
Abstract
The compaction of genomic DNA into chromatin has far-ranging consequences for almost all aspects of DNA metabolism activity. ATP-dependent chromatin remodeling complexes (CRCs) enable DNA-binding proteins access to nucelosomal DNA by altering chromatin structure through distinct mechanisms including nucleosome sliding, nucleosome assembly, and histone exchanges, in an energy-dependent manner. Consequently, CRCs play critical roles in diverse cellular processes that are dependent on chromatin template, including transcription, replication, and DNA repair. Thus, an aberration in these chromatin remodeling proteins leads to human diseases including cancer. In this chapter, we discuss the functional roles of CRCs in the regulation of gene transcription, DNA damage response, and its potential connection with cancer development as well as tumor therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Chromatin remodeling
- Transcription
- DNA damage response
- Cancer
- Cancer development and progression
- Cancer therapeutics
1 Introduction
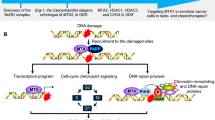
It is increasingly accepted that cancer is a genetic disease. A precise understanding of how genetic alternations contribute to tumor development and progression is the key to develop effective strategies for winning the fight against cancer. The genetic material is stored into the nucleus in the form of chromatin. The fundamental building block of chromatin is the nucleosome core particle, which is made of approximately 147 base pairs of DNA wrapped around a histone octamer consisting of two copies of each of the four core histones H2A, H2B, H3, and H4 [1, 2]. The repeating nucleosome cores are connected by 20–80 base pairs of linker DNA and further assembled into hierarchically folded higher-order structures with the linker histone H1, nonhistone proteins and divalent metal ions [1–3]. By its very nature, the highly condensed structure of chromatin generally limits the accessibility of DNA binding proteins to the DNA, thus exerting an inhibitory effect on many critical DNA metabolizing activities, such as transcription, DNA replication, recombination and repair. To counteract this repressive barrier imposed by nucleosome architecture, eukaryotes have developed multiple intricate mechanisms to remodel nucleosomes, thus allowing DNA binding proteins such as transcription factors and DNA repair proteins access to the DNA. One of such mechanisms is involved in the ATP-dependent chromatin remodeling complexes (CRCs) that hydrolyze ATP to alter histone-DNA contacts through several mechanisms including nucleosome sliding, histone exchange, and nucleosome/histone eviction [4]. To date, four families of CRCs have been characterized in eukaryotes based on their compositions and functional domains, including the SWI/SNF (switching/sucrose non-fermenting), ISWI (imitation switch), Mi-2/NuRD (nucleosome remodeling and histone deacetylase), and INO80 (inositol requiring) [5, 6]. All CRCs are multisubunit complexes that contain an ATPase subunit [6] and most of them are conserved from yeast to humans. These CRCs play essential roles in many basic biological processes, including gene expression, DNA damage repair, and cell division (Fig. 16.1) [6–11]. Consequently, aberrations in these chromatin remodeling proteins are associated with a variety of human diseases including cancer. Thus, targeting the components of chromatin remodeling signaling pathways is currently being evaluated as a major therapeutic strategy in the prevention and treatment of human cancers. In the following sections, we focus on discussing the emerging role of CRCs in gene transcription, DNA damage response (DDR), and tumor development as well as its potential implication in cancer therapeutics.

Functional role of the CRCs in transcription and DDR. The CRCs alter chromatin structure in an energy-dependent manner. Consequently, the transcriptional and DDR machinery proteins get access to nucleosomal DNA and facilitate gene transcription and efficient DNA repair
2 Characterization of the CRC Family
The first discovered CRC family is the SWI/SNF, which was initially identified in independent screens for mutants affecting mating-type switching and growth on sucrose in 1994 [12–14]. The SWI/SNF family is composed of 8–14 subunits and is characterized by a bromodomain in its ATPase catalytic subunits, BRG1 (also known as SMARCA4 or BAF190A) and BRM (also known as SMARCA2 or BAF190B) http://www.genecards.org/ [15]. The bromodomain preferentially interacts with acetylated histones, which play both positive and negative roles in regulating the activity of the SWI/SNF remodeling factors [16]. In S. cerevisiae, this family contains the founding member SWI/SNF complex as well as the highly related RSC (remodel the structure of chromatin) complex [17]. Both SWI/SNF and RSC complexes exhibit a DNA-dependent ATPase activity to perturb nucleosome structure [17, 18], and contain nuclear actin-related proteins Arp7 and Arp9 [19, 20]. Arp7 and Arp9 form a stable heterodimer relying on their actin-related regions for heterodimerization, and function with DNA binding proteins to facilitate proper chromatin architecture and complex-complex interactions [19, 20]. Human complexes of this family have also been identified, including the BRG1-associated factor (BAF) complex and the polybromo BRG1-assocaited factor (PBAF) complex [21, 22]. With regard to homology, BAF1 is similar to the yeast SWI/SNF and PBAF is more like yeast RSC complex [21, 22].
The second family of CRCs is ISWI. One distinguishing feature of this family is that its ATPase subunit contains a carboxyl-terminal SANT and a SLIDE (SANT-like ISWI domain) domain, which together form a nucleosome recognition module that binds histone tails and linker DNA [23, 24]. The founding member of this family is the Drosophila NURF (nucleosome remodeling factor) complex, which was identified in 1995 by assaying the ability of drosophila embryo extracts to generate a nuclease-hypersensitive site within an array of nucleosomes [25]. NURF is composed of four distinct subunits, including the 140-kD ISWI ATPase subunit (NURF140) [26], a 55-kD WD repeat protein (NURF55) [27], the smallest NURF38 component [28], and the large NURF301 subunit [29]. In contrast to the SWI/SNF complex, the ATPase activity of NURF requires nucleosomes rather than free DNA or histones [25]. Thus, NURF acts directly on a nucleosome to alter chromatin structure by catalyzing nucleosome sliding, thereby exposing DNA sequences associated with nucleosomes [25, 30, 31]. Interestingly, the N-terminal histone tails are functionally important for modulating nucleosome mobility and regulating ATP-dependent nucleosome sliding by NURF [30]. In addition to NURF, the drosophila ISWI complex also contains the CHRAC (chromatin remodeling and assembly complex) [32] and ACF (ATP-utilizing chromatin remodeling and assembly factor) complexes [33]. Both exhibit chromatin assembly and nucleosome sliding activity in an ATP-dependent mechanism [32, 33]. In mammals, two highly related ATPase subunits of the ISWI CRC have been identified, including SNF2L (SNF2 like) and SNF2H (SNF2 homologue) [34]. Biochemical analysis revealed that the SNF2H ATPase catalytic subunit is contained in multiple complexes including ACF, CHRAC, RSF (remodeling and spacing factor), NoRC (nucleosome-remodeling complex), WICH (WSTF-ISWI chromatin remodeling), and WCRF (WSTF-related chromatin-remodeling factor) [34–41]. In contrast, only a small number of complexes, such as the human NURF and CERF (CECR2-containing remodeling factor) complexes, contain the SNF2L ATPase subunit [35, 42].
The third family of CRCs is the Mi-2/NuRD complex, which was identified in1998 from several independent groups and processes both ATP-dependent chromatin remodeling and histone deacetylase activities [43–46]. The complex contains the histone deacetylases HDAC1/2, the histone-binding proteins RbAp46/48, the dermatomyositis-specific autoantigen Mi-2, the metastasis-associated proteins MTA1/2/3, and the methyl-CpG-binding domain proteins MBD2/3 [43, 44, 47]. Notably, Mi-2 subunit contains a chromodomain and plant homeo domain (PHD)-type zinc finger, and functions as a DNA-dependent, nucleosome-stimulated ATPase that remodels nucleosomes in an ATP-dependent manner [48]. In particular, Mi-2 lacking its chromodomains fails to bind or remodel nucleosomes [23].
The fourth family of CRCs is the evolutionarily conserved INO80 subfamily, which includes the INO80 complex and SWR1 complex [49]. The subfamily is characterized by a split ATPase domain and the presence of two RuvB-like proteins Rvb1 and Rvb2 [49]. The INO80 complex was initially purified from S. cerevisiae in 2000, consisting of about 12 subunits including Arp4, Arp5, Arp8, actin, and the Rvb1 and Rvb2 helicase proteins, and displays nucleosome-stimulated ATPase activity and ATP-dependent chromatin remodeling activities [50]. Deletion of Arp5 in yeast strains impairs INO80 ATPase activity, DNA binding, and nucleosome mobilization [51]. Similarly, Arp8 forms a complex with nucleosomes via the H3 and H4 histones [51, 52] and is essential for activity of INO80. In this context, deletion of Arp8 results in loss of INO80 function with multiple effects on cellular processes such as double-strand break (DSB) repair and chromosome alignment [52–55]. In contrast, Rvbp1p/Rvb2p is required for the complete assembly of a functional INO80 complex and for recruiting Arp5p to the INO80 complex in an ATP dependent manner [56]. The highly-related SWR1 complex was identified from S. cerevisiae in 2004 [57] and its human counterparts, termed Snf2-related CREBBP activator protein (SRCAP) and p400, were also identified afterward [58, 59]. In S. cerevisiae, this SWR1 complex contains Swr1p, a putative Swi2/Snf2-related ATPase, and 12 additional subunits. Among them, several subunits including Act1, Arp4, Rvb1 and Rvb2 are common to the INO80 complex [50]. Despite highly related to INO80, the SWR1 complex is unique in its ability to catalyze the incorporation of the histone variant H2AZ (Htz1 in S. cerevisiae) into nucleosomes [57, 60, 61], and this occurs in vitro in a stepwise and unidirectional fashion and requires dual activation with histone H2AZ and canonical nucleosome [62].
3 CRCs in Gene Transcription
The compaction of DNA into chromatin in the eukarytotic nucleus poses many obstacles to transcription [63]. The CRCs bind directly to nucleosomes and disrupt histone-DNA interactions using the energy of ATP hydrolysis, thus facilitating the access of the core transcription machinery proteins and general cofactors to nucleosomal DNA. As a result, CRCs play a fundamental role in modulating transcription in yeast and higher eukaryotes. Notably, these CRCs have a range of specific and context-dependent roles in control of gene expression depending on the circumstance.
3.1 The SWI/SNF Complex
The SWI/SNF complex is involved in a variety of functionally distinct complexes and exerts diverse roles in gene regulation and genome function [64]. One outstanding example is that the SWI/SNF CRC participates in promoting transcriptional activation by nuclear receptors. The androgen receptor (AR) is a ligand-dependent transcription factor whose activity is tightly regulated by interacting cofactors and cofactor complexes and is a key player in prostate cancer development and progression [65, 66]. Considerable evidence has pointed out that the SWI/SNF CRC directs AR-mediated transcriptional activation, and different AR targets show disparity in the requirement for SWI/SNF [65–67]. A case in point is the BAF57 (also known as SMARCE1) subunit, which directly binds to the AR and is recruited to endogenous AR targets upon ligand activation, thus regulating AR activity, coactivator function, and AR-dependent proliferation [68]. Similarly, the BAF57 subunit specifically regulates estrogen receptor alpha (ERα)-dependent gene expression and proliferation in human breast cancer cells [69, 70]. Consequently, mutations in BAF57 deregulate several oncogenic signaling pathways, thus contributing to the development of breast cancer [71, 72].
In addition to BAF57, the BRG1subunit is a critical modulator of transcriptional regulation in various tissues and pathophysiological conditions [73]. For instance, BRG1predominantly interacts with Smad2 and Smad3 and is specifically required for transforming growth factor β-induced expression of endogenous Smad2/3 target genes through recruitment to Smad-dependent promoters [74, 75]. BRG1, as well as BRM, associates with the CD44 and E-cadherin promoters and promotes their transcriptional activation in cancer cells through deceasing DNA methylation at their promoters [76]. In addition, SNF5 (also known as Ini1, BAF47, SNR1, or SMARCB1) mediates BRG1 recruitment to the p15 INK4b and p16 INK4a promoters and activates their expression through eviction of polycomb group silencing complex and extensive chromatin reprogramming [77].
Although the SWI/SNF CRC is generally associated with transcriptional activation, emerging evidence points out its additional role in transcriptional silencing pathway [78]. For instance, SWI3B, an essential subunit of the SWI/SNF complex, physically interacts with a long noncoding RNA (lncRNA)-binding protein, IDN2, and contributes to lncRNA-mediated transcriptional silencing [78]. Human BRM is functionally linked with the methyl-CpG binding protein MeCP2-depenendent transcriptional silencing [79]. Both BRG-1 and SNF5 subunits repress transcription of cyclin D1 gene through the direct recruitment of histone deacetylase (HDAC) activity to its promoter, thereby exerting their tumor suppressor functions [80, 81]. More interestingly, BRG1 and BRM can switch their mode of function at same promoter between activation and repression through the context-dependent reprogramming of the SWI/SNF complex [82].
3.2 The ISWI Complex
The ISWI complex can space nucleosomes, thus affecting a variety of nuclear processes including transcription. Genome-wide analysis demonstrates that ISWI binds both genic and intergenic regions, and remarkably, binds genes near their promoters causing specific alterations in nucleosome positioning at the level of the transcription start sites [83]. Accumulating evidence suggests that the ISWI containing NURF complex is able to facilitate transcriptional activation via remodeling of chromatin in vitro and in vivo [29, 84, 85]. However, NURF also functions as a co-repressor of a large set of JAK/STAT target genes in drosophila to regulate innate immunity network [86]. Similarly, ISWI and ACF1 directly repress Wingless transcriptional targets in drosophila [87]. In S. cerevisiae, Isw1 also functions in stress-induced gene repression under normal growth conditions [88]. In contrast, the Isw2 complex represses transcription of early meiotic genes during mitotic growth and this repressor function of lsw2 complex is largely dependent upon Ume6p, which recruits the complex to target genes [89, 90]. Subsequent studies further demonstrate that Isw2 acts as a transcriptional repressor by altering nucleosome positions, and loss of Isw2 activity results in the generation of both coding and noncoding transcripts due to inappropriate transcription [91].
3.3 The Mi-2/NuRD Complex
Accumulating evidence has uncovered a number of interesting connections between the Mi-2/NuRD complex and gene regulation [9, 92, 93]. A case in point is the metastasis-associated protein 1 (MTA1), the founding member of the MTA family, which was isolated by differential cDNA library screening using a rat mammary adenocarcinoma metastatic system [94]. MTA1 functions not only as a transcriptional repressor of estrogen receptor α [95], p21WAF1 [96], breast cancer type 1 susceptibility protein [97], RING finger protein 144A [98], phosphatase and tensin homolog [99], transforming growth factor β signaling component SMAD7 [100], guanine nucleotide-binding protein G(i) subunit alpha-2 [101], and homeobox protein SIX3 [102], but also as a transcriptional activator for certain genes, such as the breast cancer-amplified sequence 3 [103] paired box 5 [104], tumor suppressor alternative reading frame [105], cell surface oncogenic protein hyaluronan-mediated motility receptor [106], proto-oncogene protein Wnt-1 [107], and tyrosine hydroxylase [108]. One unanswered question in this field is what is the underlying mechanism for the physiologic switch between coactivator and corepressor functions of MTA1. It is becoming increasingly clear that post-translational modifications might play a role in the regulation of MTA1 function in transcription. In this context, SUMOylation and SUMO-interacting motif of MTA1 synergistically regulate its co-repressor activity on PS2 transcription [109]. Similarly, acetylation status of MTA1 might also be crucial for its corepressor function on a negative modifier of Ras activation and its oncogenic activity [101]. More interestingly, methylation of lysine 532 in MTA1 protein seemly represents a molecular switch between coactivator and corepressor [110]. In this context, methylated MTA1 is required for the NuRD repressor complex, while demethylated MTA1 recognizes the active histone mark and recruits coactivator complex onto its target gene promoters in a signaling-dependent manner [110].
3.4 The INO80 Complex
Involvement of the INO80 complex in transcription was first discovered in S. cerevisiae, in which INO80 facilitates transcription in vitro and in vivo [50, 111]. Subsequent studies further demonstrate that its mammalian orthologue also promotes transcription with transcription factor Yin-Yang-1 (YY1) [112]. In contrast, TBP-interacting protein 49b (TIP49b), a component of the INO80 complex, inhibits transcription factor 2 (ATF2) transcriptional activities in response to stress and DNA damage [113].
4 CCRs in the DDR
In response to DNA damage, chromatin undergoes a marked reorganization in an energy dependent manner, thus facilitating the DDR machinery proteins to recognize and repair the damaged DNA [114]. In addition to their putative roles in transcription, CCRs are intimately linked with the DDR.
4.1 The SWI/SNF Complex
The SWI/SNF complex is required for DNA replication [115, 116], somatic recombination [117], nucleotide excision repair (NER) [118, 119], and DSB repair [120]. SWI/SNF also regulates checkpoint activation after ultraviolet (UV) damage via regulation of the proliferating cell nuclear antigen-binding proteins Gadd45a and p21 [121]. The highly related RSC complex is also linked with efficient DSB repair [122, 123]. Interestingly, two isoforms of this complex, defined by the presence of either Rsc1 or Rsc2, play distinct roles in DDR and that at least part of the functional specificity is dictated by the bromo-adjacent homology (BAH) domains [124]. Moreover, the RSC and SWI/SNF chromatin remodelers play distinct roles in DSB repair; SWI/SNF is required during the early steps of homologous recombination (HR), while RSC is important upon the completion of the repair process [125].
4.2 The ISWI Complex
SNF2H, the catalytic subunit of ISWI complex, is rapidly recruited to DSBs in a poly(ADP-ribose) polymerase 1 (PARP1)-dependent manner and facilitates the RNF168-dependent signaling and repair of DSBs [126]. Similarly, the ACF1 chromatin remodeling factor accumulates at UV-induced DNA damage sites immediately following UV radiation [127] and promotes NER of UV-induced DNA lesions [128]. Similarly, the ACF1 complex accumulates rapidly at DSBs and is also required for non-homologous end joining (NHEJ) repair of DSBs in human cells [129]. Rsf-1 (also known as HBXAP) protein interacts with SNF2H to form an ISWI complex, RSF, and has been reported as an amplified gene in human cancers, including the highly aggressive ovarian serous carcinoma [130]. Emerging evidence shows that Rsf-1induces DNA damage and promotes genomic instability [131], and consequently, high-grade ovarian serous carcinomas, especially those with Rsf-1 overexpression, exhibit high levels of the DDR [132]. These findings highlight that increased Rsf-1 expression in tumors can induce chromosomal instability probably through DDR [131].
4.3 The Mi2/NuRD Complex
The initial link between the NuRD complex and DDR was found in 1999 [133], when Schmidt and colleagues discovered that ataxia telangiectasia and Rad3-related protein (ATR), a master regulator of the DDR, associates with multiple components of the NuRD complex, including MTA1, MTA2, HDAC1, HDAC2, and CHD4 [133]. Afterward, van Haaften G et al. in 2006 defined a role for C. elegans early growth response protein 1 (Egr-1), the homologue of human MTA2 gene, in cellular sensitivity to ionizing radiation (IR) using a genome-wide RNA interference screening [134]. In 2009, Li et al. further discovered a previously unknown role for MTA1in IR-induced DSB repair and cell survival using MTA1-knockout fibroblasts [135]. In 2010, several studies from four different groups simultaneously reported a conserved role of the NuRD complex, including MTA1, MTA2, CHD4, HDAC1, and HDAC2 in DDR and DNA repair in multiple model systems [136–140].
The PARP family of proteins has been implicated in recruitment of proteins to sites of damage and is known to localize rapidly to sites of damage [136, 141]. In support of our early findings, emerging evidence shows that MTA1 is recruited to sites of DNA damage in a PARP-dependent manner, and depletion of MTA1 by siRNAs renders cells sensitive to IR, further highlighting its importance in promoting DNA repair [136]. The human homologue of egr-1, MTA2, also protects human cells against IR, suggesting its conserved role in the DDR [139]. CHD4 is rapidly recruited to DSBs in a PARP-dependent manner [136, 138], where it promotes RNF8/RNF168-mediated histone ubiquitylation and the ubiquitin-dependent accumulation of RNF168 and BRCA1 at sites of DNA lesions [137, 139]. CHD4 also acts as an important regulator of the G1/S cell-cycle transition by controlling p53 deacetylation [138]. Consequently, loss of CHD4 causes defects in DNA repair and checkpoint activation, resulting in accumulation of spontaneous DNA damage and increased IR sensitivity [138, 139]. Furthermore, human HDAC1 and HDAC2 also function in the DDR to promote NHEJ repair [140]. Consistently, HDAC inhibitors block the activity of HDAC1 and HDAC2, resulting in defects in the DDR and hypersensitivity to the DSB-inducing agents [140]. Taken together, the NuRD chromatin–remodeling complex is a novel DDR factor that helps to preserve genome stability by regulating signaling and repair of DNA damage [11, 142]. Interestingly, recent studies pointed out that multiple NuRD components are lost during premature and normal ageing, resulting in accumulation of DNA damage during ageing [143], which could contribute to aging-related genomic instability and cancer [144].
4.4 The INO80 Complex
In addition to their well-established role in regulating transcriptional processes, accumulating evidence shows that INO80 and SWR1 chromatin remodeling components are essential for maintaining genomic integrity [10]. The INO80 complex is recruited to sites of DSBs through a specific interaction with the DNA damage-induced phosphorylated histone H2A (termed γH2AX) [145, 146], and mediates DSB repair through its role in DNA end strand resection [147]. INO80 is also recruited to sites of UV lesion repair through interactions with the NER apparatus and promotes the removal of UV lesions by the NER pathway [148, 149]. Moreover, INO80 is required for the restoration of chromatin structure after repair in response to UV-induced damage [149]. Interestingly, INO80 also shapes the DNA replication landscape. In this context, INO80 complexes are enriched at sites of replication and are required for efficient replication of late-replicating regions during replication stress through regulating S-phase checkpoint activity [4, 150]. INO80 also regulates the threshold of DNA damage during replication phase via modifying PCNA ubiquitination and Rad51-mediated processing of recombination intermediates at impeded replication forks [151–153].
5 CRCs in Cancer Development and Progression
5.1 The SWI/SNF Complex
Given its central function in epigenetic chromatin remodeling mechanisms, it is not surprising that alternation of the SWI/SNF CRC plays an important role in tumor development and progression. A substantial body of evidence indicates that several components of the SWI/SNF complexes function as tumor suppressors or negative regulator of cellular proliferation [21, 154, 155]. One such example is the SNF5 core subunit, which has been documented to be mutated or inactivated in a number of human cancers including rhabdoid, rhabdomyosarcoma, epithelioid sarcoma, chronic myeloid leukemia, medulloblastomas, choroid plexus carcinomas, and melanoma [156–161]. In support of this notion, haploinsufficiency of SNF5 predisposes to malignant rhabdoid tumors in mice, and loss of SNF5 results in highly penetrant cancer predisposition with 100 % of mice developing T cell lymphoma or rhabdoid tumors with a median onset of only 11 weeks [162]. Collective evidence establishes that the tumor suppressor activity of SNF5 depends on its regulation of cell cycle progression, cell survival and senescence [163–168]. Inactivation of the SNF5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss to accelerate oncogenic transformation and tumor growth in mice [169, 170]. The inhibition of RhoA-dependent migration is another crucial tumor suppressor function of hSNF5, and its loss-of-function may lead to increased invasiveness and metastatic potential of cancer cells [171].
Another example is the ARID1A (also known as BAF250A, SMARCF1, p270, or hOSA1), which encodes a human homolog of yeast SWI1. The significance of ARID1A loss or mutation in cancer is now subject to intensive investigation. In this context, mutation of the ARID1A gene has been widely described in a broad array of tumor types, including gynecologic ovarian and endotrial carcinomas, pediatric Burkitt lymphoma, gastric carcinoma, breast cancer, and hepatitis B virus-associated hepatocellular carcinoma [172–178]. Consistently, restoring wild-type ARID1A expression in cancer cells that harbor ARID1A mutations is sufficient to suppress cell proliferation and tumor growth in mice [172, 175]. In contrast, ARID1A knockdown significantly promotes the proliferation, migration and invasion of cancer cells [173]. Functional evidence further points out that ARID1A collaborates with p53 to regulate p21WAF1 and SMAD family member 3 [179]. Together, accumulating genomic and functional evidence strongly supports classification of ARID1A as a tumor suppressor [177]. Similar to ARID1A, ARID1B (also known as BAF250B, or hOsa2) also inhibits cell growth and regulates cell cycle arrest through differentially regulating c-myc and p21WAF1 gene expression [180].
In addition, loss or inactivation of BRG1, BRM, BAF155/SMARCC1, BAF180, and BAF200/ARID2 expression represents another mechanism for SWI/SNF complex in the development in human cancers, including hepatitis C infection-related liver cancer, melanoma, lung, pancreatic, skin, and breast cancers [21, 181–188]. Notably, BRG1 and BRM are silenced by different mechanisms. BRG1 is commonly silenced by loss-of-function mutations, whereas epigenetic silencing is a major mechanism for the loss of BRM in human cancer cells [188].
5.2 The ISWI Complex
A well-studied example is the Rsf-1, which plays an important role in cellular growth, survival, and oncogenic transformation, and its up-regulation is closely associated with disease aggressiveness and poor prognosis in patients with various types of human cancers including bladder, colon, nasopharyngeal, gallbladder, oral, and ovarian carcinomas [130, 189–197]. A mechanistic study demonstrates that Rsf-1 interacts and collaborates with cyclin E1 in neoplastic transformation and p53 mutations are a prerequisite for tumour-promoting functions of the RSF/cyclin E1 complex [194]. In contrast, overexpression of Rsf-1 is rare in breast cancer, indicating that Rsf-1 is not a critical gene in breast cancer development [130, 198]. In contrast, SNF2L, a mammalian ISWI ortholog, suppresses cell proliferation and migration in human HeLa cells by attenuating Wnt signaling [199].
5.3 The Mi2/NuRD Complex
Of all the NuRD complex subunits, the MTA family members are best studied in the context of cancer development [92, 93]. MTA1, the founding member of the MTA1 family, has been documented to be overexpressed in a variety of human cancers and is significantly associated with tumor progression and poor clinical outcome [92, 93]. In contrast, the information concerning the expression of MTA2 and MTA3 in human cancers is limited. Like MTA1, increased expression of HDAC1 and HDAC2 has been documented in a variety of human cancers and linked with therapeutic resistance [200–202]. In contrast, lysine-specific demethylase 1, a newly identified component of the Mi-2/NuRD complex, inhibits the invasion of breast cancer cells in vitro and suppresses breast cancer metastatic potential in vivo [203].
5.4 The INO80 Complex
Although the function of the INO80 complex in transcription and DDR, its connection with human cancers is rarely reported. The SRCAP, a homolog of Swr1 in human cells, modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells [204]. Similarly, p400, another Swr1 homolog, inhibits p53-mediated p21WAF1 transcription and the development of premature senescence [205]. P400 is an essential E1A transformation target that plays a major role in the E1A transforming process [206].
6 CRCs in Cancer Therapeutics
Glucocorticoids are used in the curative treatment of acute lymphoblastic leukemia (ALL) and resistance to glucocorticoids is an important adverse prognostic factor in newly diagnosed ALL patients [207]. Emerging evidence suggests that decreased expression of the BRG1, ARID1A, and SNF5 subunits appears to be associated with glucocorticoid resistance in primary ALL cells [207]. Similarly, knockdown of BRG1 and BRM enhances cellular sensitivity to chemotherapy drug cisplatin by regulating efficient repair of the cisplatin DNA lesions [208]. Thus, cisplatin chemotherapy could be more effective in BRG1- and BRM-negative or inactivated tumors (Fig. 16.2a). Consistent with these findings, depletion of CHD4 renders cell significantly hypersensitive to DSB-inducing agents and PARP inhibitors as a consequence of impaired HR repair (Fig. 16.2a) [209]. As loss or mutation of BRG1, BRM and CHD4 has been observed in a variety of human cancers [186, 188, 210, 211], it is highly interesting to examine whether these tumors are sensitive to PAPR inhibitors or other DNA-damaging agents.

Implication of CRCs in cancer therapeutics. Cancer cells with loss, mutation, or inactivation of the CRC components such as of BRG1, BRM and CHD4 are sensitive to DNA damage based radiotherapy and chemotherapy due to impaired DNA repair (a). In contrast, the CRC subunits such as HDACs are over expressed or amplified in cancer cells and promote efficient DNA repair, thus contributing to therapeutic resistance to DNA damage based radiotherapy and chemotherapy (b)
In contrast, overexpression of some components of CRCs is linked with therapeutic resistance (Fig. 16.2b). For instance, Rsf-1 overexpression confers paclitaxel resistance in ovarian cancer cells [212] and is associated with poor therapeutic response in rectal cancer patients treated with neoadjuvant chemoradiation therapy [191] and associated with incomplete response to radiotherapy in patients with nasopharyngeal carcinoma [192]. Notably, the Rsf-1-hSNF2H interaction is essential for developing resistance phenotype in tumors overexpressing Rsf-1 [212]. Thus, inhibition of Rsf-1 activity or disruption of the Rsf-1-hSNF2H interaction has the potential to sensitize cells to paclitaxel in human cancers with Rsf-1 amplication or overexpression. Similarly, HDAC2 is highly expressed in pancreatic ductal adenocarcinoma (PDAC) and confers resistance towards the topoisomerase II inhibitor etoposide in PDAC cells [201]. Consistently, selective inhibition of HDACs synergises with etoposide to induce apoptosis in PDAC cells [201]. In a broader perspective, targeting the CRC-mediated DNA repair pathways might provide unique potential therapeutic avenues for human cancers when used in combination with DNA-damaging chemotherapeutic drugs [213, 214].
7 Conclusions and Perspectives
During the past decades, it has been made great progress in our understanding of the functional roles for ATP-dependent CRCs in transcription and DDR, and it has been increasingly recognized that these CRCs show remarkable diversity and specifity in their contributions to these biological processes. However, it remains unknown why the transcription and DDR pathways need multiple CCRs and whether or how these CRCs exert their functions in these processes in an integrated manner at molecular levels. In addition, the detailed mechanisms by which these CRCs regulate transcription and DDR and drive tumorigenesis and progression are largely unclear.
From a translational perspective, the importance of the CRCs in cancer causation and progression provides new avenues to improve cancer management by targeting the chromatin remodeling machinery. One example is that the CRCs predominantly function in the DNA repair pathways, which may contribute to therapeutic resistance in patients with cancers by enabling cancer cells to survival DNA damage induced by chemotherapeutic agents and radiotherapy [214]. Thus, targeting the CRC components and related DNA repair signaling pathways in human cancers could be efficacious as monotherapy or in combination with DNA-damaging agents [213, 214]. A case in point is the HDACs, whose inhibitors are emerging as promising drugs for cancer therapy that selectively kill cancer cells and sensitize cancer cells to DSB-inducing agents [200]. On the other hand, as components of the CRCs are frequently mutated in human cancers, this unique property of cancer cells gives a great opportunity to screen appropriate patients in clinic for optimum personal therapy using DNA-damaging radiotherapy and chemotherapy. Together, further work that directs to understand the in vivo function and mechanism of action of these CRCs will definitely provide opportunities to discover new therapeutic targets and therapeutic strategies for the treatment of cancer as well as other CRC-related diseases.
References
Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
McGhee JD, Felsenfeld G (1980) Nucleosome structure. Annu Rev Biochem 49:1115–1156
Luger K (2003) Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev 13:127–135
Au TJ, Rodriguez J, Vincent JA, Tsukiyama T (2011) ATP-dependent chromatin remodeling factors tune S phase checkpoint activity. Mol Cell Biol 31:4454–4463
Bao Y, Shen X (2007) SnapShot: chromatin remodeling complexes. Cell 129:632
Tsukiyama T (2002) The in vivo functions of ATP-dependent chromatin-remodelling factors. Nat Rev Mol Cell Biol 3:422–429
Euskirchen G, Auerbach RK, Snyder M (2012) SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J Biol Chem 287:30897–30905
Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C (1999) Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev 13:686–697
Denslow SA, Wade PA (2007) The human Mi-2/NuRD complex and gene regulation. Oncogene 26:5433–5438
Morrison AJ, Shen X (2009) Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol 10:373–384
Li DQ, Kumar R (2010) Mi-2/NuRD complex making inroads into DNA-damage response pathway. Cell Cycle 9:2071–2079
Cairns BR, Kim YJ, Sayre MH, Laurent BC, Kornberg RD (1994) A multisubunit complex containing the SWI1/ADR6, SWI2/SNF2, SWI3, SNF5, and SNF6 gene products isolated from yeast. Proc Natl Acad Sci U S A 91:1950–1954
Cote J, Quinn J, Workman JL, Peterson CL (1994) Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 265:53–60
Imbalzano AN, Kwon H, Green MR, Kingston RE (1994) Facilitated binding of TATA-binding protein to nucleosomal DNA. Nature 370:481–485
Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC et al (1992) Brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68:561–572
Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K (2004) Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol Cell 13:33–43
Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H et al (1996) RSC, an essential, abundant chromatin-remodeling complex. Cell 87:1249–1260
Saha A, Wittmeyer J, Cairns BR (2002) Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev 16:2120–2134
Szerlong H, Saha A, Cairns BR (2003) The nuclear actin-related proteins Arp7 and Arp9: a dimeric module that cooperates with architectural proteins for chromatin remodeling. EMBO J 22:3175–3187
Cairns BR, Erdjument-Bromage H, Tempst P, Winston F, Kornberg RD (1998) Two actin-related proteins are shared functional components of the chromatin-remodeling complexes RSC and SWI/SNF. Mol Cell 2:639–651
Wilson BG, Roberts CW (2011) SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11:481–492
Xue Y, Canman JC, Lee CS, Nie Z, Yang D, Moreno GT et al (2000) The human SWI/SNF-B chromatin-remodeling complex is related to yeast RSC and localizes at kinetochores of mitotic chromosomes. Proc Natl Acad Sci U S A 97:13015–13020
Clapier CR, Cairns BR (2009) The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273–304
Boyer LA, Latek RR, Peterson CL (2004) The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol 5:158–163
Tsukiyama T, Wu C (1995) Purification and properties of an ATP-dependent nucleosome remodeling factor. Cell 83:1011–1020
Tsukiyama T, Daniel C, Tamkun J, Wu C (1995) ISWI, a member of the SWI2/SNF2 ATPase family, encodes the 140 kDa subunit of the nucleosome remodeling factor. Cell 83:1021–1026
Martinez Balbas MA, Tsukiyama T, Gdula D, Wu C (1998) Drosophila NURF 55, a WD repeat protein involved in histone metabolism. Proc Natl Acad Sci U S A 95:132–137
Gdula DA, Sandaltzopoulos R, Tsukiyama T, Ossipow V, Wu C (1998) Inorganic pyrophosphatase is a component of the Drosophila nucleosome remodeling factor complex. Genes Dev 12:3206–3216
Badenhorst P, Voas M, Rebay I, Wu C (2002) Biological functions of the ISWI chromatin remodeling complex NURF. Genes Dev 16:3186–3198
Hamiche A, Kang JG, Dennis C, Xiao H, Wu C (2001) Histone tails modulate nucleosome mobility and regulate ATP dependent nucleosome sliding by NURF. Proc Natl Acad Sci U S A 98:14316–14321
Hamiche A, Sandaltzopoulos R, Gdula DA, Wu C (1999) ATP dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell 97:833–842
Varga Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, Becker PB (1997) Chromatin remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 388:598–602
Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT (1997) ACF, an ISWI containing and ATP utilizing chromatin assembly and remodeling factor. Cell 90:145–155
Bozhenok L, Poot R, Collins N, Varga Weisz P (2004) Functional analysis of ISWI complexes in mammalian cells. Methods Enzymol 377:376–389
Barak O, Lazzaro MA, Lane WS, Speicher DW, Picketts DJ, Shiekhattar R (2003) Isolation of human NURF: a regulator of Engrailed gene expression. EMBO J 22:6089–6100
Poot RA, Dellaire G, Hulsmann BB, Grimaldi MA, Corona DF, Becker PB et al (2000) HuCHRAC, a human ISWI chromatin remodelling complex contains hACF1 and two novel histone fold proteins. EMBO J 19:3377–3387
Strohner R, Nemeth A, Jansa P, Hofmann Rohrer U, Santoro R, Langst G et al (2001) NoRC–a novel member of mammalian ISWI-containing chromatin remodeling machines. EMBO J 20:4892–4900
Bozhenok L, Wade PA, Varga-Weisz P (2002) WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J 21:2231–2241
LeRoy G, Orphanides G, Lane WS, Reinberg D (1998) Requirement of RSF and FACT for transcription of chromatin templates in vitro. Science 282:1900–1904
Bochar DA, Savard J, Wang W, Lafleur DW, Moore P, Cote J et al (2000) A family of chromatin remodeling factors related to Williams syndrome transcription factor. Proc Natl Acad Sci U S A 97:1038–1043
LeRoy G, Loyola A, Lane WS, Reinberg D (2000) Purification and characterization of a human factor that assembles and remodels chromatin. J Biol Chem 275:14787–14790
Banting GS, Barak O, Ames TM, Burnham AC, Kardel MD, Cooch NS et al (2005) CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum Mol Genet 14:513–524
Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D (1998) The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 95:279–289
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W (1998) NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell 2:851–861
Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL (1998) Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395:917–921
Wade PA, Jones PL, Vermaak D, Wolffe AP (1998) A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol 8:843–846
Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 13:1924–1935
Wang HB, Zhang Y (2001) Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res 29:2517–2521
Bao Y, Shen X (2007) INO80 subfamily of chromatin remodeling complexes. Mutat Res 618:18–29
Shen X, Mizuguchi G, Hamiche A, Wu C (2000) A chromatin remodelling complex involved in transcription and DNA processing. Nature 406:541–544
Shen X, Ranallo R, Choi E, Wu C (2003) Involvement of actin-related proteins in ATP-dependent chromatin remodeling. Mol Cell 12:147–155
Saravanan M, Wuerges J, Bose D, McCormack EA, Cook NJ, Zhang X et al (2012) Interactions between the nucleosome histone core and Arp8 in the INO80 chromatin remodeling complex. Proc Natl Acad Sci U S A 109:20883–20888
van Attikum H, Fritsch O, Gasser SM (2007) Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 26:4113–4125
Tsukuda T, Lo YC, Krishna S, Sterk R, Osley MA, Nickoloff JA (2009) INO80-dependent chromatin remodeling regulates early and late stages of mitotic homologous recombination. DNA Repair 8:360–369
Aoyama N, Oka A, Kitayama K, Kurumizaka H, Harata M (2008) The actin-related protein hArp8 accumulates on the mitotic chromosomes and functions in chromosome alignment. Exp Cell Res 314:859–868
Jonsson ZO, Jha S, Wohlschlegel JA, Dutta A (2004) Rvb1p/Rvb2p recruit Arp5p and assemble a functional Ino80 chromatin remodeling complex. Mol Cell 16:465–477
Mizuguchi G, Shen X, Landry J, Wu WH, Sen S, Wu C (2004) ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303:343–348
Ruhl DD, Jin J, Cai Y, Swanson S, Florens L, Washburn MP et al (2006) Purification of a human SRCAP complex that remodels chromatin by incorporating the histone variant H2A.Z into nucleosomes. Biochemistry 45:5671–5677
Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L (2007) p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev 21:1869–1881
Krogan NJ, Keogh MC, Datta N, Sawa C, Ryan OW, Ding H et al (2003) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12:1565–1576
Kobor MS, Venkatasubrahmanyam S, Meneghini MD, Gin JW, Jennings JL, Link AJ et al (2004) A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol 2:E131
Luk E, Ranjan A, Fitzgerald PC, Mizuguchi G, Huang Y, Wei D et al (2010) Stepwise histone replacement by SWR1 requires dual activation with histone H2A.Z and canonical nucleosome. Cell 143:725–736
Edmondson DG, Roth SY (1996) Chromatin and transcription. FASEB J 10:1173–1182
Euskirchen GM, Auerbach RK, Davidov E, Gianoulis TA, Zhong G, Rozowsky J et al (2011) Diverse roles and interactions of the SWI/SNF chromatin remodeling complex revealed using global approaches. PLoS Genet 7:e1002008
Marshall TW, Link KA, Petre-Draviam CE, Knudsen KE (2003) Differential requirement of SWI/SNF for androgen receptor activity. J Biol Chem 278:30605–30613
van de Wijngaart DJ, Dubbink HJ, Molier M, de Vos C, Trapman J, Jenster G (2009) Functional screening of FxxLF-like peptide motifs identifies SMARCD1/BAF60a as an androgen receptor cofactor that modulates TMPRSS2 expression. Mol Endocrinol 23:1776–1786
Inoue H, Furukawa T, Giannakopoulos S, Zhou S, King DS, Tanese N (2002) Largest subunits of the human SWI/SNF chromatin-remodeling complex promote transcriptional activation by steroid hormone receptors. J Biol Chem 277:41674–41685
Link KA, Burd CJ, Williams E, Marshall T, Rosson G, Henry E et al (2005) BAF57 governs androgen receptor action and androgen-dependent proliferation through SWI/SNF. Mol Cell Biol 25:2200–2215
Garcia-Pedrero JM, Kiskinis E, Parker MG, Belandia B (2006) The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J Biol Chem 281:22656–22664
Belandia B, Orford RL, Hurst HC, Parker MG (2002) Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J 21:4094–4103
Villaronga MA, Lopez-Mateo I, Markert L, Espinosa E, Fresno Vara JA, Belandia B (2011) Identification and characterization of novel potentially oncogenic mutations in the human BAF57 gene in a breast cancer patient. Breast Cancer Res Treat 128:891–898
Kiskinis E, Garcia-Pedrero JM, Villaronga MA, Parker MG, Belandia B (2006) Identification of BAF57 mutations in human breast cancer cell lines. Breast Cancer Res Treat 98:191–198
Trotter KW, Archer TK (2008) The BRG1 transcriptional coregulator. Nucl Recept Signal 6:e004
Xi Q, He W, Zhang XH, Le HV, Massague J (2008) Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. J Biol Chem 283:1146–1155
Ross S, Cheung E, Petrakis TG, Howell M, Kraus WL, Hill CS (2006) Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J 25:4490–4502
Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES et al (2005) SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res 65:3542–3547
Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP (2008) SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol 28:3457–3464
Zhu Y, Rowley MJ, Bohmdorfer G, Wierzbicki AT (2013) A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol Cell 49:298–309
Harikrishnan KN, Chow MZ, Baker EK, Pal S, Bassal S, Brasacchio D et al (2005) Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet 37:254–264
Rao M, Casimiro MC, Lisanti MP, D’Amico M, Wang C, Shirley LA et al (2008) Inhibition of cyclin D1 gene transcription by Brg-1. Cell Cycle 7:647–655
Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D et al (2002) Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol 22:5975–5988
Zhang B, Chambers KJ, Faller DV, Wang S (2007) Reprogramming of the SWI/SNF complex for co-activation or co-repression in prohibitin-mediated estrogen receptor regulation. Oncogene 26:7153–7157
Sala A, Toto M, Pinello L, Gabriele A, Di Benedetto V, Ingrassia AM et al (2011) Genome-wide characterization of chromatin binding and nucleosome spacing activity of the nucleosome remodelling ATPase ISWI. EMBO J 30:1766–1777
Mizuguchi G, Tsukiyama T, Wisniewski J, Wu C (1997) Role of nucleosome remodeling factor NURF in transcriptional activation of chromatin. Mol Cell 1:141–150
Song H, Spichiger-Haeusermann C, Basler K (2009) The ISWI-containing NURF complex regulates the output of the canonical Wingless pathway. EMBO Rep 10:1140–1146
Kwon SY, Xiao H, Glover BP, Tjian R, Wu C, Badenhorst P (2008) The nucleosome remodeling factor (NURF) regulates genes involved in Drosophila innate immunity. Dev Biol 316:538–547
Liu YI, Chang MV, Li HE, Barolo S, Chang JL, Blauwkamp TA et al (2008) The chromatin remodelers ISWI and ACF1 directly repress Wingless transcriptional targets. Dev Biol 323:41–52
Lindstrom KC, Vary JC Jr, Parthun MR, Delrow J, Tsukiyama T (2006) Isw1 functions in parallel with the NuA4 and Swr1 complexes in stress-induced gene repression. Mol Cell Biol 26:6117–6129
Fazzio TG, Kooperberg C, Goldmark JP, Neal C, Basom R, Delrow J et al (2001) Widespread collaboration of Isw2 and Sin3-Rpd3 chromatin remodeling complexes in transcriptional repression. Mol Cell Biol 21:6450–6460
Goldmark JP, Fazzio TG, Estep PW, Church GM, Tsukiyama T (2000) The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103:423–433
Whitehouse I, Rando OJ, Delrow J, Tsukiyama T (2007) Chromatin remodelling at promoters suppresses antisense transcription. Nature 450:1031–1035
Lai AY, Wade PA (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11:588–596
Li DQ, Pakala SB, Nair SS, Eswaran J, Kumar R (2012) Metastasis-associated protein 1/nucleosome remodeling and histone deacetylase complex in cancer. Cancer Res 72:387–394
Toh Y, Pencil SD, Nicolson GL (1994) A novel candidate metastasis-associated gene, mta1, differentially expressed in highly metastatic mammary adenocarcinoma cell lines. cDNA cloning, expression, and protein analyses. J Biol Chem 269:22958–22963
Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M et al (2001) Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol 3:30–37
Li DQ, Pakala SB, Reddy SD, Ohshiro K, Peng SH, Lian Y et al (2010) Revelation of p53-independent function of MTA1 in DNA damage response via modulation of the p21 WAF1-proliferating cell nuclear antigen pathway. J Biol Chem 285:10044–10052
Molli PR, Singh RR, Lee SW, Kumar R (2008) MTA1-mediated transcriptional repression of BRCA1 tumor suppressor gene. Oncogene 27:1971–1980
Marzook H, Li DQ, Nair VS, Mudvari P, Reddy SD, Pakala SB et al (2012) Metastasis-associated protein 1 drives tumor cell migration and invasion through transcriptional repression of RING finger protein 144A. J Biol Chem 287:5615–5626
Reddy SD, Pakala SB, Molli PR, Sahni N, Karanam NK, Mudvari P et al (2012) Metastasis-associated protein 1/histone deacetylase 4-nucleosome remodeling and deacetylase complex regulates phosphatase and tensin homolog gene expression and function. J Biol Chem 287:27843–27850
Salot S, Gude R (2013) MTA1-mediated transcriptional repression of SMAD7 in breast cancer cell lines. Eur J Cancer 49:492–499
Ohshiro K, Rayala SK, Wigerup C, Pakala SB, Natha RS, Gururaj AE et al (2010) Acetylation-dependent oncogenic activity of metastasis-associated protein 1 co-regulator. EMBO Rep 11:691–697
Manavathi B, Peng S, Rayala SK, Talukder AH, Wang MH, Wang RA et al (2007) Proc Natl Acad Sci U S A 104:13128–13133
Gururaj AE, Singh RR, Rayala SK, Holm C, den Hollander P, Zhang H et al (2006) MTA1, a transcriptional activator of breast cancer amplified sequence 3. Proc Natl Acad Sci U S A 103:6670–6675
Balasenthil S, Gururaj AE, Talukder AH, Bagheri-Yarmand R, Arrington T, Haas BJ et al (2007) Identification of Pax5 as a target of MTA1 in B-cell lymphomas. Cancer Res 67:7132–7138
Li DQ, Pakala SB, Reddy SD, Ohshiro K, Zhang JX, Wang L et al (2011) Bidirectional autoregulatory mechanism of metastasis-associated protein 1-alternative reading frame pathway in oncogenesis. Proc Natl Acad Sci U S A 108:8791–8796
Sankaran D, Pakala SB, Nair VS, Sirigiri DN, Cyanam D, Ha NH et al (2012) Mechanism of MTA1 protein overexpression-linked invasion: MTA1 regulation of hyaluronan-mediated motility receptor (HMMR) expression and function. J Biol Chem 287:5483–5491
Kumar R, Balasenthil S, Manavathi B, Rayala SK, Pakala SB (2010) Metastasis-associated protein 1 and its short form variant stimulates Wnt1 transcription through promoting its derepression from Six3 corepressor. Cancer Res 70:6649–6658
Reddy SD, Rayala SK, Ohshiro K, Pakala SB, Kobori N, Dash P et al (2011) Multiple coregulatory control of tyrosine hydroxylase gene transcription. Proc Natl Acad Sci U S A 108:4200–4205
Cong L, Pakala SB, Ohshiro K, Li DQ, Kumar R (2011) SUMOylation and SUMO-interacting motif (SIM) of metastasis tumor antigen 1 (MTA1) synergistically regulate its transcriptional repressor function. J Biol Chem 286:43793–43808
Nair SS, Li DQ, Kumar R (2013) A core chromatin remodeling factor instructs global chromatin signaling through multivalent reading of nucleosome codes. Mol Cell 49:704–718
Ebbert R, Birkmann A, Schuller HJ (1999) The product of the SNF2/SWI2 paralogue INO80 of Saccharomyces cerevisiae required for efficient expression of various yeast structural genes is part of a high-molecular-weight protein complex. Mol Microbiol 32:741–751
Cai Y, Jin J, Yao T, Gottschalk AJ, Swanson SK, Wu S et al (2007) YY1 functions with INO80 to activate transcription. Nat Struct Mol Biol 14:872–874
Cho SG, Bhoumik A, Broday L, Ivanov V, Rosenstein B, Ronai Z (2001) TIP49b, a regulator of activating transcription factor 2 response to stress and DNA damage. Mol Cell Biol 21:8398–8413
Kruhlak MJ, Celeste A, Nussenzweig A (2006) Spatio-temporal dynamics of chromatin containing DNA breaks. Cell Cycle 5:1910–1912
Flanagan JF, Peterson CL (1999) A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res 27:2022–2028
Driscoll R, Cimprich KA (2009) HARPing on about the DNA damage response during replication. Genes Dev 23:2359–2365
Kwon J, Morshead KB, Guyon JR, Kingston RE, Oettinger MA (2000) Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol Cell 6:1037–1048
Hara R, Sancar A (2002) The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol Cell Biol 22:6779–6787
Ray A, Mir SN, Wani G, Zhao Q, Battu A, Zhu Q et al (2009) Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol Cell Biol 29:6206–6219
Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN et al (2006) Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 25:3986–3997
Gong F, Fahy D, Liu H, Wang W, Smerdon MJ (2008) Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle 7:1067–1074
Liang B, Qiu J, Ratnakumar K, Laurent BC (2007) RSC functions as an early double-strand-break sensor in the cell’s response to DNA damage. Curr Biol 17:1432–1437
Oum JH, Seong C, Kwon Y, Ji JH, Sid A, Ramakrishnan S et al (2011) RSC facilitates Rad59-dependent homologous recombination between sister chromatids by promoting cohesin loading at DNA double-strand breaks. Mol Cell Biol 31:3924–3937
Chambers AL, Brownlee PM, Durley SC, Beacham T, Kent NA, Downs JA (2012) The two different isoforms of the RSC chromatin remodeling complex play distinct roles in DNA damage responses. PLoS One 7:e32016
Chai B, Huang J, Cairns BR, Laurent BC (2005) Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 19:1656–1661
Smeenk G, Wiegant WW, Marteijn JA, Luijsterburg MS, Sroczynski N, Costelloe T et al (2013) Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J Cell Sci 126:889–903
Erdel F, Rippe K (2011) Binding kinetics of human ISWI chromatin-remodelers to DNA repair sites elucidate their target location mechanism. Nucleus 2:105–112
Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S et al (2001) ATP-dependent chromatin remodeling facilitates nucleotide excision repair of UV-induced DNA lesions in synthetic dinucleosomes. EMBO J 20:2004–2014
Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R et al (2010) The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell 40:976–987
Mao TL, Hsu CY, Yen MJ, Gilks B, Sheu JJ, Gabrielson E et al (2006) Expression of Rsf-1, a chromatin-remodeling gene, in ovarian and breast carcinoma. Hum Pathol 37:1169–1175
Sheu JJ, Guan B, Choi JH, Lin A, Lee CH, Hsiao YT et al (2010) Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem 285:38260–38269
Kshirsagar M, Jiang W, Shih IM (2012) DNA damage response is prominent in ovarian high-grade serous carcinomas, especially those with Rsf-1 (HBXAP) overexpression. J Oncol 2012:621685
Schmidt DR, Schreiber SL (1999) Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry 38:14711–14717
van Haaften G, Romeijn R, Pothof J, Koole W, Mullenders LH, Pastink A et al (2006) Identification of conserved pathways of DNA-damage response and radiation protection by genome-wide RNAi. Curr Biol 16:1344–1350
Li DQ, Ohshiro K, Reddy SD, Pakala SB, Lee MH, Zhang Y et al (2009) E3 ubiquitin ligase COP1 regulates the stability and functions of MTA1. Proc Natl Acad Sci U S A 106:17493–17498
Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE et al (2010) A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 107:18475–18480
Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ et al (2010) The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol 190:731–740
Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP (2010) Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 29:3130–3139
Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H (2010) The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol 190:741–749
Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S et al (2010) Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 17:1144–1151
Huber A, Bai P, de Murcia JM, de Murcia G (2004) PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair 3:1103–1108
Smeenk G, van Attikum H (2011) NuRD alert! NuRD regulates the DNA damage response. Epigenomics 3:133–135
Pegoraro G, Kubben N, Wickert U, Gohler H, Hoffmann K, Misteli T (2009) Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol 11:1261–1267
Zhang Y (2011) Biology of the Mi-2/NuRD Complex in SLAC (Stemness, Longevity/Ageing, and Cancer). Gene Regul Syst Bio 5:1–26
Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE et al (2004) INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119:767–775
Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N et al (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16:979–990
Gospodinov A, Vaissiere T, Krastev DB, Legube G, Anachkova B, Herceg Z (2011) Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol 31:4735–4745
Jiang Y, Wang X, Bao S, Guo R, Johnson DG, Shen X et al (2010) INO80 chromatin remodeling complex promotes the removal of UV lesions by the nucleotide excision repair pathway. Proc Natl Acad Sci U S A 107:17274–17279
Sarkar S, Kiely R, McHugh PJ (2010) The Ino80 chromatin-remodeling complex restores chromatin structure during UV DNA damage repair. J Cell Biol 191:1061–1068
Vincent JA, Kwong TJ, Tsukiyama T (2008) ATP-dependent chromatin remodeling shapes the DNA replication landscape. Nat Struct Mol Biol 15:477–484
Niimi A, Chambers AL, Downs JA, Lehmann AR (2012) A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res 40:7393–7403
Kato D, Waki M, Umezawa M, Aoki Y, Utsugi T, Ohtsu M et al (2012) Phosphorylation of human INO80 is involved in DNA damage tolerance. Biochem Biophys Res Commun 417:433–438
Falbo KB, Alabert C, Katou Y, Wu S, Han J, Wehr T et al (2009) Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat Struct Mol Biol 16:1167–1172
Gunduz E, Gunduz M, Nagatsuka H, Beder L, Demircan K, Tamamura R et al (2006) Epigenetic alterations of BRG1 leads to cancer development through its nuclear-cytoplasmic shuttling abnormalities. Med Hypotheses 67:1313–1316
Reisman D, Glaros S, Thompson EA (2009) The SWI/SNF complex and cancer. Oncogene 28:1653–1668
Hornick JL, Dal Cin P, Fletcher CD (2009) Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33:542–550
Grand F, Kulkarni S, Chase A, Goldman JM, Gordon M, Cross NC (1999) Frequent deletion of hSNF5/INI1, a component of the SWI/SNF complex, in chronic myeloid leukemia. Cancer Res 59:3870–3874
Decristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE (2001) Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol 186:136–145
Lin H, Wong RP, Martinka M, Li G (2009) Loss of SNF5 expression correlates with poor patient survival in melanoma. Clin Cancer Res 15:6404–6411
Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira MR, Pilotti S et al (2005) SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res 65:4012–4019
Sevenet N, Lellouch-Tubiana A, Schofield D, Hoang-Xuan K, Gessler M, Birnbaum D et al (1999) Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Hum Mol Genet 8:2359–2368
Roberts CW, Leroux MM, Fleming MD, Orkin SH (2002) Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell 2:415–425
Sansam CG, Roberts CW (2006) Epigenetics and cancer: altered chromatin remodeling via Snf5 loss leads to aberrant cell cycle regulation. Cell Cycle 5:621–624
Xu Y, Yan W, Chen X (2010) SNF5, a core component of the SWI/SNF complex, is necessary for p53 expression and cell survival, in part through eIF4E. Oncogene 29:4090–4100
Medjkane S, Novikov E, Versteege I, Delattre O (2004) The tumor suppressor hSNF5/INI1 modulates cell growth and actin cytoskeleton organization. Cancer Res 64:3406–3413
Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LM, Mohd-Sarip A et al (2004) P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem 279:3807–3816
Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV (2011) Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res 71:3225–3235
Reincke BS, Rosson GB, Oswald BW, Wright CF (2003) INI1 expression induces cell cycle arrest and markers of senescence in malignant rhabdoid tumor cells. J Cell Physiol 194:303–313
DelBove J, Kuwahara Y, Mora-Blanco EL, Godfrey V, Funkhouser WK, Fletcher CD et al (2009) Inactivation of SNF5 cooperates with p53 loss to accelerate tumor formation in Snf5(+/−);p53(+/−) mice. Mol Carcinog 48:1139–1148
Isakoff MS, Sansam CG, Tamayo P, Subramanian A, Evans JA, Fillmore CM et al (2005) Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A 102:17745–17750
Caramel J, Quignon F, Delattre O (2008) RhoA-dependent regulation of cell migration by the tumor suppressor hSNF5/INI1. Cancer Res 68:6154–6161
Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S et al (2012) An integrated genomic approach identifies ARID1A as a candidate tumor-suppressor gene in breast cancer. Oncogene 31:2090–2100
Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N et al (2012) Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet 44:1117–1121
Abe H, Maeda D, Hino R, Otake Y, Isogai M, Ushiku AS et al (2012) ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Arch 461:367–377
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T et al (2010) ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363:1532–1543
Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW et al (2011) Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol 224:328–333
Wu JN, Roberts CW (2013) ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Dis 3:35–43
Giulino-Roth L, Wang K, Macdonald TY, Mathew S, Tam Y, Cronin MT et al (2012) Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood 120:5181–5184
Guan B, Wang TL, Shih IM (2011) ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res 71:6718–6727
Inoue H, Giannakopoulos S, Parkhurst CN, Matsumura T, Kono EA, Furukawa T et al (2011) Target genes of the largest human SWI/SNF complex subunit control cell growth. Biochem J 434:83–92
DelBove J, Rosson G, Strobeck M, Chen J, Archer TK, Wang W et al (2011) Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics 6:1444–1453
Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP et al (2012) A landscape of driver mutations in melanoma. Cell 150:251–263
Manceau G, Letouze E, Guichard C, Didelot A, Cazes A, Corte H et al (2013) Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int J Cancer 132:2217–2221
Shain AH, Giacomini CP, Matsukuma K, Karikari CA, Bashyam MD, Hidalgo M et al (2012) Convergent structural alterations define SWItch/Sucrose NonFermentable (SWI/SNF) chromatin remodeler as a central tumor suppressive complex in pancreatic cancer. Proc Natl Acad Sci U S A 109:E252–E259
Bock VL, Lyons JG, Huang XX, Jones AM, McDonald LA, Scolyer RA et al (2011) BRM and BRG1 subunits of the SWI/SNF chromatin remodelling complex are downregulated upon progression of benign skin lesions into invasive tumours. Br J Dermatol 164:1221–1227
Rodriguez-Nieto S, Canada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F et al (2011) Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Hum Mutat 32:E1999–E2017
Xia W, Nagase S, Montia AG, Kalachikov SM, Keniry M, Su T et al (2008) BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res 68:1667–1674
Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D (2007) The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene 26:7058–7066
Liang PI, Wu LC, Sheu JJ, Wu TF, Shen KH, Wang YH et al (2012) Rsf-1/HBXAP overexpression is independent of gene amplification and is associated with poor outcome in patients with urinary bladder urothelial carcinoma. J Clin Pathol 65:802–807
Fang FM, Li CF, Huang HY, Lai MT, Chen CM, Chiu IW et al (2011) Overexpression of a chromatin remodeling factor, RSF-1/HBXAP, correlates with aggressive oral squamous cell carcinoma. Am J Pathol 178:2407–2415
Lin CY, Tian YF, Wu LC, Chen LT, Lin LC, Hsing CH et al (2012) Rsf-1 expression in rectal cancer: with special emphasis on the independent prognostic value after neoadjuvant chemoradiation. J Clin Pathol 65:687–692
Tai HC, Huang HY, Lee SW, Lin CY, Sheu MJ, Chang SL et al (2012) Associations of Rsf-1 overexpression with poor therapeutic response and worse survival in patients with nasopharyngeal carcinoma. J Clin Pathol 65:248–253
Chen TJ, Huang SC, Huang HY, Wei YC, Li CF (2011) Rsf-1/HBXAP overexpression is associated with disease-specific survival of patients with gallbladder carcinoma. APMIS 119:808–814
Sheu JJ, Choi JH, Guan B, Tsai FJ, Hua CH, Lai MT et al (2013) Rsf-1, a chromatin remodelling protein, interacts with cyclin E1 and promotes tumour development. J Pathol 229:559–568
Maeda D, Chen X, Guan B, Nakagawa S, Yano T, Taketani Y et al (2011) Rsf-1 (HBXAP) expression is associated with advanced stage and lymph node metastasis in ovarian clear cell carcinoma. Int J Gynecol Pathol 30:30–35
Davidson B, Trope CG, Wang TL, Shih IM (2006) Expression of the chromatin remodeling factor Rsf-1 is upregulated in ovarian carcinoma effusions and predicts poor survival. Gynecol Oncol 103:814–819
Shih Ie M, Sheu JJ, Santillan A, Nakayama K, Yen MJ, Bristow RE et al (2005) Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci U S A 102:14004–14009
Davidson B, Wang TL, Shih Ie M, Berner A (2008) Expression of the chromatin remodeling factor Rsf-1 is down-regulated in breast carcinoma effusions. Hum Pathol 39:616–622
Eckey M, Kuphal S, Straub T, Rummele P, Kremmer E, Bosserhoff AK et al (2012) Nucleosome remodeler SNF2L suppresses cell proliferation and migration and attenuates Wnt signaling. Mol Cell Biol 32:2359–2371
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51
Fritsche P, Seidler B, Schuler S, Schnieke A, Gottlicher M, Schmid RM et al (2009) HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 58:1399–1409
Weichert W, Roske A, Niesporek S, Noske A, Buckendahl AC, Dietel M et al (2008) Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res 14:1669–1677
Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W et al (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138:660–672
Slupianek A, Yerrum S, Safadi FF, Monroy MA (2010) The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. J Cell Physiol 224:369–375
Chan HM, Narita M, Lowe SW, Livingston DM (2005) The p400 E1A-associated protein is a novel component of the p53 – > p21 senescence pathway. Genes Dev 19:196–201
Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V et al (2001) The p400 complex is an essential E1A transformation target. Cell 106:297–307
Pottier N, Yang W, Assem M, Panetta JC, Pei D, Paugh SW et al (2008) The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J Natl Cancer Inst 100:1792–1803
Kothandapani A, Gopalakrishnan K, Kahali B, Reisman D, Patrick SM (2012) Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp Cell Res 318:1973–1986
Pan MR, Hsieh HJ, Dai H, Hung WC, Li K, Peng G et al (2012) Chromodomain helicase DNA-binding protein 4 (CHD4) regulates homologous recombination DNA repair, and its deficiency sensitizes cells to poly(ADP-ribose) polymerase (PARP) inhibitor treatment. J Biol Chem 287:6764–6772
Kim MS, Chung NG, Kang MR, Yoo NJ, Lee SH (2011) Genetic and expressional alterations of CHD genes in gastric and colorectal cancers. Histopathology 58:660–668
Le Gallo M, O’Hara AJ, Rudd ML, Urick ME, Hansen NF, O’Neil NJ et al (2012) Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet 44:1310–1315
Choi JH, Sheu JJ, Guan B, Jinawath N, Markowski P, Wang TL et al (2009) Functional analysis of 11q13.5 amplicon identifies Rsf-1 (HBXAP) as a gene involved in paclitaxel resistance in ovarian cancer. Cancer Res 69:1407–1415
Curtin NJ (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 12:801–817
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8:193–204
Acknowledgement
We sincerely apologize to all those colleagues whose important work has not been cited owing to space limitations. We would like to thank all members of the Kumar laboratory for helpful discussion and comments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Li, DQ., Kumar, R. (2014). Therapeutic Significance of Chromatin Remodeling Complexes in Cancer. In: Kumar, R. (eds) Nuclear Signaling Pathways and Targeting Transcription in Cancer. Cancer Drug Discovery and Development. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-8039-6_16
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8039-6_16
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-8038-9
Online ISBN: 978-1-4614-8039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)