
Abstract
Stearoyl-CoA desaturases are a family of enzymes that play a crucial role in the endogenous synthesis of monounsaturated fatty acids, also known as de novo lipogenesis. Of the four known SCD isoforms, stearoyl-CoA desaturase 1 (SCD1) is the most predominant and most ubiquitously expressed, with the highest induction in lipogenic tissues such as liver and adipose tissue. In this chapter, we will discuss the effects of SCD1 and its substrates, saturated fatty acids, on three levels as shown in Fig. 7.4. First, we will introduce SCD1 and its role in lipid biosynthesis. Next, we will cover intermediate responses to SCD1 modulation, such as inflammation and ER stress. Finally, we will expand our discussion of SCD1 into the context of vascular diseases such as atherosclerosis and vascular calcification. The studies discussed in this chapter are significant because cardiovascular diseases are some of the leading causes of mortality worldwide. In these diseases, lipids such as fatty acids and their metabolites can accumulate and lead to cellular dysfunction and death, known as lipotoxicity. Since SCD1 plays such an important role in fatty acid formation, a deeper understanding of this pivotal enzyme may significantly contribute to further advances in the treatment of lipid-related diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Endoplasmic Reticulum Stress
- Unfold Protein Response
- Vascular Calcification
- Unfold Protein Response Activation
- SCD1 Activity
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
SCD1 and Lipid Biosynthesis
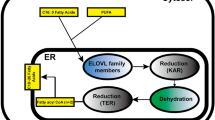
Stearoyl-CoA desaturase 1 (SCD1) is an enzyme involved in the de novo lipogenesis of fatty acids (Fig. 7.1). In the first step of this biosynthetic pathway, acetyl-CoA carboxylase converts acetyl-CoA to malonyl-CoA. Next, malonyl-CoA is converted into the saturated fatty acid (SFA) palmitate (C16:0), through a multiple-step process catalyzed by fatty acid synthase (FAS). Elongases convert palmitate into stearate (C18:0), the SFA substrate for SCD. Following elongation, SCD catalyzes the conversion of stearate into the monounsaturated fatty acid (MUFA) oleate (C18:1). MUFAs such as oleate serve as major substrates for the synthesis of complex lipids such as phospholipids, cholesterol esters, and wax esters. Since SCD1 controls the delicate balance between SFAs and MUFAs, SCD1 plays an important role in determining the composition of fatty acids and complex lipids in the cell. An SFA to MUFA ratio that is either excessive or insufficient can lead to severe metabolic consequences, including atherosclerosis, obesity, and type II diabetes (Sampath et al. 2007). To study global and tissue-specific effects of SCD1 on these diseases, researchers have developed several models that have a deletion or decreased expression of SCD1. These include conditional knockout mouse models (SCD1−/−) or transient knockdown mouse models using antisense oligonucleotide technology (SCD1 ASO). Inhibition of SCD1 in these models leads to an increase in SFAs such as stearate and a decrease in MUFAs such as oleate (Sampath et al. 2007). These are therefore very useful models to study the physiological role of SFAs and MUFAs.

SCD1 in de novo lipogenesis. Dietary carbohydrates such as glucose and fructose are major substrates of hepatic de novo lipogenesis. The carbohydrates are converted to citrate through glycolysis and citric acid cycle. Citrate is converted back to acetyl-CoA in the reaction of ATP citrate lyase (ACL). Acetyl-CoA carboxylase (ACC) catalyzes the irreversible conversion of the 2-carbon acetyl-CoA to the 3-carbon intermediate, malonyl-CoA. Malonyl-CoA serves as the precursor for the endogenous synthesis of fatty acids via the fatty acid synthase (FAS) multienzyme complex. FAS catalyzes seven cycles of sequential condensation, reduction, and dehydration reactions to form the 16-carbon saturated fatty acid (SFA), palmitate (16:0). Further elongation generally occurs through the actions of microsomal elongase (Elovl6) to form stearate (18:0). Cellular levels of stearate are regulated by a lipogenic enzyme, stearoyl-CoA desaturase, which catalyzes the conversion of stearate to oleate. More than 60 % of stearate derived from foods or de novo lipogenesis is converted into oleate. The reaction of stearoyl-CoA desaturase involves the introduction of the first cis-double bond in the ∆9 position in a spectrum of saturated fatty acyl-CoAs
SCD1 and Inflammation
Inflammation is a key event in the pathogenesis of many vascular diseases, including atherosclerosis. SCD1 may have an intriguing role in macrophage-mediated inflammation. Studies on β amyloid peptide and toll-like receptor 4 (TLR4)/NF-κB signaling demonstrate that SCD1 inhibition accelerates inflammation, whereas other studies report that SCD1 inhibition exerts no effect on inflammation (Macdonald et al. 2009a, b; Brown et al. 2008; Uryu et al. 2003; Liu et al. 2010, 2011). In studies supporting the pro-inflammatory role of fatty acids, accumulation of SFAs was shown to directly induce inflammation through TLR4 and NF-κB signaling (Lee et al. 2001, 2003a, b, 2004). In this pathway, SFAs activate TLR4, a pattern-recognition receptor that plays a role in activating innate immunity and inflammation (Fig. 7.2). TLR4 then induces NF-κB, a protein complex involved in cellular responses to harmful stimuli such as stress, free radicals, and toxic lipids. NF-κB activation increases the transcription of most enzymes in the de novo synthesis of ceramides, which are sphingosines covalently linked to a fatty acid. De novo ceramide synthesis uses SFAs, but not unsaturated fatty acids. Therefore, an increase in SFAs due to SCD1 inhibition leads to ceramide accumulation. Ceramides potently inhibit Akt, a serine/threonine kinase that upregulates nutrient storage and inhibits apoptosis. Increased ceramide and the resulting Akt inhibition lead to the activation of pro-apoptotic pathways through enzymes such as caspase-9 (Holland et al. 2007; Summers 2006). This pathway seems to be differentially activated in various tissues, since it has been shown that stearate activates inflammatory genes in macrophages but not in adipose tissue. In addition to TLR4, it has also been shown that SFAs induce macrophage inflammation through TLR2 when the receptor is dimerized with TLR6 or TLR1. In patients with atherosclerosis, TLR4 signaling is increased particularly in endothelial cells and macrophages, suggesting that the TLR4-NF-κB pathway plays an important role in atherosclerotic lesions.

SFAs in TLR4 signaling. SFAs directly bind and activate TLR4, resulting in inflammation. TLR4 toll-like receptor-4, MyD88 myeloid differentiation primary response gene-88, IRAK-1 interleukin-1 receptor-associated kinase 1, TRAF6 TNF receptor-associated factor-6, IKK IκB kinase, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells
In addition to the TLR4 studies using diets high in SFAs mentioned above, other studies actually inhibited SCD1 itself to see the effects of the resulting SFA accumulation on inflammation (Brown et al. 2008; Flowers et al. 2008). One of these early studies examining the β amyloid peptide suggested that SCD1 inhibition promotes inflammation in macrophages (Uryu et al. 2003). Using an oligonucleotide microarray analysis, SCD1 was found to be specifically and significantly upregulated by β amyloid peptide (Aβ) during Aβ-induced macrophage activation. However, this study did not propose a mechanism explaining how SCD1 expression was correlated with macrophage inflammation (Uryu et al. 2003).
The pro-inflammatory effects of SCD1 inhibition were also confirmed in related studies on toll-like receptor signaling (Brown et al. 2008). Using ASO-mediated knockdown of SCD1, a study reported that SCD1-knockdown mice demonstrated increases in SFA-enriched plasma lipoproteins and TLR4 hypersensitivity. SCD1 inhibition leads to an accumulation of SFAs, which serve as ligands for TLR4 and consequently mediate atherosclerotic progression by activating TLR4-driven pro-inflammatory responses in macrophages. In addition to activating the innate immune response, transmembrane receptor TLR4 is highly expressed in macrophages present in atherosclerotic plaques and plays an important role in vascular endothelial cell activation, inflammatory cytokine recruitment, and macrophage apoptosis (Brown et al. 2008). In another study, SCD1 inhibition similarly leads to TLR4 activation and increased atherosclerosis, but these effects appear to be reversible by dietary supplementation of ω-3 polyunsaturated fatty acids (PUFAs) from fish oil. Using a hyperlipidemic mouse model, a combination of SCD1 ASO and fish oil treatments decreased both metabolic syndrome and atherosclerosis (Brown et al. 2010).
Although the mechanisms by which SFAs induce TLR4 have yet to be elucidated, one putative explanation is that SFAs change lipid and protein composition of microdomains on raft membranes. Changes in membrane composition will affect TLR signaling since activated TLR4 is thought to translocate to these domains. SFAs will allow for easier TLR4 trafficking to domains, while PUFAs will disturb lipid composition and raft order thus interfering with TLR4 recruitment to rafts. As seen in the study using SCD1 ASO and fish oil-derived ω-3 PUFAs, dual therapy of SCD1 inhibitors and other anti-inflammatory agents may be effective in reducing both metabolic syndrome and atherosclerosis (Brown et al. 2010). Although TLR4 antagonists appear to be a putative treatment for atherosclerotic progression in mice models, the role of TLR4 in human atherosclerosis has been debated. Therefore, further studies will be required to determine whether TLR4 activation is necessary for SFA-induced atherosclerosis and whether these results can be applied clinically.
In contrast to the Aβ and TLR4 studies, others reported that SCD1 inhibition exerts no effect on inflammation. A recent study compared peritoneal macrophages from SCD1-deficient mice and wild-type mice but reported no differences between groups when the macrophages were treated with LPS (Liu et al. 2010). In a similar study using peritoneal macrophages from SCD1-deficient mice, macrophage inflammation also did not differ from the control, although inflammatory changes in the skin and plasma were observed (MacDonald et al. 2009b). One plausible explanation for this discrepancy may be due to the presence of more than one SCD isoform. SCD2 is more highly expressed in macrophages than SCD1 and therefore might be compensating for the low expression of SCD1 in macrophages. The expression of SCD1 and SCD2 in different cell types may be responsible for differences in inflammatory responses (Liu et al. 2010, 2011; Liu and Ntambi 2009). Although it is well-known that SCD1 deficiency exerts harmful pro-inflammatory effects in several cell types including skin and β-cells (Zheng et al. 1999; Flowers et al. 2007), the role of SCD1 in macrophage inflammation is still ambiguous. Further investigation will be necessary to determine the complex regulation of SCD1 in macrophage inflammation.
SCD and ER Stress
A second method by which SCD1 inhibition and SFAs promote lipid-related diseases is by inducing stress in the endoplasmic reticulum (ER), a central organelle for protein processing and lipid synthesis (Fig. 7.3). ER stress is an accumulation of misfolded or unfolded proteins in the endoplasmic reticulum due to adverse conditions such as ischemia, hypoxia, heat shock, oxidative stress, or depletion of stored ER calcium. ER stress triggers a restorative/corrective pathway known as the unfolded protein response (UPR), which attempts to restore proper ER function. The UPR alleviates ER stress through three main compensatory mechanisms: (1) decreasing the load of proteins that enter the ER by suppressing translation, (2) raising protein-folding capacity by increasing the number of available chaperones, or (3) increasing degradation of misfolded proteins through a ubiquitin-proteasome pathway. If all three compensatory mechanisms fail and homeostasis cannot be reestablished, prolonged ER stress and UPR activation can trigger apoptosis. The UPR comprises three branches mediated by ER-resident transmembrane proteins: PKR-like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor-6 (ATF6). These three ER-resident signaling proteins are activated either by binding to a protein chaperone called BiP or by directly sensing the presence of misfolded proteins in the ER lumen. Following activation, these ER-resident proteins induce downstream effectors such as XBP-1 (X-box binding protein 1) and CHOP (C/EBP-homologous protein) (Flowers et al. 2007; Tabas and Ron 2011; Walter and Ron 2011).

ER stress signaling. Upon accumulation of unfolded proteins and lipids such as SFAs in the ER, three ER stress sensors are activated and initiate signal transduction events that control cell survival or death. PERK PKR-like ER kinase, IRE1 inositol requiring enzyme 1, ATF6 activating transcription factor-6, eIF2α eukaryotic translation initiation factor 2α, ATF4 activating transcription factor-4, CHOP C/EBP-homologous protein, Ocn Osteocalcin, Osx Osterix, uXBP-1 unspliced X-box binding protein 1, sXBP-1 spliced X-box binding protein 1, TRAF2 TNF receptor-associated factor 2, JNK Jun N-terminal kinase, Bax Bcl-2-associated X protein, Bak Bcl-2 homologous antagonist/killer, S1P site-1 protease, S2P site-2 protease
Recent studies have shown that abnormal lipid metabolism activates ER stress and prolonged UPR in atherosclerotic plaques. In normal, healthy cells, macrophages transport lipoprotein-cholesterol to the ER, where the cholesterol is esterified (Scull and Tabas 2011). However, in atherosclerotic lesions, this process is interrupted and massive amounts of free cholesterol accumulate. SCD can regulate intracellular free cholesterol levels, since its product oleate is a preferred substrate for cholesteryl ester synthesis. SCD1−/− mice have higher levels of free cholesterol in the skin, liver, and aorta compared to wild-type mice. In addition to this increase in free cholesterol, lesion exposure to SFAs such as stearic acid induces ER stress, leading to macrophage apoptosis in atherosclerotic plaques (Anderson et al. 2012). Several ER stress transducers and their downstream effectors have been implicated in the development of atherosclerosis. However, better understanding of the molecular mechanisms behind this induction is required. Expanding knowledge of how lipids induce ER stress and atherosclerosis may lead to better treatment of atherosclerosis and associated vascular calcification.
The PERK branch of the UPR is activated by SCD1 inhibition and the resulting accumulation of stearate (Masuda et al. 2012). In this UPR branch, PERK oligomerizes and phosphorylates itself before phosphorylating the α subunit of eukaryotic initiation factor 2 (eIF2). Phosphorylation of this α subunit renders eIF2α inactive, resulting in the inhibition of 80S ribosomal assembly and translation as well as a reduction in the protein load entering the ER. Although most protein translation is reduced when eIF2α is phosphorylated, translation of activating transcription factor-4 (ATF4) increases because ATF4 possesses upstream open reading frames that are bypassed only when eIF2α is phosphorylated. ATF4 activates its targets including the pro-apoptotic CHOP and proteins involved in osteoblast differentiation such as Osteocalcin (Ocn) and Osterix (Osx) (Masuda et al. 2012). During prolonged ER stress, CHOP induces apoptosis through BH3-only proteins, Bcl-2-associated X protein (Bax), and Bcl-2 homologous antagonist/killer (Bak). Using in vitro models, our group has previously shown that SCD1 inhibition increases stearate, leading to increased ATF4 and CHOP expression through the PERK-eIF2α pathway. We found that stearate most potently induces vascular calcification in preference to other fatty acids. ATF4 knockdown inhibits stearate-induced vascular calcification. We therefore concluded that UPR activation of the PERK-eIF2α-ATF4 branch contributes to stearate-mediated vascular calcification (Masuda et al. 2012).
A second branch of the UPR, the IRE1 branch, has also been shown to be activated by SFA accumulation (Wei et al. 2006). In this branch, IRE1 cleaves XBP-1, which then activates the transcription of UPR genes for chaperones, lipid synthesis, and ER-associated degradation of misfolded proteins. IRE1 can also recruit TRAF2, leading to the activation of caspase-12 and Jun N-terminal kinase (JNK). Apoptosis can then be activated either by the TRAF2-JNK pathway or by pro-apoptotic proteins Bax and Bak, which are targets of IRE1 as well as CHOP (Choi et al. 2011). Our study showed that SCD1 inhibition induces the expression of other ER stress targets including spliced XBP-1 in vascular smooth muscle cells (Masuda et al. 2012). In studies on β cells, SFAs such as palmitate were shown to induce the IRE1 pathway, leading to an increase in the IRE1-dependent JNK response (Ron and Walter 2007). Other studies have also confirmed that both stearate and palmitate can induce the IRE1 branch of the UPR, leading to an increase in pro-apoptotic proteins such as caspase-3 (Wei et al. 2006). These changes were accompanied by increases in other UPR-related proteins including ATF4, CHOP, chaperone GRP78, and growth arrest and DNA damage-inducible protein (GADD34). Although β-cell ceramide synthesis has been correlated with fatty acid-induced apoptosis, this study showed that UPR activation by stearate and palmitate could occur independently of de novo ceramide synthesis. Taken together, these studies demonstrate that another UPR branch, the IRE1 pathway, can also mediate the effects of SFAs on apoptosis.
PERK and IRE1 branches of the UPR have been found to link lipotoxic signals to atherosclerosis and other diseases (Scull and Tabas 2011; Erbay et al. 2009). ER stress signaling is significantly activated in animal models of atherosclerosis and other lipid-mediated disorders and human atherosclerotic lesions (Myoishi et al. 2007; Duan et al. 2009). CHOP deficiency decreases atherosclerotic plaque lesions, cell death lesions, and vascular remodeling in hyperlipidemic ApoE−/− and LDLR−/− mice (Thorp et al. 2009; Gao et al. 2011). Chemical chaperones such as 4-phenyl butyrate and taurine-conjugated chenodeoxycholic acid were shown to reduce atherosclerotic lesion area, accompanied with reduced ER stress (Erbay et al. 2009). In a study examining macrophage ER stress, toxic lipids such as oxidized LDL and palmitate (C16:0) induced ER stress through a macrophage lipid chaperone called fatty acid-binding protein-4 (aP2). Their results suggested that toxic lipids accumulate and upregulate aP2, which then inhibits de novo lipogenesis and leads to activation of UPR-mediated apoptosis (Erbay et al. 2009). Other studies have observed the induction of ER chaperone GRP78, PERK, and CHOP, following lipid accumulation in mouse and rat models on high fat diets (Brookheart et al. 2009). In rat models of nonalcoholic fatty liver disease, high SFA diets and prolonged ER stress have also been shown to activate caspase-3, a crucial death protease that mediates apoptosis. Collectively, these studies suggest that dyslipidemia plays a pivotal role in inducing ER stress and apoptosis. Although links between dyslipidemia and ER stress have been found, mechanisms by which SFAs induce ER stress have not been fully elucidated. Several studies have proposed potential mechanisms. In a study using SCD1−/− mice on very low fat diets, SCD1 loss led to increases in spliced XBP-1, CHOP, and ATF3 (another ER stress-induced transcription factor) in the liver. In this study, Flowers et al. postulated that SCD1 inhibition activates ER stress by altering hepatic fatty acid composition of cellular membranes, leading to impaired function of membrane transport proteins (Flowers et al. 2008). Another explanation attributes the effects of SFAs on ER stress to unsaturated fatty acid depletion, which may contribute to oxidative stress (Flowers et al. 2008). Our studies have shown that the SFA stearate must be converted to its CoA conjugated form (stearoyl-CoA) in order to activate ER stress and vascular calcification (Masuda et al. 2012). We hypothesize that stearoyl-CoA is incorporated into an ER membrane lipid. This altered membrane composition is then sensed by ER stress transducers, which activate the UPR. Although current evidence links SCD1 deficiency to ER stress, the necessity for defining mechanisms behind ER stress induction warrants further investigation.
In addition to SFAs themselves, downstream products of fatty acids such as phospholipids have been shown to regulate ER stress. For example, previous studies have demonstrated that phospholipids play a role in activating ER stress (Testerink et al. 2009; Van Der Sanden et al. 2003; Fu et al. 2011). The two most abundant phospholipids in the ER membrane are phosphatidylcholine (PC) and phosphatidylethanolamine (PE). When lipogenesis or dietary lipid intake is increased, PC is the most common phospholipid component for packaging and storing lipid droplets and lipoproteins. In addition, SCD inhibition changes intracellular PC and PE levels (Dobrzyn et al. 2005). PC synthesis is catalyzed by choline-phosphate cytidylyltransferase A (PCTY1a), while PC to PE conversion is regulated by phosphatidylethanolamine N-methyl transferase (PEMT) (Fu et al. 2011). In ER samples isolated from the obese liver tissue of leptin-deficient mice, the PC/PE ratio was higher than in the control. Since the PC/PE ratio in lean controls was the same as that found in their diet, the increased PC/PE ratio in obese hepatic ER seemed to result from de novo lipogenesis rather than from dietary sources (Fu et al. 2011). Similar to free cholesterol, higher PC levels in ER membranes were shown to inhibit the activity of sarco/endoplasmic reticulum calcium ATPase (SERCA), a transport protein that maintains calcium homeostasis. Altered calcium levels caused by SERCA dysfunction impaired protein-folding chaperones such as BiP and calnexin, and thus activate ER stress. This upregulation of ER stress was evidenced by IRE1α and eIF2α phosphorylation, accompanied by increased expression of ER chaperones GRP78 and GRP94. Conversely, a decreased PC/PE ratio caused by PEMT suppression also led to IRE1α and eIF2α phosphorylation and the induction of CHOP, homocysteine-inducible ER stress-inducible protein (HERP), and Der1-like domain family member 2 (DERL2). Other studies have also implicated the role of PC in the activation of ER stress. The active spliced form of an IRE1 target, XBP-1, was found to be associated with ER expansion and increased PC synthesis in fibroblasts in vitro. Collectively, these studies seem to indicate that an overabundance of PC is integral to the development of ER stress (Testerink et al. 2009; Van der Sanden et al. 2003). These studies suggest that an alteration of PC/PE ratio in the ER membrane contributes to ER stress mediated by SCD inhibition and SFA overload.
Conclusion
In this chapter, we have discussed specific pathways by which SCD1 and its substrates, SFAs, contribute to inflammation and apoptosis (Fig. 7.4). The SCD1 activity is tightly regulated mostly at the transcription level. However, once this tight regulation is interrupted, the induction of SCD1 expression increases levels of oleate, which is a preferred substrate for triglyceride and cholesteryl ester synthesis. Conversely, substantial reduction of SCD1 increases levels of intracellular SFAs such as stearate and palmitate, leading to TLR4 and ER stress activation. ER stress is induced by abnormal ratios of lipid species such as saturated/unsaturated fatty acids, free/esterified cholesterol, and PC/PE, leading to apoptosis frequently found in atherosclerotic lesions. These ratios can be regulated through the modulation of SCD1 activity. Thus, composition and ratio of lipid species contributing to SFA-mediated ER stress remain to be determined through further investigation. Turning to the broader perspective of disease pathology, we see that the development of atherosclerosis is characterized by all of these factors: abnormal lipid accumulation, TLR4-mediated inflammation, and ER stress-mediated apoptosis. These factors clearly suggest that SCD1, its SFA substrates, and its MUFA products play a multifaceted role in the development of vascular diseases such as atherosclerosis and vascular calcification.

SCD1 in inflammation and ER stress. Loss of SCD1 and the resulting accumulation of SFAs activate TLR4 and ER stress signaling, resulting in inflammation and cell death leading to vascular diseases such as atherosclerosis and vascular calcification. TLR4 toll-like receptor-4, NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells, PERK PKR-like ER kinase, IRE1 inositol requiring enzyme 1, ATF6 activating transcription factor-6
References
Anderson EK, Hill AA, Hasty AH (2012) Stearic acid accumulation in macrophages induces toll-like receptor 4/2-independent inflammation leading to endoplasmic reticulum stress-mediated apoptosis. Arterioscler Thromb Vasc Biol 32:1687–1695
Brookheart RT, Michel CI, Schaffer JE (2009) As a matter of fat. Cell Metab 10:9–12
Brown JM, Chung S, Sawyer JK, Degirolamo C, Alger HM, Nguyen T, Zhu X, Duong MN, Wibley AL, Shah R, Davis MA, Kelley K, Wilson MD, Kent C, Parks JS, Rudel LL (2008) Inhibition of stearoyl-coenzyme A desaturase 1 dissociates insulin resistance and obesity from atherosclerosis. Circulation 118:1467–1475
Brown JM, Chung S, Sawyer JK, Degirolamo C, Alger HM, Nguyen TM, Zhu X, Duong MN, Brown AL, Lord C, Shah R, Davis MA, Kelley K, Wilson MD, Madenspacher J, Fessler MB, Parks JS, Rudel LL (2010) Combined therapy of dietary fish oil and stearoyl-CoA desaturase 1 inhibition prevents the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol 30:24–30
Choi SE, Jung IR, Lee YJ, Lee SJ, Lee JH, Kim Y, Jun HS, Lee KW, Park CB, Kang Y (2011) Stimulation of lipogenesis as well as fatty acid oxidation protects against palmitate-induced INS-1 beta-cell death. Endocrinology 152:816–827
Dobrzyn A, Dobrzyn P, Miyazaki M, Sampath H, Chu K, Ntambi JM (2005) Stearoyl-CoA desaturase 1 deficiency increases CTP:choline cytidylyltransferase translocation into the membrane and enhances phosphatidylcholine synthesis in liver. J Biol Chem 280:23356–23362, Epub 2005 Apr 13
Duan X, Zhou Y, Teng X, Tang C, Qi Y (2009) Endoplasmic reticulum stress-mediated apoptosis is activated in vascular calcification. Biochem Biophys Res Commun 387:694–699
Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, Fazio S, Wiest MM, Watkins SM, Linton MF, Hotamisligil GS (2009) Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 15:1383–1391
Flowers JB, Rabaglia ME, Schueler KL, Flowers MT, Lan H, Keller MP, Ntambi JM, Attie AD (2007) Loss of stearoyl-CoA desaturase-1 improves insulin sensitivity in lean mice but worsens diabetes in leptin-deficient obese mice. Diabetes 56:1228–1239
Flowers MT, Keller MP, Choi Y, Lan H, Kendziorski C, Ntambi JM, Attie AD (2008) Liver gene expression analysis reveals endoplasmic reticulum stress and metabolic dysfunction in SCD1-deficient mice fed a very low-fat diet. Physiol Genomics 33:361–372
Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473:528–531
Gao J, Ishigaki Y, Yamada T, Kondo K, Yamaguchi S, Imai J, Uno K, Hasegawa Y, Sawada S, Ishihara H, Oyadomari S, Mori M, Oka Y, Katagiri H (2011) Involvement of endoplasmic stress protein C/EBP homologous protein in arteriosclerosis acceleration with augmented biological stress responses. Circulation 124:830–839
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Lee JY, Sohn KH, Rhee SH, Hwang D (2001) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276:16683–16689
Lee JY, Plakidas A, Lee WH, Heikkinen A, Chanmugam P, Bray G, Hwang DH (2003a) Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res 44:479–486
Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH (2003b) Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 278:37041–37051
Lee JY, Zhao L, Youn HS, Weatherill AR, Tapping R, Feng L, Lee WH, Fitzgerald KA, Hwang DH (2004) Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J Biol Chem 279:16971–16979
Liu X, Ntambi JM (2009) Atherosclerosis: keep your macrophages in shape. Nat Med 15:1357–1358
Liu X, Miyazaki M, Flowers MT, Sampath H, Zhao M, Chu K, Paton CM, Joo DS, Ntambi JM (2010) Loss of Stearoyl-CoA desaturase-1 attenuates adipocyte inflammation: effects of adipocyte-derived oleate. Arterioscler Thromb Vasc Biol 30:31–38
Liu X, Strable MS, Ntambi JM (2011) Stearoyl CoA desaturase 1: role in cellular inflammation and stress. Adv Nutr 2:15–22
Macdonald ML, Bissada N, Vallance BA, Hayden MR (2009a) Absence of stearoyl-CoA desaturase-1 does not promote DSS-induced acute colitis. Biochim Biophys Acta 1791:1166–1172
MacDonald ML, van Eck M, Hildebrand RB, Wong BW, Bissada N, Ruddle P, Kontush A, Hussein H, Pouladi MA, Chapman MJ, Fievet C, van Berkel TJ, Staels B, McManus BM, Hayden MR (2009b) Despite antiatherogenic metabolic characteristics, SCD1-deficient mice have increased inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol 29:341–347
Masuda M, Ting TC, Levi M, Saunders SJ, Miyazaki-Anzai S, Miyazaki M (2012) Activating transcription factor 4 regulates stearate-induced vascular calcification. J Lipid Res 53:1543–1552
Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M (2007) Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation 116:1226–1233
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Sampath H, Miyazaki M, Dobrzyn A, Ntambi JM (2007) Stearoyl CoA desaturase-1 mediates the pro-lipogenic effects of dietary saturated fat. J Biol Chem 282(4):2483–93
Scull CM, Tabas I (2011) Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol 31:2792–2797
Summers SA (2006) Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res 45:42–72
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13:184–190
Testerink N, van der Sanden MH, Houweling M, Helms JB, Vaandrager AB (2009) Depletion of phosphatidylcholine affects endoplasmic reticulum morphology and protein traffic at the Golgi complex. J Lipid Res 50:2182–2192
Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I (2009) Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab 9:474–481
Uryu S, Tokuhiro S, Oda T (2003) beta-Amyloid-specific upregulation of stearoyl coenzyme A desaturase-1 in macrophages. Biochem Biophys Res Commun 303:302–305
Van der Sanden MH, Houweling M, van Golde LM, Vaandrager AB (2003) Inhibition of phosphatidylcholine synthesis induces expression of the endoplasmic reticulum stress and apoptosis-related protein CCAAT/enhancer-binding protein-homologous protein (CHOP/GADD153). Biochem J 369:643–650
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086
Wei Y, Wang D, Topczewski F, Pagliassotti MJ (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291:E275–281
Zheng Y, Eilertsen KJ, Ge L, Zhang L, Sundberg JP, Prouty SM, Stenn KS, Parimoo S (1999) Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet 23:268–270
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Ting, T.C., Miyazaki, M. (2013). Stearoyl-CoA Desaturase-1 in the Regulation of Toll-Like Receptor Signaling and Endoplasmic Reticulum Stress Signaling. In: Ntambi, Ph.D., J. (eds) Stearoyl-CoA Desaturase Genes in Lipid Metabolism. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7969-7_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7969-7_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7968-0
Online ISBN: 978-1-4614-7969-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)