
Abstract
This chapter delineates the mechanistic aspects of the NO–NH3–O2 reacting system, also known as the standard SCR reaction. The standard SCR technology was first developed in the 1970s, and thus has a long history in the research area of catalyst development as well as the associated reaction mechanisms.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Redox Site
- Apparent Reaction Order
- Reaction Kinetic Analysis
- Acid Site Amount
- Associate Reaction Mechanism
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
This chapter delineates the mechanistic aspects of the NO–NH3–O2 reacting system, also known as the standard SCR reaction. The standard SCR technology was first developed in the 1970s, and thus has a long history in the research area of catalyst development as well as the associated reaction mechanisms. Nevertheless, the reaction mechanisms do not always coincide even among the most popular systems such as the Cu or Fe ion-exchanged zeolites and vanadium-based catalysts. However, such extensive research activities conducted might be reaching a substantial agreement on several mechanistic details.
In this chapter, the reaction mechanisms of the standard SCR reaction are discussed from various perspectives including steady-state kinetics, the relations with NH3/NO oxidation ability and acid site amount, the effect of coexisting gases, and transient reaction behavior. Through these comprehensive analyses, some similarities and differences of the reaction mechanism among the conventional SCR catalysts could be extracted. Also, new perspectives on standard SCR mechanism could be suggested.
2 Steady-State Reaction Analysis
2.1 NH3/NO/O2, NH3/O2, and NO/O2 Reactions
To have a grasp of basic reaction behaviors, standard SCR as well as NH3/NO oxidation reactions were investigated under steady-state condition. For the samples, three conventional SCR catalysts, V–W/TiO2, Fe/ZSM-5, and Cu/ZSM-5, were tested. Additionally, H–ZSM-5 was also tested as a reference. Figure 8.1a–d shows the catalytic activity of standard SCR, NH3 oxidation, and NO oxidation reactions. The compositions of the feed gas were as follows: 0.05 % NO, 0.06 % NH3, 8 % O2, 10 % CO2, 8 % H2O with N2 for standard SCR; 0.1 % NH3, 8 % O2, 10 % CO2, 8 % H2O with N2 for NH3 oxidation; 0.1 % NO, 8 % O2, 10 % CO2, 8 % H2O with N2 for NO oxidation.

a NOx conversion under standard SCR reaction, b total NH3 conversion and c NH3 → NOx conversion under NH3 oxidation reaction, d NO → NO2 conversion under NO oxidation reaction. a: 0.05 % NO, 0.06 % NH3, 8 % O2, 10 % CO2, 8 % H2O with N2, b, c: 0.1 % NH3, 8 % O2, 10 % CO2, 8 % H2O with N2, d: 0.1 % NO, 8 % O2, 10 % CO2, 8 % H2O with N2
For standard SCR reaction (NH3/NO/O2 system) in Fig. 8.1a, Cu/ZSM-5 showed the highest activity in low temperature region (<300 °C), while Fe/ZSM-5 was the highest above 400 °C. On the other hand, H–ZSM-5 has little activity, and also negative conversions were seen over 400 °C due to NH3 oxidation to NOx. The conversion of V–W/TiO2 was nearly equal to that of Fe/ZSM-5 around 250 °C, though their temperature profiles differed slightly; V–W/TiO2 was more active in the lower temperature (<200 °C), while the situation was the opposite in the higher temperature (>300 °C), which is also verified by Arrhenius plots in the Sect. 8.2.2. Since N2O was not detected for all the conditions, only the standard SCR reaction (\( 2 {\text{NO}} + {\raise0.7ex\hbox{$1$} \!\mathord{\left/ {\vphantom {1 2}}\right.\kern-0pt} \!\lower0.7ex\hbox{$2$}}{\text{O}}_{ 2} + 2 {\text{NH}}_{ 3} \to 2 {\text{N}}_{ 2} + 3 {\text{H}}_{ 2} {\text{O}} \)) progresses except for unselective NH3 oxidation which was initiated at the higher temperature region (>400 °C).
From Fig. 8.1b, NH3 oxidation without inlet NO started above 300 °C for all the catalysts, which is much higher onset temperature than the SCR reaction. As by-products, N2O was not detected for all the catalysts, and NOx was produced only for H–ZSM-5 above 300 °C (Fig. 8.1c). It indicates that NH3 oxidation reaction to N2 (\( 2 {\text{NH}}_{ 3} + {\raise0.7ex\hbox{$3$} \!\mathord{\left/ {\vphantom {3 2}}\right.\kern-0pt} \!\lower0.7ex\hbox{$2$}}{\text{O}}_{ 2} \to {\text{N}}_{ 2} + 3 {\text{H}}_{ 2} {\text{O}} \)) mainly proceeds for the three SCR catalysts. As for H–ZSM-5, on the other hand, a large amount of NOx was produced from NH3, probably because the rate of NH3 oxidation to NOx should be much faster than that of SCR reaction between produced NOx and residual NH3.
Figure 8.1d shows the NO oxidation conversion to NO2 under the NH3 absent condition. For all the samples, the NO oxidation conversion was much lower than SCR and NH3 oxidation conversions, suggesting that NO is hard to be oxidized. Also, the NO oxidation ability (Cu–ZSM-5 > Fe–ZSM-5 > V–W/TiO2 ≈ H–ZSM-5 ≈ Blank) does not necessarily coincide with the trend of SCR conversion (Cu–ZSM-5 > Fe–ZSM-5 ≈ V–W/TiO2 ≫ H–ZSM-5). However, NO oxidation is believed to be a very important step in the SCR process, and thus it is discussed in more detail in another section.
2.2 Apparent Activation Energy
The apparent activation energies of the Fe/ZSM-5, Cu/ZSM-5, and V–W/TiO2 catalysts were determined from the Arrhenius plots of the logarithm of the apparent rate versus 1/T as shown in Fig. 8.2. The reaction rates were measured with maintaining pseudo-differential condition. The composition of the gas was kept as 0.05 % NO, 0.05 % NH3, 8 % O2, 8 % CO2, 10 % H2O with the remainder N2. The temperature range was varied from 120 to 200 °C because in the higher temperature region, other contributions such as diffusion limitation and/or NH3 oxidation might be affected [1, 2]. The apparent activation energies of the Fe/ZSM-5 and Cu/ZSM-5 catalysts were estimated to be 64 and 66 kJ/mol which is roughly equivalent. On the other hand, the V–W/TiO2 catalyst showed a more gradual slope than the zeolites and the activation energy was estimated to be 50 kJ/mol. This difference would be a main reason for the different temperature profiles between Fe/ZSM-5 and V–W/TiO2 in Fig. 8.1a.

Arrhenius plots of the logarithm of apparent SCR rate versus the inverse of temperature. Apparent activation energies are compared. Gas composition = 0.05 % NO, 0.05 % NH3, 8 % O2, 8 % CO2, 10 % H2O with the remainder N2
Efstathiou and Fliatoura [3] investigated the apparent activation energy of a V/TiO2 catalyst while varying the NO and NH3 concentration (0.05–0.2 %) at 150–190 °C. They reported that the change of the activation energy with the NO and NH3 concentration was small and obtained a value of 48.5 kJ/mol (11.6 kcal/mol) under 0.1 % NO and 0.05 % NH3 [3], which agrees well with the values estimated from Fig. 8.2.
2.3 Apparent Reaction Orders
Reaction orders contain important information for predicting the rate-determining step as well as the reaction mechanism. Apparent reaction orders can be estimated by measuring the reaction rates with varying gas concentration (partial pressure) under pseudo-differential condition. Figure 8.3 shows the dependence of the standard SCR rate on the NO, O2, and NH3 concentrations over Fe/ZSM-5 and Fe/BEA. Although the balance gas compositions were different for the two samples, i.e., 10 % CO2, 5 % H2O, and N2 for Fe/ZSM-5 versus only N2 for Fe/BEA, both showed identical dependence. The SCR rates increased with NO and O2 concentrations but slightly decreased with NH3 concentration. This indicates that the standard SCR reaction is promoted by NO and O2 but is inhibited by NH3. Since all the data gave linear relationships under the log–log plots, the reaction rate can be simply expressed using the power law

Log–log plots of apparent SCR rate versus NO, O2, and NH3 concentrations for Fe/ZSM-5 and Fe/BEA. Balance gas is 10 % CO2, 5 % H2O, and N2 for Fe/ZSM-5, and only N2 for Fe/BEA. Apparent activation orders are listed in Table 8.1
where r s is the standard SCR rate, k app is the apparent rate constant, and α, β, and γ are the apparent reaction orders for NO, O2, and NH3, respectively. Thus, the apparent reaction orders can be estimated from the slopes in Fig. 8.3, and are summarized in Table 8.1 along with the reported literature data [4–10]. As seen from Table 8.1, the reaction orders obtained here are comparable for the reported values for Fe/zeolites [6–8]; the NO orders are slightly lower than first order, the O2 orders are slightly lower than half, while the NH3 orders are negative.
Meanwhile, H–ZSM-5 shows approximately first order for O2 [9, 10]. The reason for the difference in the O2 orders between H-type zeolites and Fe/Cu zeolites could be due to the contribution of different forms of O2 toward the rate-determining step; for instance, undissociated molecular O2 or dissociated atomic O might participate in the elementary reaction.
If one assumes that the rate-determining step in the standard SCR is the oxidation of NO, the reaction order for the NO oxidation should have a similar value with that for the SCR reaction. To confirm this presumption, the dependence of the NO oxidation rate on the NO and O2 concentrations were investigated by using Fe/ZSM-5 and Fe/BEA. The estimated apparent orders are listed in Table 8.1 along with the relevant literature data [11]. The apparent reaction orders for the two Fe/zeolites showed similar values despite different structure and coexisting gases. Also, the reaction orders are nearly in accordance with the literature data reported by Metkar et al. [11]. They measured the NO and O2 orders in the presence of NO2 feed as an inlet gas, and found that the reaction orders for NO and O2 did not change when the inlet NO2 concentration was changed. By comparing the NO and O2 orders between the NO oxidation and the SCR reaction (Table 8.1), one can find a similarity which is nearly first order for NO and nearly half order for O2. Thus, the rate-determining step in the standard SCR might be the NO oxidation step. The same holds true when the activation energies are compared; Metkar et al. [11] reported that the activation energy for the NO oxidation (39 kJ/mol) is nearly equal to the activation energy for the standard SCR reaction (42 kJ/mol) over Fe/ZSM-5.
However, there is a crucial difference between the two reactions, which is NO2 order for the two reactions; the order is positive for the SCR reaction [4], whereas it is negative for the NO oxidation [11]. In fact, Iwasaki et al. [4] investigated the NO2 order during NO/NO2/NH3 reaction system (the so-called fast SCR condition) and found that the NO2 order is positive for Fe/ZSM-5. Also, Metkar et al. [11] investigated the NO2 order during the NO oxidation and estimated to be −0.42 to −0.49 for Fe/ZSM-5 and −0.89 to −1.00 for Cu/CHA. These differences would result from the different behavior of NO2 on the surface; the NO2 produced is consumed immediately by NH3 during the SCR reaction, whereas the NO2 and nitrate products are strongly adsorbed on the surface during the NO oxidation reaction, which is verified by in situ FT-IR in other chapter.
2.4 Relationship with NO Oxidation Activity
As mentioned in Sect. 8.2.3, the oxidation of NO would be a crucial step in the standard SCR reaction. In fact, there are many reports in the literature which suggest that the SCR activity is correlated with the NO oxidation conversion when the same type of active metal is compared [12–17]. On the other hand, some literature indicates that there is no correlation between them especially when comparing different active species, such as Fe and Cu [11, 18–20]. In this section, the reason for this inconsistency has been discussed.
Figure 8.4 shows the relationship between the standard SCR conversion and NO oxidation reaction conversion using Fe/zeolites prepared by different Fe loading methods and Fe loading amounts. The data which is used from Ref. [13] by Delahay et al. demonstrates a positive correlation between them, implying that the active sites for NO reduction are identical with the NO oxidation sites. Figure 8.5 shows the relationship between standard SCR conversion and NO oxidation reaction conversion when several Fe/zeolites with different pore structures and Si/Al2 ratios were used [16]. As can be seen from the figure, the SCR conversion in this case correlates well with the NO oxidation conversion. Thus, the activity relationship is preserved even when different types of zeolites are used.

SCR conversion versus NO oxidation conversion for Fe/zeolites prepared by different Fe loading methods and amounts of Fe. The data is used from Ref [13]. Gas composition = 0.2 % NO, 0 or 0.2 % NH3, and 2 % O2 with remainder He. The specification of the samples is described by “preparation method_Fe/Al ratio”: a sublimation with FeCl3_0.83, b sublimation with FeCl3_0.49, c ion-exchange with Fe(acac)3 followed by evaporation_0.96, d ion-exchange by Fe(acac)3 followed by washing_0.21, e ion-exchange by Fe(NO3)3 followed by precipitation_0.35

SCR conversion versus NO oxidation conversion of Fe/zeolites with different structure and Si/Al2 ratio [16]. Gas composition = 0.1 % NO, 0 or 0.12 % NH3, 8 % O2, 10 % CO2, and 8 % H2O with remainder N2
However, when the comparison is made between different kinds of active metals, the SCR conversion does not always correlate with the extent of NO oxidation. For instance, Metkar et al. [11, 19]. have reported that Fe/ZSM-5 showed a higher NO oxidation conversion than Cu/CHA, while Cu/CHA possessed a greater SCR activity than Fe/ZSM-5. Thus, it seems that the SCR activity is not dependent on the NO oxidation under the comparison between Fe and Cu. The key to solve this discrepancy is that the determining factors for the two reactions would be different. Delahay et al. [13] discussed that the difference in activity between SCR and NO oxidation can only be explained if the NO oxidation reaction is controlled by the desorption of NO2. In fact, the desorption energy of NO2 from Fe/ZSM-5 is very high (138 kJ/mol) [21], and the same goes for Cu/zeolites. Furthermore, the adsorption strength of NO2 is dependent on the metal species; for example, Fig. 8.6 shows the NO2-TPD experiment reported by Metkar et al. [11]. The NO2-TPD spectra indicate that NO2 is more strongly bound to Cu/zeolite compared to Fe/zeolite [11]. The same result has been obtained by Tronconi et al. [22]. These NO2-TPD results are in line with the reaction order results that Cu/zeolite has larger negative NO2 order than Fe/zeolite as is stated in the Sect. 8.2.3 and Table 8.1 [11].

NO2-TPD experiments carried out on Fe- and Cu-zeolite catalysts. NO2 adsorption = 150 °C; temperature ramp = 10 °C/min. Reprinted with permission from [11]
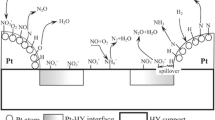
Taking the above results into consideration, we can conclude that the oxidation of NO in the absence of NH3 is strongly affected by the NO2 adsorption strength which depends on the type of active metal used. In other words, the rate-determining step of the NO oxidation under NH3 absent condition would be the NO2 desorption process. Figure 8.7 shows NO oxidation scheme over metal ion-exchanged zeolite by assuming that NO oxidation proceeds with the redox cycle of the metal ions. As the states of the metal ions, both mononuclear and binuclear states were considered. In this figure, the desorption step of the adsorbed NO2, that is the reduction step of the metal ions from Mn+ to M(n − 1)+, would probably be the slowest process, which should be determined by the type of metal.

Schematic representation of catalytic cycle for NO oxidation reaction over metal-exchanged zeolite catalysts. Redox sites are associated with oxo-metal (isolated or binuclear) ion-exchanged sites
Regarding standard SCR reaction, on the other hand, the NO2 desorption does not seem to be the rate-determining step because produced NO2 should react with NH3 immediately, which is elucidated and discussed in more detail in the other sections. Therefore, it is difficult to make a correlation between SCR activity and NO oxidation when different active metals are compared. Nevertheless, the author believes that the SCR activity is correlated with NO oxidation ability “in the presence of NH3”, simply because the fast SCR (NO/NO2/NH3 system) rate is extremely faster than the standard SCR rate. The point is that NO oxidation conversion (NO2 formation rate) under “NH3 absent condition” does not necessarily correspond to NO oxidation ability under “NH3 present condition,” i.e., standard SCR condition. Thus, the author presumes that although NO2 is hard to be desorbed from Cu sites, Cu would have a greater potential to oxidize NO to NO2 under NH3 present condition. However, quantifying this ability is very difficult because the production rate of NO2 adspecies can hardly be measured in the presence of NH3. Thus, an advanced technique which makes it possible to verify the assumption is desired.
2.5 Effect of Coexisting Gases and Poisoning
Considering the practical use of SCR catalysts for diesel engines, we have to take into account the possible effects due to the presence of other compounds such as CO, CO2, H2O, HC, and the poisoning effect from SO2 which are all present during the combustion of fuel. Figure 8.8 shows the standard SCR conversion of Fe/BEA in the presence of CO2, H2O, and C3H6. The base gas was 0.1 % NO, 0.12 % NH3, and 8 % O2 with the remainder N2. When 10 % CO2 was added to the base gas, the NOx conversion slightly declined, which indicates that CO2 has a little inhibitory effect on the SCR reaction. Next, the addition of 8 % H2O and/or 333 ppm C3H6 to the base + CO2 feed further lowered the NOx conversion. Here, the addition of 333 ppm C3H6 decreased the activity much more than 8 % H2O did, despite the low HC concentration. Additionally, the NOx conversion under the presence of both 333 ppm C3H6 and 8 % H2O was higher than that in presence of 333 ppm C3H6 only, suggesting that H2O suppresses the strong inhibition by C3H6. Thus, the degree of inhibitory effect on the SCR reaction follows the order: C3H6 > H2O > CO2.

SCR conversion of Fe/BEA in the presence of 10 % CO2, 8 % H2O, and/or 333 ppm C3H6. Base gas composition: 0.1 % NO, 0.12 % NH3, and 8 % O2 with the remainder N2
Li et al. [23] and He et al. [24] have investigated the effect of the regeneration temperature on the Fe/ZSM-5 and Fe/BEA catalysts, respectively, after C3H6 poisoning. Figure 8.9a shows the change in the NOx conversions in response to the presence (on) and absence (off) of C3H6 at several temperatures over Fe/ZSM-5 [23]. When 0.1 % C3H6 was added to the feed (10 min), all the conversions decreased, but the extent of decrease differed with the temperature. Then, after the removal of C3H6 from the feed (40 min), all the conversions except at 200 °C recovered. However, the extent of recovery depended on the temperature; with no improvement observed at 200 °C, while the conversions recovered to some extent at 300 and 400 °C. At 500 °C, the NOx conversion completely recovered to the initial conversion level before the addition of C3H6. Figure 8.8b shows the comparison of SCR activities of fresh, poisoned, and regenerated samples [23]. The term “poisoned” means that the catalyst was pretreated with C3H6 containing gas at 200 °C, while “regenerated” means that the poisoned catalyst was treated at 550 °C in presence of 10 % O2. It can be seen that the NOx conversion of the poisoned sample clearly decreased at 200–400 °C. However, the regenerated sample showed almost similar activity to the fresh one, indicating that the catalyst can be completely regenerated by the 550 °C treatment. In addition, an FT-IR study suggested that the hydrocarbon oxygenates such as formate-, acetate-, and nitrogen-containing organic compounds were created on the surface [23]. Thus, they concluded that the major cause for the deactivation was the carbonaceous deposition onto Fe3+ sites which are responsible for the oxidation of NO to NO2. In fact, they have also confirmed that the NO oxidation activity was significantly inhibited by the C3H6 poisoning [23].

a The change in NOx conversions with response to the C3H6 on and off at 200–500 °C for Fe/ZSM-5. b NOx conversions of fresh, C3H6 poisoned (at 200 °C), and regenerated (in 10 % O2 at 550 °C for 30 min) samples. Gas composition = 0.1 % NO, 0.1 % NH3, 0.1 % C3H6 (when used), 5 % O2, and 2 % H2O with remainder He. Reprinted with permission from [23]
Malpartida et al. [25] have reported the effect of HC types on the SCR reactivity using Fe/ZSM-5. Figure 8.10 shows NO conversion when different types of HCs (C3H6, C7H8, C10H22) coexisted [25]. The addition of any type of HC led to a decrease in the NO conversion, but its effect was different for different HCs; the rate of decline was limited in the case of C3H6 but was more significant in the presence of toluene (C7H8) and decane (C10H22). In situ FT-IR observed the deposition of C-species, and its quantity was dependent on the HCs; C10H22 > C7H8 > C3H6, which is in the reverse order with respect to the NO conversion [25]. Thus, they concluded that the most important effect from HC poisoning at low temperature is the competitive adsorption between the hydrocarbon molecules and NH3 onto the active Fe sites [25].

SCR conversion of Fe/ZSM-5 as a function of the temperature for different HC composition: without HC, 85 ppm C3H6, 85 ppm C7H8, 85 ppm C10H22, and 85 ppm mixed HCs (17 ppm C3H6, 25 ppm C7H8 and 43 ppm C10H22). Base gas composition: 150 ppm NO, 150 ppm NH3, 300 ppm CO, 14 % O2, 4 % CO2, and 1 % H2O with remainder Ar. Reprinted with permission from [25]
Next, the effect of SO2 poisoning and regeneration is presented. Figure 8.11 shows the influence of H2O and SO2 on the SCR conversion over Fe/ZSM-5 [26]. It is evident that SO2 exerts a poisoning influence at low temperature both in case of dry as well as moist feed. Remarkably, this effect is reversed at a high temperature, i.e., above 450 °C (723 K), the NO conversion is the highest even in the presence of SO2.

Influence of H2O and SO2 on the NO conversion over Fe/ZSM-5. Dry condition: 0.1 % NO, 0.1 % NH3, and 2 % O2 with the remainder He. Moist condition: as dry, with 2.5 % H2O added, ‘+SO2’, with 200 ppm SO2 added. Reprinted with permission from [26]
Finally, the effect of regeneration on the SO2-poisoned catalysts is presented [27]. First, hydrothermally aged samples (700 °C for 5 h) were poisoned by a feed containing 30 ppm SO2 until the adsorption saturated at 300 °C, and was then regenerated under standard SCR condition by maintaining at 550 °C for 10 min. Figure 8.12 shows the NOx conversions before the SO2 poisoning and after the SO2 regeneration for Fe/zeolites, Cu/ZSM-5, and V–W/TiO2. Obviously, all the regenerated Fe/zeolites maintained equivalent activity with those free from SO2. Meanwhile, the Cu/ZSM-5 catalyst greatly deteriorated even after the regeneration. This suggests that the major cause of the deterioration is not the zeolite structure but the type of ion-exchanged metal. Therefore, Fe/zeolites possess a higher endurance for SO2 poisoning than Cu/zeolites in this case. In addition, V–W/TiO2 shows also high endurance for SO2. Interestingly, regenerated V–W/TiO2 has greater activity than SO2 free one at 500 °C. This is probably because the SO2 poisoning inactivates aggregated V species which provoke NH3 unselective oxidation at high temperature.

NOx conversions of hydrothermally aged (700 °C for 5 h) Fe/zeolites with different structures (a, b), Cu/ZSM-5 (c), and V–W/TiO2 (d) before SO2 poisoning (filled symbols) and after SO2 poisoning and regeneration (open symbols). SCR feed: 0.1 % NO, 0.12 % NH3, 8 % O2, 10 % CO2, and 8 % H2O with remainder N2. SO2 poisoning and regeneration: poisoning under 30 ppm SO2 + SCR feed at 300 °C for 20 min, followed by regenerating under SCR feed at 550 °C for 10 min
3 Transient Reaction Analysis
3.1 Periodic NH3 Supply
As the composition and the concentration of diesel exhaust gases vary significantly depending on the engine operation, it is quite important to understand the transient behavior of NOx reduction following NH3 supply and shutoff. In this section, the effect of the periodical supply of NH3 on the NOx reduction behavior is presented.
As a first test, symmetrical NH3 on/off cycles were conducted by changing the switching time from 5/5 to 600/600 s/s at 200 and 250 °C. Figure 8.13a shows the outlet NOx concentration with the Fe/BEA zeolite catalyst under 600/600 s switching condition. The feed gas was a mixture having the composition: 0.1 % NO, 8 % O2, 10 % CO2, 8 % H2O, and the remainder N2 with a periodical supply of 0.2 % NH3. When NH3 was added to the feed, the NOx concentration quickly decreased due to the SCR reaction, went through a minimum, and then slowly approached a steady-state level. When NH3 was removed from the feed, the NOx concentration was reduced to a greater degree than the steady-state level, went through a minimum and then reached the inlet NOx value. Similar results have been reported by many researchers [4, 5, 18, 28–30]. This indicates that the SCR reaction using adsorbed NH3 continues even after the NH3 feed is shutoff, and furthermore, the reaction is accelerated in the absence of gaseous NH3. In other words, SCR is inhibited by the presence of gaseous NH3 and/or by a high coverage of adsorbed NH3. In Fig. 8.13a, one can see that the degree of transient NOx reduction is pronounced at a lower temperature. This can be due to the following two factors: First, the amount of adsorbed NH3 available for the SCR reaction increases with decrease in the temperature [4, 28, 30, 31] and second, the inhibiting effect of gaseous NH3 and/or high NH3 coverage is intensified at a lower temperature [4].

NOx concentration (a, c) and average NOx conversion (b, d) under periodic supply of NH3 with Fe/BEA (a, b) and V–W/TiO2 (c, d) catalysts. Gas composition = 0.1 % NO, 0.2 % NH3, 8 % O2, 10 % CO2, and 8 % H2O with the remainder N2; catalyst weight = 1 g; flow rate = 3.5 l/min
Figure 8.13b shows the average NOx conversion with change in the NH3 on/off cycle time. Interestingly, under certain conditions the average NOx conversion exceeded the steady-state conversion. It suggests that an intermittent supply of NH3 is more effective than a continuous supply. Additionally, because of the difference in the NH3 adsorption capacity, the maximum conversion at 200 °C is shifted to longer cycle time (smaller frequency) as compared to that at 250 °C. Thus, depending on the temperature there is an optimum NH3 on/off condition. The optimum condition would be varied with several factors such as NH3 storage capacity and W/F. Recently, the contributions of these parameters have also been elucidated by fitting procedure of simulation studies [30, 32, 33].
Figure 8.13c shows the NOx profile with a commercial V–W/TiO2 catalyst. In this case, the NOx concentration monotonically decreased and increased in response to the NH3 on/off switching. The transient NOx removal behavior was hardly observed in this case. This is probably due to the low NH3 adsorption capacity of the V-based catalyst, which has been verified later. Figure 8.13d shows the average NOx conversions with the V–W/TiO2 catalyst versus the NH3 on/off cycle time. The average conversions were mostly less than the steady-state level.
Next, asymmetric NH3 on/off switching (on/off = 60/120 s) with variation of the temperature (150–400 °C) were conducted. Figure 8.14a–c shows the average NOx conversions for the Fe/ZSM-5, Cu/ZSM-5, and V–W/TiO2 catalysts. In case of the Fe/ZSM-5 catalyst (Fig. 8.14a), the NOx conversions under the periodic condition were higher than those under the steady-state condition below 300 °C, which is in conformity with the result depicted in Fig. 8.13b. Similar enhancement in the NOx reduction in the low temperature region was observed over H–ZSM-5 by Wallin et al. [28]. They have reported that NOx reduction was enhanced by up to five times as compared to a continuous supply of NH3 by changing the NH3 pulse condition at 200–300 °C [28].

Average NOx conversion for a Fe/ZSM-5, b Cu/ZSM-5, and c V–W/TiO2 under periodic and static supply of NH3. Catalyst weight = 1 g; flow rate = 3.5 l/min. Periodic: NH3 on/off = 60/120 s/s; 0.02 % NO, 0.073 % NH3, 8 % O2, 10 % CO2, and 8 % H2O with the reminder N2. Static: 0.05 % NO, 0.06 % NH3, 8 % O2, 10 % CO2, and 8 % H2O with the remainder N2
The same observation holds true for the Cu/ZSM-5 catalyst (Fig. 8.14b) below 250 °C. However, at 400 °C the periodic conversion is less than the steady-state one. The reason for this lower activity for the periodic condition is probably due to the decrease of the reaction selectivity between NO and NH3 [34, 35]. As shown in Fig. 8.1b, Cu/ZSM-5 exhibits higher activity for NH3 oxidation than Fe/ZSM-5 and V–W/TiO2. Thus, it can be assumed that a part of the adsorbed NH3 is consumed by O2 under NH3 off condition, leading to the lower activity for the periodic measurement.
Meanwhile, the periodic conversions with V–W/TiO2 were roughly in accordance with the steady-state conversions at the low temperatures (≤250 °C). However, in the high temperature region (≥300 °C), the periodic conversions greatly decreased despite the fact that the steady-state conversions monotonically increased with the temperature. This is because the NH3 adsorption capacity available for the transient SCR reaction in case of the V–W/TiO2 catalyst is low, especially at the high temperature. To confirm this, NH3-TPD spectra are presented in Fig. 8.15. In fact, the amount of desorbed NH3 from the V–W/TiO2 catalyst is very little compared to the zeolite-based catalysts.

NH3-TPD spectra for Fe/ZSM-5, Cu/ZSM-5 and V–W/TiO2 catalysts. NH3 adsorption = 150 °C; temperature ramp = 20 °C/min; W/F = 3.3 g min/l
3.2 NO Pulse Reaction
To investigate the catalytic effect of Fe/ZSM-5 on the transient NOx reduction in more detail, the NO pulse reaction test was conducted with monitoring of the N2 production [4]; NO pulses were introduced to a continuous gas flow consisting of either 0 or 400 ppm NH3, 10 % O2, with the remainder He. Figure 8.16a shows the N2 production rates in the presence and absence of the NH3 feed. In the presence of NH3 (pulses 1–3), the N2 production rates were almost constant and increased with increasing temperature. When NH3 was removed from the feed, the amount of N2 increased (pulse 4) and then decreased (pulses 5–10), suggesting that the SCR reaction between the absorbed NH3 and pulsed NO is promoted in the absence of NH3. The total amount of N2 produced in the absence of NH3 (pulses 4–10) which corresponds to the amount of NH3 adsorbed is larger at lower temperatures [4, 31].

N2 production rate by NO pulse reaction in the presence and absence of NH3 feed over Fe/ZSM-5. Continuous gas flow = 0.1 l/min, either 0 or 0.04 % NH3, and 10 % O2 with remainder He; NO pulse = 0.34 ml; catalyst weight = 0.25 g. Reprinted with permission from [4]
Figure 8.16b shows the N2 production rates when the presence and absence of NH3 is repeated at the interval of three pulses. The N2 production rates in the first NH3 absent region (pulses 4 and 10) were higher than those in the NH3 present regions; this suggests that the promotional effect of the absence of NH3 is repeatable. This is in conformity with the behavior observed during the periodical NH3 supply test as shown in Fig. 8.13a.
3.3 In Situ FT-IR Analysis
To trace the surface-adsorbed species during the transient NOx reduction, in situ FT-IR analysis was applied. Analyzing the transient change of these adsorbates would help in understanding the SCR mechanism and identifying the rate-determining step.
Before the transient reaction analysis, the steady-state adsorption species formed when NH3/N2 or NO + O2/N2 was passed over the Fe/BEA catalyst have been presented. Figure 8.17a shows the difference spectra before and after 730 ppm NH3 flow at 150 °C. Prior to the NH3 adsorption, the sample was pretreated with 10 % O2 for 30 min at 550 °C followed by cooling it down to 150 °C. A strong band at 1,450 cm−1 and a weak band at 1,680 cm−1 can be assigned to the symmetric and asymmetric bending vibrations respectively, of chemisorbed NH4 + on the Brønsted acidic sites [12, 36, 37]. Meanwhile, weaker bands at 1,300 and 1,600 cm−1 can be assigned to the symmetric and asymmetric bending vibrations respectively, of coordinately linked NH3 to Lewis acidic sites [12, 36, 37]. The bands at 3,275 and 3,355 cm−1 can be assigned to the N–H stretching vibration of NH4 + ions with the three hydrogen atoms bonded to the three oxygen ions of the AlO4 tetrahedra (3H structure) [12, 36, 38]. The broad band between 2,600 and 2,900 cm−1 can be attributed to the N–H stretching vibration of physisorbed NH3 [12, 36, 37]. Figure 8.17b shows the NH3-adsorbed spectra after purging with 10 % O2/N2 at 150 °C. There is no change after the O2 purge, suggesting that neither oxidation nor desorption of adsorbed NH3 occurred at this temperature.

Difference FT-IR spectra over Fe/BEA in presence of 730 ppm NH3 flowing at 150 °C (a), and after the purge with 10 % O2 at 150 °C (b)
Figure 8.18a shows the difference spectra before and after NO + O2 flow. After pretreatment with 10 % O2 at 550 °C, the sample was treated with a flow of 1,000 ppm NO, 10 % O2 at 150 °C. A strong sharp band was observed at 1,635 cm−1 which can be assigned to nitro (NO2) group on the ion-exchanged Fe sites [36, 39–41]. Thus, NO was oxidized to NO2 over the Fe sites and adsorbed there. Small bands at 1,575 and 1,875 cm−1 can be assigned to nitrate and NO species, respectively, on the Fe sites [36, 39–41]. Additionally, a very weak broad band at 2,150 cm−1 could be assigned to NO+ species adsorbed on the Brønsted acidic sites [36, 39–41]. Figure 8.18b shows the NOx-adsorbed spectra after purging with 10 % O2 at 150 °C. The bands at 2,150 and 1,875 cm−1 disappeared, and only the NO2 and nitrate bands remained.

Difference FT-IR spectra over Fe/BEA in presence of 1,000 ppm NO and 10 % O2 flowing at 150 °C (a), and after the purge with 10 % O2 at 150 °C (b)
Next, a transient reaction test was carried out by introducing NO + O2 onto NH3 preadsorbed on the Fe/BEA catalyst at 150 °C. Figure 8.19a shows the peak intensity profiles of the three main bands; 1,450 cm−1 (NH4 +), 1,635 cm−1 (NO2), and 1,875 cm−1 (NO). When 0.2 % NO was added to 10 % O2 + N2 feed (7 min), the NH4 + band slightly increased and then decreased with increase in the NO and NO2 bands. After the disappearance of the NH4 + band (13 min), the NO and NO2 bands reached a steady-state level (20 min). Interestingly, the NO band went through a maximum at around 12 min, whereas the NO2 band increased sharply after the NH4 + band vanished (≥13 min). Thus, the NO2 band was lower than the NO band in the presence of the NH4 + band, but it exceeded after the disappearance of the NH4 + band. In other words, the band intensities between NO and NO2 reversed before and after the disappearance of the NH4 + band.

IR peak intensity profiles of NO, NO2, and NH4 + bands at 150 °C. 0.2 % NO was added to 10 % O2 feed in the presence of preadsorbed NH3 (a), or in the absence of preadsorbed NH3 (b)
Figure 8.19b shows the intensity profiles of the NO and NO2 bands in the absence of preadsorbed NH3. In this case, the NO2 band intensity remained constantly higher than the NO band, which is consistent with the steady-state result observed in Fig. 8.18a. Comparing Fig. 8.19a and b we can make the following hypotheses: While the SCR reaction with using adsorbed NH3 was occurring, (a) NO2 formation from the oxidation of NO is inhibited by adsorbed NH4 +, and/or (b) the NO2 produced is immediately consumed via the reaction with the adsorbed NH4 +. In any case, it could be said from this result that the rate-determining step of the surface SCR reaction would be the formation of NO2 adspecies by NO oxidation over the active Fe sites.
4 Reaction Mechanisms
4.1 Vanadium-Based Catalysts
In this section, some mechanistic implications of the SCR reaction over vanadium catalysts have been discussed based on the previous literature. The standard SCR reaction over V-based catalysts is generally considered to occur between the strongly adsorbed NH3 and gaseous or weakly adsorbed NO [42–49]. The proposed reaction mechanisms often involve two adjacent vanadium species, namely the terminal oxygen species, i.e., \(\text{V}=\text{O}\) (redox sites), and the hydroxyl group, i.e., \(\text{V-OH}\) (Brønsted acidic sites). Topsøe et al. [44–46] suggested that the reaction scheme involves the adsorption of NH3 on the Brønsted acidic sites (\(\text{V}^{5+}\text{--}\text{OH}\)) followed by activation of adsorbed NH3 via reaction at the redox sites (\(\text{V}^{5+}=\text{O}\)):
This activated form of NH3 reacts with gaseous or weakly adsorbed NO, producing N2 and H2O, and leading to partially reduced state (\(\text{V}^{4+}-\text{OH}\)). This reduced species could be reoxidized by oxygen to the \(\text{V}^{5+}\text{=O}\) species.
Topsøe et al. [44–46] reported that under high O2 concentration condition (>1 %), the NH3 activation step is fast and equilibrated, and thus the rate-limiting step is the reaction of NO with activated NH4 +.
Kamata et al. [48] estimated the ratio of the redox sites (\(\text{V}^{5+}\!\!=\!\text{O}\)) to the Brønsted acidic sites (\(\text{V}^{5+}\!\!\!-\!\!\text{OH}\)) by steady-state kinetic analysis. The relative amount of \(\text{V}^{5+}\!\!=\!\text{O}\) sites varied from ~0.1 to ~0.4 with the partial pressure of O2, indicating that the number of \(\text{V}^{5+}\!\!=\!\text{O}\) sites are less than the number of \(\text{V}^{5+}\!\!\!-\!\!\text{OH}\) sites.
Roduit et al. [1] proposed a global kinetic model for the standard SCR reaction based on V-based catalysts. The kinetic model accounts for three different reactions and intraparticle diffusion. The three reactions are Langmuir–Hinshelwood; LH-type SCR, Eley–Rideal; ER-type SCR, and direct NH3 oxidation. The main SCR pathway proceeds via the ER-type mechanism, but in the low temperature region (T ≤ 200 °C), LH-type reaction occurs. Furthermore, at high temperatures (T ≥ 300 °C), NH3 oxidation and intraparticle mass transfer also takes place [1].
Tronconi et al. [50, 51] observed a transient improvement of SCR activity just after NH3 shut off at low temperature. From this result, they pointed that the inhibitory effect of NH3 cannot be accommodated by a simple ER kinetics assuming the reaction between adsorbed NH3 and gaseous NO. They, then proposed that NO is oxidized by the V catalyst to a nitrite species, but the equilibrium is highly unfavorable and shifts to the right only in the presence of NH3 [52]. The NH3 which adsorbs on nearby acidic sites react with the nitrites to give N2 and H2O via decomposition of unstable ammonium nitrite intermediates, for example, according to
where NH3* represents adsorbed ammonia on the acidic sites. In this case, the inhibitory effect of NH3 could be more easily explained by either a competitive adsorption of NH3 onto the V sites involved in NO activation or an adverse electronic interaction of the adsorbed NH3 with the vanadium oxidizing centers [52].
Taking into account the above contributions, the SCR schemes at low temperature condition can be generalized as in Fig. 8.20. The catalytic cycle could be divided into two parts which takes place on; (a) acidic sites meant for NH3 adsorption/activation and (b) redox sites meant for adsorbed NH3 activation and NO adsorption/activation. This reaction mechanism suggests the requirement of two types of surface vanadium atoms. Many papers have suggested that the NOx conversion is correlated with the amount of \(\text{V}\!\!=\!\!\text{O}\) sites, and concluded that the redox site is indispensable to induce the SCR reaction [42, 44, 48, 49]. However, assuming the usage of transient SCR with preadsorbed NH3, the amount of acidic sites would also play a key role, because very little NH3 can be adsorbed and available for the transient SCR reaction.

Schematic representation of catalytic cycle for standard SCR reaction over vanadium-based catalyst. Acidic site and redox site are associated with \(\text{V}^{5+}\!\!\!-\!\!\text{OH}\text\;\text{and}\;\text{V}^{5+}\!\!\!=\!\!\text{O}\), respectively
4.2 Fe- or Cu-Exchanged Zeolite Catalysts
Numerous studies have been conducted to reveal the SCR reaction mechanism [4, 8, 18, 30, 35–37, 53–57] as well as active species [7, 58–66] for metal-exchanged zeolites. Recently, spectroscopic studies including EXAFS analysis revealed that a binuclear structure (M–O–M) is created on the ion-exchanged sites [60–63]. Also, it has been commonly accepted that such binuclear species play an important role as active sites for several reactions such as N2O/NO decomposition [67–70] and C6H6/CH4 oxidation by N2O [71–74]. Komatsu et al. [5] hypothesized that paired Cu2+ is the active copper species in view of the relationship between the SCR activity and the copper concentration. Similarly, Chen and Sachtler [58], and Mauvezin et al. [59] assumed that an oxygen-bridged binuclear iron complex acts as the active site for the SCR reaction because CO consumption in CO-TPR was nearly half of the amount of Fe. Schwidder et al. [29, 64] and Brandenberger et al. [7, 65] suggested that not only mononuclear Fe but also the binuclear species contribute to the SCR reaction.
For the SCR mechanism, Delahay et al. [13, 75] proposed a Fe2+/Fe3+ (or Cu+/Cu2+) redox cycle as a part of the SCR scheme; the extra-framework oxygen on Fe3+ (Cu2+) oxo/hydroxo species reacts with NO to form a surface nitrogen oxide intermediate (Fe3+–N x O y or Cu2+–N x O y ). Then, this species reacts with NH3 to form N2 and H2O with concomitant reduction of Fe3+ (Cu2+) to Fe2+ (Cu+) species. The Fe2+ (Cu+) species is reoxidized back to Fe3+ (Cu2+) oxo/hydroxo species by O2 [13, 75].
Considering these previous reports, one can assume the following general SCR mechanism (Fig. 8.21): First, the reaction sites would be composed of acidic sites and redox sites similar to the V catalyst. However, in some cases, there is a possibility that one site can possess both the properties. For the acidic sites, the ion-exchanged metal sites (Brønsted or Lewis type) or residual free proton sites (Brønsted type) can be expected. Strong NH3 adsorption on such acidic sites during the SCR reaction has been reported in the literature [36, 37, 54]. Regarding the redox sites, the ion-exchanged metal cations (mononuclear and/or binuclear oxo species) can be expected. From TPR study, these sites are known to be easily reduced compared to extra-framework bulk metal oxides [63, 73, 76–78]. Also, Nobukawa et al. [73, 74] reported that very reactive oxygen atoms can be created over the binuclear Fe sites after N2O treatment, and that it can desorb easily during a TPD run. This reactive oxygen atom in the nascent state is able to oxidize CH4 to CO/CO2 which is one of the difficult reactions to occur [73, 74].

Schematic representation of catalytic cycle for standard SCR reaction over metal-exchanged zeolite catalysts. Acid sites are associated with Lewis or Brønsted sites at ion-exchanged metal or free proton sites. Redox sites are associated with oxo-metal (isolated or binuclear) ion-exchanged sites
By comparing the SCR scheme (Fig. 8.21) with NO oxidation scheme (Fig. 8.7), one can find similarities and differences. First of all, both reactions seem to progress via similar redox cycles of the active metal. However, judging from the reaction kinetic analysis discussed in Sects. 8.2.3 and 8.2.4, the rate-determining step of NO oxidation would be NO2 desorption process. As for the SCR reaction, on the other hand, the process prior to the formation of NO2 related adspecies (e.g., nitrite or nitrate intermediate) would be the rate-determining step, as is discussed in Sect. 8.3.3. Therefore, the slowest step in the redox cycles would be different in the two reactions.
For the SCR reaction, NO activation to form NO2 species would be the key step. For a deeper understanding of the SCR mechanism, further reaction kinetic analysis combined with tracing the reversible valence change of active metal is necessary. However, it can be said from the result of Sects. 8.3.1 and 8.3.2 that gaseous and/or high coverage NH3 inhibits the rate-determining step, and thus reaction rate can be promoted by improving the reaction conditions such as adapting the periodic operation. Additionally, the acid site amount is also an important factor affecting the SCR activity especially for the periodic operation condition.
5 Conclusions
In this chapter, the mechanistic aspects of the NO–NH3–O2 reacting system were addressed by using three conventional SCR catalysts, Cu or Fe ion-exchanged zeolites and V–W/TiO2. There had been several differences among the catalysts. For instance, the temperature profiles of SCR activity were different between the zeolite-based catalysts and V–W/TiO2. Regarding the tolerance for SO2 poisoning, Fe/zeolite and V–W/TiO2 showed high durability, while Cu/zeolite greatly deteriorated in the low temperature activity due to the different affinity between the active metals and sulfur.
Also, the periodic operation (NH3 on/off cycling) had positive effect on the low temperature activity for Fe- and Cu-exchanged zeolites, because of strong inhibition by gaseous NH3 and/or high NH3 coverage. For V–W/TiO2, on the other hand, the periodic operation had not positive effect but negative especially under high temperature condition due to its lower NH3 storage capacity.
However, the reaction mechanism could be depicted as common schemes which would be composed of (a) acidic sites meant for NH3 adsorption/desorption, and (b) redox sites meant for NO oxidation and forming NOx–NH y intermediate species. Although the SCR activities of the three catalysts were not always correlated with NH3/NO oxidation abilities as well as acid site amount, each property plays an important role for explaining the reaction behaviors under high temperature and/or periodic conditions, as well as determining the reaction mechanisms. In situ FT-IR analysis of the SCR reaction between NO + O2 and preadsorbed NH3 over Fe/zeolite suggested that the rate-determining step of the surface SCR would be the formation of NO2 adspecies by NO oxidation over Fe sites, which is an important insight to clarify the detailed SCR mechanism.
References
Roduit B, Wokaun A, Baiker A (1998) Ind Eng Chem Res 37:4577–4590.
Metkar PS, Balakotaiah V, Harold MP (2011) Chem Eng Sci 66:5192–5203.
Efstathiou AM, Fliatoura K (1995) Appl Catal B: Environ 6:35–59.
Iwasaki M, Yamazaki K, Shinjoh H (2009) Appl Catal A: Gen 366:84–92.
Komatsu T, Nunokawa M, Moon IS, Takahara T, Namba S, Yashima T (1994) J Catal 148:427–437.
Huang HY, Long RQ, Yang RT (2002) Appl Catal A: Gen 235:241–251.
Brandenberger S, Kröcher O, Tissler A, Althoff R (2010) Appl Catal B: Environ 95:348–357.
Metkar PS, Salazar N, Muncrief R, Balakotaiah V, Harold MP (2011) Appl Catal B: Environ 104:110–126.
Moon IS, Namba S, Yashima T (1993) J Jpn Petrol Inst 36:339–342.
Eng J, Bartholomew CH (1997) J Catal 171:14–26.
Metkar PS, Balakotaiah V, Harold MP (2012) Catal Today 184:115–128.
Long RQ, Yang RT (2002) J Catal 207:274–285.
Delahay G, Valade D, Guzman-Vargas A, Coq B (2005) Appl Catal B: Environ 55:149–155.
Devadas M, Kröcher O, Elsener M, Wokaun A, Mitrikas G, Söger N, Pfeifer M, Demel Y, Mussmann L (2007) Catal Today 119:137–144.
Balle P, Geiger B, Kureti S (2009) Appl Catal B: Environ 85:109–119.
Iwasaki M, Yamazaki K, Shinjoh H (2011) Appl Catal B: Environ 102:302–309.
Wilken N, Wijayanti K, Kamasamudram K, Currier NW, Vedaiyan R, Yezerets A, Olsson L (2012) Appl Catal B: Environ 111–112:58–66.
Grossale A, Nova I, Tronconi E (2008) Catal Today 136:18–27.
Metkar PS, Harold MP, Balakotaiah V (2012) Appl Catal B: Environ 111–112:67–80.
Colombo M, Nova I, Tronconi E (2010) Catal Today 151:223–230.
Iwasaki M, Shinjoh H (2010) Phys Chem Chem Phys 12:2365–2372.
Tronconi E, Nova I, Colombo M (2010) Ind Eng Chem Res 49:10374–10385.
Li JH, Zhu RH, Cheng YS, Lambert CK, Yang RT (2010) Environ Sci Technol 44:1799–1805.
He CH, Wang YH, Cheng YS, Lambert CK, Yang RT (2009) Appl Catal A: Gen 368:121–126.
Malpartida I, Marie O, Bazin P, Daturi M, Jeandel X (2011) Appl Catal B: Environ 102:190–200.
Ma AZ, Grunert W (1999) Chem Commun:71–72.
Iwasaki M (2011) R&D Review of Toyota CRDL 42:21–32 http://www.tytlabs.co.jp/review/
Wallin M, Karlsson C-J, Skoglundh M, Palmqvist A (2003) J Catal 218:354–364.
Schwidder M, Heikens S, De Toni A, Geisler S, Berndt M, Bruckner A, Grunert W (2008) J Catal 259:96–103.
Sjövall H, Blint RJ, Gopinath A, Olsson L (2009) Ind Eng Chem Res 49:39–52.
Kröcher O, Devadas M, Elsener M, Wokaun A, Söger N, Pfeifer M, Demel Y, Mussmann L (2006) Appl Catal B: Environ 66:208–216.
Auvray X, Partridge WP, Choi J-S, Pihl JA, Yezerets A, Kamasamudram K, Currier NW, Olsson L (2012) Appl Catal B: Environ 126:144–152.
Colombo M, Nova I, Tronconi E, Schmeißer V, Bandl-Konrad B, Zimmermann L (2012) Appl Catal B: Environ 111–112:106–118.
Sjövall H, Olsson L, Fridell E, Blint RJ (2006) Appl Catal B: Environ 64:180–188.
Olsson L, Sjövall H, Blint RJ (2008) Appl Catal B: Environ 81:203–217.
Long RQ, Yang RT (2000) J Catal 194:80–90.
Sun Q, Gao ZX, Wen B, Sachtler WMH (2002) Catal Lett 78:1–5.
Eng J, Bartholomew CH (1997) J Catal 171:27–44.
Gao ZX, Qi S, Sachtler WMH (2001) Appl Catal B: Environ 33:9–23.
Iwasaki M, Yamazaki K, Banno K, Shinjoh H (2008) J Catal 260:205–216.
Iwasaki M, Shinjoh H (2010) J Catal 273:29–38.
Inomata M, Miyamoto A, Murakami Y (1980) J Catal 62:140–148.
Marshneva VI, Slavinskaya EM, Kalinkina OV, Odegova GV, Moroz EM, Lavrova GV, Salanov AN (1995) J Catal 155:171–183.
Topsoe NY, Dumesic JA, Topsoe H (1995) J Catal 151:241–252.
Topsoe NY, Topsoe H, Dumesic JA (1995) J Catal 151:226–240.
Dumesic JA, Topsøe NY, Topsøe H, Chen Y, Slabiak T (1996) J Catal 163:409–417.
Lietti L, Nova I, Tronconi E, Forzatti P (1998) Catal Today 45:85–92.
Kamata H, Takahashi K, Ingemar Odenbrand CU (1999) J Catal 185:106–113.
A. Centeno M, Carrizosa I, A. Odriozola J (1999) Phys Chem Chem Phys 1:349–354.
Tronconi E, Nova I, Ciardelli C, Chatterjee D, Bandl-Konrad B, Burkhardt T (2005) Catal Today 105:529–536.
Ciardelli C, Nova I, Tronconi E, Konrad B, Chatterjee D, Ecke K, Weibel M (2004) Chem Eng Sci 59:5301–5309.
Tronconi E, Nova I, Ciardelli C, Chatterjee D, Weibel M (2007) J Catal 245:1–10.
Long RQ, Yang RT (2001) J Catal 198:20–28.
Long RQ, Yang RT (2002) J Catal 207:224–231.
Sun Q, Gao ZX, Chen HY, Sachtler WMH (2001) J Catal 201:89–99.
Grossale A, Nova I, Tronconi E, Chatterjee D, Weibel M (2008) J Catal 256:312–322.
Iwasaki M, Shinjoh H (2010) Appl Catal A: Gen 390:71–77.
Chen HY, Sachtler WMH (1998) Catal Today 42:73–83.
Mauvezin M, Delahay G, Coq B, Kieger S, Jumas JC, Olivier-Fourcade J (2001) J Phys Chem B 105:928–935.
Marturano P, Drozdova L, Kogelbauer A, Prins R (2000) J Catal 192:236–247.
Marturano P, Drozdova L, Pirngruber GD, Kogelbauer A, Prins R (2001) Phys Chem Chem Phys 3:5585–5595.
Battiston AA, Bitter JH, de Groot FMF, Overweg AR, Stephan O, van Bokhoven JA, Kooyman PJ, van der Spek C, Vanko G, Koningsberger DC (2003) J Catal 213:251–271.
Battiston AA, Bitter JH, Heijboer WM, de Groot FMF, Koningsberger DC (2003) J Catal 215:279–293.
Schwidder M, Kumar MS, Klementiev K, Pohl MM, Bruckner A, Grunert W (2005) J Catal 231:314–330.
Brandenberger S, Kröcher O, Tissler A, Althoff R (2010) Appl Catal A: Gen 373:168–175.
Iwasaki M, Shinjoh H (2011) Chem Commun 47:3966–3968.
Moretti G, Ferraris G, Fierro G, Jacono ML, Morpurgo S, Faticanti M (2005) J Catal 232:476–487.
Hansen N, Heyden A, Bell AT, Keil FJ (2007) J Catal 248:213–225.
Guesmi H, Berthomieu D, Kiwi-Minsker L (2008) J Phys Chem C 112:20319–20328.
Pirngruber GD, Roy PK, Weiher N (2004) J Phys Chem B 108:13746–13754.
Li G, Pidko EA, van Santen RA, Feng Z, Li C, Hensen EJM (2011) J Catal 284:194–206.
Xia H, Sun K, Sun K, Feng Z, Li WX, Li C (2008) J Phys Chem C 112:9001–9005.
Nobukawa T, Yoshida M, Okumura K, Tomishige K, Kunimori K (2005) J Catal 229:374–388.
Nobukawa T, Sugawara K, Okumura K, Tomishige K, Kunimori K (2007) Appl Catal B: Environ 70:342–352.
Delahay G, Kieger S, Tanchoux N, Trens P, Coq B (2004) Appl Catal B: Environ 52:251–257.
Yoshida M, Nobukawa T, Ito SI, Tomishige K, Kunimori K (2004) J Catal 223:454–464.
Pérez-Ramírez J, Mul G, Kapteijn F, Moulijn JA, Overweg AR, Doménech A, Ribera A, Arends IWCE (2002) J Catal 207:113–126.
Lobree LJ, Hwang IC, Reimer JA, Bell AT (1999) J Catal 186:242–253.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Iwasaki, M. (2014). Mechanistic Aspect of NO–NH3–O2 Reacting System. In: Nova, I., Tronconi, E. (eds) Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts. Fundamental and Applied Catalysis. Springer, New York, NY. https://doi.org/10.1007/978-1-4899-8071-7_8
Download citation
DOI: https://doi.org/10.1007/978-1-4899-8071-7_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4899-8070-0
Online ISBN: 978-1-4899-8071-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)