, C. A. Angell, Electrochem. Solid-State Lett. 2001, 4, E1-E4. LiBOB and its derivatives. Weakly coordinating anions, and the exceptional conductivity of their nonaqueous solutions.
K. Xu, S. Zhang, T. R. Jow, W. Xu, C. A. Angell, Electrochem. Solid-State Lett. 2002, 5, A26-A29. LiBOB as salt for lithium-ion batteries.
K. Xu, S. Zhang, B. A. Poese, T. R. Jow, Electrochem. Solid-State Lett. 2002, 5, A259-A262. Lithium bis(oxalato)borate stabilizes graphite anode in propylene carbonate.
K. Xu, S. Zhang, T. R. Jow, Electrochem. Solid-State Lett. 2003, 6, A117-A120. Formation of the graphite/electrolyte interface by lithium bis(oxalato)borate.
K. Xu, U. Lee, S. Zhang, M. Wood, T. R. Jow, Electrochem. Solid-State Lett. 2003, 6, A144-A148. Chemical analysis of graphite/electrolyte interface formed in LiBOB-based electrolytes.
T. R. Jow, M. S. Ding, K. Xu, S. S. Zhang, J. L. Allen, K. Amine, G. L. Henriksen, J. Power Sources 2003, 119–121, 343-348. Nonaqueous electrolytes for wide-temperature-range operation of Li-ion cells.
J. Jiang, J. R. Dahn, Electrochem. Solid-State Lett. 2003, 6, A180-A182. Comparison of the thermal stability of lithiated graphite in LiBOB EC/DEC and in LiPF6 EC/DEC.
J. Jiang, J. R. Dahn, Electrochem. Commun. 2004, 6, 39-43. ARC studies of the thermal stability of three different cathode materials: LiCoO2; Li[Ni0.1Co0.8Mn0.1]O2; and LiFePO4, in LiPF6 and LiBoB EC/DEC electrolytes.
P. Y. Zavalij, S. Yang, M. S. Whittingham, Acta Crystallogr. 2003, B59, 753-759. Structures of potassium, sodium and lithium bis(oxalato)borate salts from powder diffraction data.
Y. Aihara, T. Bando, H. Nakagawa, H. Yoshida, K. Hayamizu, E. Akiba, W. S. Price, J. Electrochem. Soc. 2004, 151, A119-A122. Ion transport properties of six lithium salts dissolved in γ-butyrolactone studied by self-diffusion and ionic conductivity measurements.
Z. Chen, J. R. Dahn, Electrochim. Acta 2004, 49, 1079-1090. Methods to obtain excellent capacity retention in LiCoO2 cycled to 4.5 V.
J. Jiang, H. Fortier, J. N. Reimers, J. R. Dahn, J. Electrochem. Soc. 2004, 151, A609-A613. Thermal stability of 18650 size Li-ion cells containing LiBOB electrolyte salt.
J. Jiang, J. R. Dahn, Electrochim. Acta 2004, 49, 2661-2666. Effects of particle size and electrolyte salt on the thermal stability of Li0.5CoO2.
K. Amine, J. Liu, S. Kang, I. Belharouak, Y. Hyung, D. Vissers, G. Hendriksen, J. Power Sources 2004, 129, 14-19. Improved lithium manganese oxide spinel/graphite Li-ion cells for high-power applications.
J. Jiang, J. R. Dahn, Electrochem. Commun. 2004, 6, 724-728. ARC studies of the reaction between Li0FePO4 and LiPF6 or LiBOB EC/DEC electrolytes.
J. Jiang, J. R. Dahn, Electrochim. Acta 2004, 49, 4599-4604. Effects of solvents and salts on the thermal stability of LiC6.
G. V. Zhang, K. Xu, T. R. Jow, P. N. Ross Jr. Electrochem. Solid-State Lett. 2004, 7, A224-A227. Study of SEI layer formed on graphite anodes in PC/LiBOB electrolyte using IR spectroscopy.
K. Xu, U. Lee, S. Zhang, J. L. Allen, T. R. Jow, Electrochem. Solid-State Lett. 2004, 7, A273-A277. Graphite/electrolyte interface formed in LiBOB-based electrolytes.
T. R. Jow, K. Xu, M. S. Ding, S. S. Zhang, J. L. Allen, K. Amine, J. Electrochem. Soc. 2004, 151, A1702-A1706. LiBOB-based electrolytes for Li-ion batteries for transportation applications.
J. Jiang, J. Chen, J. R. Dahn, J. Electrochem. Soc. 2004, 151, A2082-A2087. Comparison of the reactions between Li7/3Ti5/3O4 or LiC6 and nonaqueous solvents or electrolytes using accelerating rate calorimetry.
K. Xu, U. Lee, S. Zhang, J. L. Allen, T. R. Jow, J. Electrochem. Soc. 2004, 151, A2106-A2112. Graphite/electrolyte interface formed in LiBOB-based electrolytes.
M. S. Ding, J. Electrochem. Soc. 2005, 152, A132-A140. Conductivity and viscosity of PC-DEC and PC-EC solutions of LiBOB.
H.-G. Schweiger, M. Multerer, U. Wietelmann, J.-C. Panitz, T. Burgermeister, H. J. Gores, J. Electrochem. Soc. 2005, 152, A622-A627. NMR determination of trace water in lithium salts for battery electrolytes.
K. Xu, S. Zhang, R. Jow, J. Power Sources 2005, 143, 197-202. Electrochemical impedance study of graphite/electrolyte interface formed in LiBOB/PC electrolyte.
K. Amine, J. Liu, I. Belharouak, Electrochem. Commun. 2005, 7, 669-673. High-temperature storage and cycling of C-LiFePO4/graphite Li-ion cells.
K. Xu, S. Zhang, T. R. Jow, Electrochem. Solid-State Lett. 2005, 8, A365-A368. LiBOB as additive in LiPF6-based lithium ion electrolytes.
M. S. Ding, T. R. Jow, J. Electrochem. Soc. 2005, 152, A1199-A1207. Properties of PC-EA solvent and its solution of LiBOB comparison of linear esters to linear carbonates for use in lithium batteries.
K. Amine, J. Liu, I. Belharouak, S.-H. Kang, I. Bloom, D. Vissers, G. Henriksen, J. Power Sources 2005, 146, 111-115. Advanced cathode materials for high-power applications.
H. Kaneko, K. Sekine, T. Takamura, J. Power Sources 2005, 146, 142-145. Power capability improvement of LiBOB/PC electrolyte for Li-ion batteries.
K. Striebel, J. Shim, A. Sierra, H. Yang, X. Song, R. Kostecki, K. McCarthy, J. Power Sources 2005, 146, 33-38. The development of low cost LiFePO4-based high power lithium-ion batteries.
K. Xu, S. S. Zhang, U. Lee, J. L. Allen, T. R. Jow, J. Power Sources 2005, 146, 79-85. LiBOB: Is it an alternative salt for lithium ion chemistry?
B.-T. Yu, W.-H. Qiu, F.-S. Li, G.-X. Xu, Electrochem. Solid-State Lett. 2006, 9, A1-A4. The electrochemical characterization of lithium bis(oxalato)borate synthesized by a novel method.
W. Larsson, J.-C. Panitz, A. Cedergren, Talanta 2006, 69, 276-280. Interference-free coulometric titration of water in lithium bis(oxalato)borate using Karl Fischer reagents based on Nmethylformamide.
J.-C. Panitz, U. Wietelmann, M. Wachtler, S. Stroebele, M. Wohlfahrt-Mehrens, J. Power Sources 2006, 153, 396-401. Film formation in lithium bis(oxalato)borate-containing electrolytes.
K. Amine, Q. Wang, D. R. Vissers, Z. Zhang, N. A. A. Rossi, R. West, Electrochem. Commun. 2006, 8, 429-433. Novel silane compounds as electrolyte solvents for Li-ion batteries.
S. S. Zhang, K. Xu, T. R. Jow, J. Power Sources 2006, 154, 276-280. LiBOB-based gel electrolyte Li-ion battery for high temperature operation.
Z. Chen, W. Q. Lu, J. Liu, K. Amine, Electrochim. Acta 2006, 51, 3322-3326. LiPF6/LiBOB blend salt electrolyte for high-power lithium-ion batteries.
S. S. Zhang, K. Xu. T. R. Jow, J. Power Sources 2006, 156, 629-633. Enhanced performance of Li-ion cell with LiBF4-PC based electrolyte by addition of small amount of LiBOB.
X. Zhang, T. M. Devine, J. Electrochem. Soc. 2006, 153, B365-B369. Passivation of aluminum in lithium-ion battery electrolytes with LiBOB.
S. S. Zhang, K. Xu, T. R. Jow, J. Power Sources 2006, 159, 702-707. An improved electrolyte for the LiFePO4 cathode working in a wide temperature range.
S. Wang, W. Qiu, T. Li, B. Yu, H. Zhao, Int. J. Electrochem. Sci. 2006, 1, 250-257. Properties of lithium bis(oxalato)borate (LiBOB) as a lithium salt and cycle performance in LiMn2O4 half cell.
M. Wachtler, M. Wohlfahrt-Mehrens, S. Stroebele, J.-C. Panitz, U. Wietelmann, J. Appl. Electrochem. 2006, 36, 1199-1206. The behaviour of graphite, carbon black, and Li4Ti5O12 in LiBOB-based electrolytes.
W. Lu, Z. Chen, H. Joachin, J. Prakash, J. Liu, K. Amine, J. Power Sources 2007, 163, 1074-1079. Thermal and electrochemical characterization of MCMB/LiNi1/3Co1/3Mn1/3O2 using LiBOB as an electrolyte additive.
B.-T. Yu, W.-H. Qiu, F.-S. Li, L. Cheng, J. Power Sources 2007, 166, 499-502. Comparison of the electrochemical properties of LiBOB and LiPF6 in electrolytes for LiMn2O4/Li cells.
S. Wang, W. Qiu, Y. Guan, B. Yu, H. Zhao, W. Liu, Electrochim. Acta 2007, 52, 4907-4910. Electrochemical characteristics of LiMxFe1-xPO4 cathode with LiBOB based electrolytes.
E. Zinigrad, L. Larush-Asraf, G. Salitra, M. Sprecher, D. Aurbach, Thermochim. Acta 2007, 457, 64-69. On the thermal behavior of Li bis(oxalato)borate LiBOB.
N.-S. Choi, K. H. Yew, H. Kim, S.-S. Kim, W.-U. Choi, J. Power Sources 2007, 172, 404-409. Surface layer formed on silicon thin-film electrode in lithium bis(oxalato) borate-based electrolyte.
L. Larush-Asraf, M. Bliton, H. Teller, E. Zinigrad, D. Aurbach, J. Power Sources 2007, 174, 400-407. On the electrochemical and thermal behavior of lithium bis(oxalato)borate (LiBOB) solutions.
D.-T. Shieh, P.-H. Hsieh, M.-H. Yang, J. Power Sources 2007, 174, 663-667. Effect of mixed LiBOB and LiPF6 salts on electrochemical and thermal properties in LiMn2O4 batteries.
J. Liu, Z. Chen, S. Busking, I. Belharouak, K. Amine, J. Power Sources 2007, 174, 852-855. Effect of electrolyte additives in improving the cycle and calendar life of graphite/Li1.1[Ni1/3Co1/3Mn1/3]0.9O2 Li-ion cells.
H. Xie, Z. Tang, Z. Li, Y. He, H. Wang, Y. Liu, Electrochem. Solid-State Lett. 2008, 11, C19-C22. Aluminum corrosion behavior of LiBOB in composite polymer electrolyte for Li-polymer batteries.
D. P. Abraham, M. M. Furczon, S.-H. Kang, D. W. Dees, A. N. Jansen, J. Power Sources 2008, 180, 612-620. Effect of electrolyte composition on initial cycling and impedance characteristics of lithium-ion cells.
J. C. Arrebola, A. Caballero, L. Hernan, J. Morales, J. Power Sources 2008, 183, 310-315. A high energy Li-ion battery based on nanosized LiNi0.5Mn1.5O4 cathode material.
Y. Guo, Z. Yin, Z. Tao, X. Li, Z. Wang, J. Power Sources 2008, 184, 513-516. An advanced electrolyte for improving surface characteristics of LiMn2O4 electrode.
K. Xu, J. Electrochem. Soc. 2008, 155, A733-A738. Tailoring electrolyte composition for LiBOB.
M. Amereller, M. Multerer, C. Schreiner, J. Lodermeyer, A. Scmid, J. Berthel, H. J. Gores. J. Chem. Engr. Data 2009, 54, 468-471. Investigation of the hydrolysis of lithium bis[1,2-oxalato(2-)-O,O'] borate (LiBOB) in water and acetonitrile by conductivity and NMR measurements in comparison to some other borates.
K. Xu, B. Deveney, K. Nechev, Y. Lam, T. R. Jow, J. Electrochem. Soc. 2008, 155, A959-A964. Evaluating LiBOB/lactone electrolytes in large-format lithium-ion cells based on nickelate and iron phosphate.
Y. Abu-Lebdeh, I. Davidson, J. Electrochem. Soc. 2009, 156, A60-A65. High-voltage electrolytes based on adiponitrile for Li-ion batteries.
A. Xiao, L. Yang, B. L. Lucht, S.-H. Kang, D. P. Abraham, J. Electrochem. Soc. 2009, 156, A318-A327. Examining the solid electrolyte interphase on binder-free graphite electrodes.
J.-Y. Huang, X.-J. Liu, X.-L. Kang, Z.-X. Yu, T.-T. Xu, W.-H. Qiu, J. Power Sources 2009, 189, 458-461. Study on γ-butyrolactone for LiBOB-based electrolytes.
M.-Q. Li, M.-Z. Qu, X.-Y. He, Z.-L. Yu, Electrochim. Acta 2009, 54, 4506-4513. Effects of electrolytes on the electrochemical performance of Si/graphite/disordered carbon composite anode for lithium-ion batteries.
S. Santee, A. Xiao, L. Yang, J. Gnanaraj, B. L. Lucht, J. Power Sources 2009, 194, 1053-1060. Effect of combinations of additives on the performance of lithium ion batteries.
L. Yang, M. M. Furxzon, A. Xiao, B. L. Lucht, Z. Zhang, D. P. Abraham, J. Power Sources 2010, 195, 1698-1705. Effect of impurities and moisture on lithium bisoxalatoborate (LiBOB) electrolyte performance in lithium-ion cells.
B. Markovsky, F. Amalraj, H. E. Gottlieb, Y. Gofer, S. K. Surendra, D. Aurbach. J. Electrochem. Soc. 2010, 157, A423-A429. On the electrochemical behavior of aluminum electrodes in nonaqueous electrolyte solutions of lithium salts.
Z. Yu, T. Xu, T. Xing, L.-Z. Fan, F. Lian, Q. Qiu, J. Power Sources 2010, 195, 4285-4289. A Raman spectroscopy investigation of the interactions of LiBOB with γ-BL as electrolyte for advanced lithium batteries.
C. Täubert, M. Fleischhammer, M. Wohlfahrt-Mehrens, U. Wietelmann, T. Buhrmester, J. Electrochem. Soc. 2010, 157, A721-A728. LiBOB as electrolyte salt or additive for lithium-ion batteries based on LiNi0.8Co0.15Al0.05O2/graphite.
L. Yang, M. M. Furczon, A. Xiao, B. L. Lucht, Z. Zhang, D. P. Abraham, J. Power Sources 2010, 195, 1698-1705. Effect of impurities and moisture on lithium bisoxalatoborate (LiBOB) electrolyte performance in lithium-ion cells.
S. Dalavi, M. Xu, B. Ravdel, L. Zhou, B. L. Lucht, J. Electrochem. Soc. 2010, 157, A1113-A1120. Nonflammable electrolytes for lithium-ion batteries containing dimethyl methylphosphonate.
S.-T. Myung, H. Natsui, Y.-K. Sun, H. Yashiro, J. Power Sources 2010, 195, 8297-8301. Electrochemical behavior of Al in a non-aqueous alkyl carbonate solution containing LiBOB salt.
P. Ping, Q. Wang, J. Sun, X. Feng, C. Chen, J. Power Sources 2011, 196, 776-783. Effect of sulfites on the performance of LiBOB/γ-butyrolactone electrolytes.
P. Ping, Q. Wang, J. Sun, X. Feng, C. Chen, J. Electrochem. Soc. 2010, 157, A1170-A1176. Thermal stabilities of some lithium salts and their electrolyte solutions with and without contact to a LiFePO4 electrode.
L. Yang, T. Markmaitree, B. L. Lucht, J. Power Sources 2011, 196, 2251-2254. Inorganic additives for passivation of high voltage cathode materials.
J. Dong, Z. Zhang, Y. Kusachi, K. Amine, J. Power Sources 2011, 196, 2255-2259. A study of tri(ethylene glycol)-substituted trimethylsilane (1NM3)/LiBOB as lithium battery electrolyte.
L.-Z. Fan, T. Xing, R. Awan, W. Qiu, Ionics 2011, 17, 491-494. Studies on lithium bis(oxalato)borate/propylene carbonate-based electrolytes for Li-ion batteries.
Y. Kusachi, J. Dong, Z. Zhang, K. Amine, J. Power Sources 2011, 196, 8301-8306. Tri(ethylene glycol)-substituted trimethylsilane/lithium bis(oxalate)borate electrolyte for LiMn2O4/graphite system.
K. Amine, Z. Chen, Z. Zhang, J. Liu, W. Lu, Y. Qin, J. Lu, L. Curtis, Y.-K. Sun, J. Mater. Chem. 2011, 21, 17754-17759. Mechanism of capacity fade of MCMB-Li1.1[Ni1/3Mn1/3Co1/3]0.9O2 cell at elevated temperature and additives to improve its cycle life.
Z. Chen, A. Jansen, K. Amine, Energy Environ. Sci. 2011, 4, 4567-4571. Novel functionalized electrolyte for MCMB/Li1.156Mn1.844O4 lithium-ion cells.
M. C. Smart, F. C. Krause, C. Hwang, W. C. West, J. Soler, G. K. S. Prakash, B. V. Ratnakumar, ECS Trans. 2011, 35, 1-11. The evaluation of triphenyl phosphate as a flame retardant additive to improve the safety of lithium-ion battery electrolytes.
D.-J. Lee, J. Hassoun, S. Panero, Y.-K. Sun, B. Scrosati, Electrochem. Commun. 2012, 14, 43-46. A tetraethylene glycol dimethylether-lithium bis(oxalate)borate (TEGDME-LiBOB) electrolyte for advanced lithium ion batteries.
S. Dalavi, M. Xu, B. knight, B. L. Lucht, Electrochem Solid-State Lett. 2012, 15, A28-A31. Effect of added LiBOB on high voltage (LiNi0.5Mn1.5O4) spinel cathodes.
S. Xiong, X. Kai, X. Hong, Y. Diao, Ionics 2012, 18, 249-254. Effect of LiBOB as additive on electrochemical properties of lithium-sulfur batteries.
M. Cuisinier, J.-F. Martin, P. Moreau, T. Epicier, R. Kanno, D. Guyomard, N. Dupre, Solid St. Nuc. Mag. Res. 2012, 42, 51-61. Quantitative MAS NMR characterization of the LiMn1/2Ni1/2O2 electrode/electrolyte interphase.
S. Li, Y. Zhao, X. Shi, B. Li, X. Xu, W. Zhao, X. Cui, Electrochim. Acta 2012, 65, 221-227. Effect of sulfolane on the performance of lithium bis(oxalato)borate-based electrolytes for advanced lithium ion batteries.
S. Li, B. Li, X. Xu, X. Shi, Y. Zhao, L. Mao, X. Cui, J. Power Sources 2012, 209, 295-300. Electrochemical performances of two kinds of electrolytes based on lithium bis(oxalate)borate and sulfolane for advanced lithium ion batteries.
S. Y. Li, X. L. Cui, X. L. Xu, X. M. Shi, G. X. Li, Russ. J. Electrochem. 2012, 48, 518-524. Quantitative MAS NMR characterization of the LiMn1/2Ni1/2O2 electrode/electrolyte interphase.
M. C. Smart, B. L. Lucht, S. Dalavi, F. C. Krause, B. V. Ratnakumar, J. Electrochem. Soc. 2012, 159, A739-A751. The effect of additives upon the performance of MCMB/LiNixCo1-xO2 Li-ion cells containing methyl butyrate-based wide operating temperature range electrolytes.
M. Matsui, K. Dokko, Y. Akita, H. Munakata, K. Kanamura, J. Power Sources 2012, 210, 60-66. Surface layer formation of LiCoO2thin film electrodes in non-aqueous electrolyte containing lithium bis(oxalate)borate.
V. Aravindan, Y. L. Cheah, W. C. Ling, S. Madhavi, J. Electrochem. Soc. 2012, 159, A1435-A1439. Effect of LiBOB additive on the electrochemical performance of LiCoPO4.
C. Li, Y. Zhao, H. Zhang, J. Liu, J. Jing, X. Cui, S. Li, Electrochim. Acta 2013, 104, 134-139. Compatibility between LiNi0.5Mn1.5O4 and electrolyte based upon lithium bis(oxalate)borate and sulfolane for high voltage lithium-ion batteries.
X. Cui, H. Zhang, S. Li, Y. Zhao, L. Mao, W. Zhao, Y. Li, X. Ye, J. Power Sources 2013, 240, 476-485. Electrochemical performances of a novel high-voltage electrolyte based upon sulfolane and γ-butyrolactone.
Y. Zhu, Y. Li, M. Bettge, D. P. Abraham, Electrochim. Acta 2013, 110, 191–199. Electrolyte additive combinations that enhance performance of high-capacity Li1.2Ni0.15Mn0.55Co0.1O2-graphite cells.
W. Xu, A. J. Shuysterman, M. Videa, V. Velikov, R. Marzke, C. A. Angell, J. Electrochem. Soc. 2003, 150, E74-E80. Structures of orthoborate anions and physical properties of their lithium salt nonaqueous solutions.
Z.-M. Xue, B.-B. Sun, W. Zhou, C.-H. Chen, J. Power Sources 2011, 196, 8710-8713. A new lithium salt with dihydroxybenzene and lithium tetrafluoroborate for lithium battery electrolytes.
X.-Y. Li, Z.-M. Xue, J.-F. Zhao, C.-H. Chen, J. Power Sources 2013, 235, 274-279. A new lithium salt with tetrafluoro-1,2-benzenediolato and oxalato complexes of boron for lithium battery electrolytes.
R. Pizer, P. J. Ricatto, Inorg. Chem. 1994, 33, 2402-2406. Thermodynamics of several 1:1 and 1:2 complexation reactions of the borate ion with bidentate ligands. 11B NMR spectroscopic studies.
S. Dhanuskodi, P. A. Angeli Mary, S. Thamotharan, V. Parthasarathi, Acta Crystallogr. 2002, E58, m212-m214. Lithium borodilactate.
V. G. Kalacheva, E. Svarcs, V. G. Ben’kovskii, I. D. Leonov, Zh. Neorg. Khim. 1970, 15, 401-403. Dipinacolborates of alkali metals.
J. Dale, J. Chem. Soc. 1961, 922-930. The stereochemistry of polyborate anions and of borate complexes of diols and certain polyols.
S. S. Zhang, Electrochem. Commun. 2006, 8, 1423-1428. An unique lithium salt for the improved electrolyte of Li-ion battery.
S. S. Zhang, J. Power Sources 2006, 162, 1379-1394. A review on electrolyte additives for lithium-ion batteries.
Z. Chen, J. Liu, K. Amine, Electrochem. Solid-State Lett. 2007, 10, A45-A47. Lithium difluoro(oxalato)borate as salt for lithium-ion batteries.
J. Liu, Z. Chen, S. Busking, K. Amine, Electrochem. Commun. 2007, 9, 475-479. Lithium difluoro(oxalato)borate as a functional additive for lithium-ion batteries.
S. S. Zhang, J. Power Sources 2007, 163, 713-718. Electrochemical study of the formation of a solid electrolyte interface on graphite in a LiBC2O4F2-based electrolyte.
V. Aravindan, P. Vickraman, Solid State Sci. 2007, 9, 1069-1073. A novel gel electrolyte with lithium difluoro(oxalato)borate salt and Sb2O3 nanoparticles for lithium ion batteries.
J. Liu, Z. Chen, S. Busking, I. Belharouak, K. Amine, J. Power Sources 2007, 174, 852-855. Effect of electrolyte additives in improving the cycle and calendar life of graphite/Li1.1[Ni1/3Co1/3Mn1/3]0.9O2 Li-ion cells.
H.-Q. Gao, Z.-A. Zhang, Y.-Q. Lai, J. Li, Y.-X. Liu, J. Cent. South Univ. Technol. 2008, 15, 830-834. Structure characterization and electrochemical properties of new lithium salt LiODFB for electrolyte of lithium ion batteries.
V. Aravindan, P. Vickraman, K. Krishnaraj, Polym. Int. 2008, 57, 932-938. Lithium difluoro(oxalate)borate-based novel nanocomposite polymer electrolytes for lithium ion batteries.
S.-H. Kang, D. P. Abraham, A. Xiao, B. L. Lucht, J. Power Sources 2008, 175, 526-532. Investigating the solid electrolyte interphase using binder-free graphite electrodes.
D.P. Abraham, M. M. Furczon, S.-H. Kang, D. W. Dees, A. N. Jansen, J. Power Sources 2008, 180, 612-620. Effect of electrolyte composition on initial cycling and impedance characteristics of lithium-ion cells.
V. Aravindan, P. Vickraman, K. Krishnaraj, Curr. Appl. Phys. 2009, 9, 1474-1479. Li+ ion conduction in TiO2 filled polyvinylidenefluoride-co-hexafluoropropylene based novel nanocomposite polymer electrolyte membranes with LiDFOB.
M. Amereller, M. Multerer, C. Schreiner, J. Lodermeyer, A. Schmid, J. Barthel, H. J. Gores, J. Chem. Eng. Data 2009, 54, 468-471. Investigation of the hydrolysis of lithium bis[1,2-oxalato(2-)-O,O′] borate (LiBOB) in water and acetonitrile by conductivity and NMR measurements in comparison to some other borates.
A. Xiao, L. Yang, B. L. Lucht, S.-H. Kang, D. P. Abraham, J. Electrochem. Soc. 2009, 156, A318-A327. Examining the solid electrolyte interphase on binder-free graphite electrodes.
Z. Chen, Y. Qin, J. Liu, K. Amine, Electrochem. Solid-State Lett. 2009, 12, A69-A72. Lithium difluoro(oxalato)borate as additive to improve the thermal stability of lithiated graphite.
J. Li, K. Xie, Y. Lai, Z. Zhang, F. Li, X. Hao, X. Chen, Y. Liu, J. Power Sources 2010, 195, 5344-5350. Lithium oxalyldifluoroborate/carbonate electrolytes for LiFePO4/artificial graphite lithium-ion cells.
Z. Zhang, X. Chen, F. Li, Y. Lai, J. Li, P. Liu, X. Wang, J. Power Sources 2010, 195, 7397-7402. LiPF6 and lithium oxalydifluoroborate blend salts electrolyte for LiFePO4/artifical graphite lithium-ion cells.
E. Zygadło-Monikowska, Z. Florjańczyk, P. Kubisa, T. Biedroń, A. Tomaszewska, J. Ostrowska, N. Langwald, J. Power Sources 2010, 195, 6202-6206. Mixture of LiBF4 and lithium difluoro(oxalato)borate for application as a new electrolyte for lithium-ion batteries.
M. H. Fu, K. L. Huang, S. Q. Liu, J. S. Liu, Y. K. Li, J. Power Sources 2010, 195, 862-866. Lithium difluoro(oxalato)borate/ethylene carbonate + propylene carbonate + ethyl (methyl) carbonate electrolyte for LiMn2O4 cathode.
D. Moosbauer, S. Zugmann, M. Amereller, H. J. Gores, J. Chem. Eng. Data 2010, 55, 1794-1798. Effect of ionic liquids as additives on lithium electrolytes: Conductivity, electrochemical stability, and aluminum corrosion.
J. Huang, L.-Z. Fan, B. Yu, T. Xing, W. Qiu, Ionics 2010, 16, 509-513. Density functional theory studies on the B-containing lithium salts.
S. Zugmann, D. Moosbauer, M. Amereller, C. Schreiner, F. Wudy, R. Schmitz, R. Schmitz, P. Isken, C. Dippel, R. Müller, M. Kunze, A. Lex-Balducci, M. Winter, H. J. Gores, J. Power Sources 2011, 196, 1417-1424. Electrochemical characterization of electrolytes for lithium-ion batteries based on lithium difluoromono(oxalato)borate.
J. L. Allen, S.-D. Han, P. D. Boyle, W. A. Henderson, J. Power Sources 2011, 196, 9737-9742. Crystal structure and physical properties of lithium difluoro(oxalato)borate (LiDFOB or LiBF2Ox).
S. Li, X. Xu, X. Shi, X. Cui, Adv. Mater. Res. 2011, 197–198, 1121-1124. Electrochemical performance of lithium difluoro(oxalate)borate synthesized by a novel method.
S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, M. Winter, H. J. Gores, J. Chem. Eng. Data 2011, 56, 4786-4789. Salt diffusion coefficients, concentration dependence of cell potentials, and transference numbers of lithium difluoromono(oxalato)borate-based solutions.
S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer, H. J. Gores, Electrochim. Acta 2011, 56, 3926-3933. Measurement of transference numbers for lithium ion electrolytes via four different methods, a comparative study.
L. Zhou, W. Li, M. Xu, B. Lucht, Electrochem. Solid-State Lett. 2011, 14, A161-A164. Investigation of the disproportionation reactions and equilibrium of lithium difluoro(oxalato) borate (LiDFOB).
J. L. Allen, P. D. Boyle, W. A. Henderson, Acta Crystallogr. 2011, E67, m533. Lithium difluoro(oxalato) borate tetramethylene sulfone disolvate.
K. Amine, Z. Chen, Z. Zhang, J. Liu, W. Lu, Y. Qin, J. Lu, L. Curtis, Y.-K. Sun, J. Mater. Chem. 2011, 21, 17754-17759. Mechanism of capacity fade of MCMB/Li1.1[Ni1/3Mn1/3Co1/3]0.9O2 cell at elevated temperature and additives to improve its cycle life.
M. Xu, L. Zhou, L. Hao, L. Xing, W. Li, B. L. Lucht, J. Power Sources 2011, 196, 6794-6801. Investigation and application of lithium difluoro(oxalate)borate (LiDFOB) as additive to improve the thermal stability of electrolyte for lithium-ion batteries.
L. Yang, T. Markmaitree, B. L. Lucht, J. Power Sources 2011, 196, 2251-2254. Inorganic additives for passivation of high voltage cathode materials.
Y. Zhu, Y. Li, M. Bettge, D. P. Abraham, J. Electrochem. Soc. 2012, 159, A2109-A2118. Positive electrode passivation by LiDFOB electrolyte additive in high-capacity lithium-ion cells batteries and energy storage.
S. Li, X. Xu, X. Shi, B. Li, Y. Zhao, H. Zhang, Y. Li, W. Zhao, X. Cui, L. Mao, J. Power Sources 2012, 217, 503-508. Composition analysis of the solid electrolyte interphase film on carbon electrode of lithium-ion battery based on lithium difluoro(oxalate)borate and sulfolane.
H. Zhou, F. Liu, J. Li, Appl. Mech. Mater. 2012, 152–154, 1106-1111. Preparation, thermal stability and electrochemical properties of LiODFB.
X. Wu, Z. Wang, X. Li, H. Guo, Y. Zhang, W. Xiao, J. Power Sources 2012, 204, 133-138. Effect of lithium difluoro(oxalato)borate and heptamethyldisilazane with different concentrations on cycling performance of LiMn2O4.
C. Yang, Y. Ren, B. Wu, F. Wu, Adv. Mater. Res. 2012, 455–456, 258-264. Formulation of a new type of electrolytes for LiNi1/3Co1/3Mn1/3O2 cathodes working in an ultra-low temperature range.
V. Aravindan, P. Vickaman, Ind. J. Phys. 2012, 86, 341-344. Effect of aging on the ionic conductivity of polyvinylidenefluoride–hexafluoropropylene (PVdF–HFP) membrane impregnated with different lithium salts.
M. Hu, J. Wei, L. Xing, Z. Zhou, J. Appl. Electrochem. 2012, 42, 291-296. Effect of lithium difluoro(oxalate) borate (LiDFOB) additive on the performance of high-voltage lithium-ion batteries.
S. Dalavi, P. Guduru, B. L. Lucht, J. Electrochem. Soc. 2012, 159, A642-A646. Performance enhancing electrolyte additives for lithium ion batteries with silicon anodes.
Q. Wu, W. Lu, M. Miranda, T. K. Honaker-Schroeder, K. Y. Lakhsassi, D. Dees, Electrochem. Commun. 2012, 24, 78-81. Effects of lithium difluoro(oxalate)borate on the performance of Li-rich composite cathode in Li-ion battery.
T. Schedlbauer, S. Krueger, R. Schmitz, R. W. Schmitz, C. Schreiner, H. J. Gores, S. Passerini, M. Winter, Electrochim. Acta 2013, 92, 102-107. Lithium difluoro(oxalato)borate: A promising salt for lithium metal based secondary batteries?
F. Wu, Q. Zhu, L. Li, R. Chen, S. Chen, J. Mater. Chem. A 2013, 1, 3659-3666. A diisocyanate/sulfone binary electrolyte based on lithium difluoro(oxalate)borate for lithium batteries.
S. Li, W. Zhao, X. Cui, Y. Zhao, B. Li, H. Zhang, Y. Li, G. Li, X. Ye, Y. Luo, Electrochim. Acta 2013, 91, 282-292. An improved method for synthesis of lithium difluoro(oxalato)borate and effects of sulfolane on the electrochemical performances of lithium-ion batteries.
H. Zhou, Z. Fang, J. Li, J. Power Sources 2013, 230, 148-154. LiPF6 and lithium difluoro(oxalato)borate/ethylene carbonate + dimethyl carbonate + ethyl(methyl)carbonate electrolyte for Li4Ti5O12.
J. Xiang, F. Wu, R. Chen, L. Li, H. Yu, J. Power Sources 2013, 233, 115-120. High voltage and safe electrolytes based on ionic liquid and sulfone for lithium-ion batteries.
Z.-M. Xue, B.-H. Zhao, C. H. Chen, J. Power Sources 2011, 196, 6478-6482. A new lithium salt with 3-fluoro-1,2-benzenediolato and lithium tetrafluoroborate for lithium battery electrolytes.
Z.-M. Xue, X.-F. Zhang, W. Zhou, C.-H. Chen, J. Power Sources 2012, 202, 336-340. A new lithium salt with tetrafluoro-1,2-benzenediolato and lithium tetrafluoroborate for lithium battery electrolytes.
Y.-N. Tang, Z.-M. Xue, J. Ding, C.-H. Chen, J. Power Sources 2012, 218, 134-139. Two unsymmetrical lithium organoborates with mixed-ligand of croconato and oxalicdiolato or benzenediolato for lithium battery electrolytes.
L. Yang, H. Zhang, P. F. Driscoll, B. Lucht, J. B. Kerr, ECS Trans. 2011, 33, 57-69. Six-membered-ring malonatoborate-based lithium salts as electrolytes for lithium ion batteries.
W. Xu, A. J. Shusterman, R. Marzke, C. A. Angell, J. Electrochem. Soc. 2004, 151, A632-A638. LiMOB, an unsymmetrical nonaromatic orthoborate salt for nonaqueous solution electrochemical applications.
M. Videa, W. Xu, B. Geil, R. Marzke, C. A. Angell, J. Electrochem. Soc. 2001, 148, A1352-A1356. High Li+ self-diffusivity and transport number in novel electrolyte solutions.
S. Tsujioka, H. Takase, M. Takahashi, H. Sugimoto, M. Koide, U.S. Patent 20020061450 A1, 2002. Electrolyte for electrochemical device.
S. Tsujioka, H. Takase, M. Takahashi, H. Sugimoto, M. Koide, Eur. Patent EP 1195834 A2, 2002. Electrolyte containing ionic metal complex for electrochemical device.
M. Schmidt, N. Ignatiev, W.-R. Pitner, J. Eichhorn, Ger. Patent DE 102008021271 A1, 2010. Reactive ionic liquids.
S. Tsujioka, H. Takase, M. Takahashi, Eur. Patent EP 1075036 A2, 2001. Electrolyte for electrochemical device.
S. Tsujioka, H. Takase, M. Takahashi, Eur. Patent EP 1074555 A2, 2001. Ionic metal complex and process for synthesizing same.
W. Xu, C. A. Angell, Electrochem. Solid-State Lett. 2000, 3, 366-368. A fusible orthoborate lithium salt with high conductivity in solutions.
U. Lischka, U. Wietelmann, M. Wegner, Ger. Patent DE 19829030 C1, 1999. Lithium bis(oxalato)borate, method for its production and application.
M. Ue, K. Shima, S. Mori, Electrochim. Acta 1994, 39, 2751-2756. Electrochemical properties of quaternary ammonium borodiglycolates and borodioxalates.
M. Finkelstein, E. A. Mayeda, S. D. Ross, J. Org. Chem. 1975, 40, 804-805. Preparation and anodic peak potentials of salts of coordination compounds derived from boric acid and polyhydric phenols.
B. G. Nolan, S. H. Strauss, J. Electrochem. Soc. 2003, 150, A1726-A1734. Nonaqueous lithium battery electrolytes based on bis(polyfluorodiolato)borates.
B. G. Nolan, S. Tsujioka, S. H. Strauss, in Fluorinated Materials for Energy Conversion, Chap. 9 (T. Nakajima, H. Groult, Eds.), Elsevier, New York, 2005. Electrochemical properties of lithium electrolytes based on bis(polyfluorodiolato)borate and tetrakis(polyfluoroalkoxy) aluminate superweak anions.
H. Yamaguchi, H. Takahashi, M. Kato, J. Arai, J. Electrochem. Soc. 2003, 150, A312-A315. Lithium tetrakis(haloacyloxy)borate: An easily soluble and electrochemically stable electrolyte for lithium batteries.
J. Arai, Eur. Patent Appl. EP1197494 A2, 2002. New organic borate compounds and the nonaqueous electrolytes and lithium secondary batteries using the compounds.
M. Drache, B. Vandorpe, J. Heubel, Bull. Soc. Chim. Fr. 1976, 1749-1754. Reactions of alkaline tetrachloroborates with sulfur trioxide in liquid sulfur dioxide. Mixed alkali metal boron chlorosulfates.
G. Mairesse, M. Drache, Acta Crystallogr. 1980, B36, 2767-2768. Lithium tetrakis(chlorosulfato)borate.
G. A. Olah, K. Laali, O. Farooq, J. Org. Chem. 1984, 49, 4591-4594. Chemistry in superacids. 6. Perfluoroalkanesulfonic acid-boron perfluoroalkanesulfonates: New superacid systems for generation of carbocations and catalysts for electrophilic transformations of hydrocarbons.
E. Zygadło-Monikowska, Z. Florjańczyk, K. Służewska, J. Ostrowska, N. Langwald, A. Tomaszewska, J. Power Sources 2010, 195, 6055-6061. Lithium conducting ionic liquids based on lithium borate salts.
R. Tao, T. Fujinami, J. Power Sources 2005, 146, 407-411. Application of mixed-salts composed of lithium borate and lithium aluminate in PEO-based polymer electrolytes.
N. Y. Adonin, V. V. Bardin, U. Floerke, H.-J. Hermann, Z. Anorg. Allg. Chem. 2005, 631, 2638-2646. Polyfluoroorganoboron-oxygen compounds. 4. Lithium pentafluorophenyltrimethoxyborate, Li[C6F5B(OMe)3], reactions with selected electrophiles and nucleophiles.
M. G. Harriss, J. B. Milne, Can. J. Chem. 1971, 49, 3612-3616. Trifluoroacetic acid solvent system. III. Acid, HB(OOCCF3)4, and the solvent autoprotolysis constant.
M. G. Harriss, J. B. Milne, Can. J. Chem. 1972, 50, 3789-3798. Trifluoroacetic acid solvent system. IV. Triple ions.
H. R. Allcock, E. C. Bissell, J. Am. Chem. Soc. 1973, 95, 3154-3157. Triethylammonium tris(o-phenylenedioxy)phosphate. Crystal and molecular structure.
N. Nanbu, K. Tsuchiya, T. Shibazaki, Y. Sasaki, Electrochem. Solid-State Lett. 2002, 5, A202-A205. Lithium tris[3-fluoro-1,2-benzenediolato(2-)-O,O']phosphate as a novel lithium salt for lithium battery electrolytes.
N. Nanbu, K. Tsuchiya, Y. Sasaki, J. Power Sources 2005, 142, 333-338. Electrolytic properties of lithium chelatophosphates and application to lithium batteries. A new lithium salt with a chelate complex of phosphorus for lithium battery electrolytes.
N. Nanbu, T. Shibazaki, Y. Sasaki, Chem. Lett. 2001, 30, 862-863. A novel lithium salt with a chelate complex of phosphorus for lithium battery electrolytes.
M. Eberwein, A. Schmid, M. Schmidt, M. Zabel, T. Burgemeister, J. Barthel, W. Kunz, H. J. Gores, J. Electrochem. Soc. 2003, 150, A994-A999. Synthesis and electrochemical properties of some lithium chelatophosphates.
S. Zhang, C. Xu, T. R. Jow, U.S. Patent 2010/0267984 A1, 2010. Oxyfluorophosphate synthesis process and compound therefrom.
U. Wietelmann, W. Bonrath, T. Netscher, H. Nöth, J.-C. Panitz, M. Wohlfahrt-Mehrens, Chem. Eur. J. 2004, 10, 2451-2458. Tris(oxalato)phosphorus acid and its lithium salt.
L. Zhou, B. L. Lucht, J. Power Sources 2012, 205, 439-448. Performance of lithium tetrafluorooxalatophosphate (LiFOP) electrolyte with propylene carbonate (PC).
L. Zhou, S. Dalavi, M. Xu, B. L. Lucht, J. Power Sources 2011, 196, 8073-8084. Effects of different electrode materials on the performance of lithium tetrafluorooxalatophosphate (LiFOP) electrolyte.
M. Xu, L. Zhou, D. Chalasani, S. Dalavi, B. L. Lucht, J. Electrochem. Soc. 2011, 158, A1202-A1206. Investigation of the solid electrolyte interphase on MCMB and NG electrodes in lithium tetrafluorooxalatophosphate [LiPF4C2O4] based electrolyte.
Y. Qin, Z. Chen, J. Liu, K. Amine, Electrochem. Solid-State Lett. 2010, 13, A11-A14. Lithium tetrafluoro oxalato phosphate as electrolyte additive for lithium-ion cells.
M. Xu, A. Xiao, W. Li, B. L. Lucht, J. Electrochem. Soc. 2010, 157, A115-A120. Investigation of lithium tetrafluorooxalatophosphate [LiPF4(C2O4)] as a lithium-ion battery electrolyte for elevated temperature performance.
M. Xu, A. Xiao, W. Li, B. L. Lucht, Electrochem. Solid-State Lett. 2009, 12, A155-A158. Investigation of lithium tetrafluorooxalatophosphate as a lithium-ion battery electrolyte.
A. Xiao, L. Yang, B. L. Lucht, Electrochem. Solid-State Lett. 2007, 10, A241-A244. Thermal reactions of LiPF6 with added LiBOB. Electrolyte stabilization and generation of LiF4OP.
U. Heider, M. Schmidt, A. Kuehner, D. Petigk, Eur. Patent Appl. 1143548 A2, 2001. Method of preparation of lithium salts for nonaqueous electrolyte batteries.
D. Hellwinkel, Chem. Ber. 1965, 98, 576-587. at-Complexes with six-coordinated phosphorus.
A. Chandrasekaran, Roberta O. Day, Robert R. Holmes, Inorg. Chem. 2002, 41, 1645-1651. P-O donor action from carboxylate anions with phosphorus in the presence of hydrogen bonding. A model for phosphoryl-transfer enzymes.
S. Tsujioka, B. G. Nolan, H. Takase, B. P. Fauber, S. H. Strauss, J. Electrochem. Soc. 2004, 151, A1418-A1423. Conductivities and electrochemical stabilities of lithium salts of polyfluoroalkoxyaluminate superweak anions.
A. K. Powell, S. L. Heath, Coord. Chem. Rev. 1996, 149, 59-80. X-ray structural analysis of biologically relevant aluminium(III) complexes.
S. N. Mhatre, R. K. Iyer, P. N. Moorthy, Magn. Res. Chem. 1993, 31, 169-175. Characterization of aluminum complexes in tea extract.
A. J. A. Aquino, D. Tunega, G. Haberhauer, M. Gerzabek, H. Lischka, Phys. Chem. Chem. Phys. 2000, 2, 2845-2850. A density functional theoretical study on solvated Al 3 +-oxalate complexes: Structures and thermodynamic properties.
X. Jin, Y. Yan, W. Shi, S. Bi, Environ. Sci. Technol. 2011, 45, 10082-10090. Density functional theory studies on the structures and water-exchange reactions of aqueous Al(III)oxalate complexes.
K. E. Peterman, J. M. Shreeve, Inorg. Chem. 1975, 14, 1223-1228. Reactions and photochemistry of some fluorinated amines and imines.
C. W. Tullock, U.S. Patent 3077499, 1963. Preparation of bis(trifluoromethyl)amine.
J. A. Young, S. N. Tsoukalas, R. D. Dresdner, J. Am. Chem. Soc. 1958, 80, 3604-3606. Fluorocarbon nitrogen compounds. III. Some reactions of bis(trifluoromethyl)amine.
D. A. Barr, R. N. Haszeldine, J. Chem. Soc. 1955, 2532-2534. Perfluoroalkyl derivatives of nitrogen. II. Bis(perfluoroalkyl)amines.
N. Ignatiev, U. Welz-Biermann, M. Schmidt, M. Weiden, U. Heider, H. Willner, A. Kucheryna, Ger. Patent DE 10258671 A1, 2004. Procedure for the production and use of ionic liquids with [N(CF3)2]- anion.
A. Kennedy, C. B. Colburn, J. Am. Chem. Soc. 1959, 81, 2906-2907. Difluoroamine.
E. A. Lawton, J. Q. Weber, J. Am. Chem. Soc. 1959, 81, 4755. Direct fluorination of urea: Synthesis and properties of difluoramine.
A. M. Sapse, D. C. Jain, Chem. Phys. Lett. 1984, 110, 251-255. Ab initio studies of the lithium difluoroamide (NF2Li) and dilithium imide (NHLi2) dimers.
A. M. Sapse, D. C. Jain, J. Phys. Chem. 1987, 91, 3923-3925. Solvent effect on the dimerization of some lithium compounds.
R. Minkwitz, A. Liedtke, Inorg. Chem. 1989, 28, 1627-1630. Preparation and spectroscopic characterization of CF3-substituted amides, phosphides, and arsenides, M(CF3)2 - (M = N, P, As).
A. Guerrero, R. Herrero, J. Z. Dávalos, I. Koppel, J.-L. M. Abboud, A. Chana, I. A. Koppel, J. Phys. Chem. A 2009, 113, 6422-6429. Hydrogen-bonding interactions of (CF3)3CH and (CF3)3C- in the gas phase. An experimental (FT-ICR) and computational study.
P. Marshall, M. Schwartz, J. Phys. Chem. A 1997, 101, 2906-2909. Computational study of C-H bond strengths in polyfluoralkanes.
J. S. Francisco, I. H. Williams, Chem. Phys. 1985, 98, 105-114. Structural and spectral consequences of ion pairing: A theoretical study of CF3O-Li+ and CF3O-Na+.
M. E. Redwood, C. J. Willis, Can. J. Chem. 1967, 45, 389-395. Fully fluorinated alkoxides. II. Ethoxides, propoxides, and butoxides.
F. Seel, R. Budenz, W. Gombler, Angew. Chem. Int. Ed. 1967, 6, 256. Cesium and rubidium pentafluoroethoxide.
C. T. Ratcliffe, J. M. Shreeve, Chem. Commun. 1966, 674-675. A simple method for the preparation of nitrosyl fluoride.
R. E. Dear, W. B. Fox, R. J. Fredericks, E. E. Gilbert, D. K. Hubbins, Inorg. Chem. 1970, 9, 2590-2591. Volatile fluorinated alkoxides of the alkali metals.
L. O. Müller, R. Scopelliti, I. Krossing, Chimia 2006, 60, 220-223. Chasing an elusive alkoxide: Attempts to synthesize [OC(tBu)(CF3)2]-.
R. Francke, D. Cericola, R. Kötz, D. Weingarth, S. R. Waldvogel, Electrochim. Acta 2012, 62, 372-380. Novel electrolytes for electrochemical double layer capacitors based on 1,1,1,3,3,3-hexafluoropropan-2-ol.
H. Bürger, G. Pawelke, J. Chem. Soc. Chem. Commun. 1988, 105-106. Difluoromethanimine, F2C=NH, a novel unstable molecule.
G. Pawelke, R. Dammel, W. Poll, Z. Naturforsch. B 1992, 47, 351-357. Preparation of difluoromethanimine F2C:NH and F2C:ND by hydrolysis of CF3NCO.
Y. V. Zeifman, N. P. Gambaryan, I. L. Knunyants, Dokl. Akad. Nauk. SSSR, Ser. Khim. 1963, 153, 1334-1337. Hexafluoroacetonimines.
W. J. Middleton, C. G. Krespan, J. Org. Chem. 1965, 30, 1398-1402. Fluorimines.
K. Niedenzu, K. E. Blick, C. D. Miller, Inorg. Chem. 1970, 9, 975-977. Boron-nitrogen compounds. XXXI. The interaction of boron trihalides with hexafluoroisopropylidenimine.
R. F. Swindell, T. J. Oullette, D. P. Babb, J. M. Shreeve, Inorg. Nucl. Chem. Lett. 1971, 7, 239-241. Reactions of LiN=C(CF3)2 with halides.
R. F. Swindell, D. P. Babb, T. J. Oullette, J. M. Shreeve, Inorg Chem. 1972, 11, 242-245. Hexafluoropropylideniminolithium reacts with halides.
S. G. Metcalf, J. M. Shreeve, Inorg. Chem. 1972, 11, 1631-1634. Bis(hexafluoroisopropylidenimino) disulfide, chloro(hexafluoroisopropylidenimino)sulfur(II), and some derivatives.
G. W. Parshall, R. Cramer, R. E. Foster, Inorg. Chem. 1962, 1, 677-679. Iminosulfur oxydifluoride and poly(oxofluorosulfur nitride).
H. W. Roesky, O. Glemser, A. Hoff, W. Koch, Inorg. Nucl. Chem. Lett. 1967, 3, 39-42. Über das azyldifluorosulfation NSF2O-.
R. D. Cramer, U.S. Patent 3,410,669, 1968. Iminosulfur oxydifluoroides and the process for their preparation.
M. Feser, R. Hoefer, O. Glemser, Z. Naturforsch. B 1974, 29, 716-718. Preparation and properties of sulfur oxide difluoride imide salts and their reaction with dimethyltin dihalides.
D. T. Sauer, J. M. Shreeve, Inorg. Chem. 1972, 11, 238-242. Bis(perfluoroalkyl)sulfur oxyimines and silver bis(trifluoromethyl)sulfur oxyimine.
T. Kitazume, J. M. Shreeve, Inorg. Chem. 1978, 17, 2173-2176. Stable fluorinated sulfuranes and sulfurane oxides. Synthesis and reactions.
V. K. Seppelt, Angew. Chem. 1977, 89, 325. Trifluoromethanol, CF3OH.
G. Klöter, K. Seppelt, J. Am. Chem. Soc. 1979, 101, 347-349. Trifluoromethanol (CF3OH) and trifluoromethylamine (CF3NH2).
M. E. Redwood, C. J. Willis, Can. J. Chem. 1965, 43, 1893-1898. Fully fluorinated alkoxides. part I. Trifluoromethoxides of alkali metals.
J. Arlt, M. Jansen, Chem. Ber. 1991, 124, 321-327. The crystal structures of potassium, rubidium and cesium trifluoroorthocarbonates with a similarity to barium sulfate type.
T. J. Barbarich, P. F. Driscoll, Electrochem. Solid-State Lett. 2003, 6, A113-A116. A lithium salt of a Lewis acid-base complex of imidazolide for lithium-ion batteries.
T. J. Barbarich, P. F. Driscoll, S. Izquierdo, L. N. Zakharov, C. D. Incarvito, A. L. Rheingold, Inorg. Chem. 2004, 43, 7764-7773. New family of lithium salts for highly conductive nonaqueous electrolytes.
T. J. Barbarich, U.S. Patent Appl. US 2003/0108800 A1, 2003. Non-aqueous electrolytes for lithium electrochemical cells.
T. J. Barbarich, U.S. Patent Appl. US 2009/0269676 A1, 2009. Non-aqueous electrolytes for lithium electrochemical cells.
M. Ihara, T. Kubota, U.S. Patent Appl. US 2011/0214895 A1, 2011. Lithium secondary battery, electrolytic solution for lithium secondary battery, electric power tool, electric vehicle, and electric power storage system.
H. S. Lee, X. Q. Yang, C. L. Xiang, J. McBreen, J. Electrochem. Soc. 1998, 145, 2813-2818. The synthesis of a new family of boron-based anion receptors and the study of their effect on ion pair dissociation and conductivily of lithium salts in nonaqueous solutions.
X. Sun, H. S. Lee, S. Lee, X. Q. Yang, J. McBreen, Electrochem. Solid-State Lett. 1998, 1, 239-240. A novel lithium battery electrolyte based on lithium fluoride and a tris(pentafluorophenyl) borane anion receptor in DME.
X. Sun, H. S. Lee, X. Q. Yang, J. McBreen, J. Electrochem. Soc. 1999, 146, 3655-3659. Comparative studies of the electrochemical and thermal stability of two types of composite lithium battery electrolytes using boron-based anion receptors.
J. McBreen, H. S. Lee, X. Q. Yang, X. Sun, J. Power Sources 2000, 89, 163-167. New approaches to the design of polymer and liquid electrolytes for lithium batteries.
H. S. Lee, X. Q. Yang, X. Sun, J. McBreen, J. Power Sources 2001, 97–98, 566-569. Synthesis of a new family of fluorinated boronate compounds as anion receptors and studies of their use as additives in lithium battery electrolytes.
X. Sun, H. S. Lee, X. Q. Yang, J. McBreen, J. Electrochem. Soc. 2002, 149, A335-A359. A new additive for lithium battery electrolytes based on an alkyl borate compound.
X. Sun, H. S. Lee, X.-Q. Yang, J. McBreen, Electrochem. Solid-State Lett. 2002, 5, A248-A251. Using a boron-based anion receptor additive to improve the thermal stability of LiPF6-based electrolyte for lithium batteries.
H. S. Lee, Z. F. Ma, X. Q. Yang, X. Sun, J. McBreen, J. Electrochem. Soc. 2004, 151, A1429-A1435. Synthesis of a series of fluorinated boronate compounds and their use as additives in lithium battery electrolytes.
B. Xie, H. S. Lee, H. Li, X. Q. Yang, J. McBreen, L. Q. Chen, J. Power Sources 2008, 10, 1195-1197. New electrolytes using Li2O or Li2O2 oxides and tris(pentafluorophenyl) borane as boron based anion receptor for lithium batteries.
L. F. Li, H. S. Lee, H. Li, X. Q. Yang, K. W. Nam, W. S. Yoon, J. McBreen, X. J. Huang, J. Power Sources 2008, 184, 517-521. New electrolytes for lithium ion batteries using LiF salt and boron based anion receptors.
S. Brownstein, Inorg. Chem. 1973, 12, 584-589. Complex fluoro anions in solution, I. Homo- and heteropolyfluoro anions of niobium and tantalum.
S. Brownstein, J. Inorg. Nucl. Chem. 1973, 35, 3567-3574. Complex fluoroanions in solution—II. Reactions of boron trifluoride and phosphorous pentafluoride with group VB hexafluoroanions.
S. Brownstein, Can. J. Chem. 1978, 56, 343-347. Complex fluoroanions in solution. VII. Replacement of fluoride by trifluoroacetate.
S. Brownstein, J. Bornais, G. Latremouille, Can. J. Chem. 1978, 56, 1419-1422. Complex fluoroanions in solution. VIII. Mixed anions containing fluoride and trifluoromethanesulfonate or fluorosulfate.
S. Brownstein, G. Latremouille, Can. J. Chem. 1978, 56, 2764-2767. Complex fluoroanions in solution. IX. BF3-anion complexes and their disproportionation.
P. J. Chevrier, S. Brownstein, J. Inorg. Nucl. Chem. 1980, 42, 1397-1405. Complex fluoroanions in solution—X. Complexes of phosphorus and arsenic fluorides with simple anions.
A. Alijah, E. S. Kryachko, J. Mol. Struct. 2007, 844–845, 193-199. On the N3O2 - paradigm.
H. Brand, P. Mayer, K. Polborn, A. Schulz, J. J. Weigand, J. Am. Chem. Soc. 2005, 127, 1360-1361. Blue alkali dinitrosomethanides: Synthesis, structure, and bonding.
A. Gorkovenko, S. Jaffe, U.S. Patent Appl. 2006/0154144 A1, 2006. Novel enhanced electrochemical cells with solid-electrolyte interphase promoters.
H. Brand, J. F. Liebman, A. Schulz, P. Mayer, A. Villinger, Eur. J. Inorg. Chem. 2006, 4294-4308. Nonlinear, resonance-stabilized pseudohalides: From alkali methanides to ionic liquids of methanides.
A. P. Purdy, E. Houser, C. F. George, Polyhedron 1997, 16, 3671-3679. Lithium dicyanamide, its reactions with cyanuric chloride, and the crystal structures of LiN(CN)2(MeCN)2 and LiCN(C5H5N)2.
H. Yoon, G. H. Lane, Y. Shekibi, P. C. Howlett, M. Forsyth, A. S. Best, D. R. MacFarlane, Energy Environ. Sci. 2013, 6, 979-986. Lithium electrochemistry and cycling behaviour of ionic liquids using cyano based anions.
S. A. Forsyth, S. R. Batten, Q. Dai, D. R. MacFarlane, Aust. J. Chem. 2004, 57, 121-124. Ionic liquids based on imidazolium and pyrrolidinium salts of the tricyanomethanide anion.
J. Scheers, P. Johansson, P. Jacobsson, J. Electrochem. Soc. 2008, 155, A628-A634. Anions for lithium battery electrolytes: A spectroscopic and theoretical study of the B(CN)4 - anion of the ionic liquid C2mim[B(CN)4].
T. Küppers, E. Bernhardt, H. Willner, H. W. Rohm, M. Köckerling, Inorg. Chem. 2005, 44, 1015-1022. Tetracyanoborate salts M[B(CN)4] with M = singly charged cations: Properties and structures.
R. Younesi, M. Hahlin, M. Treskow, J. Scheers, P. Johansson, K. Edström, J. Phys. Chem. C 2012, 116, 18597-18604. Ether based electrolyte, LiB(CN)4 salt and binder degradation in the Li−O2 battery studied by hard X-ray photoelectron spectroscopy (HAXPES).
T. Küppers, E. Bernhardt, H. Willner, H. W. Rohm, M. Köckerling, Inorg. Chem. 2005, 44, 1015-1022. Tetracyanoborate salts M[B(CN)4] with M = singly charged cations: Properties and structures.
J. Scheers, J. Pitawala, F. Thebault, J.-K. Kim, J.-H. Ahn, A. Matic, P. Johansson, P. Jacobson, Phys. Chem. Chem. Phys. 2011, 13, 14953-14959. Ionic liquids and oligomer electrolytes based on the B(CN)4 - anion; Ion association, physical and electrochemical properties.
W. J. Middleton, U.S. Patent US 2766243, 1956. Tetracyanoethylene and 1,1,2,3,3-pentacyanopropene.
W. J. Middleton, E. L. Little, D. D. Coffman, V. A. Engelhardt, J. Am. Chem. Soc. 1958, 80, 2795-2806. Cyanocarbon chemistry. V. Cyanocarbon acids and their salts.
C. E. Looney, J. R. Downing, J. Am. Chem. Soc. 1958, 80, 2840-2844. Cyanocarbon chemistry. XII. Some physical characteristics of cyanocarbon derivatives.
R. H. Boyd, J. Am. Chem. Soc. 1961, 83, 4288-4290. Strengths of cyanocarbon acids and a H-acidity scale for concentrated acid solutions.
R. H. Boyd, J. Phys. Chem. 1963, 67, 737-744. Cyanocarbon chemistry. XXIII. The ionization behavior of cyanocarbon acids.
C. Richardson, C. A. Reed, Chem. Commun. 2004, 706-707. Exploration of the pentacyano-cyclopentadienide ion, C5(CN)5 -, as a weakly coordinating anion and potential superacid conjugate base. Silylation and protonation.
P. Johansson, H. Nilsson, P. Jacobsson, M. Armand, Phys. Chem. Chem. Phys. 2004, 6, 895-899. Novel Hückel stabilised azole ring-based lithium salts studied by ab initio Gaussian-3 theory.
R. Vianello, J. F. Liebman, Z. B. Maksić, Chem. Eur. J. 2004, 10, 5751-5760. In search of ultrastrong Brønsted neutral organic superacids: A DFT study on some cyclopentadiene derivatives.
A. Kütt, I. Koppel, I. A. Koppel, I. Leito, ChemPhysChem 2009, 10, 499-502. Boratabenzene anions C5B(CN)6 - and C5B(CF3)6 - and the superacidic properties of their conjugate acids.
P. Johansson, S. Beranger, M. Armand, H. Nilsson, P. Jacobsson, Solid State Ionics 2003, 156, 129-139. Spectroscopic and theoretical study of the 1,2,3-triazole-4,5-dicarbonitrile anion and its lithium ion pairs.
M. Egashira, B. Scrosati, M. Armand, S. Beranger, C. Michot, Electrochem. Solid-State Lett. 2003, 6, A71-A73. Lithium dicyanotriazolate as a lithium salt for poly(ethylene oxide) based polymer electrolytes.
M. J. Crawford, K. Karaghiosoff, T. M. Klapötke, F. A. Martin, Inorg. Chem. 2009, 48, 1731-1743. Synthesis and characterization of 4,5-dicyano-2H-1,2,3-triazole and its sodium, ammonium, and guanidinium salts.
C. M. Sabaté, E. Jeanneau, H. Delalu, Dalton Trans. 2012, 41, 3817-3825. Metal salts of the 4,5-dicyano-2H-1,2,3-triazole anion ([C4N5]-).
C. M. Sabaté, H. Delalu, New Trends in Research of Energetic Materials, Proceedings (Pardubice, Czech Republic, Apr. 18-20, 2012) 2012, 747-750. Study of metal salts of the 4,5-dicyano-2H-1,2,3-triazole anion as pyrotechnic ingredients.
C. D. Weiss, J. Org. Chem. 1962, 27, 3695-3696. 3,4,5-Tricyanopyrazole.
H. Markusson, S. Béranger, P. Johansson, M. Armand, P. Jacobsson, J. Phys. Chem. A 2003, 107, 10177-10183. Lithium-pyrazole-3,4,5-tricarbonitrile: Ion pairing and lithium ion affinity studies.
O. W. Webster, Ger. Patent DE 2317453 A1, 1973. 4,5-Dicyanoimidazoles.
H. E. Simmons, U.S. Patent 1962-223825, 1962. Cyanopyrroles.
H. E. Simmons, R. D. Vest, S. A. Vladuchick, O. W. Webster, J. Org. Chem. 1980, 45, 5113-5121. Thiacyanocarbons. 5. Reactions of tetracyano-l,4-dithiin and tetracyanothiophene with nucleophiles: Synthesis of tetracyanopyrrole and tetracyanocyclopentadiene salts.
M. Becker, J. Harloff, T. Jantz, A. Schulz, A. Villinger, Eur. J. Inorg. Chem. 2012, 5658-5667. Structure and bonding of tetracyanopyrrolides.
R. W. Begland, D. R. Hartter, F. N. Jones, D. J. Sam, W. A. Sheppard, O. W. Webster, F. J. Weigert, J. Org. Chem. 1974, 39, 2341-2350. Hydrogen cyanide chemistry. VIII. New chemistry of diaminomaleonitrile. Heterocyclic synthesis.
M. Bukowska, J. Prejzner, P. Szczecinski, Pol. J. Chem. 2004, 78, 417-422. Synthesis of 4,5-dicyanoimidazoles.
L. Niedzicki, M. Kasprzyk, K. Kuziak, G. Z. Żukowska, M. Armand, M. Bukowska, M. Marcinek, P. Szczecinski, W. Wieczorek, J. Power Sources 2009, 192, 612-617. Modern generation of polymer electrolytes based on lithium conductive imidazole salts.
M. Bukowska, P. Szczecinski, W. Wieczorek, L. Nidzicki, B. Scrosati, S. Panero, P. Reale, M. Armand, S. Laruelle, S. Grugeon, Int. Patent Appl. WO 2010023413 A1, 2010. Pentacyclic anion salt used as electrolyte for batteries.
L. Niedzicki, G. Z. Żukowska, M. Bukowska, P. Szczeciński, S. Grugeon, S. Laruelle, M. Armand, S. Panero, B. Scrosati, M. Marcinek, W. Wieczorek, Electrochim. Acta 2010, 55, 1450-1454. New type of imidazole based salts designed specifically for lithium ion batteries.
J. Scheers, P. Johansson, P. Szczeciński ,W. Wieczorek, M. Armand, P. Jacobsson, J. Power Sources 2010, 195, 6081-6087. Benzimidazole and imidazole lithium salts for battery electrolytes.
L. Niedzicki, S. Grugeon, S. Laruelle, P. Judeinstein, M. Bukowska, J. Prejzner, P. Szczeciñski, W. Wieczorek, M. Armand, J. Power Sources 2011, 196, 8696-9700. New covalent salts of the 4+ V class for Li batteries.
L. Niedzicki, M. Kasprzyk, K. Kuziak, G. Z. Żukowska, M. Marcinek, W. Wieczorek, M. Armand, J. Power Sources 2011, 196, 1386-1391. Liquid electrolytes based on new lithium conductive imidazole salts.
J. Scheers, L. Niedzicki, G. Z. Żukowska, P. Johansson, W. Wieczorek, P. Jacobsson, Phys. Chem. Chem. Phys. 2011, 13, 11136-11147. Ion–ion and ion–solvent interactions in lithium imidazolide electrolytes studied by Raman spectroscopy and DFT models.
M. Dranka, L. Niedzicki, K. Kasprzyk, M. Marcinek, W. Wieczorek, J. Zachara, Polyhedron 2013, 51, 111-116. An insight into coordination ability of dicyanoimidazolato anions toward lithium in presence of acetonitrile. Crystal structures of novel lithium battery electrolyte salts.
G. Schmidt, M. Flasque, Int. Patent Appl. WO 2013072591 A1, 2013. Method for the preparation of a salt of a pentacyclic anion, particularly lithium 4,5-dicyano-2-fluoroalkylimidazolates, useful for manufacturing of an electrolyte composition for batteries.
S. Schmidt, Int. Patent Appl. WO 2013083894 A1, 2013. Use of lithium salt mixtures as electrolytes for lithium batteries.
Y. Kamitori, J. Heterocyclic. Chem. 2001, 38, 773-776. A facile synthesis of fluorine-containing heterocycles - Use of 1,1,1-trifluoro-2-alkanones as a convenient synthetic intermediate.
E. D. Laganis, D. M. Lemal, J. Am. Chem. Soc. 1980, 102, 6634-6636. 5H-Perfluoropentamethylcyclopentadiene, an extraordinary carbon acid.
Y. Kobayashi, A. Ando, K. Kaeada, A. Ohsawa, I. Kumadaki, J. Org. Chem. 1980, 45, 2962-2966. Organic fluorine compounds. 32.' A 1,3-dipolar cycloaddition reaction of tetrakis(trifluoromethyl)(dewar thiophene) and some reactions of the cycloadducts.
R. D. Chambers, S. J. Mullins, A. J. Roche, J. F. S. Vaughan, J. Chem. Soc., Chem. Commun. 1995, 841-842. Direct syntheses of pentakis(trifluoromethyl)cyclopentadienide salts and related dienes.
R. D. Chambers, A. J. Roche, J. F. S. Vaughan, Can. J. Chem. 1996, 74, 1925-1929. Direct syntheses of pentakis(trifluoromethyl)cyclopentadienide salts and related systems.
R. D. Chambers, W. K. Gray, J. F. S. Vaughan, S. R. Korn, M. Médebielle, A. S. Batsanov, C. W. Lehmann, J. A. K. Howard, J. Chem. Soc., Perkin Trans. 1997, 1, 135-145. Reactions involving fluoride ion. Part 41. Synthesis of hexakis(trifluoromethyl)cyclopentadiene and derived cyclopentadienide salts.
H. Brand, J. F. Liebman, A. Schulz, Eur. J. Chem. 2008, 4665-4675. Cyano-, nitro- and nitrosomethane derivatives: Structures and gas-phase acidities.
V. G. Kiselev, N. P. Gritsan, J. Phys. Chem. A 2008, 112, 4458-4464. Theoretical study of the nitroalkane thermolysis. 1. Computation of the formation enthalpy of the nitroalkanes, their isomers and radical products.
M. D. Cliff, M. W. Smith, J. Energetic Mater. 1999, 17, 69-86. Thermal characteristics of alkali metal dinitramide salts.
S. B. Babkin, A. N. Pavlov, G. M. Nazin, Russ. Chem. Bull. 1997, 46, 1844-1847. Anomalous decomposition of dinitramide metal salts in the solid phase.
R. Gilardi, J. Flippen-Anderson, C. George, R. J. Burcher, J. Am. Chem. Soc. 1997, 119, 9411-9416. A new class of flexible energetic salts: The crystal structures of the ammonium, lithium, potassium, and cesium salts of dinitramide.
R. J. Less, T. C. Wilson, M. McPartlin, P. T. Wood, D. S. Wright, Chem. Commun. 2011, 47, 10007-10009. Transition metal complexes of the pentacyanocyclopentadienide anion.
R. J. Less, B. Guan, N. M. Muresan, M. McPartlin, E. Reisner, T. C. Wilson, D. S. Wright, Dalton Trans. 2012, 41, 5919-5924. Group 11 complexes containing the [C5(CN)5]- ligand; ‘Coordination-analogues’ of molecular organometallic systems.
R. J. Less, T. C. Wilson, B. Guan, M. McPartlin, A. Steiner, P. T. Wood, D. S. Wright, Eur. J. Inorg. Chem. 2013, 1161-1169. Solvent direction of molecular architectures in Group 1 metal pentacyanocyclopentadienides.
C. Michot, PhD Thesis, Institut National Polytechnique de Grenoble Publisher, 1995.
N. Chaix, F. Alloin, J.-P. Bélières, J. Saunier, J.-Y. Sanchez, Electrochim. Acta 2002, 47, 1327-1334. Electrochemical study of polymethacrylonitrile electrolytes Comparative investigation of polynitriles electrochemical stability through model molecules. Part II.
L. Jäger, B. Freude, R. Skirl, K. Schubert, H. Köhler, Z. Anorg. Allg. Chem. 1993, 619, 711-717. Pseudoelement compounds. IV. Modification of the ions sulfite [SO2Y]2 -, sulfate [SO4 -nYn]2 -, and sulfonate [RSO2Y]- by introducing pseudochalcogen groups NCN and C(CN)2.
A. D. Fanta, P. T. Pham, W. M. Lamanna, Int. Patent WO 00/11742, 2000. Cyano-substituted methide and amide salts.
Y. L. Yagupolskii, N. V. Pavlenko, E. Lork, Heteroatom Chem. 1998, 9, 565-570. Trifluoromethylsulfonylmethanides functionalized by a CN group.
M. Armand, Y. Choquette, M. Gauthier, C. Michot, Eur. Patent, EP 0 850 921 A1, 1998. Sels d'anions dérivés du malonitrile, et leurs utilisations comme matériaux à conduction ionique.
H. W. Roesky, Angew. Chem. Int. Ed. 1968, 7, 63-64. Preparation of N-(trifluoromethyl)fluorosulfonamide and its salts.
B. Huber, T. Linder, K. Hormann, T. Frömling, J. Sundermeyer, B. Roling, Z. Phys. Chem. 2012, 226, 377-390. Synthesis of novel lithium salts containing pentafluorophenylamido-based anions and investigation of their thermal and electrochemical properties.
S. Maeda, S. Hayakawa, S. Kawada, Y. Aoki, Jap. Patent JP 2008251530 A, 2008. Electrolyte solution and secondary lithium battery using the electrolyte solution.
H. Yamamoto, M. Matsui, Int. Patent Appl. WO 2008069207, 2008. Preparation of lithium salts as supporting salts for electrolyte solutions.
V. Geis, K. Guttsche, C. Knapp, H. Scherer, R. Uzun, Dalton Trans. 2009, 2687-2694. Synthesis and characterization of synthetically useful salts of the weakly-coordinating dianion [B12Cl12]2 -.
J. W. Johnson, M. S. Whittingham, J. Electrochem. Soc. 1980, 127, 1653-1654. Lithium closoboranes as electrolytes in solid cathode lithium cells.
J. W. Johnson, A. H. Thompson, J. Electrochem. Soc. 1981, 128, 932-933. Lithium closoboranes. II. Stable nonaqueous electrolytes for elevated temperature lithium cells.
J. W. Johnson, J. F. Brady, J. Electrochem. Soc. 1982, 129, 2213-2219. Lithium closoborane electrolytes. III. Preparation and characterization.
G. GirishKumar, W. H. Bailey III, B. K. Peterson, W. J. Casteel Jr., J. Electrochem. Soc. 2011, 158, A146-A153. Electrochemical and spectroscopic investigations of the overcharge behavior of StabiLife electrolyte salts in lithium-ion batteries.
Z. Chen, A. N. Jansen, K. Amine, Energy Environ. Sci. 2011, 4, 4567-4571. Novel functionalized electrolyte for MCMB/Li1.156Mn1.844O4 lithium-ion cells.
Z. Chen, J. Liu, A. N. Jansen, G. GirishKumar, B. Casteel, K. Amine, Electrochem. Solid-State Lett. 2010, 13, A39-A42. Lithium borate cluster salts as redox shuttles for overcharge protection of lithium-ion cells.
S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev, S. H. Strauss, J. Am. Chem. Soc. 2003, 125, 4694-4695. Synthesis and stability of reactive salts of dodecafluoro-closo-dodecaborate(2-).
D.V. Peryshkov, S.H. Strauss, J. Fluor. Chem. 2010, 131, 1252-1256. K2B12F12: A rare A2X structure for an ionic compound at ambient conditions.
D. V. Peryshkov, R. Friedemann, E. Goreshnik, Z. Mazej, K. Seppelt, S. H. Strauss, J. Fluor. Chem. 2013, 145, 118-127. Anion packing, hole filling, and HF solvation in A2(HF)nB12F12and K2(HF)TiF6 (A = K, Cs).
J. Derendorf, M. Kessler, C. Knapp, M. Ruhle, C. Schulz, Dalton Trans. 2010, 39, 8671-8678. Alkali metal-sulfur dioxide complexes stabilized by halogenated closo-dodecaborate anions.
K. Hayamizu, A. Matsuo, J. Arai, J. Electrochem. Soc. 2009, 156, A744-A750. A divalent lithium salt Li2B12F12 dissolved in propylene carbonate studied by NMR methods.
J. Arai, A. Matsuo, T. Fujisaki, K. Ozawa, J. Power Sources 2009, 193, 851-854. A novel high temperature stable lithium salt (Li2B12F12) for lithium ion batteries.
Z. Chen, Y. Qin, K. Amine, Electrochim. Acta 2009, 54, 5605-5613. Redox shuttles for safer lithium-ion batteries.
Z. Chen, Y. Ren, A. N. Jansen, C. Lin, W. Weng, K. Amine, Nat. Commun. 2013, 4, 1513-1520. New class of nonaqueous electrolytes for long-life and safe lithium-ion batteries.
S. V. Ivanov, W. J. Casreel Jr., G. P. Pez, M. Ulman, U.S. Patent US 2005/0064288, 2005. Polyfluorinated boron cluster anions for lithium electrolytes.
C. M. Ionica-Bousquet, D. Muñoz-Rojas, W. J. Casteel, R. M. Pearlstein, G. GirishKumar, G. P. Pez, M. R. Palacín, J. Power Sources 2010, 195, 1479-1485. Polyfluorinated boron cluster-based salts: A new electrolyte for application in Li4Ti5O12/LiMn2O4 rechargeable lithium-ion batteries.
L. Lipping, I. Leito, I. Koppel, I. A. Koppel, J. Phys. Chem. A 2009, 113, 12972-12978. Gas-phase Brønsted superacidity of some derivatives of monocarba-closo-borates: A computational study.
A. N. Dey, J. Miller, J. Electrochem. Soc. 1979, 126, 1445-1451. Primary Li/SOCl2 cells. VII. Effect of Li2B10Cl10 and Li2B12Cl12 electrolyte salts on the performance.
A. Hammami, N. Raymond, M. Armand, Nature 2003, 424, 635-636. Runaway risk of forming toxic compounds.
T. Ohzuku, R. J. Brodd, J. Power Sources 2007, 174, 449-456. An overview of positive-electrode materials for advanced lithium-ion batteries.
J. Choi, A. Manthiram, J. Electrochem. Soc. 2005, 152, A1714-A1718. Role of chemical and structural stabilities on the electrochemical properties of layered LiNi1/3Mn1/3Co1/3O2cathodes.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Henderson, W.A. (2014). Nonaqueous Electrolytes: Advances in Lithium Salts. In: Jow, T., Xu, K., Borodin, O., Ue, M. (eds) Electrolytes for Lithium and Lithium-Ion Batteries. Modern Aspects of Electrochemistry, vol 58. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0302-3_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0302-3_1
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0301-6
Online ISBN: 978-1-4939-0302-3
eBook Packages: EnergyEnergy (R0)