
Abstract
RUNX2, a member of the Runt family of transcription factors, plays important roles in embryonic development to promote osteogenesis and angiogenesis. RUNX2 has been implicated in the promotion of disease, including cleidocranial dysplasia, in cancer progression, and in metastasis of breast and prostate tumors. Its aberrant expression in disease states may be the result of several mechanisms such as haploinsufficiency, mutation, or amplification. In osteogenesis and cancer progression, interactions with core-binding factor-β (Cbf-β) and other cofactors are responsible for the regulation of target gene expression including, but not limited to, VEGF, osteopontin, osteocalcin, MMPs, and BMPs. RUNX2 transcriptional function within cells is regulated by signal transduction events leading to activation of ERK, Smads, cdks, and Akt, which result in phosphorylation, DNA binding, and transcriptional activation or repression of target genes. Constitutive activation of signaling pathways in tumor cells results in aberrant expression and activation of RUNX2. Specific RUNX2 targeting agents, therefore, may bypass the effects of redundant signal transduction pathways within cancer cells and be an effective therapeutic strategy for treatment of RUNX2-positive cancer patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The RUNX genes are a family of transcription factors originally identified in Drosophila [1, 2]. There are three mammalian RUNX genes encoding the proteins RUNX1, RUNX2, and RUNX3. Historically, the literature contains many different nomenclatures for the mammalian RUNX family proteins. RUNX2(Runt related transcription factor 2) is also AML-3 (acute myeloid leukemia-3), PEBP2α (polyoma enhancer binding protein 2α), and CBFα (core binding factor α) [2–5]. The RUNX genes control many normal cellular processes including hematopoiesis (RUNX1), osteogenesis (RUNX2), and epithelial and neuronal development (RUNX3) [1]. RUNX genes encode several evolutionarily conserved proteins. There are four RUNX genes in zebra fish, four in drosophila, one in sea urchins, and one in C. elegans to name a few [2]. High conservation of proteins through evolutionary history generally suggests an extremely essential biological function.
The RUNX proteins are members of a heterodimeric complex composed of an α and β subunit. The runt domain genes contain the DNA binding α subunit [1]. The β subunit, consisting of core-binding factor-β (Cbf-β), binds to the runt domain within RUNX proteins (Fig. 3.1) to help stabilize RUNX-DNA interactions [1]. In addition, Cbf-β protects RUNX proteins from phosphorylation and degradation via the proteasome [1]. The RUNX proteins share many common protein domains (Fig. 3.1). The runt domain, which is responsible for binding DNA, is located in the N-terminus of the protein and is composed of 128 amino acids [3]. The runt domain is the most highly conserved domain among members of the RUNX family including the Drosophila ortholog runt. Located C-terminal to the runt domain is the nuclear matrix targeting signal (NMTS) which is responsible for sub nuclear localization of RUNX proteins [1, 6]. This domain is comprised of 38 amino acids [5] folding into a loop-turn-loop tertiary structure [6]. Mutations within this domain reveal the essential nature of its function: without a functional NMTS, RUNX proteins cannot be transactivated or localized in foci within the nucleus [5]. Mutations within the second loop of the NMTS have been shown to inhibit RUNX2 interaction with the nuclear matrix [6] leading to compromised gene regulation. The nuclear localization signal/sequence (NLS) is comprised of 9 amino acids [5] and maintains RUNX localization to the nucleus. Furthermore, the C-terminus of RUNX proteins contains binding sites for corepressors and coactivators [1], which modulate RUNX activity. Depending on the cell stimulus or the cell type, different modulators of transcription are able to bind the RUNX proteins and either enhance or repress transcription.

Mammalian RUNX isoforms protein structure. The three mammalian RUNX proteins share domains with drosophila runt. N terminal (purple) are the P1 and P2 promoters. The Runt domain (red) is the DNA binding and Cbf-β interaction domain. C-terminal of the Runt domain (orange) is the nuclear localization sequence (NLS) controlling nuclear translocation of RUNX proteins. C-terminus (light blue) are a variety of sequences mediating co-factor binding to either activate or repress RUNX transcription of target genes. The nuclear matrix target sequence (NMTS; yellow) controls sub-nuclear localization of RUNX proteins
All of the RUNX proteins have been implicated in disease (see Fig. 3.2 for diseases in which RUNX2 has been shown to play a role). Their normal functions are altered via mutations, epigenetic silencing, chromosomal translocation, cellular mislocalization, or by gene amplification. There is evidence that the RUNX proteins function as both tumor suppressors and as oncogenes depending on the disease context.

RUNX2 in development and disease. Normal functions of RUNX2 (yellow) and diseases in which RUNX2 has been shown to play a role (blue)
Although RUNX1 is not the largest protein isoform in mammals, it does encompass the largest genomic coding and regulatory region of 260 kb of DNA, comprised of 11 exons [4] encoding for 453 amino acids [1]. Within cells, RUNX1 functions to maintain normal hematopoiesis [1]. It is the target of numerous mutations and chromosomal translocations in hematological malignancies such as leukemia. In most reported cases of leukemia (specifically acute myeloid leukemia) where RUNX1 translocations are discovered, there appears to be a dominant negative function resulting from the new RUNX1-fusion protein. RUNX1 translocations and mutations are seen in acute myeloid leukemia (AML), blast crisis of chronic myeloid leukemia (CML), and acute lymphoblastic leukemia (ALL) [4]. There have been a few reports of gain of function mutations in RUNX1 as a result of an extra copy of the RUNX1 gene. This has been reported in Down Syndrome-related acute megakaryoblastic leukemia [4]. Knockout mice display a wide range of defects including, but not limited to, megakaryocyte defects, T-cell defects, myeloproliferative diseases, as well as T-cell lymphomas [2]. New studies have suggested a role for RUNX1 in endochondral ossification to mediate fracture healing in bone [7].
The smallest of the three mammalian RUNX proteins, RUNX3, is also thought to be the most primitive in evolutionary history, spanning 67 kb of DNA and composed of six exons [4] translating to 415 amino acids [1]. Expressed ubiquitously throughout the body, it can be found within the epithelia, mesenchyma, blood cells, dorsal root ganglion neurons, and predominantly in the gut epithelia [4]. RUNX3 has been shown to be essential for proper gut epithelial and neuronal development [8]. RUNX3 is essential for proprioceptive neuron axon path finding in the spinal cord [2] and there have also been reports of RUNX3 regulating CD4 silencing in T-cells [2]. Within the gut epithelia, research demonstrates that inactivation of RUNX3 leads to hyperplasia with a loss of response to transforming growth factor-β (TGF-β) inhibition [4] resulting in gastric cancers [8]. Inactivation has been described to occur through mutation, epigenetic silencing, hemizygous deletion, or cytoplasmic mislocalization.
Human RUNX2, located on the short arm of chromosome 6 at position 21 (6p21) [3], is the largest family member containing 513 amino acids [1] and has unique domains not present in the other mammalian isoforms (RUNX1 and RUNX3); one in the N-terminus and one in the C-terminus (see Fig. 3.1). RUNX2 is expressed early in embryonic development in mesenchymal stem cells [5]. During mouse embryogenesis, RUNX2 mRNA has been detected as early as E11.5 in the limb buds and the condensation of the humerus [5]. There is very weak expression observed as early as E9.5 in the notochord [5]. RUNX2 is the master regulator of osteoblast differentiation and chondrocyte maturation in a process called osteogenesis [1, 5]. RUNX2 controls the commitment of mesenchymal stem cells to the osteoblast lineage and has been shown to be abnormally expressed in adult tissues, leading to disease. Haploinsufficiency of RUNX2 promotes cleidocranial dysplasia (CCD) [9, 10]. In addition, there have been a few reports of RUNX2 mutations occurring within the runt domain which also result in CCD [5]. The oncogenic potential of RUNX2 was first identified from its ability to synergize with c-myc in T-cell lymphoma development [8, 127]. Aberrantly expressed RUNX2, normally at non-detectable to low levels in epithelial tissue, is thought to promote bone metastasis through activation of genes in malignancies such as breast and prostate cancer [8]. These target genes include, but are not limited to, vascular endothelial growth factor (VEGF), osteopontin (OPN), osteocalcin (OC), and matrix metalloproteinases (MMP’s) [8]. The rest of this review will focus on RUNX2.
2 RUNX2: A Master Transcription Factor
2.1 Function in Osteogenesis and Angiogenesis
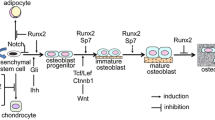
Osteogenesis consists of intramembranous ossification (bone) and endochondral ossification (cartilage) [5]. Bone homeostasis is an important process that requires a balance between bone formation (osteoblasts) and resorption (osteoclasts). Osteoblasts are responsible for laying down new bone matrix in addition to the mineralization of the new bone matrix [11]. Osteoblasts also stimulate the differentiation of osteoclasts while osteoclasts produce factors which digest the mineralized bone matrix [11]. RUNX2 is the master regulator of osteoblast differentiation and osteogenesis [132]. RUNX2 expression is controlled by two promoters: P1 and P2 early in osteoblast differentiation [12]. As differentiation progresses RUNX2 protein levels do not increase, but rather, the transcriptional activity level increases [11]. Experiments in knockout mice show the essential role of RUNX2 in bone formation: knockout mice die soon after birth because of asphyxiation as a consequence of a lack of skeletal formation [5, 11]. In addition, analyses reveal that these mice lack mature osteoblasts thereby inhibiting the formation of any bone matrix or osteoclast differentiation.
Since osteoblasts lay down bone matrix to form mineralized bone and osteoclasts break down the matrix to resorb bone; RUNX2 indirectly controls osteoclast differentiation. Receptor activator of nuclear factor kappa-b ligand (RANKL) promotes osteoclast maturation and is also a RUNX2 target gene [5]. Cells lacking RUNX2 express less RANKL and, therefore, there is less osteoclast maturation [5] and less bone resorption. Research has further shown that while endochondral ossification is delayed in RUNX2 knockout mice, it does eventually occur [5, 130, 134]. Therefore, there is redundancy and other factors are able to compensate for the lack of RUNX2 during endochondral ossification.
The RUNX2 transcription factor is also a regulator of angiogenesis in bone development [13, 14], is expressed in vascularizing adult tissues [15], and promotes tumor metastasis [16, 17]. It interacts with its heterodimeric partner, Cbf-β, and with hypoxia-inducible factor 1-α (Hif1α) to activate the major angiogenic factor, VEGF [18]. RUNX2 is a transcriptional activator of specific target genes that promote angiogenesis, such as MMPs [19]. Conversely, it represses the cell cycle inhibitor p21Cip1 and increases endothelial cell (EC) or cancer cell proliferation [20, 21]. Our laboratory has found that glucose metabolism, autocrine IGF-1 signaling, and phosphorylation by cyclin-dependent kinases, regulate RUNX2 DNA-binding activity, angiogenic target genes, EC proliferation, tube formation, and wound healing [20, 22–25, 138]. However, exposure of EC to hyperglycemia (HG) activated the aldose reductase (AR) polyol pathway, which increased oxidative stress and inhibited RUNX2 DNA binding [22].
2.2 RUNX2 in Disease
Although RUNX2 regulates osteogenesis and angiogenesis [1, 12], aberrant expression of RUNX2 can lead to disease (Fig. 3.2). Cleidocranial dysplasia (CCD) is a disease in which there are abnormalities in bone and dental development [5, 9, 26, 27]. Haploinsufficiency [1, 9] of RUNX2 has been shown to be a leading cause of CCD. In addition, there are mutations in RUNX2 that lead to CCD [26]. A heterozygous single-base deletion resulting in a premature stop codon in the runt domain produces a truncated form of RUNX2 [26] that is unable to bind DNA and control transcription of essential target genes. This mutation was not found in normal individuals or non-CCD subjects.
RUNX2 has been proposed as a biomarker in numerous cancers [1–4, 16, 17, 28, 29, 36–38, 41, 43, 45–49, 53, 54, 61, 94, 127–129, 131, 135] to evaluate the promotion of cancer cell metastasis to the bone. Sase et al. showed that RUNX2 expression was significantly associated with human colon carcinoma progression [28, 29]. In colorectal cancer RUNX2 is not only amplified, but the RUNX2 gene also contains genetic variations termed single nucleotide polymorphisms or SNPs. In cases of colorectal carcinoma, Slattery et al. found a total of 19 SNPs in RUNX2 [29].
One tumor in which both mRNA and protein levels of RUNX2 have been shown to be elevated is osteosarcoma [24, 30–35]. Osteosarcoma is a very aggressive pediatric cancer of the bone with a highly heterogeneous phenotype [32, 33, 35]. In osteosarcoma and chondrosarcomas, RUNX2 expression was found to positively correlate with bone morphogenetic protein-2 (BMP-2) mRNA levels [27]. Cell cycle deregulation of RUNX2 led to osteosarcoma pathogenesis [24]. High levels of RUNX2 in osteosarcoma resulted in high rates of metastasis and a poor survival rate, supporting the notion of RUNX2 as a good prognostic marker [35]. RUNX2 was also the only upregulated marker in osteosarcoma that exhibited a positive correlation with chemotherapeutic resistance [33].
RUNX2 expression correlates with unfavorable prognoses in prostate cancer [36] and has been found to be upregulated in both breast and prostate cancers [8, 16, 25, 37–46, 128, 129]. In breast and prostate cancer cell lines RUNX2 was shown to enhance cell motility [41]. Unfortunately, many of the RUNX2 functions are cell type specific making it difficult to discern a universal function in disease progression. In MDA-MB-231 breast cancer cells, knockdown of RUNX2 had no effect on cell growth and proliferation whereas in MCF7 breast cancer cells RUNX2 enhanced cell proliferation upon growth factor deprivation [41]. In MCF-10A cells RUNX2 disrupts normal mammary acini formation in suspension culture [47]. Using electron microscopy, an absence of lumen formation was noted, possibly due to an increase in cell proliferation, decreased apoptosis, and a loss of basement membrane formation which is dependent on RUNX2 expression [47]. These phenotypes can be reversed in MDA-MB-231 cells using siRNA to deplete RUNX2 [47]. The consensus of data supports an oncogenic function for RUNX2 in breast cancer. However, there is a report suggesting RUNX2 functions as a tumor suppressor in breast cancer [48]. There have been a few reports implicating RUNX2 in promoting the formation of hematological malignancies [131] such as myeloid leukemia [49]. For example, RUNX2 cooperates with the fusion protein, Cbf-β-SMMHC, to promote leukemia development [49] and haploinsufficiency of RUNX2 delays the onset of acute myeloid leukemia [49].
3 Transcriptional Regulation: Target Genes and Cofactors
3.1 RUNX2 Target Genes
As a DNA-binding factor RUNX2 controls the transcription (through activation and repression) of numerous genes important in normal tissue homeostasis (Table 3.1). Many of these genes are abnormally activated in disease states, including cancers, and enable cancer cells to survive and metastasize to distant niches.
In osteoblasts, RUNX2 activates expression of the essential osteogenesis signaling factor: bone morphogenetic protein-2 (BMP-2) [50]. BMP-2 is a member of the transforming growth factor-β (TGF-β) superfamily of signaling molecules. It binds to TGF-β receptors, leading to activation of intracellular signaling via Smad-dependent and/or Smad-independent pathways ultimately resulting in further RUNX2 activation and promotion of a feed-forward loop. In MC3T3-E1 cells, BMP-2 was shown to enhance RUNX2 association with the promoter of Atf6 [51]. Atf6 is another transcription factor which is known to mediate osteoblast differentiation in a RUNX2 dependent manner [51]. Specifically, BMP-2-stimulated RUNX2 activation of Atf6 enables the bone extracellular matrix, osteocalcin, gene to be expressed. A dominant negative construct of Atf6 was shown to inhibit RUNX2 activation of the osteocalcin promoter [51]. However, by restoring wild type Atf6, osteoblasts were able to differentiate and the expression of osteocalcin was restored.
During cancer progression, cancer cells express many bone specific proteins that mediate metastasis to the bone. Some of these proteins are regulated by RUNX2 transcriptional activity and mediate migration/motility and adhesion. Bone sialoprotein (BSP) and osteopontin (OPN) are two factors which mediate breast cancer metastasis to the bone and are activated by RUNX2 in breast cancer cells [45]. Using siRNA technology, Reufsteck et al. were able to demonstrate that targeting BSP and OPN drastically inhibits migration of MDA-MB-231 breast cancer cells when injected into athymic nude mice [45]. In prostate cancer, RUNX2 has been shown to upregulate genes not only associated with increased migration but genes associated with angiogenesis, epithelial-mesenchymal-transition (EMT), membrane trafficking/secretion, and osteolysis [36]. RUNX2 upregulates secretion factors including PTHrP, IL8, CSF2, and SDF-1 (See Table 3.1 for a description of protein function) [37, 38]. RUNX2 also upregulates MMP9, MMP13, VEGF, osteopontin, CST7, Sox9, SNAI2, Smad3, SDC2, Twinfilin, and SH3PXD2A [37, 38]. RUNX2 promoter occupancy in osteosarcoma was examined to determine potential RUNX2 target genes enabling progression of osteosarcoma. In SAOS-2 osteosarcoma cells, knockdown of RUNX2 resulted in an inhibition of motility [34]. RUNX2 upregulated matrix metalloproteinases during cancer progression to promote cell migration from primary tumor sites. MMP13 and MMP9 are two common MMP’s upregulated by RUNX2 to mediate cancer cell invasion and metastasis [1, 52–55].
In addition to epithelial cells, RUNX2 has been shown to modulate gene expression in vascular cells. In human aortic smooth muscle cells and C3H10T1/2 cells RUNX2 inhibited the expression of connective tissue growth factor (CTGF) [56]. This is important for endothelial cells since CTGF has been shown to be a contributing factor to the development of atherosclerosis. Therefore, RUNX2 may protect the vasculature from development of atherosclerosis. Knockdown of RUNX2 enhanced CTGF expression [56] in a TGF-β dependent manner. Further analysis of the CTGF promoter revealed Smad binding elements that were able to interact with RUNX2/Smad3 heterodimers upon stimulation of the TGF-β receptors to inhibit transcription [56].
3.2 RUNX2 Cofactors
Regulation of gene expression requires a transcriptional complex composed of RNA polymerase, transcription factors, and corepressors or coactivators. RUNX2 C-terminal domain contains many binding sites for both corepressors and coactivators (Table 3.2).
One group of proteins that function as strong corepressors of RUNX2 are histone deacetylases (HDACs). In osseous cells, HDAC1 has been shown to interact with RUNX2 to inhibit ribosomal RNA (rRNA) gene expression [57] thus inhibiting cellular proliferation and protein synthesis. Knockout of HDAC1 was shown to alleviate the RUNX2-mediated repression of rRNA expression resulting in an increase in cell proliferation and overall protein synthesis [57]. Transducin Like Enhancer-1 (TLE-1), functions to also promote RUNX2 inhibition of rRNA gene synthesis during mitosis [58]. In C3H10T1/2 cells HDAC1 bound to RUNX2 inhibited expression of osteopontin, thus inhibiting both proliferation and differentiation of the osteoblast cell [59]. HDAC7 was also shown to be a potent inhibitor of RUNX2 transcriptional activity [60] and HDAC5 was shown to repress RUNX2 expression [5].
In prostate cancer the forkhead box O (FoxO1) protein was found to be a corepressor of RUNX2 [61]. Inhibition of RUNX2 by upregulation of PTEN or FoxO1 protein inhibited prostate cancer cell migration and invasion. In prostate cancer specimens, immunohistochemistry revealed an inverse relationship between RUNX2 and FoxO1 nuclear localization [61]. PTEN inactivating mutations are often seen in prostate cancer [61] and therefore would potentiate RUNX2 activity to promote prostate cancer cell migration and invasion.
While some cofactors function as coactivators and corepressors a few are also able to function as both in a gene-dependent manner. One group used a doxycycline-regulated RUNX2 expression system in C4-2B prostate cancer cells to show that G9a (histone methyltransferase) is able to function as a corepressor for RUNX2 target genes MMP9, CSF2, SDF1, and CST7 [44]. However, G9a functions as a coactivator for RUNX2 transcription of MMP13 and PIP in the C4-2B prostate cancer cell line [44].
4 Transcriptional Regulation: Activation and Repression
4.1 Regulation of RUNX2 Activity
Several intracellular signaling pathways have been shown to modulate RUNX2 activity (Fig. 3.3). When RUNX2 becomes activated it is able to bind DNA and either promote or inhibit the transcription of its target genes (Refer to Table 3.1). Mitogen-activated protein kinase (MAPK) and TGF-β intracellular signaling are both essential to RUNX2 activation to promote osteoblast differentiation or tumor progression. TGF-β/BMP signaling is a highly important signaling axis used by numerous cells to inhibit cell growth and proliferation under normal conditions. In cancer, tumor cells escape the inhibitory effects of TGF-β resulting in unrestricted proliferation. TGF-β/BMP intracellular signaling can occur via canonical Smad-dependent or non-canonical Smad-independent pathways. Upon stimulation, TGF-β receptors initiate intracellular signaling events leading to Smad activation (canonical TGF-β pathway), nuclear translocation, and Smad interaction with RUNX2 [6, 56, 62–64]. In osteogenesis, TGF-β/BMP signaling is responsible for activating RUNX2 to promote osteoblast differentiation [62, 65]. Conversely, in PC3 prostate cancer cells TGF-β cooperates with RUNX2 to promote cellular growth [46].

RUNX2 and signal transduction. Many signaling pathways lead to activation or repression of RUNX2 transcriptional activity. These Pathways converge onto the MAPK pathway, the TGF-β/Smad pathway, and the PI3K/Akt pathway
A potent regulator of RUNX2 activity is the extracellular signal-regulated kinase, ERK [66, 67, 138, 139]. ERK is a classical MAPK activated upstream in response to numerous extracellular factors including growth factors, cytokines, and G-protein coupled receptor ligands. MAPK signaling is essential for normal bone development. In many cancers where RUNX2 is aberrantly expressed, MAPK signaling is also altered generally through inactivating or activating mutations to components within the signaling cascade. In preosteoblast MC3T3-E1 cells it was demonstrated that S301 and S319 [66], two ERK phosphorylation sites on RUNX2, are phosphorylated upon nuclear translocation of ERK [67]. MAP3K mixed-lineage kinase 3 (MLK3) has been shown to activate ERK and p38 (another classical MAP kinase). Activation of ERK via MLK3 results in RUNX2 phosphorylation and subsequent osteoblast differentiation [68]. Insulin-like growth factor-1 (IGF-1) binding to IGF-1R (IGF-1 receptor tyrosine kinase) has been shown to be essential for normal skeletal development [69]. IGF-1 regulates the activation of RUNX2 via activation of ERK resulting in phosphorylation of RUNX2 [22, 69, 138]. p38, activated via several extracellular factors, has been implicated in modulating RUNX2 activity levels. p38 activation from TGF-β/BMP signaling (non-canonical TGF-β pathway) has been shown to mediate RUNX2 phosphorylation to promote osteoblast differentiation [62].
Fibroblast growth factor 2 (FGF2) is another extracellular growth factor implicated in promoting osteoblast differentiation through activation of RUNX2 [70–72]. In activating RUNX2, FGF2 functions as a double-edge sword. FGF2 activates protein kinase C (PKC), which in turn increases ERK activity, leading to RUNX2 phosphorylation in MC3T3 osteoblasts [71]. FGF2 also activates ERK through PKC-independent mechanisms (refer to Fig. 3.3). The gap junction protein, connexin-43 (Cx43), enhances this activation via stabilization of FGF2 to FGF2R (refer to Fig. 3.3). An inhibitor of gap junctions, 18β-glycyrrhetinic acid, attenuated the enhancement in RUNX2 transcriptional activity in MC3T3 osteoblasts [71]. In breast cancer MCF7 cells, FGF2 was able to increase BSP expression, which in turn upregulated RUNX2 mRNA [70].
Not all intracellular signaling pathways that lead to RUNX2 phosphorylation result in RUNX2 activation. The c-jun-N-terminal kinase (JNK) is activated by BMP-2 signaling in both C2C12 multipotent cells and MC3T3-E1 preosteoblastic cells [73]. JNK is able to phosphorylate RUNX2 at S104 [73] resulting in inhibition of RUNX2 activity and thus preventing osteoblast differentiation. Inhibition of JNK via a dominant negative JNK1, JNK knockdown, or treatment with a JNK inhibitor counteracted this inhibition to enable osteoblast differentiation.
RUNX2 has also been shown to play a role in endothelial signaling and cell cycle progression in response to physiological levels of glucose [22]. Euglycemic conditions were able to restore RUNX2 DNA binding through autocrine IGF1/IGFR signaling to promote endothelial cell migration, proliferation, and angiogenesis [22] indicating glucose has a function in modulating RUNX2 activity. Hyperglycemic conditions, however, inhibited RUNX2 activity through the aldose reductase polyol pathway [22]. Inhibition of RUNX2 by hyperglycemia inhibited endothelial cell migration, proliferation, and angiogenesis [20, 22]. Treatment with 2-deoxyglucose (inhibitor of glucose metabolism) under euglycemic growth conditions in endothelial cells resulted in a delayed exit from G1/S into G2 phases of the cell cycle with subsequent lower levels of RUNX2 DNA binding activity [20]. Similarly, nutrient and serum deprivation blocked endothelial cell exit from G1 [20]. Using shRNA lentiviral knockdown to reduce RUNX2 levels, exit from G1 and progression through the cell cycle was found to be dependent upon RUNX2 [20].
4.2 Regulation of RUNX2 Expression
RUNX2 expression in osteoblasts promotes differentiation through tight control of RUNX2 activity to maintain the balance of bone formation and remodeling. Similarly, during tumorigenesis many of the negative regulators of RUNX2 are inhibited, thus allowing sustained expression and activity. In general, RUNX2 expression and DNA-binding activity depend on transcriptional mechanisms of activation versus repression and on post-translational protein modifications that include phosphorylation, acetylation, and ubiquitination [74].
4.2.1 Transcriptional Activation and Repression
Osteoblast differentiation requires activation of RUNX2, but what regulates RUNX2 expression in pre-osteoblasts has remained an enigma for quite some time. Tu et al. determined that activation of RUNX2 expression is dependent upon Indian hedgehog (Ihh) signaling [75]. In Ihh null mice even the forced expression of RUNX2 failed to induce osteoblast differentiation [75] confirming that Ihh is not only important for activation of RUNX2 expression but also for RUNX2 activity. In osteoblasts, it was found that BMP-2 increased RUNX2 induction and expression [76]. Shu et al. generated BMP-2 knockout mice and found a dramatic reduction in RUNX2 expression resulting in severe chondrodysplasia [76]. BMP-2 was also able to upregulate PlexinA2 (PlxnA2) in pre-osteoblastic cells [77]. The upregulation of PlxnA2 was associated with increased RUNX2 expression, osteoblast differentiation, and bone mineralization [77]. This upregulation of RUNX2 was thought to be a result of PlxnA2 stabilization of BMP-2 binding to BMP-2 receptors.
TWIST, a transcription factor implicated in epithelial-mesenchymal-transition (EMT) and metastasis in many types of cancers is also an essential transcription factor in development. TWIST was shown to be an inhibitor of RUNX2 expression in bone marrow-derived mesenchymal stem cells [78]. Under low oxygen conditions (hypoxia), Hif1α upregulated expression of TWIST, which directly inhibited RUNX2 expression [78] and osteoblast differentiation. Conversely, studies in PC3 prostate cancer cells showed that TWIST enhances RUNX2 expression levels to promote metastasis to the bone [79]. Glucocorticoid receptor binding to the P2 promoter of RUNX2 was shown to inhibit RUNX2 expression [80]. This resulted in adipocyte differentiation [80] instead of osteoblast differentiation and reveals how signaling events can lead to cell fate determination by regulating RUNX2 expression.
RUNX2 expression is increased in breast tumors where it has been implicated in mediating metastasis to the bone. How RUNX2 expression is enhanced in breast cancer tissue is poorly understood. Using breast cancer cells, one group was able to show that serotonin induced parathyroid hormone related protein (PTHrP), which in turn increased transcription of RUNX2 [40]. Since PTHrP is also a RUNX2 target gene, it fuels a feed forward loop potentiating maximal RUNX2 expression to promote progression of breast cancer to a metastatic stage. In multiple myeloma (MM) it has been shown that Gfi1 is upregulated and represses RUNX2 expression [81]. This inhibition of RUNX2 then results in inhibition of osteoblast differentiation.
4.2.2 Post-translational Regulation
Intracellular signaling is able to modulate RUNX2 post-translational modifications to regulate DNA-binding activity and RUNX2 levels. Using antibodies to detect phosphoserine sites on RUNX2 in endothelial cells, it was shown that RUNX2 is phosphorylated under euglycemic growth conditions by cyclin dependent kinase 4 (cdk4). This phosphorylation was abrogated by mutation of the cdk site S451 [20]. A RUNX2-S451A mutant showed inhibition of DNA binding in endothelial cells as well as a reduction in wound healing activity [20]. Inhibition of cdk4 produced similar results demonstrating that cdk4 can activate RUNX2 through phosphorylation of S451 in response to glucose. In addition to glucose modulation, RUNX2 activity was cell cycle regulated [136, 137]. RUNX2 was associated with DNA when cells were proliferating but was sequestered to subnuclear loci when cells were quiescent [20, 23, 136]. RUNX2 protein levels were maximal in endothelial cells in late G2 and M phases of the cell cycle [23]. Using RNA interference to knockdown RUNX2, endothelial cell exit from G2/M phases of the cell cycle was delayed [23] resulting in a decrease in cell proliferation. In vitro kinase assays showed S451 must be phosphorylated to allow RUNX2 to function in promoting progression through the cell cycle [23]. However, in osteoblasts, RUNX2 inhibited osteoblast proliferation and RUNX2 protein levels were maximal in G1 [24], suggesting that RUNX2 regulation may be cell type dependent.
BMP2 is able to regulate RUNX2 protein levels via inhibition of cdk4 [76]. This inhibition leads to protection from proteasomal degradation thus maintaining cellular protein levels. Recent research has focused on how micro-RNA’s (miRNAs) regulate RUNX2 protein levels. MiRNAs modulate protein levels through an RNA interference pathway ultimately leading to mRNA degradation or reduced protein translation. MiRNAs are 18–25 nucleotide RNAs that repress translational activity of mRNAs [82]. Wu et al. have suggested that the miR-30 family of miRNAs may play an essential role in osteogenesis. Their data show that miR-30 was able to negatively regulate both Smad1 and RUNX2 [82]. Alkaline phosphatase (marker of osteoblast differentiation) was shown to be dramatically decreased after exogenous miR-30 expression [82]. Furthermore, miR-30 family miRNAs were able to bind to the 3′-untranslated region of both Smad1 and RUNX2 mRNA [82] thus inhibiting the effects of BMP-2-stimulated osteoblast differentiation pathways. MiR-203 is a known tumor suppressor miRNA which is downregulated in prostate cancer [83, 84]. It has been shown to bind RUNX2 mRNA resulting in a mesenchymal to epithelial transition (MET), inhibition of cell proliferation, and inhibition of cell migration and invasion [83, 84]. In addition to the miRNAs already described, it has been shown that miR-23a, miR-34 cluster, miR-133a, miR-135a, miR-137, miR-204, miR-205, miR-217, miR-218, and miR-338 all regulate RUNX2 expression [85, 86]. In addition to miRNA regulation of RUNX2, the proteasomal degradation pathway is implicated in regulating RUNX2 levels in the cell. For example, WWP1 (WW domain-containing E3 ubiquitin protein ligase 1) has been shown to function as the E3 ubiquitin ligase responsible for ubiquitinating RUNX2 and targeting it for proteasomal degradation [87].
5 RUNX2 as a Therapeutic Target
RUNX2 is a transcriptional regulator of gene expression. Mutations, amplification, or inappropriate expression of RUNX2 has the potential to amplify the expression or repression of a variety of target genes. This could regulate global changes in gene regulatory networks and lead to a process called transformation amplification. Therefore, RUNX2 may be a prime target for therapeutic intervention [88, 128] to treat disease because multiple transformation pathways could be inhibited. Many cancers develop resistance to therapies due to signaling pathway redundancy allowing signaling switches to occur. Being able to target transcription factors allows therapies to bypass the redundancy of signaling.
Prostate cancer bone metastases form osteolytic lesions before the development of osteoblastic lesions. Li et al. found that isoflavone and 3, 3′-diindolylmethone (BR-DIM) are able to multifunctionally inhibit these metastases from forming [88]. This combination therapy may inhibit not only osteoblast differentiation but also osteoclast differentiation. Their research showed that one of the ways in which this combination treatment functions is to inhibit signaling of RANKL [88]. However, further cellular analysis revealed isoflavone and BR-DIM combination therapy inhibited signaling from the Akt, AR (androgen receptor), PSA, and p27 signaling axis as well as blocking the RNA interference pathway by inhibiting miR-92a which is associated with RANKL signaling [88]. This is one example of inhibiting the effects of RUNX2 through targeting of its target genes.
Natural compounds have been shown to have potent medicinal benefits. Astragaloside II a compound from the plant, Radix astragalus, was tested on rat primary osteoblasts to determine its effects on viability, proliferation, differentiation and maturation [89]. Astragaloside II promoted proliferation, differentiation and mineralization of primary rat osteoblasts [89]. The effects of this drug on post-menopausal women could potentially prevent osteoporosis. One benefit in cancer patients could be prevention of bone fractures associated with osteolytic metastases by promoting osteoblast differentiation to prevent bone degradation caused by cancer cells. However, use of this drug for patients with metastatic cancers could increase the incidence of bone metastases and lead to early death by stimulating RUNX2 expression within tumor cells. A second compound, Neobavaisoflavone (NBIF), was isolated from the plant Psoralea corylifolia L and was shown to have a similar effect on RUNX2 in osteoblasts as Astragaloside II [90]. NBIF was shown to upregulate RUNX2 expression in MC3T3-E1 cells while also activating its gene regulatory functions [90]. NBIF was shown to upregulate osteocalcin, bone sialoprotein, and type 1 collagen [90]. While this drug may have pro-bone forming functions and could potentially be a way to restore bone loss as a result of bone degradative diseases, one must also take into consideration the dosing regimen that would make this drug specific for osteoblasts while not further stabilizing RUNX2 positive cancer cells.
An understanding of upstream signaling pathways that activate RUNX2 and how cofactors regulate RUNX2 activity in disease would improve the development of therapeutics. In MM it was shown that the Gfi1 targeted drug, Trichostatin-A, was able to block the inhibition that Gfi1 imposes on RUNX2 [81]. Bortezomib (Velcade) is a proteasomal inhibitor and is also used in the treatment of MM because it was shown to induce osteoblast differentiation [91]. Bortezomib inhibited FGF2 induced TAZ (a RUNX2 binding coactivator) protein degradation [91], thus allowing TAZ to interact with RUNX2 and promote osteoblast differentiation in MC3T3-E1 cells. Therefore, restoration of RUNX2 expression enabled osteoblast differentiation and disabled MM progression in the bone microenvironment.
Studies that show targeting upstream signaling pathways that activate RUNX2 or cofactors have been reported, but the research for direct RUNX2 inhibition is limited. Our laboratory showed for the first time that cholecalciferol (inactive Vitamin D3 precursor) directly modulates RUNX2 DNA-binding activity [25]. Cholecalciferol is produced in the skin as a result of exposure to UV or it can be absorbed in the digestive system through the diet [25]. Cholecalciferol is normally converted to active Vitamin D3 in the body to 1,25OH-D3, which interacts with Vitamin D Receptor (VDR) to promote calcium absorption in the gut and increase bone formation. However, Vitamin D3 also exhibits paracrine and autocrine activity by regulating epithelial cell differentiation and modulating immune system function [25]. Using a quantitative DNA binding assay (D-ELISA) it was shown that cholecalciferol was able to modulate RUNX2 DNA binding in a VDR-independent manner. Analysis of RUNX2-positive breast tumor cells (MCF7), endothelial cells (HBME), and osteosarcoma cells (SaOs2) showed that cholecalciferol was able to inhibit cellular proliferation [25], suggesting a RUNX2-specific function. Further research needs to be conducted to study the effects in animal models before this strategy could be modified for clinical trials in the treatment of RUNX2 positive tumors.
Direct targeting of epithelial or bone cell RUNX2 pathways and cofactors that modulate RUNX2 expression or activity is another therapeutic strategy. However, targeting the microenvironment is also another option. Angiogenesis is an essential process that must occur for many solid tumors to metastasize to bone. Endothelial cells express RUNX2, which mediates wound healing by stimulating new blood vessel formation. However, tumors use angiogenesis to vascularize and provide nutrients to tumors that also allow cancer cells to intravasate. Inhibition of angiogenesis using VEGF inhibitors has been a therapeutic strategy for many years. In theory if one starves tumors of their nutrient supply, it should lead to tumor regression or necrosis. Alternatively, by stabilizing blood vessels feeding the tumors then in theory chemotherapeutic agents would be able to get to sites of tumor growth more effectively. However, these therapies have been less successful in the clinic. LGD1069 is a selective retinoid X receptor ligand used to treat T-cell lymphoma but has also been shown to inhibit angiogenesis in lung cancer [92]. LGD1069 inhibited activation of the TGF-β/Smad pathway thus reducing both activation and expression of RUNX2 in human umbilical vein endothelial cells (HUVECs) [92]. However, endothelial cells are not the only cells that use the TGF-β/Smad pathway to activate the expression and activity of RUNX2. Therefore, LGD1069 activity in other cell lines (breast, prostate, and osteosarcoma) should be tested to determine whether it decreases RUNX2 protein or activity. In melanoma, it was shown that SD-208 (a TGF-β receptor I kinase inhibitor) blocked expression of RUNX2 through downregulation of TGF-β/Smad signaling [93], supporting the notion that targeting of the TGF-β/Smad axis could have therapeutic benefit in RUNX2 positive tumors by inhibiting expression within the tumor itself or by preventing angiogenesis.
6 Future Directions
The biological understanding of disease will be essential to creating new therapeutics to treat disease. Signaling pathways are difficult to ablate because of pathway redundancy. Therefore, it is essential to target upstream signaling as well as downstream signaling, especially of transcription factors. Understanding the role that RUNX2 plays in cancer progression will be essential to be able to use it as a therapeutic target to inhibit metastasis to the bone. RUNX2 has been shown to be highly upregulated in the cancer stem cell (CSC) populations of prostate cancer, breast cancer, and osteosarcoma [94–98]. Therefore, with the emergence of the CSC as a key contributor to cancer development, progression, and resistance to modern therapies, it will be important to understand the role that RUNX2 is playing in CSC regulation so that appropriate therapeutic strategies can be developed.
Abbreviations
- 1,25-OH D3:
-
1,25-OH Vitamin D3
- AA:
-
Amino acid
- Akt:
-
v-akt murine thymomaviral oncogene homolog 1 (aka: protein kinase B or PKB)
- AML:
-
Acute myeloid leukemia
- AR:
-
Aldose reductase (Context dependent. Not to be confused with androgen receptor)
- AR:
-
Androgen receptor (Context dependent. Not to be confused with aldose reductase)
- Atf6:
-
Activating transcription factor 6
- BMP:
-
Bone morphogenetic protein
- BMP-2:
-
Bone morphogenetic protein-2
- BR-DIM:
-
3,3′-diindolylmethane
- BSP:
-
Bone sialoprotein (aka: OPN)
- C/EBPβ:
-
CCAAT/enhancer binding protein beta
- CBFα:
-
Core-binding factor alpha
- Cbf-β:
-
Core-binding factor subunit beta
- Cbfβ-SMMHC:
-
Core binding factor β – smooth muscle myosin heavy chain
- CBP:
-
CREB-binding protein
- CCD:
-
Cleidocranial dysplasia
- Cdk:
-
Cyclin-dependent kinase
- CML:
-
Chronic myeloid leukemia
- c-myc:
-
v-myc avian myelocytomatosis viral oncogene homolog
- CoAA:
-
Co-activator activator
- CSC:
-
Cancer stem cell
- CSF2:
-
Colony stimulating factor 2
- CST7:
-
Cystatin-7 (aka: cystatin-F or leukocystatin)
- CTGF:
-
Connective tissue growth factor
- Cx43:
-
Connexin 43
- D-ELISA:
-
Deoxyribonucleic acid binding-enzyme-linked immunosorbent assay
- DNA:
-
Deoxyribonucleic acid
- EC:
-
Endothelial cell
- EMT:
-
Epithelial mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinase
- ERα:
-
Estrogen receptor alpha
- EWS-FLI:
-
Ewing’s sarcoma breakpoint region 1 t(1122)(q24q12)
- FGF2:
-
Fibroblast growth factor 2 (basic)
- FGF2R:
-
Fibroblast growth factor 2 receptor
- FoxO1:
-
Forkhead box O1
- G9a:
-
Histone-lysine N-methyltransferase H3 lysine 9 specific 3 (aka: euchromatic histone-lysine N-methyltransferase)
- GPCR:
-
G-protein coupled receptor
- HBME:
-
Human bone marrow endothelial cell
- HDAC:
-
Histone deacetylase
- HG:
-
Hyperglycemia
- HIF1-α:
-
Hypoxia inducible factor 1 alpha subunit
- HUVEC:
-
Human umbilical vein endothelial cell
- IGF-1:
-
Insulin-like growth factor-1
- IGF-1R:
-
Insulin-like growth factor 1 receptor
- Ihh:
-
Indian hedgehog
- IL8:
-
Interleukin 8
- JNK:
-
c-jun-N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- MET:
-
Mesenchymal epithelial transition
- miRNA:
-
Micro-ribonucleic acid
- MLK3:
-
MAP3K mixed-lineage kinase 3
- MM:
-
Multiple myeloma
- MMP:
-
Matrix metalloproteinase
- MORF:
-
Monocyte leukemia zinc finger related factor
- MOZ:
-
Monocyte leukemia zinc finger protein
- mRNA:
-
Messenger ribonucleic acid
- mSin3a:
-
Mammalian transcriptional corepressors Sin3a
- NBIF:
-
Neobavaisoflavone
- NLS:
-
Nuclear localization signal
- NMTS:
-
Nuclear matrix targeting signal
- OPN:
-
Osteopontin (aka: BSP)
- p21CIP1 :
-
Cyclin-dependent kinase inhibitor 1A
- p27:
-
Cyclin-dependent kinase inhibitor 1B
- p300:
-
E1A binding protein p300
- p38:
-
p38 mitogen activated protein kinase
- PEBP2α:
-
Polyomavirus enhancer-binding protein 2 alpha
- PI3K:
-
Phosphoinositide-3-kinase
- PIP:
-
Prolactin-induced protein
- PKC:
-
Protein kinase C
- PlxnA2:
-
Plexin A2
- PSA:
-
Prostate specific antigen
- PTEN:
-
Phosphatase and tensin homolog
- PTHrP:
-
Parathyroid hormone-related protein
- RANKL:
-
Receptor activator of nuclear factor kappa-B ligand
- RNA:
-
Ribonucleic acid
- Rorβ:
-
Retinoid-related orphan receptor beta
- rRNA:
-
Ribosomal ribonucleic acid
- RUNX1/2/3:
-
Runt-related transcription factor 1/2/3
- SDC2:
-
Syndecan 2
- SDF1:
-
Stromal cell-derived factor 1
- SH3PXD2A:
-
SH3 and PX domain-containing protein 2A
- shRNA:
-
Small hairpin ribonucleic acid
- siRNA:
-
Small interfering ribonucleic acid
- Smad:
-
Sma and mad related proteins
- Smurf:
-
Smad specific E3 ubiquitin protein ligase
- SNAI2:
-
Snail homolog 2/slug
- SNP:
-
Single nucleotide polymorphism
- Sox9:
-
Sex determining region Y-box 9 transcription factor
- Suv39h1:
-
Suppressor of variegation 3–9 homolog 1
- TAZ:
-
Transcriptional coactivator with a PDZ-binding motif
- TGF-β:
-
Transforming growth factor beta
- TLE-1:
-
Transducin-like enhancer protein 1
- TLE2/3:
-
Transducin-like enhancer protein 2/3
- UV:
-
Ultraviolet
- VDR:
-
Vitamin D receptor
- VEGF:
-
Vascular endothelial growth factor
- Wip1:
-
Protein phosphatase magnesium-dependent 1 delta
- WWOX:
-
WW domain containing oxidoreductase
- WWP1:
-
WW domaincontaining E3 ubiquitin protein ligase 1
- YAP:
-
Yes-associated protein
References
Anglin I, Passaniti A (2004) Runx protein signaling in human cancers. Cancer Treat Res 119:189–215
Ito Y (2008) RUNX genes in development and cancer: regulation of viral gene expression and the discovery of RUNX family genes. Adv Cancer Res 99:33–76
Blyth K, Cameron ER, Neil JC (2005) The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 5(5):376–387
Ito Y (2004) Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene 23(24):4198–4208
Jonason JH, Xiao G, Zhang M, Xing L, Chen D (2009) Post-translational regulation of Runx2 in bone and cartilage. J Dent Res 88(8):693–703
Zaidi SK, Javed A, Pratap J, Schroeder TM, Westendorf JJ, Lian JB et al (2006) Alterations in intranuclear localization of Runx2 affect biological activity. J Cell Physiol 209(3):935–942
Soung DY, Talebian L, Matheny CJ, Guzzo R, Speck ME, Lieberman JR et al (2012) Runx1 dose-dependently regulates endochondral ossification during skeletal development and fracture healing. J Bone Miner Res 27(7):1585–1597
Pratap J, Lian JB, Stein GS (2011) Metastatic bone disease: role of transcription factors and future targets. Bone 48(1):30–36
Lou Y, Javed A, Hussain S, Colby J, Frederick D, Pratap J et al (2009) A Runx2 threshold for the cleidocranial dysplasia phenotype. Hum Mol Genet 18(3):556–568
Zhang X, Ting K, Pathmanathan D, Ko T, Chen W, Chen F et al (2012) Calvarial cleidocraniodysplasia-like defects with ENU-induced Nell-1 deficiency. J Craniofac Surg 23(1):61–66
Dalle Carbonare L, Innamorati G, Valenti MT (2011) Transcription factor Runx2 and its application to bone tissue engineering. Stem Cell Rev 8(3):891–897
Liu JC, Lengner CJ, Gaur T, Lou Y, Hussain S, Jones MD et al (2011) Runx2 protein expression utilizes the Runx2 P1 promoter to establish osteoprogenitor cell number for normal bone formation. J Biol Chem 286(34):30057–30070
Stein GS, Lian JB, van Wijnen AJ, Stein JL, Montecino M, Javed A et al (2004) Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene 23(24):4315–4329
Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S et al (2001) Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev 106(1–2):97–106
Bronckers AL, Sasaguri K, Cavender AC, D’Souza RN, Engelse MA (2005) Expression of Runx2/Cbfa1/Pebp2alphaA during angiogenesis in postnatal rodent and fetal human orofacial tissues. J Bone Miner Res 20(3):428–437
Barnes GL, Hebert KE, Kamal M, Javed A, Einhorn TA, Lian JB et al (2004) Fidelity of Runx2 activity in breast cancer cells is required for the generation of metastases-associated osteolytic disease. Cancer Res 64(13):4506–4513
Pratap J, Lian JB, Javed A, Barnes GL, van Wijnen AJ, Stein JL et al (2006) Regulatory roles of Runx2 in metastatic tumor and cancer cell interactions with bone. Cancer Metastasis Rev 25(4):589–600
Kwon TG, Zhao X, Yang Q, Li Y, Ge C, Zhao G et al (2011) Physical and functional interactions between Runx2 and HIF-1alpha induce vascular endothelial growth factor gene expression. J Cell Biochem 112(12):3582–3593
Sun L, Vitolo M, Passaniti A (2001) Runt-related gene 2 in endothelial cells: inducible expression and specific regulation of cell migration and invasion. Cancer Res 61(13):4994–5001
Pierce AD, Anglin IE, Vitolo MI, Mochin MT, Underwood KF, Goldblum SE et al (2012) Glucose-activated RUNX2 phosphorylation promotes endothelial cell proliferation and an angiogenic phenotype. J Cell Biochem 113(1):282–292
Vitolo MI, Anglin IE, Mahoney WM Jr, Renoud KJ, Gartenhaus RB, Bachman KE et al (2007) The RUNX2 transcription factor cooperates with the YES-associated protein, YAP65, to promote cell transformation. Cancer Biol Ther 6(6):856–863
D’Souza DR, Salib MM, Bennett J, Mochin-Peters M, Asrani K, Goldblum SE et al (2009) Hyperglycemia regulates RUNX2 activation and cellular wound healing through the aldose reductase polyol pathway. J Biol Chem 284(27):17947–17955
Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A (2006) Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem 281(11):7118–7128
San Martin IA, Varela N, Gaete M, Villegas K, Osorio M, Tapia JC et al (2009) Impaired cell cycle regulation of the osteoblast-related heterodimeric transcription factor Runx2-Cbfbeta in osteosarcoma cells. J Cell Physiol 221(3):560–571
Underwood KF, D’Souza DR, Mochin-Peters M, Pierce AD, Kommineni S, Choe M et al (2012) Regulation of RUNX2 transcription factor-DNA interactions and cell proliferation by vitamin D3 (cholecalciferol) prohormone activity. J Bone Miner Res 27(4):913–925
Fang CY, Xue JJ, Tan L, Jiang CH, Gao QP, Liang DS et al (2011) A novel single-base deletion mutation of the RUNX2 gene in a Chinese family with cleidocranial dysplasia. Genet Mol Res 10(4):3539–3544
Sugawara M, Kato N, Tsuchiya T, Motoyama T (2011) RUNX2 expression in developing human bones and various bone tumors. Pathol Int 61(10):565–571
Sase T, Suzuki T, Miura K, Shiiba K, Sato I, Nakamura Y et al (2012) Runt-related transcription factor 2 in human colon carcinoma: a potent prognostic factor associated with estrogen receptor. Int J Cancer 131(10):2284–2293
Slattery ML, Lundgreen A, Herrick JS, Caan BJ, Potter JD, Wolff RK (2011) Associations between genetic variation in RUNX1, RUNX2, RUNX3, MAPK1 and eIF4E and risk of colon and rectal cancer: additional support for a TGF-beta-signaling pathway. Carcinogenesis 32(3):318–326
Eliseev RA, Dong YF, Sampson E, Zuscik MJ, Schwarz EM, O’Keefe RJ et al (2008) Runx2-mediated activation of the Bax gene increases osteosarcoma cell sensitivity to apoptosis. Oncogene 27(25):3605–3614
Kurek KC, Del Mare S, Salah Z, Abdeen S, Sadiq H, Lee SH et al (2010) Frequent attenuation of the WWOX tumor suppressor in osteosarcoma is associated with increased tumorigenicity and aberrant RUNX2 expression. Cancer Res 70(13):5577–5586
Martin JW, Zielenska M, Stein GS, van Wijnen AJ, Squire JA (2011) The role of RUNX2 in osteosarcoma oncogenesis. Sarcoma 2011:282745
Sadikovic B, Thorner P, Chilton-Macneill S, Martin JW, Cervigne NK, Squire J et al (2010) Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer 10:202
van der Deen M, Akech J, Lapointe D, Gupta S, Young DW, Montecino MA et al (2012) Genomic promoter occupancy of runt-related transcription factor RUNX2 in osteosarcoma cells identifies genes involved in cell adhesion and motility. J Biol Chem 287(7):4503–4517
Won KY, Park HR, Park YK (2009) Prognostic implication of immunohistochemical Runx2 expression in osteosarcoma. Tumori 95(3):311–316
Little GH, Noushmehr H, Baniwal SK, Berman BP, Coetzee GA, Frenkel B (2012) Genome-wide Runx2 occupancy in prostate cancer cells suggests a role in regulating secretion. Nucleic Acids Res 40(8):3538–3547
Akech J, Wixted JJ, Bedard K, van der Deen M, Hussain S, Guise TA et al (2010) Runx2 association with progression of prostate cancer in patients: mechanisms mediating bone osteolysis and osteoblastic metastatic lesions. Oncogene 29(6):811–821
Baniwal SK, Khalid O, Gabet Y, Shah RR, Purcell DJ, Mav D et al (2010) Runx2 transcriptome of prostate cancer cells: insights into invasiveness and bone metastasis. Mol Cancer 9:258
Baniwal SK, Little GH, Chimge NO, Frenkel B (2012) Runx2 controls a feed-forward loop between androgen and prolactin-induced protein (PIP) in stimulating T47D cell proliferation. J Cell Physiol 227(5):2276–2282
Hernandez LL, Gregerson KA, Horseman ND (2012) Mammary gland serotonin regulates parathyroid hormone-related protein and other bone-related signals. Am J Physiol Endocrinol Metab 302(8):E1009–E1015
Leong DT, Lim J, Goh X, Pratap J, Pereira BP, Kwok HS et al (2010) Cancer-related ectopic expression of the bone-related transcription factor RUNX2 in non-osseous metastatic tumor cells is linked to cell proliferation and motility. Breast Cancer Res 12(5):R89
Mendoza-Villanueva D, Zeef L, Shore P (2011) Metastatic breast cancer cells inhibit osteoblast differentiation through the Runx2/CBFbeta-dependent expression of the Wnt antagonist, sclerostin. Breast Cancer Res 13(5):R106
Onodera Y, Miki Y, Suzuki T, Takagi K, Akahira J, Sakyu T et al (2010) Runx2 in human breast carcinoma: its potential roles in cancer progression. Cancer Sci 101(12):2670–2675
Purcell DJ, Khalid O, Ou CY, Little GH, Frenkel B, Baniwal SK et al (2012) Recruitment of coregulator G9a by Runx2 for selective enhancement or suppression of transcription. J Cell Biochem 113(7):2406–2414
Reufsteck C, Lifshitz-Shovali R, Zepp M, Bauerle T, Kubler D, Golomb G et al (2012 Jun) Silencing of skeletal metastasis-associated genes impairs migration of breast cancer cells and reduces osteolytic bone lesions. Clin Exp Metastasis 29(5):441–456
van der Deen M, Akech J, Wang T, FitzGerald TJ, Altieri DC, Languino LR et al (2010) The cancer-related Runx2 protein enhances cell growth and responses to androgen and TGFbeta in prostate cancer cells. J Cell Biochem 109(4):828–837
Pratap J, Imbalzano KM, Underwood JM, Cohet N, Gokul K, Akech J et al (2009) Ectopic runx2 expression in mammary epithelial cells disrupts formation of normal acini structure: implications for breast cancer progression. Cancer Res 69(17):6807–6814
Chimge NO, Baniwal SK, Luo J, Coetzee S, Khalid O, Berman BP et al (2012) Opposing effects of Runx2 and estradiol on breast cancer cell proliferation: in vitro identification of reciprocally regulated gene signature related to clinical letrozole responsiveness. Clin Cancer Res 18(3):901–911
Kuo YH, Zaidi SK, Gornostaeva S, Komori T, Stein GS, Castilla LH (2009) Runx2 induces acute myeloid leukemia in cooperation with Cbfbeta-SMMHC in mice. Blood 113(14):3323–3332
Shim JH, Greenblatt MB, Singh A, Brady N, Hu D, Drapp R et al (2012) Administration of BMP2/7 in utero partially reverses Rubinstein-Taybi syndrome-like skeletal defects induced by Pdk1 or Cbp mutations in mice. J Clin Invest 122(1):91–106
Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB, Kim SH et al (2012) BMP2 protein regulates osteocalcin expression via Runx2-mediated Atf6 gene transcription. J Biol Chem 287(2):905–915
Hirata M, Kugimiya F, Fukai A, Saito T, Yano F, Ikeda T et al (2012) C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum Mol Genet 21(5):1111–1123
Mak IW, Cowan RW, Popovic S, Colterjohn N, Singh G, Ghert M (2009) Upregulation of MMP-13 via Runx2 in the stromal cell of giant cell tumor of bone. Bone 45(2):377–386
Singh S, Mak IW, Cowan RW, Turcotte R, Singh G, Ghert M (2011) The role of TWIST as a regulator in giant cell tumor of bone. J Cell Biochem 112(9):2287–2295
Takarada T, Yoneda Y (2009) Transactivation by Runt related factor-2 of matrix metalloproteinase-13 in astrocytes. Neurosci Lett 451(2):99–104
Ohyama Y, Tanaka T, Shimizu T, Matsui H, Sato H, Koitabashi N et al (2012) Runx2/Smad3 complex negatively regulates TGF-beta-induced connective tissue growth factor gene expression in vascular smooth muscle cells. J Atheroscler Thromb 19(1):23–35
Ali SA, Dobson JR, Lian JB, Stein JL, van Wijnen AJ, Zaidi SK et al (2012) A Runx2-HDAC1 co-repressor complex regulates rRNA gene expression by modulating UBF acetylation. J Cell Sci 125(Pt 11):2732–2739
Ali SA, Zaidi SK, Dobson JR, Shakoori AR, Lian JB, Stein JL et al (2010) Transcriptional corepressor TLE1 functions with Runx2 in epigenetic repression of ribosomal RNA genes. Proc Natl Acad Sci USA 107(9):4165–4169
Zhang Z, Deepak V, Meng L, Wang L, Li Y, Jiang Q et al (2012) Analysis of HDAC1-mediated regulation of Runx2-induced osteopontin gene expression in C3h10t1/2 cells. Biotechnol Lett 34(2):197–203
Jensen ED, Schroeder TM, Bailey J, Gopalakrishnan R, Westendorf JJ (2008) Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J Bone Miner Res 23(3):361–372
Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, Jensen ED et al (2011) FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res 71(9):3257–3267
Chen G, Deng C, Li YP (2012) TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8(2):272–288
Javed A, Bae JS, Afzal F, Gutierrez S, Pratap J, Zaidi SK et al (2008) Structural coupling of Smad and Runx2 for execution of the BMP2 osteogenic signal. J Biol Chem 283(13):8412–8422
Pratap J, Wixted JJ, Gaur T, Zaidi SK, Dobson J, Gokul KD et al (2008) Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res 68(19):7795–7802
Nishimura R, Hata K, Matsubara T, Wakabayashi M, Yoneda T (2012) Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J Biochem 151(3):247–254
Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT (2012) Interactions between extracellular signal-regulated kinase 1/2 and P38 map kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res 27(3):538–551
Li Y, Ge C, Long JP, Begun DL, Rodriguez JA, Goldstein SA et al (2012) Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. J Bone Miner Res 27(6):1263–1274
Zou W, Greenblatt MB, Shim JH, Kant S, Zhai B, Lotinun S et al (2011) MLK3 regulates bone development downstream of the faciogenital dysplasia protein FGD1 in mice. J Clin Invest 121(11):4383–4392
Chen J, Yuan K, Mao X, Miano JM, Wu H, Chen Y (2012) Serum response factor regulates bone formation via IGF-1 and Runx2 signals. J Bone Miner Res 27(8):1659–1668
Li Z, Wang Z, Yang L, Li X, Sasaki Y, Wang S et al (2010) Fibroblast growth factor 2 regulates bone sialoprotein gene transcription in human breast cancer cells. J Oral Sci 52(1):125–132
Niger C, Buo AM, Hebert C, Duggan BT, Williams MS, Stains JP (2012) ERK acts in parallel to PKCdelta to mediate the connexin43-dependent potentiation of Runx2 activity by FGF2 in MC3T3 osteoblasts. Am J Physiol Cell Physiol 302(7):C1035–C1044
Teplyuk NM, Haupt LM, Ling L, Dombrowski C, Mun FK, Nathan SS et al (2009) The osteogenic transcription factor Runx2 regulates components of the fibroblast growth factor/proteoglycan signaling axis in osteoblasts. J Cell Biochem 107(1):144–154
Huang YF, Lin JJ, Lin CH, Su Y, Hung SC (2012) c-Jun N-terminal kinase 1 negatively regulates osteoblastic differentiation induced by BMP-2 via phosphorylation of Runx2 at Ser104. J Bone Miner Res 27(5):1093–1105
Bae SC, Lee YH (2006) Phosphorylation, acetylation and ubiquitination: the molecular basis of RUNX regulation. Gene 366(1):58–66
Tu X, Joeng KS, Long F (2012) Indian hedgehog requires additional effectors besides Runx2 to induce osteoblast differentiation. Dev Biol 362(1):76–82
Shu B, Zhang M, Xie R, Wang M, Jin H, Hou W et al (2011) BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J Cell Sci 124(Pt 20):3428–3440
Oh JE, Kim HJ, Kim WS, Lee ZH, Ryoo HM, Hwang SJ et al (2012) PlexinA2 mediates osteoblast differentiation via regulation of Runx2. J Bone Miner Res 27(3):552–562
Yang DC, Yang MH, Tsai CC, Huang TF, Chen YH, Hung SC (2011) Hypoxia inhibits osteogenesis in human mesenchymal stem cells through direct regulation of RUNX2 by TWIST. PLoS One 6(9):e23965
Yuen HF, Kwok WK, Chan KK, Chua CW, Chan YP, Chu YY et al (2008) TWIST modulates prostate cancer cell-mediated bone cell activity and is upregulated by osteogenic induction. Carcinogenesis 29(8):1509–1518
Zhang YY, Li X, Qian SW, Guo L, Huang HY, He Q et al (2012) Down-regulation of type I Runx2 mediated by dexamethasone is required for 3T3-L1 adipogenesis. Mol Endocrinol 26(5):798–808
D’Souza S, del Prete D, Jin S, Sun Q, Huston AJ, Kostov FE et al (2011) Gfi1 expressed in bone marrow stromal cells is a novel osteoblast suppressor in patients with multiple myeloma bone disease. Blood 118(26):6871–6880
Wu T, Zhou H, Hong Y, Li J, Jiang X, Huang H (2012) miR-30 family members negatively regulate osteoblast differentiation. J Biol Chem 287(10):7503–7511
Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V et al (2011) Regulatory role of mir-203 in prostate cancer progression and metastasis. Clin Cancer Res 17(16):5287–5298
Viticchie G, Lena AM, Latina A, Formosa A, Gregersen LH, Lund AH et al (2011) MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle 10(7):1121–1131
Hassan MQ, Gordon JA, Beloti MM, Croce CM, van Wijnen AJ, Stein JL et al (2010) A network connecting Runx2, SATB2, and the miR-23a~27a~24-2 cluster regulates the osteoblast differentiation program. Proc Natl Acad Sci USA 107(46):19879–19884
Zhang Y, Xie RL, Croce CM, Stein JL, Lian JB, van Wijnen AJ et al (2011) A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc Natl Acad Sci USA 108(24):9863–9868
Zhi X, Chen C (2012) WWP1: a versatile ubiquitin E3 ligase in signaling and diseases. Cell Mol Life Sci 69(9):1425–1434
Li Y, Kong D, Ahmad A, Bao B, Sarkar FH (2012) Targeting bone remodeling by isoflavone and 3,3’-diindolylmethane in the context of prostate cancer bone metastasis. PLoS One 7(3):e33011
Kong XH, Niu YB, Song XM, Zhao DD, Wang J, Wu XL et al (2012) Astragaloside II induces osteogenic activities of osteoblasts through the bone morphogenetic protein-2/MAPK and Smad1/5/8 pathways. Int J Mol Med 29(6):1090–1098
Don MJ, Lin LC, Chiou WF (2012) Neobavaisoflavone stimulates osteogenesis via p38-mediated up-regulation of transcription factors and osteoid genes expression in MC3T3-E1 cells. Phytomedicine 19(6):551–561
Eda H, Aoki K, Kato S, Okawa Y, Takada K, Tanaka T et al (2010) The proteasome inhibitor bortezomib inhibits FGF-2-induced reduction of TAZ levels in osteoblast-like cells. Eur J Haematol 85(1):68–75
Fu J, Wang W, Liu YH, Lu H, Luo Y (2011) In vitro anti-angiogenic properties of LGD1069, a selective retinoid X-receptor agonist through down-regulating Runx2 expression on human endothelial cells. BMC Cancer 11:227
Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH et al (2011) TGF-beta-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res 71(1):175–184
Dalle Carbonare L, Frigo A, Francia G, Davi MV, Donatelli L, Stranieri C et al (2012) Runx2 mRNA expression in the tissue, serum, and circulating non-hematopoietic cells of patients with thyroid cancer. J Clin Endocrinol Metab 97(7):E1249–E1256
Liao CP, Adisetiyo H, Liang M, Roy-Burman P (2010) Cancer stem cells and microenvironment in prostate cancer progression. Horm Cancer 1(6):297–305
Liao CP, Adisetiyo H, Liang M, Roy-Burman P (2010) Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res 70(18):7294–7303
Wang L, Park P, Zhang H, La Marca F, Claeson A, Valdivia J et al (2011) BMP-2 inhibits the tumorigenicity of cancer stem cells in human osteosarcoma OS99-1 cell line. Cancer Biol Ther 11(5):457–463
Zhang Y, Xie RL, Gordon J, Leblanc K, Stein JL, Lian JB et al (2012) Control of mesenchymal lineage progression by microRNAs targeting the skeletal gene regulators Trps1 and Runx2. J Biol Chem 287(26):21926–21935
Hsu YL, Huang MS, Yang CJ, Hung JY, Wu LY, Kuo PL (2011) Lung tumor-associated osteoblast-derived bone morphogenetic protein-2 increased epithelial-to-mesenchymal transition of cancer by Runx2/Snail signaling pathway. J Biol Chem 286(43):37335–37346
Jeong JH, Choi JY (2011) Interrelationship of Runx2 and estrogen pathway in skeletal tissues. BMB Rep 44(10):613–618
Pratap J, Javed A, Languino LR, van Wijnen AJ, Stein JL, Stein GS et al (2005) The Runx2 osteogenic transcription factor regulates matrix metalloproteinase 9 in bone metastatic cancer cells and controls cell invasion. Mol Cell Biol 25(19):8581–8591
Lee SH, Che X, Jeong JH, Choi JY, Lee YJ, Lee YH et al (2012) Runx2 protein stabilizes hypoxia-inducible factor-1alpha through competition with von hippel-lindau protein (pVHL) and stimulates angiogenesis in growth plate hypertrophic chondrocytes. J Biol Chem 287(18):14760–14771
Sun X, Wei L, Chen Q, Terek RM (2009) HDAC4 represses vascular endothelial growth factor expression in chondrosarcoma by modulating RUNX2 activity. J Biol Chem 284(33):21881–21890
Peruzzi B, Cappariello A, Del Fattore A, Rucci N, De Benedetti F, Teti A (2012) c-Src and IL-6 inhibit osteoblast differentiation and integrate IGFBP5 signalling. Nat Commun 3:630
Ding M, Lu Y, Abbassi S, Li F, Li X, Song Y et al (2012) Targeting Runx2 expression in hypertrophic chondrocytes impairs endochondral ossification during early skeletal development. J Cell Physiol 227(10):3446–3456
Chimge NO, Baniwal SK, Little GH, Chen YB, Kahn M, Tripathy D et al (2011) Regulation of breast cancer metastasis by Runx2 and estrogen signaling: the role of SNAI2. Breast Cancer Res 13(6):R127
Goloudina AR, Tanoue K, Hammann A, Fourmaux E, Le Guezennec X, Bulavin DV et al (2012) Wip1 promotes RUNX2-dependent apoptosis in p53-negative tumors and protects normal tissues during treatment with anticancer agents. Proc Natl Acad Sci USA 109(2):E68–E75
Tandon M, Gokul K, Ali SA, Chen Z, Lian J, Stein GS et al (2012) Runx2 mediates epigenetic silencing of the bone morphogenetic protein-3B (BMP-3B/GDF10) in lung cancer cells. Mol Cancer 11(1):27
Endo T, Kobayashi T (2010) Runx2 deficiency in mice causes decreased thyroglobulin expression and hypothyroidism. Mol Endocrinol 24(6):1267–1273
Teplyuk NM, Zhang Y, Lou Y, Hawse JR, Hassan MQ, Teplyuk VI et al (2009) The osteogenic transcription factor runx2 controls genes involved in sterol/steroid metabolism, including CYP11A1 in osteoblasts. Mol Endocrinol 23(6):849–861
Teplyuk NM, Galindo M, Teplyuk VI, Pratap J, Young DW, Lapointe D et al (2008) Runx2 regulates G protein-coupled signaling pathways to control growth of osteoblast progenitors. J Biol Chem 283(41):27585–27597
Bond SR, Lau A, Penuela S, Sampaio AV, Underhill TM, Laird DW et al (2011) Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J Bone Miner Res 26(12):2911–2922
Lim M, Zhong C, Yang S, Bell AM, Cohen MB, Roy-Burman P (2010) Runx2 regulates survivin expression in prostate cancer cells. Lab Invest 90(2):222–233
Vladimirova V, Waha A, Luckerath K, Pesheva P, Probstmeier R (2008) Runx2 is expressed in human glioma cells and mediates the expression of galectin-3. J Neurosci Res 86(11):2450–2461
Zhang HY, Jin L, Stilling GA, Ruebel KH, Coonse K, Tanizaki Y et al (2009) RUNX1 and RUNX2 upregulate Galectin-3 expression in human pituitary tumors. Endocrine 35(1):101–111
Hoeppner LH, Secreto F, Jensen ED, Li X, Kahler RA, Westendorf JJ (2009) Runx2 and bone morphogenic protein 2 regulate the expression of an alternative Lef1 transcript during osteoblast maturation. J Cell Physiol 221(2):480–489
Endo T, Ohta K, Kobayashi T (2008) Expression and function of Cbfa-1/Runx2 in thyroid papillary carcinoma cells. J Clin Endocrinol Metab 93(6):2409–2412
Makita N, Suzuki M, Asami S, Takahata R, Kohzaki D, Kobayashi S et al (2008) Two of four alternatively spliced isoforms of RUNX2 control osteocalcin gene expression in human osteoblast cells. Gene 413(1–2):8–17
Li X, Hoeppner LH, Jensen ED, Gopalakrishnan R, Westendorf JJ (2009) Co-activator activator (CoAA) prevents the transcriptional activity of Runt domain transcription factors. J Cell Biochem 108(2):378–387
Mendoza-Villanueva D, Deng W, Lopez-Camacho C, Shore P (2010) The Runx transcriptional co-activator, CBFbeta, is essential for invasion of breast cancer cells. Mol Cancer 9:171
Roforth MM, Liu G, Khosla S, Monroe DG (2012) Examination of nuclear receptor expression in osteoblasts reveals Rorbeta as an important regulator of osteogenesis. J Bone Miner Res 27(4):891–901
Westendorf JJ (2006) Transcriptional co-repressors of Runx2. J Cell Biochem 98(1):54–64
Byun MR, Jeong H, Bae SJ, Kim AR, Hwang ES, Hong JH (2012) TAZ is required for the osteogenic and anti-adipogenic activities of kaempferol. Bone 50(1):364–372
Baniwal SK, Khalid O, Sir D, Buchanan G, Coetzee GA, Frenkel B (2009) Repression of Runx2 by androgen receptor (AR) in osteoblasts and prostate cancer cells: AR binds Runx2 and abrogates its recruitment to DNA. Mol Endocrinol 23(8):1203–1214
Del Mare S, Kurek KC, Stein GS, Lian JB, Aqeilan RI (2011) Role of the WWOX tumor suppressor gene in bone homeostasis and the pathogenesis of osteosarcoma. Am J Cancer Res 1(5):585–594
Li X, McGee-Lawrence ME, Decker M, Westendorf JJ (2010) The Ewing’s sarcoma fusion protein, EWS-FLI, binds Runx2 and blocks osteoblast differentiation. J Cell Biochem 111(4):933–943
Blyth K, Vaillant F, Hanlon L, Mackay N, Bell M, Jenkins A et al (2006) Runx2 and MYC collaborate in lymphoma development by suppressing apoptotic and growth arrest pathways in vivo. Cancer Res 66(4):2195–2201
Chua CW, Chiu YT, Yuen HF, Chan KW, Man K, Wang X et al (2009) Suppression of androgen-independent prostate cancer cell aggressiveness by FTY720: validating Runx2 as a potential antimetastatic drug screening platform. Clin Cancer Res 15(13):4322–4335
Das K, Leong DT, Gupta A, Shen L, Putti T, Stein GS et al (2009) Positive association between nuclear Runx2 and oestrogen-progesterone receptor gene expression characterises a biological subtype of breast cancer. Eur J Cancer 45(13):2239–2248
Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT et al (2012) Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell 22(3):597–609
Giuliani N, Mangoni M, Rizzoli V (2009) Osteogenic differentiation of mesenchymal stem cells in multiple myeloma: identification of potential therapeutic targets. Exp Hematol 37(8):879–886
Hassan MQ, Saini S, Gordon JA, van Wijnen AJ, Montecino M, Stein JL et al (2009) Molecular switches involving homeodomain proteins, HOXA10 and RUNX2 regulate osteoblastogenesis. Cells Tissues Organs 189(1–4):122–125
Jonsson S, Hjorth-Hansen H, Olsson B, Wadenvik H, Sundan A, Standal T (2012) Imatinib inhibits proliferation of human mesenchymal stem cells and promotes early but not late osteoblast differentiation in vitro. J Bone Miner Metab 30(1):119–123
Kawato Y, Hirao M, Ebina K, Tamai N, Shi K, Hashimoto J et al (2011) Nkx3.2-induced suppression of Runx2 is a crucial mediator of hypoxia-dependent maintenance of chondrocyte phenotypes. Biochem Biophys Res Commun 416(1–2):205–210
Kayed H, Jiang X, Keleg S, Jesnowski R, Giese T, Berger MR et al (2007) Regulation and functional role of the Runt-related transcription factor-2 in pancreatic cancer. Br J Cancer 97(8):1106–1115
Pockwinse SM, Kota KP, Quaresma AJ, Imbalzano AN, Lian JB, van Wijnen AJ et al (2011) Live cell imaging of the cancer-related transcription factor RUNX2 during mitotic progression. J Cell Physiol 226(5):1383–1389
Pockwinse SM, Rajgopal A, Young DW, Mujeeb KA, Nickerson J, Javed A et al (2006) Microtubule-dependent nuclear-cytoplasmic shuttling of Runx2. J Cell Physiol 206(2): 354–362
Qiao M, Shapiro P, Kumar R, Passaniti A (2004) Insulin-like growth factor-1 regulates endogenous RUNX2 activity in endothelial cells through a phosphatidylinositol 3-kinase/ERK-dependent and Akt-independent signaling pathway. J Biol Chem 279(41):42709–42718
Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X et al (2011) An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res 26(3):604–617
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Brusgard, J.L., Passaniti, A. (2014). RUNX2 Transcriptional Regulation in Development and Disease. In: Kumar, R. (eds) Nuclear Signaling Pathways and Targeting Transcription in Cancer. Cancer Drug Discovery and Development. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8039-6_3
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8039-6_3
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8038-9
Online ISBN: 978-1-4614-8039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)