
Abstract
The regulation of the Jak/STAT pathways involves multiple mechanisms, some of them exclusive to individual cascades, and other shared amongst them. Additionally, there is increasing evidence to support the interconnection of these cascades with other intracellular signaling pathways, which are regulated by independent factors. Thus, the understanding of the Jak/STAT pathway regulation is still growing and recently being revisited to account for the participation of non-conventional regulators. Acetylation of proteins is an emerging regulatory mechanism with demonstrated involvement in the regulation of several intracellular pathways. Specifically, the participation of histone deacetylases (HDACs) is garnering attention due to the feasibility of inhibition of their activity and the subsequent control over the cellular processes being modulated by them. This chapter will explore and present the current knowledge about the role of HDACs and their inhibitors in the control of members of the Jak/STAT pathways and direct activators/modulators of their signaling.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Cellular responses to the environment are mediated by numerous signaling pathways. The proper reception and translation of these signals allows integration of single cells into a multicellular system where every member has its own function. The Janus Kinase/signal transducers and activators of the transcription (JAK/STAT) signaling pathway is one of the most well studied yet enigmatic integrated signaling systems. This family of pathways’ major responsibility is allowing for cellular adaptation to an ever-changing microenvironment. Despite the name, the JAK/STAT pathway contains multiple signaling cascades with a plethora of proteins in addition to the Jaks and STATs.
The initial step in decoding the Jak/STAT pathway was the early finding of inducible genes by IFN treatment achieved almost simultaneously by the Darnell and Stark groups [1, 2]. In the following years these two groups consolidated their findings, identifying and characterizing several components of the Jak/STAT pathway, including several STATs and Jaks [3–5]. By 1994, these reports as well as findings from different groups, lead to the description of the Jak/STATs pathways. Further details about this fascinating race can be found in the article “The JAK-STAT Pathway at Twenty” [6].
The evolutionary conserved Jak/STAT pathway comprises several combinatorial subsets of functional units activated differentially by dozens of cytokines and growth factors. The final cellular responses triggered by these pathways depend on the signal, tissue, and cellular context, and include proliferation, differentiation, migration, apoptosis, and cell survival. Moreover, the JAK/STAT signaling is essential for numerous developmental and homeostatic processes, including hematopoiesis, immune cell development, stem cell maintenance, organism growth, and mammary gland development [7–9].
Some of the regulatory mechanisms affecting the Jak/STAT pathway functionality are those involving post-translational modifications, such as phosphorylation, acetylation, methylation and sumoylation [6]. In particular, this chapter will focus on the current knowledge of protein acetylation as a major modulator of the Jak/STAT pathway. A special emphasis will be assigned to the role of histone deacetylases (HDACs) and their inhibitors in this emerging and expanding field. The protagonists of these pathways will be introduced, and then the participation of HDACs at different levels during the Jak/STAT pathway activation, including their direct participation in formal Jak/STAT pathway members. In addition, their indirect role as regulators of activators of these pathways will be discussed.
2 Jak and STATs at a Glance
In a general description, activation of the Jak/STAT pathway begins with the binding of cytokines and growth factors to their corresponding receptors. This, in turn activates JAK proteins, which then introduce specific phosphorylation to either, receptors or STATs proteins on specific residues. The next step involves the homo- or heterodimerization of STATs accordingly with the respective signal sensed by the membrane receptors. This dimerization allows the translocation of STATs to the nucleus, binding to the consensus DNA sequence of 5’-TT(N4–6)AA-3’ and initiates the transcription of specific target genes [10].
The mammalian JAK family of kinases is composed by four members: JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2) [11]. Each of these proteins contains a conserved kinase domain (JH1) and a pseudo-kinase domain at the carboxyl terminus catalytically inactivated (JH2), which has been proposed as a regulator of the former kinase domain [12]. JAKs also have a Src homology 2 (SH2) domain and an N-terminal domain (FERM), which allows its association with cytokine receptors. Also, all members of this family distribute five blocks of sequence similarity throughout the amino-terminal region [13]. JAK1, JAK2, and TYK2 appear to be ubiquitously expressed, while JAK3 expression is normally limited to lymphoid cells.
There are seven mammalian STATs: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6. These proteins are highly homologous in several regions, including a N-terminal domain (NTD), a coiled-coil domain (CC), a beta-barrel DNA binding domain (DBD), a linker domain (LD), a SRC homology 2 (SH2) domain, and a C-terminal transactivation domain (TAD), which is located at the carboxyl terminus (Fig. 7.1) [12, 14]. However, the amino acid sequence diversity and their tissue-specific distributions account for the diverse roles of STATs in response to extracellular cytokines.

Activation of Jak/STAT pathways by cytokines
Abnormal activation of JAK-STAT pathways has been described in various cancers and immune disorders [8, 15]. These oncogenic processes have been largely described for either Jaks or STATs, and most of them involve constitutive activation. For example, the identification of an activating point mutation in the JAK2 kinase, JAK2V617F, in a vast majority of patients with BCR-ABL myeloproliferative neoplasms (MPNs), provided a significant advance in the knowledge of the molecular pathogenesis of these disorders [16]. This JAK2 mutant activates downstream signaling through the STATs, RAS/mitogen-activated protein kinase (MAPK), and phosphatidyl-inositol 3 (PI3). Noteworthy, treatment with the pan-HDACi LBH589, also known as panobinostat, seems to counteract the constitutive activation of JAK2V617F by inhibition of its autophosphorylation and promotion of its proteasomal degradation. Additionally, panobinostat has shown inhibition in other tumor cell lines such as the human erythroleukemia HEL92.1.7 and Ba/F3-JAK2V617F cells [17].
In most malignancies, it has been found that oncogenic mutations in STATs 1, 3, and 5 lead to persistent tyrosine phosphorylation [18]. Furthermore, abnormal activation of STAT3 has been detected in many cancers, including glioma, melanoma, non-small cell lung carcinoma, head and neck, breast, cervical, ovarian and prostate cancers [19]. The oncogenic property of STAT3 is mainly due to its function as a modulator of cellular processes such as cell cycle, cell proliferation, and apoptosis by regulation of cyclin D1, Bcl2 and Bcl-xl, genes that ultimately control tumor growth and metastasis [20]. Interestingly, STAT3 can also be abnormally induced by uncontrolled activation of its pathway. For example, paracrine sources of IL-6 can induce autocrine production of IL-6 and pStat3 expression in tumor cells, leading to heterogeneous levels of pStat3 [18].
3 Histone Deacetylases and Their Inhibitors
One of the most studied posttranslational protein modifications is the acetylation of lysine amino acids. Initially, these modifications were found at the N-terminal end of histones as a transcriptional regulatory mechanism. Briefly, in a non-modified steady-state, the highly positive N-terminal ends of histones wrap around histones, generating an obstacle for the binding of transcription factors and the recruitment of other proteins that need to read the “writing pattern” on nucleosomes to exert their transcriptional functions. In this context, acetylation of histones neutralizes these positive charges promoting a relaxed nucleosome conformation, and allowing the binding of transcription factors and “writer” proteins. Acetyl modifications are introduced by a heterogeneous group of proteins named histone acetyltransferases (HATs), most of them forming multi-protein complexes that can be selectively recruited to DNA sequences upon exogenous or endogenous cellular stimuli [21]. In opposition, these acetyl modifications can be removed by another group of proteins, HDACs. The 18 HDACs identified in humans are subdivided in two families; the classical HDAC family of zinc dependent metalloproteins, composed by Classes I, II and IV, and the Class III NAD+-dependent proteins belonging to the sirtuin family of HDACs. The Class I HDACs (HDAC1, 2, 3 and 8) are most closely related to the yeast deacetylase RPD3, and the Class II HDACs are subdivided into Class IIa (HDAC4, 5, 7 and 9) and Class IIb (HDAC6 and 10), both subclasses sharing homology with the yeast deacetylase HDA1 [22]. Finally, the newest HDAC discovered, HDAC11, comprises its own Class IV, and does not share homology with either RPD3 or HDA1 yeast deacetylases. HDAC knock-out mice have severe malfunctions at multiple cellular processes and are in some cases embryonically lethal (HDAC1, 3 and 7) or lethal at the perinatal stage (HDAC2, 4 and 8) [23].
HDACs, originally described as histone modifiers have more recently been demonstrated to modify a variety of other proteins involved in diverse cellular processes non-related to the chromatin environment. This includes deacetylation of multiple non-histone targets, including several proteins involved in cell cycle/apoptosis and immune regulation [23, 24]. The expanded role opens the possibility that the effects of HDACs and HDACi may affect non-epigenetic regulatory pathways, including the Jak/STAT pathway. Indeed, as will be shown throughout this chapter, multiple studies show that this is the case.
A logical approach to elucidating the precise mechanisms of action and functions of HDACs is the use of specific inhibitors that block HDAC enzymatic activity. Some of the compounds that inhibit HDAC activity include butyrates, TSA, trapoxin, SAHA, apicidin, depsipeptide, depudecin, CBHA, MS-275, CI-994, oxamflatin, pyroxamide, scriptaid, CHAP, and valproic acid [25, 26]. Early works indicated that extremely low concentrations of an HDAC inhibitor, TSA, effectively induced cell differentiation and G1/G2 cell cycle arrest. Many studies followed to show that cells cultured with an HDAC inhibitor result in a change in gene expression patterns. A large number of studies have shown that HDAC inhibitors can effectively arrest and counteract transformation of some cells and inhibit the growth of cancers in tumor-bearing animals. In fact, two HDAC inhibitors, Vorinostat and Romidepsin, are FDA approved for treatment of cutaneous T cell lymphoma, and many others HDACi are being pursued in clinical trials at different stages.
4 Signal Transducer and Activator of Transcription
4.1 STAT1
STAT1 is the principal transcriptional mediator of interferon (IFN) signaling and plays a central role in the regulation of innate and adaptive immune responses. In addition to IFNs, many other stimuli are able to activate STAT1, including cytokines belonging to the gp130 family such as IL-6, LIF, and OSM and growth factors such as EGF and PDGF (Fig. 7.2) [27]. All of these stimuli can also lead to STAT1 phosphorylation in association with other STATs (STAT3 and STAT5) accordingly, with the availability of other STATs and the combination of stimuli received from the environment. Probably the most well documented immunological function of STAT1 is its participation in T cell responses. It is important to mention its positive role in the induction of Th1 differentiation of T-cells, while repressing regulatory T-cells [28]. Additionally, STAT1 is considered as an anti-oncogenic protein. The mechanism by which STAT1 modulates cell death appears to involve both transcription-dependent and independent processes. At the transcriptional level, STAT1 has been described as master regulator of genes involved in the cell cycle and apoptosis such as Bcl-xL [29], p21 [30], iNOS [31], caspases [32], and death receptors [32]. Among its indirect non-transcriptional functions, its association with several pro-apoptotic mediators such as TRADD [33] and p53 [34] are well characterized. An example of the participation of STAT1 in cell death occurs in breast carcinomas, where the p21 activation by STAT1 also involves BRCA1, the most important susceptibility gene known in hereditary breast carcinoma. In fact, BRCA1 and STAT1 synergize to activate p21/waf1 transcription by means of an interaction between the BRCA1 and STAT1 TAD domain. This domain contains serine 727, whose phosphorylation is crucial for the transcriptional activity of STAT1 and which is directly implicated in the recruitment of STAT1 transcriptional coactivators; its mutation causes defective STAT1-BRCA1 binding [35].

Conserved domains of STATs: Distribution of phosphorylated and acetylated residues
The binding of IFN-γ allows the dimerization of its receptors and the reciprocal phosphorylation of Jaks. Once phosphorylated, Jaks will in turn phosphorylate the IFN receptors and provide a suitable STAT1 docking site. While associated with the receptors, STAT1 proteins are phosphorylated on conserved tyrosine 701 by JAKs, allowing the final dimerization of STAT1. There are two well described phosphorylation sites in STAT1; tyrosine 701 (Y701) and serine 727(S727), both occurring at the TAD domain and triggered by IFN stimulation (Fig. 7.1) [32]. The Y701 phosphorylation within the TAD is required for STAT1 dimerization, nuclear translocation, and DNA binding [36]. The second phosphorylation, S727, seems to be crucial for its transactivational activity, since STAT1 protein mutation at serine 727 has its transcriptional activity reduced by 20 % [37] and fails to recruit transcriptional coactivators [38]. The C-terminal domain TAD has been described as essential for its interaction with other regulatory proteins such as BRCA1 [38], CREB-binding protein (CBP)/p300 family of histone acetyl transferases [39], and MCM5 [40]. The same domain is also a target for the acetylation mediated by CBP, which also has been demonstrated as a STAT1 partner [41].
The magnitude and duration of the events downstream of STAT1 activation are influenced by several intracellular mechanisms and posttranscriptional modifications. In addition to the regulation mediated by phosphorylation, STAT1 can be regulated by acetylation. The first evidence of this mechanism came from early studies in melanoma cells, demonstrating that STAT1 was acetylated in the presence of HDACi [42]. This posttranslational modification was demonstrated to have a negative effect over STAT1 phosphorylation and its subsequent transcriptional activity [43]. The same group also demonstrated that cycles of phosphorylation/acetylation occur after IFN, in addition to other stimuli, treatment. A further hypothesis based on these observations proposes a “pulsing” mechanism of control. Essentially, the sequence of acetylation/phosphorylation observed in activated cells generates a brake for the action of STAT1 over its gene targets. Thus, the acetylation of STAT1 allows its interaction with the phosphatase TCP45 and its subsequent dephosphorylation (Fig. 7.3). This regulatory mechanism promotes a discrete action for STAT1, allowing the cell to escape from death [43].

Points of regulation in five classical Jak/STAT pathways
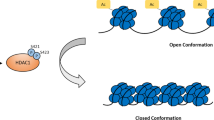
Several acetylated residues have been described for STAT1 (Fig. 7.1) [44]; however, only a few sites appear to have an impact over STAT1 function. The use of STAT1 mutants has helped to identify two specific residues undergoing acetylation, Lys410 and Lys413 [42]. Interestingly, both residues are located at the DNA-binding domain, suggesting a potential influence in the DNA-binding capability of STAT1 mediated by acetylation/deacetylation.
The first attempts to identify the role of acetylation over the functionality of STAT1 included the identification of the acetyltransferase CBP as responsible for this covalent modification. As well, experiments using HDACi and specific RNAi against HDAC1, 2, 3 helped to identify these HDACs as the counteracting enzymes in this process (Fig. 7.3) [41, 45, 46]. Additionally, a recent report indicates that HDAC4 is also involved in the deacetylation of STAT1 in ovarian cancer cells [47]. The well documented differential tissue distribution of HDAC4 [48] is opening a new perspective about the protagonists in the deacetylation/acetylation of STAT1. It is possible that cell/tissues with low levels of a specific HDAC are less susceptible to the effect of HDACi against STAT1 activity, a hypothesis that is amenable to testing in the near future.
Other groups have demonstrated the role of HDACs over the STAT1 function. For example, Klampfer et al. showed that HDACs are required for signaling by STAT1, and that HDAC inhibitors, as well as small interfering RNA specific for HDAC1, 2 and 3, are able to prevent IFN-γ induced JAK-1 activation, STAT1 phosphorylation and STAT1 dependent gene activation in a colorectal carcinoma cell line [46]. Similar outcomes were observed by TSA treatment in several myeloid cell and tumor cells [49]. Additionally, in colon cancer cells lines treated with the HDACi butyrate, TSA and SAHA, a preferential induction of apoptosis in transformed cells bearing k-RAS mutations was observed [50].
A more detailed functional role of HDACs over the STAT1 functionality and preference for DNA targets has been provided in a recent report [45]. Briefly, the treatment of different cell lines with HDACi counteracts the phosphorylation of STAT1 mediated by IFN. Interestingly, a mutant form of STAT1 replacing both lysines 410 and 413 for glutamine, but not individual mutations, mimicked the effect of a constitutive acetylated STAT1 and the subsequent inactivation of STAT1, suggesting that the number of deacetylated residues controls the functionality of STAT1. The same work also demonstrated that cells treated with IFNα can rescue the negative effect of the double mutant STAT1. A plausible explanation proposed is that IFNα promotes the phosphorylation of other STATs, such as STAT3, that in turn will heterodimerize with STAT1 and allows its recruitment to target DNAs [45].
4.2 STAT3
STAT3 modulates the expression of important genes involved in the regulation of a variety of physiological and non-physiological cellular functions, including cell cycle control, cellular differentiation, apoptosis, angiogenesis, metastasis, and innate and acquired immune responses [51, 52]. These genes include IL-17, IL-23, Bcl-xL, Bcl-2, MCL1, CCDN1, VEGF, c-Myc, p53, in addition to others [15, 27]. The Stat3 pathway is activated in response to a wide variety of cytokines such as the IL-6 and IL-10 family of cytokines, GCSF, leptin, IL-21, IL-23, etc. Moreover, STAT3 can be activated by other receptors and signals, including growth factors like PDGFR and EGFR, among others (Fig. 7.2) [15].
In the conventional STAT3 activation pathway (Fig. 7.1), the activation of cell surface receptors by growth factors or cytokines induces the phosphorylation of receptor tyrosine residues allowing the interaction of the STAT3 SH2 domain with the receptor and its subsequent phosphorylation. Phosphorylation of STAT3 can be mediated by the intrinsic tyrosine kinase activity of the activated growth factor receptor or by the Janus kinase (JAK) that associates with activated cytokine receptors, including Gp130. The final activation of STAT3 attained by its phosphorylation at the Tyrosine 705 (Y705) allows its dimerization (hetero- or homo-) and the subsequent translocation to the nucleus to modulate the expression of target genes. Additionally, a second phosphorylation at serine 727 (S727) in the C-terminal transactivation domain allows for the maximal activation of STAT3 target genes [51]. However, some evidence gathered from melanoma models suggests that the phosphorylation of Ser727 is not merely a secondary event and has a role in the survival activity and nuclear translocation of STAT3 in melanocytic cells, and its mechanism of action is independent from the Tyr705 phosphorylation [53].
It is accepted that constitutive activation of STAT3 mediates tumor-promoting inflammation. STAT3 has a dual effect in tumor inflammation and immunity by enhancing pro-oncogenic inflammatory pathways, including nuclear factor-kB (NF-KB) and interleukin-6 (IL-6)-GP130-JAK pathways, and by adding a brake to the STAT1 anti-tumor immune responses, mainly mediated by T cells. Thus, STAT3 has emerged as an attractive target for controlling oncogenic processes due to its critical role in tumor cell survival, proliferation, angiogenesis, metastasis, and immune modulation.
The important role of STAT3 in malignant processes was established after initial studies showed that STAT3 was constitutively activated during v-Src transformation [54], and that its downstream pathway was required for oncogenic transformation by v-Src [55]. Later, other transforming tyrosine kinases, such as v-Eyk, v-Ros, v-Fps, Etk/BMX, and Lck were found to constitutively activate STAT3 in the context of oncogenesis [56]. Moreover, early studies show that the constitutive activation of STAT3 in human breast cancer cells correlates with EGF receptors family kinase signaling and also with aberrant JAK and s-SRC activity [57]. These findings laid the groundwork to understanding the heterogeneous network involved in the activation of STAT3, leading to the discovery of other autocrine and paracrine stimuli influencing the aberrant functionality of STAT3. One of the most well understood signals triggering the oncogenic properties of STAT3 is the cytokine IL-6, which is particularly relevant in multiple myeloma and prostate cancer as IL-6-mediated activation of STAT3 enhances the production of key regulators of cell cycle and apoptosis such as Bcl-2, MCL-1, cyclin D1 and c-myc, which in turn prevents apoptosis and stimulates growth in these tumors [58]. In this context, in a recent report it was demonstrated that STAT3 plays a key role in G1-to S-phase cell-cycle transition through the up-regulation of cyclins D2, D3, A, and cdc25A and the concomitant down regulation of p21 and p27. Thus, constitutive Stat3 activation may lead to a growth advantage of the malignant counterpart [59].
Additionally, STAT3 tumorigenic activation has also been linked to other non-cytokine stimuli, such as EGFR, an event particularly important in lung cancer [60]. Noteworthy, it has been found that STAT3 is a common factor in many oncogenic signaling pathways, and is constitutively activated at a frequency of 50–90 % in diverse human cancers, including multiple-myeloma cells, chronic lymphocytic leukemia, head and neck cancer and many solid tumors such a breast, lung, prostate, ovarian cancers and malignant melanomas [15, 19, 61, 62]. The STAT3 activity is necessary for proliferation and/or survival of many different types of established or primary tumor cells bearing constitutive STAT3 activity, and its inhibition impairs tumor growth in vivo in different types of tumor [56]. Moreover, in murine models it has been reported that STAT3 represses p53 expression by directly binding to its promoter, inhibiting p53 pro-apoptotic activity and contributing to cell survival [63]. In a mouse model of melanoma, it was found that the alternative splice variant STAT3β with dominant negative properties suppressed tumor growth of B16 melanoma cells, supporting the role of STAT3 in these malignancies.
In addition to the well-characterized regulation mediated by phosphorylation, STAT3 can be regulated by acetylation. Interestingly, this mechanism seems to operate in the opposite direction compared to that observed in STAT1 (Fig. 7.3). Thus, like tyrosine phosphorylation, acetylation is necessary for the STAT3 activation [27]. In this context, persistent acetylation of STAT3 is generally observed in diverse human cancers. For this reason both phosphorylation and acetylation of STAT3 are crucial for STAT3-mediated up-regulation of oncogenic genes [64]. Cytokine-dependent acetylation of STAT3 at the C-terminal lysine-685 (K685) was reported by two independent groups [64, 65]. Both showed that STAT3 undergoes acetylation in various adherent cancer-derived cell lines treated with the related cytokines IL6 or OSM and with the Class I interferon IFNα. In addition to the opposite effect of acetylation observed between STAT1 and STAT3, several other discrepancies have been described. For example, STAT1 has been demonstrated to be acetylated only by CBP [42], and STAT3 can be acetylated by either CBP or p300 [65]. Moreover, the acetylated residues in both STATs are located in different structural domains; STAT1 is mainly acetylated at the residues K410 and K413 located at the DNA binding domain, and STAT3 is acetylated at the K685 located at the transactivation domain. These divergences suggest that the regulatory mechanisms behind these covalent modifications do not affect the same functions.
Early studies showed that HDAC1, HDAC2 and HDAC3 associate with STAT3 and the phosphatase PP2A. Among them; HDAC3 showed the highest deacetylase activity over STAT3. The mechanism proposed points to a facilitation of dephosphorylation of STAT3 mediated by the recruitment of PP2A through the interaction with HDACs [66]. This is further supported by the finding of hyperacetylation of STAT3 in cells treated with the Class I selective HDACi MS-275 [67], and the persistent phosphorylation of STAT3 in cells treated with HDACi [66]. Another body of evidence relating acetylation with the transcriptional regulation mediated by STAT3 involves a potential role in the control of the methylation status of gene promoters. It has been demonstrated that STAT3 interacts with the DNA methyltransferase 1 (DNMT1) [68]. In this context, a recent report showed that the impairment of the K685 acetylation is accompanied by demethylation and reactivation of several tumor-suppressor genes [69]. The same report also identified the potential role of sirtuins in this process through observations that the treatment of cells with the sirtuin activator resveratrol is directly linked to the acetylation status of STAT3 in melanoma cells, and this can reverse aberrant CpG island methylation in melanoma and several other malignancies. The participation of sirtuins in the deacetylation of STAT3 has been also reported by other groups [70, 71], positioning these deacetylases as potential targets in the modulation of the STAT3 activity.
4.3 STAT5
Two highly related STAT5 proteins exist: STAT5a and STAT5b. These proteins are encoded by two distinct but chromosomally linked genes with high similarity, although there are functional differences between them, including the DNA binding affinities [72]. STAT5a and STAT5b are activated in response to a variety of cytokines as well as tyrosine kinase receptors [73]. These include prolactin (PRL), growth hormone (GH), erythropoietin (Epo), trombopoietin (Tpo), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-2, IL-12, IL-3, IL-5, IL-7, IL-9, and IL-15, which are involved in a great many functions regarding cell growth regulation (Fig. 7.2) [74]. The canonical JAK2/STAT5 pathway is one of the most widely studied cellular signaling cascades and is critical for normal hematopoiesis [75]. Moreover, STAT5 has been reported to have oncogenic properties, mainly through its promotion of cell survival and proliferation by tightly controlling the expression of genes involved in cell cycle progression and survival, such as c-myc, bcl-2 and bcl-XL, cyclin D1, thus intervening in the growth control of these cells [76]. Since STAT5 regulates the transcription of cyclin D1/D2 and c-myc in some cell types [77], the constitutive activation of STAT5a/b, probably promotes tumorigenesis by deregulating the cyclin complexes D/CDK4-6, which is responsible for the control progression from the G1 to the S-phase of the cell cycle [56]. Thus, the constitutive activation of STAT5 is a hallmark of hematopoietic malignancies, chronic myelogenous leukemia, erythro-leukemia, acute lymphocytic leukemia, myeloproliferative neoplasms such as polycythemia vera, essential thrombocytopenia [78] and other types of cancer, including breast cancer [79], prostate cancer [80], squamous cell carcinoma of the head and neck, melanoma and hepatocellular carcinoma [81]. Its constitutive activation may also be triggered off by the expression of fusion proteins causing persistent PTK activation, such as JAK2, PDGF-R or ABL [82].
A widely pursued question is whether the STAT5a/b proteins are critical for cellular transformation, as suggested by the presence of activated STATs in a variety of tumor types and the observation that over expression of various forms of dominant-negative STAT5 proteins can partially suppress cell growth. For example, in chronic myelogenous leukemia (CML) the presence of activated STAT5a/b, in the presence of dominant negative can suppress the transformed phenotype [83]. Additionally, considerable attention has been assigned to STAT5a/b as transcriptional regulators of anti-apoptotic genes such as Bcl-X, a characteristic shared with other STATs such as STAT1. Particularly, STAT5a/b dominant negatives have a considerable reduction in Bcl-X levels, which leads to the suppression of cell growth [73].
One of the most studied roles of STAT5 is its participation in the prolactic receptor (PRLR) pathway. The PRLR is an essential type I cytokine receptor involved in mammary gland development during pregnancy and lactation. JAK2 phosphorylates PRLR on multiple tyrosine sites in a cytoplasmatic loop, participating also in the phosphorylation of STAT5a and STAT5b on a conserved tyrosine residue within the C-terminal SH2-dimerization domain. The phosphorylation of STAT5 triggers its dissociation from the receptor and the subsequent dimerization, which activates STAT5 and facilitates its translocation into the nucleus where it regulates gene expression associated with the functions of the ligand prolactin (PRL) [84].
As with other STATs, the activity of STAT5 can be regulated by phosphorylation. Additionally, recent studies in human breast cancer TD47 cells, reported that CBP induces acetylation of STAT5 and, simultaneously, PRLR. The dimerization and subsequent activation of the STAT5 pathway is also enhanced by the pan-HDACi TSA and the sirtuin inhibitor nicotinamide. These observations were further validated by inhibition of STAT5 after over expression of SIRT2 or HDAC6 [84]. This study also reported the existence of specific sites for acetylation in STAT5b, K359, K694, and K701, as reflected from mass spec analysis and site directed mutagenesis [84]. Interestingly, the residues K694 and K701 are located in close proximity to the phosphorylation site of STAT5, however, their acetylation by CBP was found to occur independently of the phosphorylation on Y699.
In a recent study it was reported that the selective class HDAC6 inhibitors NQN-1 decrease levels of constitutively active STAT5 and attenuated Erk phosphorylation by the acetylation of Hsp90 in human acute myeloid leukemia cell line MV4-11. This demonstrated that inhibition of HDAC6 and the subsequent interference with the Hsp90 chaperone function resulting in the degradation of critical proteins like STAT5, and attenuation of signaling cascades promoting leukemic cell growth [85]. Apparently, STAT5 seems to interact with a variety of HDACs. Thus, another recent report using co-immunoprecipitation and ChIP assays demonstrated that STAT5 is responsible for the recruitment of HDAC1 to the Id-1 gene through direct recruitment to the pro-B-cell enhancer (PBE) regulatory region. This recruitment induces deacetylation of the promoter region of the Id-1 gene as well as deacetylation of the transcription factor C/EBPbeta, whose acetylation diminishes its DNA-binding activity. Therefore, this new function for STAT5 enhances the transcriptional activity of Id-1 [86].
4.4 STAT2
STAT2 is mainly phosphorylated and activated by stimuli mediated by IFNα/β, cytokines that activate the ISGF3 complex, which consists of STAT1, STAT2 and IRF9 (Fig. 7.3) [73]. The tyrosine residue Tyr690 of STAT2, required for SH-phosphotyrosine interaction and thus STAT activation, is located near the SH2 domains. Contrarily to other STATs, STAT2 is the only one that does not act as a homodimer, and is the only member of the STAT family that does not bind GAS elements as homodimers [87].
IFNα/β stimulation induces the dimerization of the IFNARs and the subsequent cross-phosphorylation of Tyk2 and of JAK1. Once activated, JAKs phosphorylate the IFNAR2 receptor subunit to generate a docking site for STAT2 and promote its phosphorylation. Nevertheless, IFNAR2 phosphorylation only generates the docking site for STAT2; the subsequent phosphorylation of STAT2 in tyrosine 690, once again mediated by JAKs, will favor binding with STAT1. The binding of STAT1 to IFNAR2 depends on STAT2, but not vice versa [35]. The recruitment of STAT1 to the receptor allows its phosphorylation by JAKs at the tyrosine 701, this permits the release of the heterodimer STAT1/STAT2, which associates with the IRF9 nuclear factor and forms the ISGF3 factor [88], that in turn activates specific target genes within the nucleus recognizing promoter sequences called IFN-stimulated response elements (IRSE) [89, 90]. STAT2 deficient mice are viable and develop normally. However similar to STAT1 deficient mice, STAT2 null mice are susceptible to viral infections, and cells from these mice are unresponsive to IFNα/β, supporting the essential role that Stat2 plays in the ISGF-3 complex induced by IFNα. In addition, the absence of reduced activation of STAT1 is consistent with the concept that STAT2 facilitates recruitment to, or activation by, the IFNα/β receptor complex [73].
Like many eukaryotic transcription factors, the carboxy-terminal TADs of STAT1, STAT2, STAT3, STAT5 and STAT6 interact with co-activators histone acetyl transferases (HATs) especially p300/CBP. In this context, it has been reported that STAT2 recruits the acetyltransferase protein GCN5 (general control non-repressed), which lead to acetylation of histones in the promoters of IFN-α−regulated genes [13].
All subunits of the ISG3 complex, STAT1, IRF9 and STAT2, are highly acetylated, a process mediated mainly by CBP in IFNα stimulated Hela and 293T cells [87]. The Lys390 within the STAT2 DBD is poorly conserved among other STATs. Although it is unclear whether Lys390 is an evolutionary mistake requiring acetylation for correction or it has a function evolved specially for human cells, STAT2 acetylation on this site may warrant a more flexible interaction between STAT2 and STAT1, allowing STAT1 to interact with IRF9 and DNA within the ISGF3 complex [87]. This potential mechanism has been shown for other transcription factors such as p53, where the acetylation within DBD mediates regulatory interaction with other cofactors without affecting its DNA-binding activity [91]. Whereas acetylation of STAT2 at K390 appears to be required to allow association of STAT2 with IRF9, STAT2K390R constitutively binds STAT1 but fails to recruit IRF9 in IFNα treated cells [44]. The importance of the acetylation of STAT2 was also demonstrated by experiments using the HDACi TSA, which prevent its association with IRF9 and the subsequent recruitment of RNA polymerase II to the ISG54 promoter. Interestingly, ectopic expression of IRF9 reverses the inhibitory actions of TSA, suggesting that IRF9 functions to recruit RNA polymerase II to the promoter of interferon-stimulated genes [92].
4.5 STAT6
STAT6 is activated by IL-4 and IL-13 and regulates Th2 differentiation of lymphocytes [73], playing important roles in asthma and other inflammatory lung disease [93]. The absence of STAT6 blocks the differentiation of TH2 cells, and lack of STAT4 impairs IFNγ production by T cells and development of natural killer cells during bacterial and viral infections [13]. STAT6 has been shown to be persistently activated in various hematopoietic malignancies, such as lymphomas and leukemias [94], and upregulates genes important for hematopoietic tumor survival and proliferation when persistently activated in tumor cells [15]. As expected, STAT6 deficient mice lack most of the physiological functions associated with IL-4. In particular, the ability of IL-4 to induce the in vitro differentiation of Th2 cell is lost. This phenotype has provided support for the hypothesis that IL-4, through the activation of STAT6, induces the transcription of the IgE constant region of the heavy chain locus and thereby makes it accessible for the recombinase system [73].
A recent study showed that in several breast cell lines, STAT6 may also be involved in oncogenesis, like STAT1, STAT3 and STAT5. The inhibition of cell growth and induction of apoptosis, reported by other authors in human breast cancer cells after treatment with IL-4 seems to be mediated by STAT6 activation [95]. STAT6 is phosphorylated at low levels on the S707 and 756 serine residues in non-stimulated cells, and IL-4 stimulation triggers higher levels of phosphorylation. These serine phosphorylations occur independently of the Y641 tyrosine phosphorylation in STAT6, which is required for the STAT2 dimerization and subsequent nuclear translocation [35].
In addition to serine phosphorylation, STAT6 can be regulated by acetylation. In fact, STAT6 was the first member of the STAT family to be recognized as being acetylated [96]. However, the real contribution of its acetylation is not completely understood [44].
4.6 STAT4
STAT4 is the only STAT that has not been reported to be regulated by acetylation/deacetylation. STAT4 expression is restricted to myeloid cells [97] and is predominantly activated by IL-12, a cytokine produced mainly by APCs that is involved in the differentiation of naive T cells into Th1 cells. In fact, STAT4 and STAT6 are therefore involved in maintaining the equilibrium of the TH1 and TH2 responses, alterations in these proteins cause severe immune disorders, like autoimmune diseases for TH1 and allergic diseases for TH2 [98]. IL-12 is also important in the activation and growth of T cells. It stimulates the production of IFN-γ TNF-α from T and NK cells [99]. The IFNα/β can also activate STAT4 [100] but in a lesser magnitude.
4.7 STAT1 and STAT3 Acetylation; Opposing Roles in Survival and Immunogenicity
Although STAT3 and STAT1 control several common targets and share some characteristics, the majority of the cellular events unchained by their activation are opposite; STAT1 inhibits proliferation and enhances both, innate and adaptive immune responses. In the other hand, STAT3 promotes proliferation, survival, and immune tolerance (Fig. 7.4). The activation of these two pathways by cytokine/growth factors is indeed finely tuned by regulatory mechanisms present on each specific cell type, evidencing the intrinsic differences between cells to read and translate signals from the environment. Therefore, the fate of the final cellular outcome will depend on the composition, magnitude and duration during the activation of these two pathways, having a tremendous importance in pathological and pathophysiological conditions such as cancer, autoimmune diseases and immune response against foreign insults.

Opposite roles of STAT1 and STAT3 in survival and immunogenicity
As detailed before, the activation of STAT1 is primarily mediated by IFNs. The main function of STAT1 is triggering the expression of pro-apoptotic and anti-proliferative genes [101], and is considered a major barrier in neoplastic proliferation and expansion [27]. In addition to this function, STAT1 is considered a pro-inflammatory mediator due to its capacity to promote the recruitment and activation of immune cells during inflammatory processes. This function is in large part mediated for its capacity to promote antigen presentation by increasing the expression of MHC I and MHC II [27] and its positive regulatory role in the production of chemokines and adhesion molecules such as Mig [102], ICAM-1 [103], CXCL10 [104] and VCAM-1 [102]. A direct proof of the participation of STAT1 in antigenic processes came from early studies using STAT1 -/- and IRF-1 -/- hepatic cells, where the expression of all these mediators was diminished compared with wild type hepatic cells [102]. Additionally, STAT1 modulates the production of COX2 and nitric oxide, important mediators of vasodilatation during inflammatory processes [105].
In the opposite direction, STAT3 is a major regulator of anti-inflammatory events. This role is mainly attributed to its capacity to enhance the production of the anti-inflammatory cytokine IL-10, which in turn will counteract the inflammatory response. Additionally, by recent genome-wide analysis reports, several other target genes potentially responsible for the anti-inflammatory role of STAT3 [106, 107], including Bcl3, Il4ra and Socs3 [9, 10] have been identified. Stat3 can also indirectly ameliorate inflammation by down-regulation of STAT1 target genes, with function principally mediated by IL-10 which interferes with STAT1 phosphorylation [108]. STAT3 also promotes proliferation and inhibits apoptosis by direct regulation of several genes, including the anti-apoptotic factors Bcl-xL [27, 58], Survivin [109], MCL1 [110], and the cell cycle regulators Cyclin D1 [111] and MYC [77]. Additionally, STAT3 facilitates angiogenic events by induction of VEGF [112], HGF [113] and bFGF [114]. Interestingly, STAT1 seems to counteract the effect of STAT3 in angiogenesis by inhibiting the action of VEGF and bFGF [104].
The opposite effects of STAT1 and STAT3 in proliferation and immunogenicity have been explored in different contexts, and outstanding advances have been made by studies using selective inhibitors and direct genetic abrogation or silencing of their respective expressions [27]. However, new approaches are pointing to dual strategies to control differentially both pathways subsets, for example by manipulation of SOCS regulators [115].
As presented in this review, acetylation/deacetylation is emerging as a regulatory mechanism to control the activation of STATs. Particularly for STAT1 and STAT3 there is evidence indicating that their acetylation status has opposite outcomes in their functions; while STAT3 acetylation is necessary for its activation [64], this covalent modification impairs the transcriptional activation mediated by STAT1 [41]. This duality in the effect obtained by their acetylation status is diagrammed in Fig. 7.4, indicating the opposite outcomes in survival and immunogenicity attained upon acetylation of either, STAT1 and STAT3. This diagram also suggests that finding a common outcome in the function of STAT1 and STAT3 after either acetylation or deacetylation should be expected. Specifically, acetylation of both STATs will tip the balance toward the survival and anti-inflammatory phenotype, and their deacetylation will promote cell cycle arrest/apoptosis and stronger inflammatory responses (Fig. 7.4). This scenario is suggesting that pan-HDACi will favor survival and tolerance; however, in practice, these compounds are promoting exactly the opposite in several malignancies, indicating that the effect of pan-HDACi over other intracellular regulatory processes predominates in the final outcome. Another explanation could be that transformed cells are resistant to this putative regulatory mechanism. This resistance is supported by the fact that several malignancies bear aberrant activation of STATs [8, 15], and perhaps the regulatory mechanisms triggered by acetylation/deacetylation of STATs is bypassed by the hyper-activation of Jak/STATs pathways. Additionally, it is well documented that HDACi are able to decrease proliferation and modify other signaling pathways preferentially in oncogenic cells rather than non-transformed cells [25].
As described before, these two STATs can be acetylated by CBP [44]. However, only STAT3 can be acetylated by p300 [65]. In spite of the well characterized role of HAT complexes in the acetylation of STAT1 and STAT3, only a few HDACs have been tested for the removal of acetyl groups on STAT1 and STAT3. Interestingly, it seems like class I HDACs are able to deacetylate both proteins equally, and only a few reports describe the participation of other HDACs in this process. Noteworthy is the finding that Sirt1 deacetylates STAT3 to inhibit its function [70], and HDAC4 has the same effect over STAT1 [47]. These differences in substrate preferences by HDACs suggests that the differential inhibition of them could be used to differentially inhibit or activate STAT1 and STAT3, a possibility that must be explored in the future.
5 Regulators of Cytokine Signal Transduction
As with all biological processes, cytokine signal transduction must be tightly regulated. This regulation in the JAK/STAT signaling pathways is accomplished through multiple families of proteins, which include SH2-containing phosphatases (SHP), protein inhibitors of activated STATs (PIAS), and suppressors of cytokine signaling (SOCS). An overview of these three families of proteins, their role in regulating the JAK/STAT pathways, and the known interactions of HDACs and their inhibitors with these negative regulatory pathways is given below.
5.1 Negative Regulation of JAK/STAT Pathway by SOCs Proteins
The SOCS family of proteins includes SOCS1 through SOCS7 as well as the cytokine-inducible SH2-containing protein (CIS). In addition to the SOCS family proteins listed above, several other protein subgroups that contain the SOCS box, but lack the SH2 domain are sometimes categorizes as SOCS proteins. These include the subgroups: ankryin-repeat-containing proteins, WD-40 repeat containing proteins, SPRY domain-containing proteins, and the RAR-like GTPases. All members of the SOCS family contain a conserved sequence of 40 amino acids referred to as the SOCS box [116]. In unstimulated cells, SOCS proteins are generally expressed at low levels, and become rapidly induced by cytokines, thereby inhibiting JAK-STAT signaling, forming a classic negative-feedback loop [12]. The SOCS proteins are known to achieve signaling suppression through multiple mechanisms. SOC2, SOCS3 and CIS can bind to phosphotyrosine residues on cytokine receptors, thereby competing with STATs for binding sites. SOCS1 binds to phosphotyrosine residues on JAKs, directly inhibiting JAK activity. As well, SOCS can target bound proteins for proteasomal degradation by formation of an E3 ubiquitin ligase complex and subsequent ubiquitination of the bound protein.
SOCS members appear to have overlaps in function, thereby making exact determination of the individual roles difficult. However, knockout studies of SOCS1 mice show increased STAT1 activation and sensitivity to IFN-γ [117]. The SOCS1 promoter is also known to contain an STAT1 binding region; however, it also contains binding sites for STAT3 and STAT6 [116]. In regards to other SOCS: SOCS2 knockout mice have increased signaling by insulin-like growth factor 1, while SOCS3 knockout mice die due to placental insufficiency, a phenotype also seen in STAT3 knockout mice. In macrophages with SOCS3 knockdown, prolonged activation of STAT1 and STAT3 by IL-6 is seen. In addition, an induction of genes associated with IFN-γ signaling is observed [117].
An increase in SOCS expression in cells is dependent on tissue type and the type of cytokine or growth factor stimulation. For example IL-6 stimulation increases mRNA levels of SOCS1, SOCS2, SOCS3, and CIS in liver tissue, while growth hormone stimulation induces expression of only SOCS3 and CIS. In mammary tissue, SOCS2 and CIS are instead induced in response to growth hormone stimulation. As well as tissue and stimulus influencing SOCS expression, expression dynamics are variable. While both SOCS1 and SOCS3 expression liver levels are apparent as little as 20 min after stimulation with IL-6, SOCS1 expression returns to basal levels in 4 h, while SOCS3 take about 8 h. As well, SOCS2 and CIS levels last for 24 h [116].
Currently, little is known about the roles that histone deacetylases play in regulating SHPs, PIASs and SOCs. Indeed, only a few studies to date have been published evaluating the role of HDACi or individual HDACs on these families of negative regulators of cytokine signal transduction. Of these few studies, all of them address SOCS.
The HDACi TSA has been shown to suppress JAK2/STAT3 signaling through its effects on SOCS. In colorectal cancer cells, treatment with TSA led to hyperacetylation of the promoter regions of both SOCS1 and SOCS3, with no effect on the SHP1 promoter. This led to an increase in SOCS1 and SOCS3 expression and a decrease in JAK2/STAT3 signaling. Ultimately, TSA treatment lead to cell cycle arrest and apoptosis in colorectal cancer cells through downstream targets of JAK2/STAT3 signaling such as BCL-2, survivin, and p16 [118]. Treatment with TSA has also been reported to increase SOCS3 expression in N-1 neurons. A similar increase in SOCS3 expression is seen with valproic acid treatment. In contrast, the same study showed that TSA suppressed SOCS3 expression in 3T3-L1 adipocytes [119].
In RAW264.7 macrophages, LPS induces the phosphorylation of the transcription factor ATF-2 as well as the expression of SOCS3. However, when ATF-2 is silenced by siRNA, SOCS3 expression is significantly reduced. Further investigation showed that HDAC1 was found to interact with ATF2 after, but not before, LPS treatment. When treated with TSA prior to LPS stimulation, SOCS3 expression was inhibited. These results indicate that HDAC1 positively regulates AFT2 expression, and thereby positively regulates the expression of SOCS3 [120].
5.2 Regulation of STATs by PTPs and PIAS
Several protein tyrosine phosphatases (PTP) have been indicated to regulate JAKs, including SHP1, SHP2, CD45, PTP1B and T-cell PTP (TCPTP). SHP1 and SHP2 are SH2-domain-containing PTPs. Genetic studies indicate that SHP2 is involved in the negative regulation of JAK1 tyrosine phosphorylation after IFN-gamma stimulation is increased [121]. STATs can be negatively regulated by PTPs (such as PTP1B and TCPTP) in the cytoplasm and the nucleus [12], for example the protein tyrosine phosphatase TC45 is responsible for the dephosphorylation of STAT1 in the nucleus [122].
The PIAS family consists of PIAS1, PIAS3, PIASx, and PIASy. As well, there are two known splice variants of PIASx, the alpha and beta forms. PIAS proteins interact with STATs in response to cytokine stimulation, inhibiting the transcriptional activity of STATs. The mechanisms of STAT inhibition vary between the PIAS family members. PIAS1 and PIAS3 are known to directly bind STAT1 and STAT3 respectively, preventing association with DNA. As well, PIAS1, PIAS3, and PIASx can sumoylate STAT1 at Lys-703, close to the site of JAK phosphorylation. However, the impact of sumoylation on STAT1 activity is not understood. Interestingly, PIASx mediated STAT4 repression can be disrupted by HDACi [117]. PIAS proteins have also been implicated in various processes that have no apparent connection to STAT proteins, including induction of apoptosis, modulation of ion channels, interaction with androgen receptors and interaction with RNA Helicase [13].
The SHP family contains two members, SHP-1 and SHP-2. Both of these proteins contain two N-terminal SH2 domains in addition to a C-terminal protein-tyrosine phosphatase domain. These proteins are constitutively expressed and act by dephosphorylating signaling molecules. To achieve this, the SH2 domain of SHPs binds to phosphotyrosine residues on various cytokine receptors or other target molecules. SHP-1 is known to desphosphorylate the IL-4 receptor, c-kit, erythropoietin receptor and JAK2. Conversely, SHP-2 appears to act as a positive regulator of signaling. As well, activation of SHP2 through gp130 requires Jak1. The proliferation depends on STAT3 and SHP2 activation. In addition, SHP2 might be involved in the induction of serine phosphorylation of STAT1 and STAT3 via activation of the MAPK pathway [123]. Interestingly, upwards of 25 % of sporadic juvenile myelomonocytic leukemia patients have mutations in SHP-2 [124].
6 Cytokine Regulation by Histone Deacetylases
As cytokine signaling is the initiator of, and cytokine production is often the consequence of the JAK/STAT signaling pathways, discussing the role of HDACs in modulating cytokine expression is essential to understanding their effects on signaling pathways. Histone deacetylases are known to influence expression of a wide variety of cytokines. This knowledge came from several reports utilizing manipulation of specific HDACs and/or pharmacological inhibition of them. A list of the current effects by individual HDACs and HDACi in specific cell types is detailed in Table 7.1. Generally, inhibition of HDACs results in repression of pro-inflammatory cytokines, making histone deacetylase inhibitors (HDACi) attractive therapeutic agents in inflammatory diseases. However, in some cell types and contexts, HDAC inhibition can instead favor inflammatory cytokine production while repressing anti-inflammatory cytokine production [24, 125]. This is the result of the dynamic nature of HDACs with regards to cell type, stages of differentiation and disease context. Most studies to date conducted on histone deacetylase control of cytokine production address the effects of one or more HDACi. The majority of HDACi currently utilized target multiple HDACs, making determination of the role of individual HDACs difficult. However, using recently developed isotype specific HDACi or other experimental approaches to manipulating expression of individual HDACs, a small, but increasing number of studies have identified the role of individual HDACs in the regulation of cytokine production.
6.1 Innate Immune Cells
A large number of studies over the past decade have addressed the effect of HDAC inhibitors on leukocyte cytokine production. In a 2002 study, it was demonstrated that administration of the pan-HDACi suberoylanilide hydroamic acid (SAHA), also known as vorinostat, to mice results in a reduction of TNF, IL-1β, IL-6 and IFN-γ serum levels after induction by lipopolysaccharide (LPS). Similarly, when human monocytes are stimulated with LPS in the presence of SAHA, there is a pronounced reduction in production of TNF, IL-1β, IL-12 and IFN-γ. As well, IL-18 plus IL-12 stimulation of these cells in the presence of SAHA results in drastic reduction in IFN-γ production. Additionally, IFN-γ and TNF mRNA levels are reduced by SAHA administration, but IL-8 and IL-1β levels are unaffected. Intriguingly, IFN-γ production by CD3 stimulation is not affected by SAHA treatment [126].
In dendritic cells, pretreatment with SAHA results in a dose dependent reduction in expression of TNF, IL-12 and IL-6 after LPS stimulation [127]. These results are similar with the use of another HDACi, ITF 2,357, also known as givinostat. Furthermore, these reductions are the result of increased STAT3 acetylation [128]. Other studies in dendritic cells show that both SAHA and TSA are also able to modulate the expression of IL-12 and IL-23 [129].
Trichostatin-A (TSA), an HDACi with activity against Class I and II HDACs, has been demonstrated to increase expression of IL-10 in bone marrow derived macrophages. TSA treatment also reduces levels of TNF, IL-6 and IL-1β production in these cells [130]. A separate study demonstrated in both human and murine cells, that treatment of dendritic cells with TSA or SAHA also inhibits production of IL-12p40 post TLR stimulation. Intriguingly, despite decreased expression of protein, HDACi treatment results in increased acetylation of the IL-12p40 locus [131].
In LPS stimulated macrophages, treatment with sodium valproate represses expression of IL-12 and TNF-α while increasing IL-10 expression [132]. As well, in bone marrow derived dendritic cells, apicidin, a fungal metabolite with HDACi properties, inhibits production of IL-12, along with IL-6 and TNF production. Resultantly, treatment of dendritic cells abrogates their ability to produce a Th1 response in T-cells [133]. In agreement, a separate study shows that treatment of LPS stimulated human monocyte derived dendritic cells with butyrate results in a reduction in IL-12p40 as well as IL-6 production. As with apicidin treatment, this impairment results in a deficiency in generating Th1 responses by T-cells [134]. Treatment of LPS stimulated human dendritic cells with the pan-HDACi LBH589 also reduces expression of a variety of cytokines including IL-10, IL-12p70, IL-6, IL-23 and TNF-alpha. Again, the functional consequence of which is impaired activation of T-cells [135].
Interleukin 23 is a heterodimeric cytokine produced by both dendritic cells as well as macrophages. The p40 subunit of IL-23 is shared with IL-12, and like IL-12, the IL-23 receptor signals through the STAT4 pathway. Functionally, IL-23 along with other cytokines is responsible for directing Th17 CD4+ T-cell differentiation. Its role as a pro-inflammatory cytokine and involvement in autoimmunity is becoming increasingly clear. As with various studies in IL-12, IL-23 production is abrogated with HDACi treatment. A study using TSA and SAHA showed human and murine dendritic cells have reduced production of IL-12 and IL-23 when treated with HDACi. Intriguingly, the results are similar when HDACi was given concomitantly with LPS or IFN-γ stimulation as well as administered after stimulation [136].
The Type I interferons are composed of IFN-α and IFN-β. IFN-α is a pleiotropic protein with over twenty known variants. It is mainly produced by innate immune cells in the context of viral infection. There is also evidence of IFN-α having anti-tumor effects. Like IFN-α, IFN-β is generally associated with viral infection and anti-tumor effects. IFN-β upregulates TRAIL expression on CD8+ T-cells. TRAIL upregulation is associated with both anti-viral and anti-tumor responses. Plasmacytoid dendritic cells are the main producers of type I interferons. In a recent study, a reduction in type I interferons, TRAIL, IL-6 and TNFα was observed after TSA treatment of these cells [137].
While many studies have demonstrated the effects of pan-HDACi or class specific HDACi on cytokine production, more recently the role of individual HDACs in regulating cytokine production has become subject to experimentation. In 2008, HDAC11, the newest discovered HDAC, was discovered to negatively regulate the production of IL-10 in both human and mouse antigen presenting cells. An increase in both IL-10 and IL-12 production by LPS stimulated HDAC11-knockout macrophages is seen, while a decrease in both cytokines is seen in HDAC11-overexpressing macrophages. Interaction of HDAC11 with the proximal promoter region of the IL-10 gene, resulting in deacetylation activity, and thus transcriptional regulation was determined as the mechanism for this regulation [138].
The results of one study show that treatment of mouse macrophages with the pan-HDACi LAQ824 induces chromatin changes at the IL-10 promoter leading to repressed expression of IL-10. Importantly, this study also reveals that LAQ824 treatment results in enhanced recruitment of HDAC11, a negative regulator of IL-10 production, as well as the transcriptional repressor PU.1 to the IL-10 promoter region. Furthermore, an increase in IL-12 production by treated macrophages concomitant to the decrease in IL-10 production is seen. Functionally, in contrast to the described results with other HDACi, this results in increased Th1 activation of T-cells. Additionally, LAQ824 treated macrophages are able to restore function to anergized T-cells [139].
Another study shows that in intestinal macrophages, IL-10 regulates IL12p40 through HDAC3. In these cells, inhibition of HDAC3 by MS275, which target only Class I HDACs, results in increased histone 4 acetylation at the IL-12p40 promoter after IL-10 induction by LPS. As well, shRNA silencing of HDAC3 results in a diminished IL-12p40 repression by LPS induced IL-10 [140]. As in studies described above, another study demonstrated downregulation of IL-12p40 mRNA levels in mouse macrophages after treatment with TSA or SAHA. Additionally, when these cells are treated with the HDACi MS-275, this downregulation is not seen. However, when treated with the HDAC6 specific inhibitor referred to as 17a, a similar reduction in IL-12p40 expression is seen as when treated with TSA [132]. In dendritic cells, it has been recently demonstrated that treatment with valproic acid prior to LPS stimulation decreases the production of IL-10 as well as IL-12p70. These differences are also seen under IFN-γ stimulation. However, the roles of HDAC3 or other specific HDACs were not addressed in this study. Additionally, another study has also demonstrated that HDAC1 mediates repression of IL-12 expression in macrophages [141].
6.2 T-Cells
While the above studies have investigated the effects of various HDACi and roles HDACs in leukocytes, several reports have also addressed the direct effects of HDACi, or the role of specific HDACs, in T-cell cytokine production. In T-cells, as with leukocytes, HDACi treatment often, but not always, decreases expression of pro-inflammatory cytokines.
Results from a study in murine T-cells show that treatment with low dose of TSA and other HDACi results in marked reduction in the production of IL-2. However, sodium butylate treatment does not result in changes in IL-2 production. In addition to its effects on IL-2 production, TSA treatment also results in dramatic decreases in IL-4, IL-13 and IFN-γ production by T-cells. Treatment with both scriptaid and sodium butylate results in changes in only IFN-γ, and to a lesser degree than those seen with TSA. As well, the hyperacetylation associated with HDACi treatment is pronouncedly longer lasting with TSA treatment [142]. Conversely, a study published near the same time reported a TSA induced increase in IL-4 production by T-cells in a murine, collagen induced, rheumatoid arthritis model. This difference may be partially explained by an increase in Th1 cell apoptosis. However, Th2 cells also display an increase in IL-4 gene expression and an associated increase in histone 4 acetylation following TSA treatment [142]. As well, in regulatory T-cells TSA drastically reduces the formation of IL-17 producing cells [143].
Treatment of Jurkat cells with TSA or sodium butyrate elevates production of IL-5, a cytokine responsible for stimulating B-cell growth and antibody secretion as well as the maturation and differentiation of eosinophils. The increase in IL-5 production is associated with increased IL-5 promoter activity and hyperacetylation of both histone 3 and histone 4 at the IL-5 promoter region [144].
A study of whole human peripheral blood mononuclear cells (PBMC) showed ex vivo treatment with TSA results in decreased levels of IFN-γ and IL-2 production concomitant with increases in IL-4 and IL-13 production. When these cells are stimulated with phytohemagglutinin, an activator of T-cells, the cytokine differences resultant from TSA treatment are exacerbated [145]. A separate study also showed that Th1 T-cells when treated with various HDACi including TSA and butyrate have reduced levels of both IL-2 and IFNγ production. Resultantly, treatment with TSA is able to induce tolerance in OT-II ovalbumin-specific naïve CD4+ T-cells [146].
In addition to inhibiting IFNγ production in T-cells and PBMCs, HDACi treatment inhibits production in NK cells. When treated with TSA, valproic acid, or sodium butyrate, NK cells produce lower levels of IFNγ, even upon stimulation with exogenous cytokines such as IL-2 [147].
Class I HDACs are known to complex with Switch independent (SIN3) or Nucleosome-Remodeling protein (NuRD) in T-cells. In proliferating CD4+ T-cells, Sin3-HDAC complexes are recruited to the IFN-γ locus. This complex is displaced by the transcription factor T-bet in Th1 polarization of these cells. Therefore, treatment with HDACi is capable of augmenting IFNγ production by preventing the deacetylation of the IFNγ promoter by the Sin3-HDAC complex. However, as shown above, several HDACi reduce both IFNγ and IL-2 levels. This reduction of IL-2 has been reported to be characteristic of HDACi induced anergy in Th1 T-cells. Furthermore, T-bet has also been reported to indirectly interact with HDAC3 and HDAC5 in respect to GATA-3 binding, a transcription factor driving Th2 polarization [129].
Another study demonstrated that HDAC1 and HDAC2 in complex with Sin3A are displaced from IFN-γ locus in T-cells in Th1 differentiation. When non-stimulated, Th0 CD4+ cells were treated with TSA, there was an acquisition of histone 4 acetylation at the IFN-γ locus not seen in non-treated cells. When these cells were polarized to Th1 cells, no differences in histone 4 acetylation are seen. This increase in histone acetylation was shown to be independent of T-bet and Stat4. Sin3A\HDAC complex is found to associate with several areas of the IFNγ locus, including a known binding site of T-bet, a positive transcriptional regulator of IFNγ, in Th0 cells. When polarized to a Th1 phenotype, association of this complex at the promoter region of IFNγ is lost. However, in both Stat4 and T-bet deficient mice, when T-cells were polarized in Th1 conditions, no loss in Sin3A\HDAC complex at the IFNγ promoter is seen, indicating that dissociation of the complex is dependent on T-bet and STAT4 [148].
A separate group has also reported HDAC1 as a regulator of IL-10 production though E26 transformation-specific 1 (Ets-1). Th1 cells with Ets-1 deficiency had lower levels of HDAC1 recruitment to the IL-10 gene regulatory region and increased IL-10 production. A physical interaction between Ets-1 and HDAC1 was also demonstrated, indicating an Ets-1/HDAC1 synergism in IL-10 transcriptional repression [149].
6.3 Disease State Cells
As HDACi are potent modulators of cytokine production, and have a proclivity to down-regulate inflammatory cytokine responses, their use in autoimmune disease contexts is under continued investigation. Highlighting the complex and dynamic roles of HDACs, HDACi are utilized in the seemingly opposite setting as cancer therapeutics, due to their anti-proliferative and sometimes apoptotic effects. Indeed, two HDACi, SAHA and romidepsin, are FDA approved for the treatment of cutaneous T-cell lymphoma. Given this dynamic nature of HDACi, the effects on cytokine production in diseased cells can vary from those seen in healthy cells.
Rheumatoid arthritis is a chronic autoimmune disease with progressive destruction of the joints. Recently, a group has demonstrated that peripheral blood mononuclear cells (PBMCs) from rheumatoid arthritis patients have significantly increased HDAC activity when compared to healthy PBMCs. As well, treatment with TSA reduces levels of TNF and IL-6 production in both healthy and rheumatoid arthritis PBMCs, while treatment with an HDAC3 specific inhibitor, MI192, reduces production of IL-6 in rheumatoid arthritis PBMCs and having no effect on healthy PBMCs [150]. In a separate study, E11 cells, a cell line derived from human rheumatoid arthritis synovial fibroblasts, shows a dose-dependent reduction in IL-6, IL-18 and VEGF production when treated with either MS-275 or SAHA. In addition, treatment with either HDACi results in dose-dependent reduction in IL-1β, IL-6, IL-18 and TNF- α production by an LPS stimulated human monocyte cell line [151]. A similar study demonstrated that PBMCs or lamina propria cells from Crohn’s disease patients produced less TNF mRNA as well as protein when cultured with butyrate and stimulated with LPS. This reduction results from decreased NFkB activation [152].
In a Type 1 diabetes mouse model, the HDACi ITF2357, now known as givinostat, reduces production of IFNγ as well as TNF in peritoneal macrophages and spleenocytes. As well, when insulin-producing cells are challenged with IL-1β plus IFNγ, cells treated with givinostat have a marked reduction in apoptosis [153].
In systemic lupus erythematous, CD4+ T-cells produce high levels of IL-10 and reduced levels of IFNγ. When these cells are treated with the HDACi trichostatin A (TSA), a recovery of cytokine phenotype is seen [154]. In spleenocytes from MRL-lpr / lpr mice, a systemic lupus erythematous model, treatment with either TSA or SAHA downregulates expression of IL-10, IL-12, IFNγ, and IL-6. In vivo, TSA treatment of these mice results in a reduction in disease associated symptoms (e.g. proteinuria) [135].
Treatment of the human fibrosarcoma cell line 2fTGH with TSA reduces IFNβ production in response to viral challenge. Using a luciferase report assay and RNA interference, TSA treatment was found to interfere with the transcription factor interferon regulatory factor 3 (IRF3). Utilizing siRNA against individual HDACs, it was found that HDAC1 and HDAC8 both repressed IFNβ expression. However, these results did not explain the reduction in IFNβ production with TSA treatment. Further investigation revealed that HDAC6 enhances IFNβ expression by way of the IRF3 complex [155].
7 Conclusions
The Jak/STAT pathways represent one of the most well studied cellular signaling systems. From the initial description of IFN inducible genes, to the describing of the Jak/STAT pathways, the knowledge of these pathways has grown by a staggering amount in the last two decades. Indeed, recently regulation of the Jak/STAT pathway by mechanism beyond phosphorylation, including acetylation, has become evident. At the same time, it is becoming increasingly clear that the Jak/STAT pathways are highly intricate and that much work remains to be done. Additionally, it is also becoming increasingly evident that deregulation of the Jak/STAT pathways plays an important role in disease states, particularly cancer. In the disease context, understanding of not only the Jak/STAT pathways, but also its regulation by cytokines, HDACs, and the influence of HDAC inhibitors are essential to developing more effective and precise therapies. Overall, the Jak/STAT pathways represent a complex family of signaling pathways with equally complex regulation. Future studies will no doubt further unravel these complexities.
References
Larner AC et al (1984) Transcriptional induction of two genes in human cells by beta interferon. Proc Natl Acad Sci USA 81:6733–6737
Friedman RL, Manly SP, McMahon M, Kerr IM, Stark GR (1984) Transcriptional and posttranscriptional regulation of interferon-induced gene expression in human cells. Cell 38:745–755
Shuai K, Schindler C, Prezioso VR, Darnell JE Jr (1992) Activation of transcription by IFN-gamma: tyrosine phosphorylation of a 91-kD DNA binding protein. Science 258: 1808–1812
Schindler C, Shuai K, Prezioso VR, Darnell JE Jr (1992) Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science 257:809–813
Velazquez L, Fellous M, Stark GR, Pellegrini S (1992) A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 70:313–322
Stark, George R, Darnell Jr, James E (2012) The JAK-STAT pathway at twenty. Immunity 36:503–514
Ghoreschi K, Laurence A, O’Shea JJ (2009) Janus kinases in immune cell signaling. Immunol Rev 228:273–287
O’Shea JJ, Plenge R (2012) JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 36:542–550
Aaronson DS, Horvath CM (2002) A road map for those who don’t know JAK-STAT. Science 296:1653–1655
Murray PJ (2007) The JAK-STAT signaling pathway: input and output integration. J Immunol 178:2623–2629
Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD (1998) How cells respond to interferons. Ann Rev Biochem 67:227–264
Shuai K, Liu B (2003) Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 3:900–911
Levy DE, Darnell JE (2002) STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662
Darnell JE Jr (1997) STATs and gene regulation. Science 277:1630–1635
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Pahl HL (2012) A JAK-in-the-cell cycle. Blood 119:1096–1097
Wang Y et al (2009) Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 114:5024–5033
Sansone P, Bromberg J (2012) Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 30:1005–1014
Darnell JE (2005) Validating Stat3 in cancer therapy. Nat Med 11:595–596
Levy DE, Inghirami G (2006) STAT3: a multifaceted oncogene. Proc Natl Acad Sci USA 103:10151–10152
Lee KK, Workman JL (2007) Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol 8:284–295
Yang XJ, Seto E (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9:206–218
Woan KV, Sahakian E, Sotomayor EM, Seto E, Villagra A (2012) Modulation of antigen-presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol Cell Biol 90:55–65
Villagra A, Sotomayor EM, Seto E (2010) Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene 29:157–173
Marks PA (2010) The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs 19:1049–1066
Philippe B (2010) Inside HDAC with HDAC inhibitors. Eur J Med Chem 45:2095–2116
Regis G, Pensa S, Boselli D, Novelli F, Poli V (2008) Ups and downs: the STAT1: STAT3 seesaw of Interferon and gp130 receptor signalling. Semin Cell Dev Biol 19:351–359
Caretto D, Katzman SD, Villarino AV, Gallo E, Abbas AK (2010) Cutting edge: the Th1 response inhibits the generation of peripheral regulatory T cells. J Immunol 184:30–34
Stephanou A, Brar BK, Knight RA, Latchman DS (2000) Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x promoters. Cell Death Differ 7:329–330
Agrawal S, Agarwal ML, Chatterjee-Kishore M, Stark GR, Chisolm GM (2002) Stat1-dependent, p53-independent expression of p21(waf1) modulates oxysterol-induced apoptosis. Mol Cell Biol 22:1981–1992
Ohmori Y, Hamilton TA (2001) Requirement for STAT1 in LPS-induced gene expression in macrophages. J Leukoc Biol 69:598–604
Kim HS, Lee M-S (2007) STAT1 as a key modulator of cell death. Cell Signal 19:454–465
Wang Y, Wu TR, Cai S, Welte T, Chin YE (2000) Stat1 as a component of tumor necrosis factor alpha receptor 1-TRADD signaling complex to inhibit NF-kappaB activation. Mol Cell Biol 20:4505–4512
Townsend PA et al (2004) STAT-1 interacts with p53 to enhance DNA damage-induced apoptosis. J Biol Chem 279:5811–5820
Calò V et al (2003) STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol 197:157–168
O’Shea JJ, Gadina M, Schreiber RD (2002) Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell 109(Suppl):S121–S131
Imada K, Leonard WJ (2000) The Jak-STAT pathway. Mol Immunol 37:1–11
Ouchi T, Lee SW, Ouchi M, Aaronson SA, Horvath CM (2000) Collaboration of signal transducer and activator of transcription 1 (STAT1) and BRCA1 in differential regulation of IFN-gamma target genes. Proc Natl Acad Sci USA 97:5208–5213
Zhang JJ et al (1996) Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc Natl Acad Sci USA 93:15092–15096
Zhang JJ et al (1998) Ser727-dependent recruitment of MCM5 by Stat1alpha in IFN-gamma-induced transcriptional activation. EMBO J 17:6963–6971
KrÃmer OH et al (2009) A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev 23:223–235
Kramer OH et al (2006) Acetylation of Stat1 modulates NF-kappaB activity. Genes Dev 20:473–485
Krämer OH, Heinzel T (2010) Phosphorylation–acetylation switch in the regulation of STAT1 signaling. Mol Cell Endocrinol 315:40–48
Wieczorek M, Ginter T, Brand P, Heinzel T, Kramer OH (2012) Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev 23(6):293–305
Ginter T et al (2012) Histone deacetylase inhibitors block IFNgamma-induced STAT1 phosphorylation. Cell Signal 24:1453–1460
Klampfer L, Huang J, Swaby L-A, Augenlicht L (2004) Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem 279:30358–30368
Stronach EA et al (2011) HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res 71:4412–4422
Paul AM (2010) Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim Biophys Acta (BBA) Gene Regu Mech 1799:717–725
Tomasi TB, Magner WJ, Khan AN (2006) Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother CII 55:1159–1184
Klampfer L, Huang J, Shirasawa S, Sasazuki T, Augenlicht L (2007) Histone deacetylase inhibitors induce cell death selectively in cells that harbor activated kRasV12: the role of signal transducers and activators of transcription 1 and p21. Cancer Res 67:8477–8485
Frank DA (2007) STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett 251:199–210
Johnston PA, Grandis JR (2011) STAT3 signaling: anticancer strategies and challenges. Mol Interv 11:18–26
Sakaguchi M, Oka M, Iwasaki T, Fukami Y, Nishigori C (2012) Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma Cells. J Invest Dermatol 132:1877–1885
Yu CL et al (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269:81–83
Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE Jr (1998) Stat3 activation is required for cellular transformation by v-src. Mol Cell Biol 18:2553–2558
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488
Garcia R et al (1997) Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ 8:1267–1276
Catlett-Falcone R et al (1999) Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10:105–115
Schuringa J-J, Wierenga ATJ, Kruijer W, Vellenga E (2000) Constitutive Stat3, Tyr705, and Ser727 phosphorylation in acute myeloid leukemia cells caused by the autocrine secretion of interleukin-6. Blood 95:3765–3770
Gao SP et al (2007) Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest 117:3846–3856
Messina JL et al (2008) Activated stat-3 in melanoma. Cancer Control 15:196–201
Kortylewski M, Jove R, Yu H (2005) Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev 24:315–327
Gritsko T et al (2006) Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res 12:11–19
Yuan Z-L, Guan Y-J, Chatterjee D, Chin YE (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307:269–273
Wang R, Cherukuri P, Luo J (2005) Activation of stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem 280: 11528–11534
Togi S et al (2009) HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem Biophys Res Commun 379:616–620
Shen L et al (2012) Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One 7:e30815
Zhang Q et al (2005) STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA 102:6948–6953
Lee H et al (2012) Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc Natl Acad Sci USA 109:7765–7769
Nie Y et al (2009) STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol 11:492–500
Sestito R et al (2011) STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J 25:916–927
Boucheron C et al (1998) A single amino acid in the DNA binding regions of STAT5A and STAT5B confers distinct DNA binding specificities. J Biol Chem 273:33936–33941
Ihle JN (2001) The Stat family in cytokine signaling. Curr Opin Cell Biol 13:211–217
Takeda K, Akira S (2000) STAT family of transcription factors in cytokine-mediated biological responses. Cytokine Growth Factor Rev 11:199–207
Dawson, Mark A et al (2012) Three distinct patterns of histone H3Y41 phosphorylation mark active genes. Cell Reports 2:470–477
Matsumura I et al (1999) Transcriptional regulation of the cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J 18:1367–1377
Bowman T et al (2001) Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci USA 98:7319–7324
Buettner R, Mora LB, Jove R (2002) Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res 8:945–954
Walker SR et al (2009) Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol Cancer Res 7:966–976
Li H et al (2005) Activation of signal transducer and activator of transcription-5 in prostate cancer predicts early recurrence. Clin Cancer Res 11:5863–5868
Leong PL et al (2002) Differential function of STAT5 isoforms in head and neck cancer growth control. Oncogene 21:2846–2853
Lin JX, Leonard WJ (2000) The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 19:2566–2576
Danial NN, Rothman P (2000) JAK-STAT signaling activated by Abl oncogenes. Oncogene 19:2523–2531
Ma L et al (2010) Acetylation modulates prolactin receptor dimerization. Proc Natl Acad Sci USA 107:19314–19319
Inks ES, Josey BJ, Jesinkey SR, Chou CJ (2012) A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol 7:331–339
Xu M, Nie L, Kim SH, Sun XH (2003) STAT5-induced Id-1 transcription involves recruitment of HDAC1 and deacetylation of C/EBPbeta. EMBO J 22:893–904
Tang X et al (2007) Acetylation-dependent signal transduction for type I interferon receptor. Cell 131:93–105
Horvath CM, Stark GR, Kerr IM, Darnell JE Jr (1996) Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex. Mol Cell Biol 16:6957–6964
Li X, Leung S, Burns C, Stark GR (1998) Cooperative binding of Stat1-2 heterodimers and ISGF3 to tandem DNA elements. Biochimie 80:703–710
Darnell J, Kerr IM, Stark GR (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421
Sykes SM et al (2006) Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 24:841–851
Sakamoto S, Potla R, Larner AC (2004) Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes. J Biol Chem 279:40362–40367
Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A (2006) Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev 17:173–188
Yu CR et al (2004) Cell proliferation and STAT6 pathways are negatively regulated in T cells by STAT1 and suppressors of cytokine signaling. J Immunol 173:737–746
Gooch JL, Christy B, Yee D (2002) STAT6 mediates interleukin-4 growth inhibition in human breast cancer cells. Neoplasia 4:324–331
Shankaranarayanan P, Chaitidis P, Kühn H, Nigam S (2001) Acetylation by histone acetyltransferase CREB-binding protein/p300 of STAT6 is required for transcriptional activation of the 15-lipoxygenase-1 gene. J Biol Chem 276:42753–42760
Yamamoto K et al (1994) Stat4, a novel gamma interferon activation site-binding protein expressed in early myeloid differentiation. Mol Cell Biol 14:4342–4349
Hemmi H et al (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745
Kaplan MH, Sun YL, Hoey T, Grusby MJ (1996) Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382:174–177
Brierley MM, Fish EN (2002) Review: IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J Interferon Cytokine Res 22:835–845
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75:163–189
Jaruga B, Hong F, Kim W-H, Gao B (2004) IFN-γ/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am J Physiol Gastrointest Liver Physiol 287:G1044–G1052
Dustin ML, Singer KH, Tuck DT, Springer TA (1988) Adhesion of T lymphoblasts to epidermal keratinocytes is regulated by interferon gamma and is mediated by intercellular adhesion molecule 1 (ICAM-1). J Exp Med 167:1323–1340
Strieter RM, Kunkel SL, Arenberg DA, Burdick MD, Polverini PJ (1995) Interferon gamma-inducible protein 10 (IP-10), a member of the C-X-C chemokine family, is an inhibitor of angiogenesis. Biochem Biophys Res Commun 210:51–57
Yoshimura A (2006) Signal transduction of inflammatory cytokines and tumor development. Cancer Sci 97:439–447
Hutchins AP, Poulain S, Miranda-Saavedra D (2012) Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood 119:e110–e119
Vallania F et al (2009) Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc Natl Acad Sci USA 106:5117–5122
Ito S et al (1999) Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood 93:1456–1463
Aoki Y, Feldman GM, Tosato G (2003) Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 101:1535–1542
Epling-Burnette PK et al (2001) Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest 107:351–362
Sinibaldi D et al (2000) Induction of p21WAF1/CIP1 and cyclin D1 expression by the Src oncoprotein in mouse fibroblasts: role of activated STAT3 signaling. Oncogene 19: 5419–5427
Niu G et al (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000–2008
Hung W, Elliott B (2001) Co-operative effect of c-Src tyrosine kinase and Stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J Biol Chem 276:12395–12403
Xie TX et al (2006) Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res 66:3188–3196
Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H (2012) SOCS, inflammation, and autoimmunity. Front Immunol 3:20
Cooney RN (2002) Suppressors of cytokine signaling (SOCS): inhibitors of the JAK/STAT pathway. Shock 17:83–90
Wormald S, Hilton DJ (2004) Inhibitors of cytokine signal transduction. J Biol Chem 279:821–824
Xiong H et al (2012) Trichostatin A, a histone deacetylase inhibitor, suppresses JAK2/STAT3 signaling via inducing the promoter-associated histone acetylation of SOCS1 and SOCS3 in human colorectal cancer cells. Mol Carcinog 51:174–184
Brown R, Imran SA, Ur E, Wilkinson M (2008) Valproic acid and CEBPalpha-mediated regulation of adipokine gene expression in hypothalamic neurons and 3T3-L1 adipocytes. Neuroendocrinology 88:25–34
Hirose N, Maekawa T, Shinagawa T, Ishii S (2009) ATF-2 regulates lipopolysaccharide-induced transcription in macrophage cells. Biochem Biophys Res Commun 385:72–77
You M, Yu D-H, Feng G-S (1999) Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol Cell Biol 19:2416–2424
Shuai K (2006) Regulation of cytokine signaling pathways by PIAS proteins. Cell Res 16:196–202
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334(Pt 2):297–314
Neel BG, Gu H, Pao L (2003) The ’Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28:284–293
Wang H et al (2011) Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol 186:11
Leoni F et al (2002) The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci USA 99:2995–3000
Reddy P et al (2008) Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest 118:2562–2573
Sun Y et al (2009) Cutting edge: negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J Immunol 182:5899–5903
Glauben R, Siegmund B (2011) Inhibition of histone deacetylases in inflammatory bowel diseases. Mol Med 17:426–433
Han SB, Lee JK (2009) Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch Pharm Res 32:613–624
Bode KA et al (2007) Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 122:596–606
Wu C, Li A, Leng Y, Li Y, Kang J (2012) Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol 31:592–599
Jung ID et al (2009) Apicidin, the histone deacetylase inhibitor, suppresses Th1 polarization of murine bone marrow-derived dendritic cells. Int J Immunopathol Pharmacol 22:501–515
Wang B, Morinobu A, Horiuchi M, Liu J, Kumagai S (2008) Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cell Immunol 253:54–58
Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS (2003) Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest 111:539–552
Bosisio D et al (2008) Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J Leukoc Biol 84:1540–1548
Salvi V et al (2010) Trichostatin A blocks type I interferon production by activated plasmacytoid dendritic cells. Immunobiology 215:756–761
Villagra A et al (2009) The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol 10:92–100
Wang H et al (2011) Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol 186:3986–3996
Kobayashi T et al (2012) IL-10 regulates Il12b expression via histone deacetylation: implications for intestinal macrophage homeostasis. J Immunol 189:1792–1799
Frikeche J et al (2012) Impact of valproic acid on dendritic cells function. Immunobiology 217:704–710
Zhou X, Hua X, Ding X, Bian Y, Wang X (2011) Trichostatin differentially regulates Th1 and Th2 responses and alleviates rheumatoid arthritis in mice. J Clin Immunol 31:395–405
Koenen HJ SR, Vink PM, van Rijssen E, Boots AM, Joosten I (2008) Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 112:2340–2352
Han S et al (2007) HDAC inhibitors TSA and sodium butyrate enhanced the human IL-5 expression by altering histone acetylation status at its promoter region. Immunol Lett 108:143–150
Su RC, Becker AB, Kozyrskyj AL, Hayglass KT (2008) Epigenetic regulation of established human type 1 versus type 2 cytokine responses. J Allergy Clin Immunol 121:57–63 e53
Edens RE, Dagtas S, Gilbert KM (2006) Histone deacetylase inhibitors induce antigen specific anergy in lymphocytes: a comparative study. Int Immunopharmacol 6:1673–1681
Rossi LE et al (2012) Histone deacetylase inhibitors impair NK cell viability and effector functions through inhibition of activation and receptor expression. J Leukoc Biol 91:321–331
Chang S, Collins PL, Aune TM (2008) T-bet dependent removal of Sin3A-histone deacetylase complexes at the Ifng locus drives Th1 differentiation. J Immunol 181:8372–8381
Lee CG et al (2012) Interaction of Ets-1 with HDAC1 represses IL-10 expression in Th1 cells. J Immunol 188:2244–2253
Gillespie J et al (2012) Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum 64:418–422
Choo QY, Ho PC, Tanaka Y, Lin HS (2010) Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (Oxford) 49:1447–1460
Segain JP, Raingeard de la Blétière D, Bourreille A, Leray V, Gervois N, Rosales C, Ferrier L, Bonnet C, Blottière HM, Galmiche JP (2000) Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn’s disease. Gut 47:397–403
Lewis EC et al (2011) The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol Med 17:369–377
Mishra N, Brown DR, Olorenshaw IM, Kammer GM (2001) Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-gamma gene and protein expression in lupus T cells. Proc Natl Acad Sci USA 98:2628–2633
Nusinzon I, Horvath CM (2006) Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol 26:3106–3113
Song W et al (2011) HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia 25:161–168
Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS (2003) Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest 111:539–552
Lu J et al (2005) Interleukin-12 p40 promoter activity is regulated by the reversible acetylation mediated by HDAC1 and p300. Cytokine 31:46–51
Halili MA et al (2010) Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol 87:1103–1114
Gillespie J et al (2012) Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum 64:418–422
Choo QY, Ho PC, Tanaka Y, Lin HS (2010) Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (Oxford) 49:1447–1460
Chang S, Collins PL, Aune TM (2008) T-bet dependent removal of Sin3a-histone deacetylase complexes at the ifng locus drives Th1 differentiation. J Immunol 181:8372–8381
Nusinzon I, Horvath CM (2006) Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation. Mol Cell Biol 26:3106–3113
Kim YH, Lee JK (2012) Suppression of T cell functions by hydroxamic acid-based histone deacetylase inhibitors. Arch Pharm Res 35:929–936
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Woods, D.M., Lienlaf-Moreno, M., Sotomayor, E., Seto, E., Villagra, A. (2014). Regulation of the Jak/STATs Pathways by Histone Deacetylases. In: Kumar, R. (eds) Nuclear Signaling Pathways and Targeting Transcription in Cancer. Cancer Drug Discovery and Development. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8039-6_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8039-6_7
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8038-9
Online ISBN: 978-1-4614-8039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)