
Abstract
Cardiac remodeling at the macroscopic level occurs as the heart fails and is accompanied by changes in size, shape, and performance of its cardiomyocytes. Therefore, an understanding of the mechanisms by which cardiomyocytes sense and respond to biomechanical stress is of prime importance to the development of a molecular basis of heart failure. The cardiomyocyte experiences changes in stress and strain throughout every cardiac cycle, which is generated both internally by contractile proteins and externally through cell–cell and cell–matrix interactions. Cells within the heart are constantly remodeling through processes of gene transcription, protein translation, posttranslational modification, and the assembly of complex organelles. Because the heart is constantly challenged mechanically, it relies upon the near instantaneous posttranslational modifications of existing proteins to sense and respond rapidly to acute external stressors. Transcription and translation takes longer but are important to the overall response to chronic stress and strain. However, the mechanisms by which cardiomyocytes sense and respond to chronic mechanical strains remain largely unknown. Our team and many others are testing hypotheses that mechanical forces drive the growth of heart cells in healthy exercise but become maladaptive in disease. In this chapter we review how the biophysical forces in normal cardiomyocytes drive sarcomere remodeling in heart failure via two key structural components—namely the costamere at the cell membrane that is connected to the Z-disc of the myofibril. Each of these structural complexes has numerous proteins, many of which sense mechanical forces and are also involved in filament assembly. A diagram of the costamere and Z-disc indicates many of the key proteins (Fig. 1). In this chapter, we review how the costamere and specific components of the Z-disc may accomplish both functions of stress detection and sarcomere remodeling during adaptation to hemodynamic overload.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Cardiac remodeling at the macroscopic level occurs as the heart fails and is accompanied by changes in size, shape, and performance of its cardiomyocytes. Therefore, an understanding of the mechanisms by which cardiomyocytes sense and respond to biomechanical stress is of prime importance to the development of a molecular basis of heart failure. The cardiomyocyte experiences changes in stress and strain throughout every cardiac cycle, which is generated both internally by contractile proteins and externally through cell–cell and cell–matrix interactions. Cells within the heart are constantly remodeling through processes of gene transcription, protein translation, posttranslational modification, and the assembly of complex organelles. Because the heart is constantly challenged mechanically, it relies upon the near instantaneous posttranslational modifications of existing proteins to sense and respond rapidly to acute external stressors. Transcription and translation takes longer but are important to the overall response to chronic stress and strain. However, the mechanisms by which cardiomyocytes sense and respond to chronic mechanical strains remain largely unknown. Our team and many others are testing hypotheses that mechanical forces drive the growth of heart cells in healthy exercise but become maladaptive in disease. In this chapter we review how the biophysical forces in normal cardiomyocytes drive sarcomere remodeling in heart failure via two key structural components—namely the costamere at the cell membrane that is connected to the Z-disc of the myofibril. Each of these structural complexes has numerous proteins, many of which sense mechanical forces and are also involved in filament assembly. A diagram of the costamere and Z-disc indicates many of the key proteins (Fig. 1). In this chapter, we review how the costamere and specific components of the Z-disc may accomplish both functions of stress detection and sarcomere remodeling during adaptation to hemodynamic overload.

Cytoskeletal protein complexes within the cardiomyocyte costamere and Z-disc. Structural and signaling proteins within the costamere and Z-disc are depicted. Many of these molecules have been implicated in either mechanosensing or in sarcomere assembly. MYOZ2 myozenin 2, Cn calcineurin, PDZ-3LIM one-PDZ and three-LIM domain protein, PDZ-1LIM one-PDZ and one-LIM domain protein, MLP/CRP3 muscle-specific LIM protein/cysteine-rich protein 3, FHL2 four-and-a-half LIM protein 2, MAPRs muscle ankyrin repeat proteins, MURFs muscle-specific ring-finger proteins. Reprinted from Hoshijima et al. [10] with permission
Biophysics of Cardiomyocyte Mechanotransduction
Mechanotransduction is the process by which load-bearing cells sense physical forces, transduce the forces into biochemical signals, and generate adaptive or maladaptive responses that lead to alterations in cell structure and function. Mechanical forces regulate gain and loss of adhesion, membrane and cytoskeletal stretch, and cellular compression due to changes in pressure. Mechanical perturbations then activate intracellular signal transduction pathways that have profound effects on cellular phenotype. Mechanotransduction in the heart affects the beat-to-beat regulation of cardiac performance, but also profoundly affects the growth, phenotype, and survival of cardiomyocytes [1]. Conversely, injury to the cardiomyocyte can affect the structural integrity of components of the mechanosensory apparatus, and impair force generation, growth regulation, and cell survival [2–6]. Mechanical strain physically deforms a protein, and changes the affinity of binding sites, which alters the association between signaling molecules and their effectors [7]. Strain to a cell alters the external physical properties of the matrix molecules, which propagate to the internal networks in a secondary wave of mechano-regulated outside-in and subsequent internal cell signal changes. In many cell types, these processes are best studied at the single molecule level. One example is fibronectin, a crucial extracellular matrix (ECM) protein, but internal proteins like p130Cas and talin also respond to strain [8]. The sequences of events sensing stress and strain are similar for the protein complexes of the costamere and Z-disc in muscle. A mechanical perturbation deforms one or more proteins in the sensor complex, thus triggering changes in binding partners enabling posttranslational modifications. In some cases, specific proteins are released and translocate to the nucleus to affect transcription.
Cardiomyocytes rely on several intracellular components to sense mechanical load and convert mechanical stimuli into biochemical events that cause sarcomere remodeling during heart failure. The mechanosensors include integrins and other membrane-associated proteins that link the ECM to the cytoskeleton, protein components within the myofilaments and Z-discs, and stretch-activated ion channels. As described in a number of recently published reviews [1, 9–11], information regarding each component’s role in modulating sarcomere addition and remodeling, and their complex interactions in stress detection and cytoskeletal assembly remain limited. In fact, it is likely that multiple mechanosensors are primarily responsible for sarcomere remodeling in response to increased wall stress.
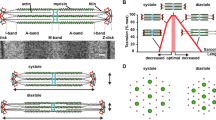
Let us consider how forces are transmitted in muscle. Muscle cells are unique in that they both respond to externally applied mechanical forces, as well as generate large internal loads that are transmitted to adjacent cells and their surrounding ECM. Externally, forces that are generated outside the cardiomyocytes are transmitted from the ECM to the interior through costameres, the general cell mechanosensor of the focal adhesion complex [12]. The anisotropic geometry of the cardiomyocyte with its longitudinal and lateral structures may allow for distinct pathways of force recognition and transmittance [13–15]. When a sarcomere is lengthened by strain, both the costameric complex and the Z-disc are extended in the longitudinal direction (Fig. 2). However, what is not generally appreciated is that normal cell shortening causes a widening of the cell, thus providing a stretch in the transverse direction which increases the distance between the thick and thin filaments (Fig. 2). Under passive tension, the filaments are closer together and the Z-disc in cross section has a small lattice. With active tension as the sarcomeres contract, the filaments move further apart and the Z-lattice becomes basket weave in pattern (Fig. 3) [16]. Cardiac muscle is usually under tension, so the basket weave pattern is generally seen. The repetitive features in the sarcomere have permitted this detailed analysis of protein complexes to be studied, which is not possible for the focal adhesion complex. Nonetheless, separate directional pathways are implicated by static transverse and longitudinal loading to activate stress in muscle for costameric proteins, such as the activation of the ERK cascade [17], or different levels of phosphorylation of focal adhesion kinase (FAK) [18] with transverse vs. longitudinal strain.

Force is distributed externally from the costameres and internally throughout the myocyte by the Z-discs. Both the longitudinal and transverse directions are deformed by cell lengthening with passive tension or during contraction by active tension, as shown by directions of arrows, respectively. Width of cell decreases with lengthening and increases with shortening. A-band (grey) length stays constant; I-band length varies (white) with shortening. Dimensions are exaggerated for clarity

Z-disc lattice is deformed with tension. Upper panels show a longitudinal section and lower panels in the transverse plane through the Z-disc. Left side is under passive tension (rest); right active tension (contraction). Thin filaments (black); to distinguish polarity the open circles enter Z-disc from below and closed from above. α-actinin (red) has small square lattice with passive but basket weave with active tension. CapZ (blue) the α/β heterodimer caps the barbed end of the thin filament. Thin filament lattice spacing is 24 nm at rest. Redrawn after Goldstein et al. [16]
Costameres in Cardiomyocyte Force Transmission and Remodeling
Peripheral myofibrils are physically attached to the sarcolemmal membrane by costameres, which are components of the subsarcolemmal cytoskeleton (Fig. 1). This structure was first identified by Pardo and colleagues [19] as vinculin-positive, circumferentially oriented bands that mechanically couple the sarcolemma and its invaginations to the contractile machinery of the cardiomyocyte during fiber lengthening and shortening. As outlined below, this structure is critically important for both force transmission and sarcomere remodeling during adaptation to hemodynamic overload.
Costameres Are Important Sites for Cell Attachment to the Extracellular Matrix
Attachment of myofibrils at costameres resembles the attachment sites of the subcortical actin cytoskeleton of nonmuscle cells to the plasma membrane at focal adhesions. Indeed, costameres are now considered striated muscle-specific extensions of focal adhesions, where they form an interface between the intracellular and extracellular environment. Like their focal adhesion counterparts in nonmuscle cells, costameres are also critically important adhesive structures that serve to physically attach cardiomyocytes to their surrounding ECM. Thus, they form a stress-tolerant linkage between the cardiac ECM and the myofibrillar apparatus of adjacent muscle cells. As described below, force generated by one cardiomyocyte can then be transmitted laterally to adjacent cells via attachment to the ECM, allowing for coordinate contraction and relaxation of the functional syncytium.
Costameres Are Sites for Outside-In and Inside-Out Force Transmission
In addition to their role in cell attachment, Danowski and colleagues first demonstrated that costameres were sites where contractile forces generated within the cardiomyocyte are directly transmitted to the surrounding ECM [20]. They showed that cultured adult rat ventricular myocytes maintained on a flexible, silicone membrane generated pleat-like wrinkles of the surrounding ECM each time the firmly attached cells contracted. The pleats were spaced 1.8–2.0 μm apart, coinciding with the distribution of α-actinin-containing Z-discs, and sites of close approximation of the cell membrane to the silicone substratum as determined by interference-reflection microscopy. Costameres are also sites where longitudinal displacement of the ECM is transmitted directly to the contractile machinery of the cell. A 10 % static, linear stretch of aligned neonatal rat ventricular myocytes maintained on laminin-coated, microtextured silicone membranes resulted in an immediate, uniform increase in sarcomere length of ~10 % throughout the entire length of the longitudinally oriented, rod-shaped cell [21]. In this case, cell attachment via circumferentially organized costameres alone was sufficient to transmit the externally applied longitudinal strain directly to the underlying myofibrils, indicating that both externally applied and intrinsically generated mechanical loads are transmitted through costameres both outside-in and inside-out.
Integrins Attach the Cardiomyocyte Cytoskeleton to the Cardiac ECM
ECM attachment at costameres is accomplished by specific, integral membrane components within two major protein complexes: the dystrophin-glycoprotein complex containing α-dystroglycan that binds laminin-2, perlecan, and other ECM proteins in the cardiomyocyte basement membrane [22], and β1-integrins, that bind laminin, fibronectin, fibrillar collagens, and a variety of other extracellular proteins to the cardiac ECM [23]. Membrane-associated proteins on the cytoplasmic face of both types of adhesion complexes collectively form a linkage to the myofibrillar apparatus via protein–protein interactions that ultimately terminate at the Z-disc of peripheral myofibrils [24]. These costamere-associated linker proteins provide both structural and signaling roles in initiating and maintaining sarcomeric organization and remodeling during hemodynamic stress [1]. Disruption of either protein complex produces left ventricular dysfunction, and combined defects act synergistically to reduce cardiac function more than disruption of either adhesive complex alone [25].
Integrin receptors are heterodimeric transmembrane proteins that are responsible for initiating cell–matrix attachment by adherens junctions in a variety of cell types. In cardiomyocytes, integrins are not randomly distributed on the cell surface, but rather are found embedded within the sarcolemmal membrane directly adjacent to costameres [26]. Thus, the ECM-integrin-costameric protein network can be viewed as a highly specialized form of lateral adherens junction, in which cell-to-matrix attachment is mediated by direct interaction of cardiomyocyte integrins with specific ECM protein sequences within the cardiac interstitium. The physical interaction between integrin cytoplasmic domains and adaptor proteins within the cytoskeleton generate a submembrane adhesion plaque that is critical for transmitting mechanical force between the ECM and the actin cytoskeleton. Furthermore, the cytoplasmic domains of integrin subunits play a direct role in these connections, as several cytoskeletal adaptor proteins can link integrins directly to actin filaments [27]. With the inclusion of additional protein–protein interactions, it is apparent that integrins provide a critical transmembrane linkage between the cardiac ECM and the actin-based cardiomyocyte cytoskeleton.
All integrin receptors consist of noncovalently associated α and β subunits that combine to form a single receptor for various ECM components. At least 18 α and 8 β subunits have been identified in mammals, and they combine to form at least 24 different paired integrin receptors in various cell types [28]. However, myocardial integrins are relatively restricted to integrins of the β1-type, including the β1D splice variant isoform, which is the predominant integrin expressed in the postnatal heart [29]. In contrast, various α subunits are expressed throughout cardiomyocyte development, with α1, α3, and α5 subunits identified in immature cardiomyocytes, whereas α6 and α7 isoforms predominate in adult ventricular myocytes [30, 31]. Nevertheless, a single integrin receptor is capable of binding to several different ECM proteins, whereas a single ECM ligand can engage several different integrin heterodimers. The promiscuous nature of ECM attachment allows for subtle differences in ECM composition to occur during development and in response to biomechanical overload.
β1-Integrin Cytoplasmic Domains Form Cytoskeletal Attachments Through Talin Dimers
Unlike growth factor receptors, the cytoplasmic tails of integrin α and β chains possess no intrinsic catalytic activity. However, they interact with other cytoskeletal proteins to transmit extrinsically applied and internally generated mechanical force. A major binding partner of the β1-integrin cytoplasmic domain is the cytoskeletal protein talin, which is a rod-shaped, multidomain protein involved in bidirectional activation of integrins [32]. Cell-surface integrins can exist in either low- or high-affinity states, and cellular modulation of integrin affinity is in part accomplished by reversible binding of the N-terminal, globular head domain of talin to the C-terminal, β1-integrin cytoplasmic tail [33]. Cardiomyocytes express predominantly talin-2 and integrin β1D isoforms, which have the highest binding affinities of various β-integrin cytoplasmic domains, suggesting that integrin engagement to the cardiac ECM favors a mechanically strong, activated integrin binding conformation [34].
The mammalian genome contains two genes for talin encoding two structurally similar proteins (talin-1 and talin-2) that share 74 % sequence identity [35]. The specific function of each talin isoform in mechanotransduction and sarcomere remodeling is not known, but it appears that the talin-2 isoform plays a unique role in muscle development and disease. Localization of talin-2 is restricted to costameres and intercalated discs in striated muscle [36], but talin-2 appears dispensable in the presence of talin-1 for normal costamere and sarcomere assembly. However, deletion of both genes produced a severe defect in sarcomere assembly in striated muscle, with profound defects in the assembly of adhesion complexes and sarcomeres by cultured myoblasts isolated from double knockout embryos [37]. The failure of normal sarcomere development in these immature muscle cells also highlights the importance of talin (and perhaps other cytoskeletal linker proteins) in myofibrillar assembly.
Cardiomyocyte adhesion to ECM proteins via β1-integrins causes the recruitment of talin dimers to the cytoplasmic face, leading to integrin transition to their high affinity state. This process is an important, early step in “outside-in” signaling during cell attachment. Conversely, talin activation by a number of intracellular signaling pathways causes the physical displacement of α-integrin subunits, thereby allowing for high-affinity ECM engagement during “inside-out” signaling [34, 38]. Intracellular talin binding alone is sufficient to alter the conformation of integrin extracellular domains and promote their attachment to ECM proteins [39]. The recruitment of talin involves the Src-dependent tyrosine phosphorylation of the β-integrin tail, and its recognition by the talin head region [40]. Thus, intracellular stimuli that cause Src-dependent integrin phosphorylation promote the activation of integrins via talin recruitment to costameres, and stimulate costamere formation during inside-out signaling. Subsequent recruitment of additional cytoplasmic linker proteins (such as paxillin and vinculin) to the costamere may also be required for Z-disc assembly and premyofibril formation during myofibrillogenesis [41, 42].
Focal Adhesion Proteins Are Critical Regulators of Costamere Assembly
Vinculin is another cytoskeletal linker protein that is recruited to focal adhesions and costameres during integrin engagement and clustering [43]. It is a cytoskeletal adaptor protein consisting of three functional domains: an N-terminal head, a flexible, proline-rich hinge region, and a C-terminal tail domain. Intramolecular association between the head and tail domains constrains vinculin in an inactive conformation, but vinculin unfolds during activation, thereby allowing vinculin to interact with a variety of other cytoskeletal proteins, including α-actinin and paxillin [44]. As vinculin unfolding and activation is driven by talin binding to the head region of the molecule, and activated vinculin binds α-actinin, then talin binding to the β1-integrin cytoplasmic tail thereby indirectly provides a physical linkage to the actin-based cytoskeleton.
Vinculin localization to costameres is accomplished by two distinct mechanisms. As indicated above, the head region binds to talin during integrin activation, whereas the C-terminal tail region can bind to paxillin, another cytoskeletal adaptor protein that is targeted to focal adhesions and costameres via its LIM3 domain [45]. The C-terminal region of vinculin also contains a four-helix bundle that is structurally similar to the focal adhesion targeting (FAT) sequence of FAK [46]. The FAT domain of FAK binds to paxillin during integrin engagement and clustering and targets the protein kinase to the growing subsarcolemmal adhesion plaque. Thus, the combinatorial interactions of all four proteins (talin, vinculin, paxillin, and FAK) suggest both structural and signaling roles for each in costamere formation and turnover [44]. Based on structural studies of focal adhesions in nonmuscle cells, it is likely that individual costameric proteins are also organized both horizontally and vertically into specific layers responsible for signaling, force transduction, and cytoskeletal attachment [47].
Costameres Are Components of the Mechanosensory Apparatus of Cardiomyocytes
Attachment is clearly one way in which costameres contribute to mechanotransduction, but there is also substantial evidence to indicate that mechanical forces (generated by passive stretch and active tension development) are “sensed” by costameres, or their focal adhesion counterparts in cultured cardiomyocytes. Biochemical signals are then transmitted internally, leading to sarcomere assembly and altered gene expression characteristic of cardiomyocyte hypertrophy. Stretch-induced deformation of cardiomyocyte integrins triggers the recruitment and activation of several signaling kinases (such as FAK, proline-rich tyrosine kinase 2 (PYK2), Src, Rho kinase (ROCK), and ERKs) to the cytoplasmic face of the adhesion complex, where they participate in downstream signaling to the nucleus and other organelles [12]. Results obtained in mechanically stressed, cultured cardiomyocytes complement elegant studies performed in pressure-overloaded, intact myocardium [48–52], and also support the close interaction between the ECM-integrin-cytoskeletal complex and growth factor receptor signaling during cardiomyocyte hypertrophy and sarcomere remodeling [53–59].
FAK and Cardiomyocyte Mechanotransduction
Exactly how components of the focal adhesion complex sense mechanical stimuli remains unclear. Seminal observations by Ingber and colleagues [60] using a magnetic twisting device to transfer force directly from integrins to the local cytoskeleton suggest that mechanical deformation of one or more adhesion plaque proteins is the proximal step in an intracellular signaling cascade that leads to global cytoskeletal rearrangements and mechanotransduction at multiple, distant sites within the cell. As integrin subunits are devoid of any catalytic activity, transmission of mechanical signals from integrins to the cytoskeleton requires the “stress activation” of one or more signaling molecules capable of transmitting biochemical signals to the internal cellular environment. Talin localization during integrin engagement and clustering may initiate outside-in signaling, but talin has no intrinsic catalytic activity capable of relaying biochemical signals into the cell interior. However, secondary recruitment and activation of protein tyrosine and serine/threonine kinases to the cytoplasmic adhesion plaque may accomplish this function. FAK is clearly one candidate enzyme that is responsible for integrin-mediated mechanotransduction within cardiomyocyte focal adhesions and costameres. As indicated above, FAK is a nonreceptor protein tyrosine kinase that functions as an “activatable scaffold” [61] in integrin-dependent signal transduction (Fig. 4). An autoinhibitory FERM domain, located within the N-terminal region of FAK, associates with the plasma membrane via its interaction with several different growth factor receptors. The C-terminal region of FAK comprises the FAT domain, which binds directly to paxillin and talin, which in turn bind to the cytoplasmic tail of β1-integrins at sites of integrin clustering. Once localized, FAK phosphorylates itself at a single tyrosine residue (Y397). This autophosphorylation site serves as a high-affinity binding domain (pYAEI motif) for the SH2 domain of Src-family protein tyrosine kinases [62] (Fig. 5). Once bound to FAK, active Src then phosphorylates FAK at residues Y576 and Y577 within its catalytic domain (which augments FAK kinase activity toward exogenous substrates), and at Y861 and Y925 near its C-terminus [63]. The Y861 phosphorylation site promotes the binding of p130Cas to FAK [64]. The Y925 phosphorylation site promotes the binding of Grb2 to FAK, and other adaptor proteins and kinases containing SH2 domains. The FAK-Src complex also phosphorylates paxillin, p130Cas, and other cytoskeletal proteins involved in costamere and cytoskeletal assembly.

FAK binding partners in cardiomyocytes mechanotransduction. The N-terminal, autoinhibitory FAK FERM domain is important for signal integration from growth factor receptors such as the Eph-family, EGF and PDGF receptor protein tyrosine kinases, and the cytoplasmic Etk protein tyrosine kinase. The FAK C-terminal focal adhesion targeting (FAT) domain binds the cytoskeletal adaptor proteins paxillin and talin, and mediates FAK localization to integrin-enriched focal adhesions and costameres. The FAT domain and proline-rich (PR) regions bind molecules involved in Rho-GTPase activation and deactivation. Numerous tyrosine and serine phosphorylation sites on FAK are also depicted. JSAP1 scaffolding protein of the JNK kinase pathway, ASAP1 130-kDa phosphatidylinositol 4,5-biphosphate (PIP2)-dependent Arf1 GTPase-activating protein (GAP), GRB2 growth factor receptor-bound protein 2, GRAF GAP for Rho associated with FAK, SOCS suppressors of cytokine signaling, SUMO small ubiquitin-like modifier protein. Reprinted from Schlaepfer et al. [61] with permission

Model for the autoactivation of focal adhesion kinase. The “clover leaf” structure of the autoinhibitory FERM domain of FAK is depicted. The FERM domain binds directly to the kinase C-lobe when FAK is auto-inhibited, impeding access to the active site and protecting FAK’s activation loop from phosphorylation by Src. FAK is activated when it is released from the auto-inhibited “closed” state by the binding of a protein or lipid partner to its FERM domain. Binding of partners to regions in the F1 or F2 subdomains of the FERM domain is likely to “open” the protein, permitting auto-phosphorylation and Src binding. This leads to Src-mediated Tyr phosphorylation of the acceptor Tyr residues Y576 and Y577 in the kinase loop of FAK and full catalytic activation, and Src-mediated Tyr phosphorylation at Y861 and Y925 to promote binding of p130Cas and GRB2, respectively. Reprinted from Frame et al. [63] with permission
In addition to multiple tyrosine phosphorylation sites, FAK contains several serine residues (S722, S843, S846, and S910) that undergo reversible phosphorylation in response to hypertrophic stimuli. These serine residues are in close proximity to critical protein–protein interaction sites within the C-terminal region of FAK, such as the binding site for p130Cas and the adjacent FAT domain. The functional role of FAK serine phosphorylation in cardiomyocytes is largely unknown, but one report indicates that serine (and tyrosine) phosphorylation of FAK increases dramatically in hypertensive rats, with different sites of phosphorylation appearing to regulate FAK subcellular localization [65]. Our group [66] recently demonstrated that endothelin-1 and other hypertrophic factors induced a time- and dose-dependent increase in FAK-S910 phosphorylation. Endothelin-induced FAK-S910 phosphorylation required endothelin Type A receptor-dependent activation of PKCδ and Src via parallel Raf-1→MEK1/2→ERK1/2 and MEK5→ERK5 signaling pathways. Replication-deficient adenoviruses expressing wild-type FAK and a non-phosphorylatable, S910A-FAK mutant were then used to examine the functional significance of FAK-S910 phosphorylation. Unlike wild-type FAK, S910A-FAK increased the half-life of GFP-tagged paxillin within costameres (as determined by total internal reflection fluorescence microscopy and fluorescence recovery after photobleaching) and increased the steady-state FAK-paxillin interaction (as determined by co-immunoprecipitation and Western blotting). These alterations resulted in reduced NRVM sarcomere reorganization and cell spreading. Finally, we found that FAK was serine-phosphorylated at multiple sites in non-failing, human left ventricular tissue, and FAK-S910 phosphorylation and ERK5 expression were both dramatically reduced in patients undergoing heart transplantation for end-stage dilated cardiomyopathy (DCM). These results suggest that reduced FAK-S910 phosphorylation may contribute to sarcomere disorganization that is frequently observed in these heart failure patients [66].
We and others have shown that FAK and the highly homologous protein tyrosine kinase PYK2 are both expressed in neonatal and adult cardiomyocytes, where they are activated in response to mechanical loading [12, 67, 68] and agonists (e.g., phenylephrine, angiotensin II, endothelin-1) that stimulate Gq-coupled receptors [53–59, 69]. Thus, FAKs, and other protein kinases bound to FAK and PYK2 during integrin clustering, can activate downstream signaling pathways that regulate integrin-mediated mechanotransduction at local as well as distant sites within the cell.
Further support for an important role for FAK in cardiomyocyte mechanotransduction has come from studies of global and cardiomyocyte-specific FAK knockout mice. Global FAK deletion led to lethality at embryonic day 8.5, and the mutant embryos displayed a profound defect in development of all mesodermal structures, including the heart and vasculature [70, 71]. Interestingly, the developmental defects found in FAK−/− embryos were phenotypically very similar in timing and phenotype to the morphological defects observed in fibronectin-null mice, suggesting an important relationship between fibronectin- and FAK-dependent signaling, especially with respect to development of the cardiovascular system [61]. In both cases, the developmental defects were attributed to the inability of mesodermal cells to migrate normally. Indeed, fibroblasts isolated from FAK−/− embryos displayed markedly reduced mobility and abnormally large focal adhesions, indicating a defect in focal adhesion turnover. However, cardiomyocyte-restricted FAK knockout mice have a variable cardiac phenotype depending on when during development FAK is deleted. Embryonic deletion of FAK in cardiomyocytes caused perinatal lethality due to the presence of large ventricular septal defects and abnormalities in outflow tract alignment, indicating again that FAK predominantly regulates mammalian cardiomyocyte migration during early cardiac development [72]. However, cardiomyocyte FAK deletion during later prenatal development was not associated with any congenital heart defects, but led to spontaneous DCM in aged animals [73], and a blunted hypertrophic response to angiotensin II infusion [73] or transverse aortic constriction [74] in the adult heart. The inability to respond normally to hypertrophic stimuli supported earlier cell culture studies that demonstrated impaired hypertrophic responses of cultured cardiomyocytes overexpressing FAK-related nonkinase (FRNK, a naturally occurring inhibitor of FAK), Y397F-FAK (a FAK autophosphorylation mutant), FAK antisense RNA, or just the FAK-FAT domain [12, 21, 54, 56–58, 75].
Other Protein Kinases Involved in Cardiomyocyte Mechanotransduction
In addition to FAK, cardiomyocytes express a structurally related kinase known as PYK2 (also known as cell adhesion kinase-β (CAK-β), related adhesion focal tyrosine kinase (RAFTK), or cell adhesion tyrosine kinase (CADTK)) [76]. PYK2 is a Ca2+-dependent nonreceptor protein tyrosine kinase that undergoes bimolecular transphosphorylation [77] in response to integrin engagement, increased intracellular Ca2+, and activation of PKCs in many cell types, including cardiomyocytes [69, 78–83]. Although PYK2 is predominantly localized to the cytoplasm [69], a minor component of the enzyme co-localizes with paxillin in focal adhesions of cultured neonatal rat ventricular myocytes [83]. Like FAK, PYK2 acts as an important scaffolding protein, and transduces signals from G-protein-coupled receptors to downstream MAPK signaling pathways depending upon which signaling kinases and adaptor proteins bind to the phosphorylated enzyme [84, 85]. PYK2 has also been shown to link a variety of stressful stimuli, including Ca2+ overload, UV irradiation, and TNF-α treatment to MAPK activation in several cell types [86]. Recently, Hirotani et al. [82] demonstrated that PYK2 is an essential signaling component in endothelin- and phenylephrine-induced cardiomyocyte hypertrophy, perhaps acting via the Ca2+- and/or PKC-dependent activation of Rac1.
Bayer et al. [51] have demonstrated that PYK2 expression and phosphorylation were significantly increased in adult rat ventricular myocytes in vivo in response to acute left ventricular pressure overload. Similarly, Melendez et al. [78] showed that PYK2 expression and phosphorylation were increased in a mouse model of DCM, but its exact role in these conditions has not been elucidated. Nevertheless, recent studies have confirmed that PYK2 is an important upstream regulator of the stress-activated protein kinases (p38MAPK and JNK1/2) in cardiomyocytes [81, 83]. Inhibition of PYK2 in vivo (by direct gene transfer of its C-terminal FAT domain) reduced activation of the fetal gene program and reduced LV remodeling in a rat model of myocardial infarction [87]. Thus PYK2 activation has been implicated in hypertrophic gene expression changes during pathological cardiomyocyte hypertrophy [83] and in the induction of apoptosis [81].
Integrin-linked kinase (ILK) is a third protein kinase that may be involved in integrin-dependent mechanotransduction in cardiomyocytes. ILK has a sequence homology to serine-threonine protein kinases, but its kinase domain is nonfunctional, thus indicating that ILK is a pseudokinase [88, 89]. However, ILK’s pseudokinase domain binds tightly to α-parvin and the complex can directly bind to the cytoplasmic tail of β1-integrins as well as other focal adhesion adaptor proteins [90, 91]. One of these interacting proteins, PINCH1, is essential to early embryonic development, but appears dispensable when its expression is specifically reduced in cardiomyocytes [92]. However, targeted ablation of ILK in cardiomyocytes caused a rapidly progressive, DCM [93]. The ILK-PINCH-parvin complex is involved in regulating Akt activity and cell survival signaling in many cell types, and despite its lack of kinase activity, ILK appears to serve these roles in cardiomyocytes. ILK is an important upstream regulator of Akt phosphorylation at S473 [94], which is essential for Akt activity and may explain ILK’s ability to suppress apoptosis [95]. ILK also interacts with thymosin β4, an actin-binding peptide that stimulates cardiomyocyte and endothelial cell migration. ILK activation by expression of thymosin β4 led to activation of Akt and improved cardiomyocyte cell survival following coronary artery ligation, further implicating ILK and the focal adhesion complex in integrin-dependent cell survival signaling [96]. Nevertheless, it remains unclear whether ILK undergoes translocation and activation in response to mechanical loading of cardiomyocytes in a manner similar to FAK and PYK2.
There is also some evidence to indicate that PKCε, the major novel PKC isoenzyme expressed in cardiomyocytes, is directly involved in integrin-dependent mechanotransduction. Three families of PKCs have been identified to date, containing a total of 11 isoenzymes. The “classical” isoforms (α, βI, βII, and γ) are regulated by calcium, diacylglycerol (DAG), and phosphatidylserine; the “novel” isoforms (δ, ε, η, φ, and μ) are regulated by DAG and phosphatidylserine; and the “atypical” isoforms (ζ and λ) only require phosphatidylserine for activation. The major PKC phorbol ester-sensitive isoenzymes found in adult cardiomyocytes are PKCα, PKCβ, PKCδ, and PKCε. Using standard immunofluorescent microscopy, Disatnik et al. [97] first localized PKCε in a striated pattern within myofibrillar structures of cultured neonatal rat cardiomyocytes following stimulation with norepinephrine or phorbol myristate acetate. Subsequently, Huang et al. [98] demonstrated that PKCε translocated in response to arachidonic acid treatment of adult rat ventricular myocytes to a region adjacent to the Z-line where actin filaments are anchored, and where transverse tubules are closely apposed to the myofilaments. This site of translocation was specific for PKCε, as PKCδ, the other novel PKC expressed in rat ventricular myocytes, translocated to the nucleus in response to arachidonic acid. Borg et al. [99] then showed that PKCε localized to the cytoplasmic side of the sarcolemma directly adjacent to the Z-disc, and Heidkamp et al. [59] demonstrated that the kinase co-localized with FAK in typical focal adhesions in cultured neonatal cardiomyocytes. These morphological results are complemented by biochemical data demonstrating that PKCε forms functional signaling complexes with PYK2 [100] and Src-family protein kinases [101–103] in tissue homogenates of left ventricular myocardium from PKCε-overexpressing mice. PKCε, in turn, is involved in the endothelin-induced activation of both FAK [59] and PYK2 [80], via signaling pathways that may regulate local changes in the actin cytoskeleton [104]. Thus, there is ample evidence to indicate that at least a portion of cardiomyocyte PKCε is found in costameres and focal adhesions, where it may regulate focal adhesion and costamere formation [105] and sarcomeric assembly [21] in response to mechanical loading and growth factor stimulation.
One way that PKCε may localize to costameres and focal adhesions is via binding to RACK1 (Receptor for Activated C-Kinase-1). RACK1 is a seven WD-domain-containing protein that binds to the cytoplasmic tail of β-integrins [106], and anchors PKC isoenzymes [107], Src family protein kinases [108], and other proteins to focal adhesions. Although originally described as a selective receptor for activated, Ca2+-dependent PKCs [109, 110], RACK1 also binds active PKCε, and increases focal adhesion formation, integrin clustering, and lamellipodia formation in human glioma cells. These responses are quite similar to those produced by overexpression of constitutively active PKCε in cultured neonatal cardiomyocytes [105]. Once localized, PKCε can phosphorylate a number of membrane-anchored and cytoskeletal proteins and participate in the activation of Rac1, which is required for cell spreading and cytoskeletal assembly [111]. Alternatively, active PKCε binds directly to the Z-disc [112] via an interaction with a RACK2-like protein [113], thus creating a situation in which PKCε may locally shuttle between costameres and Z-discs to regulate contractile function in response to changing hemodynamic loads.
It remains unknown exactly how PKCε is locally activated in response to mechanical loading. Vuori and Ruoslahti [114] showed that PKC activity in the cell membrane fraction transiently increases preceding cell spreading on fibronectin but not on polylysine, and PKC activation is required for FAK activation in response to cell spreading on fibronectin. These results would suggest that a membrane phospholipase within integrin-dependent cell attachment sites provides a local source of DAG sufficient to activate PKCε. Phospholipase C (PLC)-γ can bind to the Y397 phosphorylation site of FAK [115], and Ruwhof et al. [116] have shown that PLC (but not PLD) activity rapidly increases in neonatal cardiomyocytes in response to cyclic stretch, suggesting that PKC activation may be both upstream and downstream of FAK activation in response to mechanical loading.
Costameres Are Sites for the Earliest Steps in New Sarcomere Assembly
In addition to their important role in cell attachment, costameres may be an important site for the initial events in new sarcomere formation [41]. In cultured cardiomyocytes, costameres reorganize to form adhesive structures that are similar to typical focal adhesions found in nonmuscle cells [117], and their formation precedes the assembly of newly synthesized myofibrillar proteins into sarcomeres [118]. Because of the similarities between costamere formation in cardiomyocytes and focal adhesion formation in nonmuscle cells, we and others have proposed that FAK, and other FAKs play an important role in both processes [1]. Cardiomyocytes isolated from mice with tissue-specific deletion of FAK showed increased length but not width, and displayed disorganized myofibrils with increased nonmyofibrillar space filled with swollen mitochondria [73]. FAK deletion also causes DCM with aging and produces eccentric, rather than concentric, LV hypertrophy with angiotensin II infusion or transverse aortic coarctation, suggesting an intrinsic abnormality in sarcomere assembly [73, 74, 119, 120]. Overexpression of FRNK, or “knocking-down” FAK by siRNA prevented normal costamerogenesis and myofibrillogenesis during skeletal muscle differentiation [121]. Furthermore, overexpression of FRNK in cardiomyocytes also prevented the endothelin-induced increase in total protein/DNA, and the assembly of newly synthesized myofibrillar proteins into sarcomeres [54]. As discussed above, we recently proposed that FAK serine phosphorylation is critical for regulating the conformation of the FAK-FAT domain and, therefore, its interaction with other focal adhesion proteins required for new sarcomere addition [66]. Stabilizing the open conformation of the FAT domain may secondarily decrease FAK-paxillin interaction and promote its exit from newly forming costameres, thereby enhancing the vinculin–paxillin interaction [122, 123]. These events should strengthen the costamere and promote sarcomere formation and reorganization [37, 124] Thus, these findings suggest that FAK, and other proteins reversibly bound to FAK during costamere formation, are required for the normal assembly of sarcomeres in response to both biomechanical and neurohormonal stimuli that induce cardiomyocyte hypertrophy.
FAK, and it C-terminal binding partners p130Cas and paxillin, all co-localize to costameres of cultured neonatal rat ventricular myocytes, where they participate in sarcomere assembly induced by endothelin-1 [54, 57]. Displacement of FAK from these structures (by overexpressing FRNK, or just the FAK-FAT domain) prevented endothelin-induced assembly of newly synthesized contractile proteins into sarcomeres, and coincidently prevented the phosphorylation of paxillin [54] and p130Cas [57] by the FAK/Src complex. Conversely, preventing p130Cas binding to the C-terminal region of FAK (by overexpressing FAK residues 638–841, containing only the proline-rich, C-terminal p130Cas binding site) also substantially reduced endothelin-induced sarcomeric assembly, indicating a critical role for p130Cas binding and phosphorylation by FAK/Src in this process [57]. FAK also reduced steady-state adhesive force by recruiting vinculin to focal adhesions. FAK knockdown increased vinculin recruitment to focal adhesions, resulting in cells more firmly attached to their ECM [43]. Indeed, these cell culture results are consistent with studies of both paxillin [125] and p130Cas [126] knockout mice, which demonstrated early embryonic lethality due to severe defects in the development of the cardiovascular system. In the case of paxillin, the overall phenotype closely resembled that observed in both fibronectin- and FAK-knockout mice. In the case of p130Cas-deficient embryos (which died at a somewhat later developmental stage), cardiomyocytes displayed disorganized myofibrils and disrupted Z-discs, indicating important structural and mechanochemical signaling functions for these adaptor proteins.
Z-Disc Structure in Mechanotransduction and Filament Assembly
Internally, cardiomyocytes generate force through their contractile filaments that are anchored together by an abundance of proteins organized at the Z-disc (Fig. 1). The Z-disc anchors parallel, actin containing thin-filaments, which were observed over 30 years ago by electron microscopy to be bundled by struts assumed to be α-actinin [127]. Interestingly, cell tension extensively deforms this Z-disc lattice complex [16]. Many other proteins have subsequently been shown to form the architectural complexity of the Z-disc, which functions not only as a mechanical joint but also as a signaling complex for mechanotransduction containing proteins such as titin-T-cap, as reviewed in [128–130]. Another signaling protein is the muscle LIM protein (MLP), also known as cysteine rich protein 3 (CSRP3, CRP3). It is a mechanosensor found at the Z-disc with interactions to histone deacetylases (HDAC4) and acetylases (PCAF) and at the costamere where it interacts with ILK, zyxin, α1-spectrin, and α-actinin [131]. MLP in mechanically stimulated neonatal rat myocytes can shuttle to the nucleus where it is involved in regulating the hypertrophic gene program. Furthermore, MLP subcellular localization is sensitive to directional strain. Using microtopography, aligned cardiomyocytes were strained either transversely or longitudinally to determine nuclear translocation and myofibrillar distribution. MLP did not translocate in response to uniaxial, but only to biaxial strain. With transverse strain, MLP aligned along the fiber axis; i.e., perpendicular to the axis. But after longitudinal strain, MLP was more striated [132].
Hypertrophy in response to biophysical forces is achieved by increasing the number of thick and thin filaments in the myocytes, of which the actin filaments are thought to be assembled first into premyofibrils and the myosin thick filaments are spliced in later [133]. Since actin is one of the oldest and most conserved proteins in all living cells, there is an abundance of information about actin filament nucleation, assembly, and severing. All actin filament assembly requires numerous partnering proteins, with specialized variants found in the heart, such as CapZ, formin, and cipher. It seems likely that the filaments are built to serve the functional work being demanded at a subcellular region and that local mechanical conditions ultimately regulate how many filaments are assembled or degraded and where this occurs inside the cell. Nature is efficient in this regard, since muscle is a very high consumer of energy. Strain to the Z-disc modifies the function of the F-actin capping protein (CapZ) via mechanotransduction signaling pathways, such as those involving PKCε [97, 129, 134]. Interestingly, in transgenic models of CapZ downregulation, PKC-dependent regulation of myofilament function is abolished, and activated PKCε and PKCβ binding to the myofilament is diminished [135]. In neonatal myocytes, the additional sarcomeres are added after strain at the Z-disc [136], and this process is also regulated by PKCε [21]. Furthermore, dynamics of actin capping by CapZ is increased by hypertrophic stimuli, and regulated by PKCε activity [137]. These processes are discussed further below.
CapZ Structure and Function
Because of its major role in actin cytoskeletal remodeling, CapZ needs further discussion. CapZ was first discovered in the 1960s by Maruyama and colleagues and was called β-actinin [138–140]. An increasing concentration of CapZ decreased the length of F-actin polymers after sonication and prevented polymerization of G-actin. A fast recovery to the original state could be slowed or prevented by addition of CapZ at various time points [141]. CapZ was finally purified to homogeneity in 1980 which led to the discovery that it is composed of two major polypeptides approximately 30 kDa in size that pair to cap the barbed end of actin filaments [142]. CapZ is a heterodimer composed of an α and a β subunit. Vertebrates contain three α isoforms that are encoded from three individual genes, and three β isoforms that are generated through alternative splicing. The α1 and α2 isoforms are found throughout many tissues though their expression varies widely, and the α3 isoform is specific to the testis. In cardiac tissue, the ratio of α1 to α2 is approximately 1.2:1 [143]. The different biochemical, cellular, and functional roles of each α isoform continues to be elucidated.
The CapZ β1 and β2 isoforms are also found throughout various tissues, and similarly to the α3 subunit, the β3 isoform is specific to the testis. Because each β isoform is generated through alternative splicing, they contain great sequence similarity. The β1 and β2 isoforms vary only in their COOH-terminal ends (β1—31 amino acids, β2—26 amino acids) and β3 is identical to β2 with the addition of 29 amino acids at the NH2 terminal end [144–146]. Although the protein sequences are extremely similar, the β1 and β2 isoforms have distinct functions and biochemical roles. The evidence for their differing roles includes high conservation among vertebrate species, tissue specificity, and well-defined subcellular location [145]. The β1 isoform has been shown to be highly expressed in muscle tissue with a ratio of β1 to β2 approximately 2:1, whereas the β2 isoform is predominately expressed in non-muscle tissue [143, 145, 147]. Furthermore, the β1 isoform localizes to the Z-disc in striated muscle, whereas the β2 isoform localizes to the intercalated discs and plasma membrane [145]. Overexpression of the β1 isoform in cardiac tissue causes disruptions in the intercalated discs, and overexpression of the β2 isoform causes severe malformations of the myofibril architecture [147].
The CapZ α/β complex is shaped liked a mushroom [148]. Both subunits have similar secondary structure and are arranged such that the molecule has a pseudo-twofold axis of rotational symmetry [148]. The COOH-terminal ends of both of the α and β subunits have amphiphilic α-helices which bind actin on the hydrophobic side [148–150]. The hydrophilic side of the COOH-terminal ends has isoform-specific charge distributions and may be involved in isoform-specific recognition of target ligands [148]. Thus, the tentacles present on CapZ are hypothesized to allow binding of both actin and a specific target protein. The separation of the two C-terminal ends ensures that each tentacle binds a single actin monomer at the end of filamentous actin (F-actin). This property explains the inability of CapZ to bind monomeric actin (G-actin) and its strong association with F-actin. Furthermore, studies have shown that when CapZ is only bound to F-actin by its β-tentacle, the molecule is able to “wobble” and thus exposes additional actin and target binding sites to other molecules [151]. Although it was initially believed that the β tentacle was the only mobile region on CapZ [152], new data suggest that CapZ is intrinsically flexible allowing it to interact with the barbed-end of actin in both a high- and low-affinity state [153].
The abundance of PKC anchoring proteins at the cardiac Z-disc indicates a significant role for both the Z-disc and CapZ in PKC-dependent signaling pathways. Wild-type CapZ binds F-actin with sub-nanomolar affinity (~0.1 nM) and it has been shown through multiple studies that the C-terminal domain is necessary for high-affinity actin binding [150, 154]. Any modifications or mutations that affect the C-terminal ends may greatly reduce the affinity of CapZ to F-actin [154]. CapZ was believed to bind to actin in a two-step model whereby electrostatic interactions with the α subunit dictate the on-rate and hydrophobic interactions of actin with the β subunit dictate the off-rate [155]. Based on affinity and X-ray crystallography studies, an improved mechanism by which CapZ binds actin is as follows: (1) basic residues on the α tentacle interact electrostatically with the barbed end of actin, (2) a conformational change of CapZ to a high-affinity form occurs, and (3) supportive binding of the β tentacle strengthens the association [154]. Thus, a factor or mechanism that disturbs any of the binding or unbinding steps may inhibit the capping ability of CapZ or promote uncapping. It takes nearly 10 min to translocate PKCε to the Z-disc, suggesting intramolecular rearrangement of its binding partners and surfaces at the Z-disc [112]. It has also been suggested that the regulatory binding region of PKCε is obscured until stimulation [156]. The treatment of cardiomyocytes with the hypertrophic cytokine endothelin-1 or the α1-adrenergic receptor agonist phenylephrine caused an alteration in CapZ dynamics through a PKC- and PIP2-dependent pathway that decreased the affinity of CapZ to F-actin [137]. Recent work from our group is focused on mechanical stimulation and CapZ dynamics [157]. The alteration of CapZ dynamics and posttranslational profile in response to mechanical strain is dependent upon both the subunit isoform of CapZβ and the activity of PKCε. The dynamics data suggest that CapZβ is only able to respond to mechanical strain in a PKCε-dependent manner, and the proteomic data corroborates this finding by showing all modifications of CapZ are abrogated when cells are infected with an adenovirus expressing dominant-negative PKCε. The mechanism through which strain and PKCε are integrated on CapZ remains unknown but could be due to RACK binding, HDAC binding, or other unknown interactions [113, 158–161]. Ultimately, the increase in the actin uncapping rate may be adaptive in that it disrupts the structure of the Z-disc allowing new actin filaments to polymerize or insert, which leads to a global remodeling that decreases the localized load, and therefore strain, at the Z-disc. This feedback loop would support clinical findings of cardiomyopathies in situations of increased load and constitutively active PKCε overexpression in transgenic animals [105, 162, 163].
CapZ is also regulated by the phospholipid signaling pathway, with protein dynamics affected by PIP2 for neonatal myocytes undergoing hypertrophy in culture [137]. PIP2 is the most abundant of the phosphoinositides known to bind cellular proteins, though it accounts for approximately 1 % of lipid in the plasma membrane of a typical mammalian cell [164]. It has been reported that PIP2 might regulate the calcium-insensitive CapZ [142, 165–168]. PIP2 has been assumed to be the binding partner of CapZ and yet this lipid dissociates the CapZ-actin complex [169, 170]. PIP2 binding of CapZ results in a reduction in binding affinity of the COOH-terminal extensions of the CapZ dimer subunits for the actin filament [137, 171]. Addition of PIP2 to capped actin filaments in a polymerization reaction leads to an increase in the polymerization rate consistent with complete and rapid uncapping [151, 172, 173]. According to the computational analysis, the C-terminal half of CapZ β-subunit could contribute to lipid interaction/insertion [174]. CapZ may bind up to four PIP2 molecules, through direct hydrogen-bonding interactions with the binding sites [174]. Furthermore, treatment of cardiac myofilaments (in which only the CapZ β1-subunit is present) with PIP2 extracts CapZ and depresses myofilament tension generation [175]. PIP2 pathways increase uncapping when myocytes are growing [137].
Other Z-Disc Proteins Involved in Cardiac Remodeling and Hypertrophy
Much has been learned about important proteins relevant to the biophysics of heart failure from studies of humans with DCM, which is characterized primarily by left ventricular dilation and systolic dysfunction. This literature has been extensively reviewed elsewhere [176–179]. The recognition that genetic defects may play pivotal roles in the pathogenesis of DCM, especially familial DCM, has received increasing attention during the past decade. Mutations in multiple cytoskeletal and sarcomeric genes (dystrophin, vinculin, desmin, titin, actin, β-myosin heavy chain (MHC), troponin T, and so on) have been linked to the pathogenesis of familial DCM in both human and mouse models.
Mutations in Z-line alternatively spliced PDZ-motif protein (ZASP), a human orthologue of Cypher, have been identified in patients with isolated non-compaction of the left ventricular myocardium (INLVM), DCM, hypertrophic cardiomyopathy (HCM), as well as skeletal myopathy [180–182]. Cypher is a member of the PDZ-LIM domain family and complexes directly with Z-line-associated proteins, such as α-actinin (actinin-2), thus playing a critical role in muscle ultrastructure and function by maintaining Z-disc integrity. Global Cypher-null mice are postnatal-lethal with severe defects in striated muscle, including a congenital form of DCM [183]. In addition to its developmental roles, Cypher also plays a critical role in the adult heart, as cardiac-specific deletion in mice causes a severe form of DCM resulting in premature adult lethality [184]. Cardiac and skeletal muscle each contain two long isoforms, which have three C-terminal LIM domains and may have a signaling role in addition to a structural role at the Z-disc, and one short isoform without LIM domains, which is primarily localized to the Z-disc. Cypher has two major isoforms. Cypher short (CypherS) isoforms (2c, 2s) are barely detectable during embryogenesis but are strikingly induced postnatally in both cardiac and skeletal muscles. In contrast, Cypher long (CypherL) isoforms (1c, 1s, 3c, 3s) exhibit consistent expression patterns from embryonic stages to adulthood. CypherL and CypherS isoforms contain a common PDZ domain, which recognizes the C-terminus of α-actinin, calsarcin-1, and myotilin. In humans, a ZASP mutation (R268C) that affects only short isoforms is associated with skeletal myopathies, while ZASP mutations (I352M, D626N, T350I D366N, Y468S, Q519P, P615L) that affect only long isoforms are primarily associated with cardiomyopathies, some of which affect specific signaling pathways such as those activating PKCs (PKCα, β, and ε). Selective deletion of long but not short Cypher isoforms leads to late-onset DCM with PKC activation [184]. The long isoform of Cypher directly interacts with PKC isoforms (α, β, ζ, γ, and ε) via their LIM domains and can be phosphorylated by PKCβ1. As discussed above, PKC signaling plays a critical role in the development of cardiac hypertrophy, and in vivo models of pressure overload-induced hypertrophy and human heart failure are associated with increased activity of a number of PKC isoenzymes. Studies in patients with Cypher/ZASP mutations identified a particular gain-of-function mutation within the Cypher/ZASP third LIM domain (D626N) that is causative for DCM and enhances the affinity of PKCs for Cypher/ZASP. The identities of all the binding partners of PKCs at the Z-disc remain unknown, though some have suggested Cypher-1 and enigma homologue protein, PDZ domain containing proteins that also bind α-actinin, several RACKs, and F-actin [159, 185–187].
Formin is another protein involved in actin capping and filament assembly. The localization of the cardiac-specific formin, FHOD3, appears to vary with filament maintenance or during rapid filament assembly that is correlated with mechanical work the cell is able to do [188–191]. The Ehler group proposes that FHOD3 is at the Z-disc in the resting state but is found from the adjacent of Z-disc through the overlap region of the thick and thin filament during growth [189]. Precedent for this is found in drosophila muscle, where an actin-associated protein (SALS) also localizes closer to the pointed end of the thin filament during development of the body wall muscles, but re-localizes to Z-disc in the adult [192]. However, there is still some dispute about this explanation, in which a Japanese group claimed that FHOD3 mainly localizes to a region distant from the Z-disc in either the developing or resting stage. This is probably because of the antibodies directed to different regions of FHOD3. The antibodies of the Japanese group are against 650–802,873–974 and C-20, but the Ehler antibody recognizes 1–339, the N-terminal region of FHOD3. FH2 is the assumed catalytic domain of formin, which is much closer to C-terminus (~1,100–1,483), and associates with F-actin at the barbed end [188]. Perhaps, altered protein posttranslational modification results in different accessibility of the different antibodies along the length of the filament. More research is needed to understand how formin regulates actin assembly induced by mechanical stimulation.
The formin family of proteins stimulates actin assembly directly at the barbed end both in vivo and in vitro [193]. Formins are relatively large (100–200 kDa) multi-domain proteins, compared to CapZ (32–33 kDa). Formins usually dimerize to be functional with two FH2 domains forming a donut-like structure that wraps around the actin filament and controls its processive elongation. The FH1 domain recruits profilin-bound actin and drives rapid actin assembly from the profilin-actin pool [194]. Importantly, many formins can be autoinhibited. The mechanism of autoinhibition is most studied in the diaphanous-related formins (DRFs) where interaction between the diaphanous autoregulatory domain (DAD) and the diaphanous inhibitory domain (DID) is sufficient for autoinhibition [195]. FH1/FH2 domain-containing proteins, FHOD1 and FHOD3, are sometimes classified as DRFs, with FHOD1 first discovered in the spleen [196] and FHOD3 in heart, kidney, and brain [197]. Two isoforms of FHOD3 exist, and the heart contains only the larger one [189, 198]. FHOD1 is phosphorylated at three specific sites within C-terminal DAD by the ROCK [199], which releases the autoinhibition of FHOD1 and leads to F-actin stress fiber formation in endothelial cells [188]. Using an actin polymerization assay in vitro, formin blunted the capping property [200] but, as of yet, there is no evidence showing these proteins directly interact [201].
Conclusions with Respect to the Biophysics of the Failing Heart
Individual myocytes in the human ventricle remodel their sarcomeres over time in response to the mechanical forces experienced. This chapter reviews how biophysical forces modulate the costamere and Z-disc for sarcomere remodeling in heart failure. At each subcellular location, it is the sum of the internally generated force of the contractile filaments and the externally imposed by the stresses and strains of the other working myocytes. Mechanotransduction converts these local forces into chemical signals for myocyte assembly to begin. Since, in the failing heart, the local forces vary from one region of the ventricle to another, sarcomere remodeling also varies from cell to cell resulting in populations of myocytes in one region becoming longer or stronger over time. It is this slow remodeling process that eventually becomes maladaptive exacerbating rather than resolving the ability of the heart to pump blood effectively.
The biophysics of cardiomyocyte mechanotransduction is discussed in detail for the two major structures that drive sarcomere remodeling in heart failure, namely the costamere and the Z-disc. The muscle is highly anisotropic so biophysical force transmission depends on both the longitudinal or transverse direction of the force as well as the balance between outside-in and inside-out force generation. For the costamere, the earliest steps in new sarcomere assembly are described in terms of the attachment to the ECM and local force transmission. The most important molecules in the mechanotransduction and assembly processes of the costamere are the integrins, talin dimers, focal adhesion proteins, and other protein kinases. The forces are transmitted from the costamere internally to the Z-disc permitting an extensive amplification of filament assembly throughout the width and length of the myocyte. Therefore, the specialized structure and proteins of the Z-disc are discussed, especially the actin capping protein CapZ.
Despite the vast amount of knowledge of the multi-protein complexes of the costamere and Z-disc, there is currently no clinical strategy to reduce or prevent the maladaptive cardiac remodeling that occurs at the subcellular level of each myocyte in the heart. Most of these same local responses of the myocyte to force are advantageous in remodeling to exercise making it challenging to determine the differences between the two in order to redirect a failing heart down a healthier path.
References
Samarel, A. M. (2005). Costameres, focal adhesions, and cardiomyocyte mechanotransduction. American Journal of Physiology. Heart and Circulatory Physiology, 289, H2291–H2301.
Ganote, C. E., & Vander Heide, R. S. (1987). Cytoskeletal lesions in anoxic myocardial injury. A conventional and high-voltage electron-microscopic and immunofluorescence study. The American Journal of Pathology, 129, 327–344.
Ganote, C., & Armstrong, S. (1993). Ischaemia and the myocyte cytoskeleton: Review and speculation. Cardiovascular Research, 27, 1387–1403.
Hakim, Z. S., DiMichele, L. A., Rojas, M., Meredith, D., Mack, C. P., & Taylor, J. M. (2009). FAK regulates cardiomyocyte survival following ischemia/reperfusion. Journal of Molecular and Cellular Cardiology, 46, 241–248.
Wei, H., & Vander Heide, R. S. (2008). Heat stress activates AKT via focal adhesion kinase-mediated pathway in neonatal rat ventricular myocytes. American Journal of Physiology. Heart and Circulatory Physiology, 295, H561–H568.
Wei, H., & Vander Heide, R. S. (2010). Ischemic preconditioning and heat shock activate Akt via a focal adhesion kinase-mediated pathway in Langendorff-perfused adult rat hearts. American Journal of Physiology. Heart and Circulatory Physiology, 298, H152–H157.
Riveline, D., Zamir, E., Balaban, N. Q., Schwarz, U. S., Ishizaki, T., Narumiya, S., et al. (2001). Focal contacts as mechanosensors: Externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. The Journal of Cell Biology, 153, 1175–1186.
Vogel, V., & Sheetz, M. P. (2009). Cell fate regulation by coupling mechanical cycles to biochemical signaling pathways. Current Opinion in Cell Biology, 21, 38–46.
Lammerding, J., Kamm, R. D., & Lee, R. T. (2004). Mechanotransduction in cardiac myocytes. Annals of the New York Academy of Sciences, 1015, 53–70.
Hoshijima, M. (2006). Mechanical stress–strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. American Journal of Physiology. Heart and Circulatory Physiology, 290, H1313–H1325.
Sharif-Naeini, R., Folgering, J. H., Bichet, D., Duprat, F., Delmas, P., Patel, A., et al. (2010). Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. Journal of Molecular and Cellular Cardiology, 48, 83–89.
Torsoni, A. S., Constancio, S. S., Nadruz, W., Jr., Hanks, S. K., & Franchini, K. G. (2003). Focal adhesion kinase is activated and mediates the early hypertrophic response to stretch in cardiac myocytes. Circulation Research, 93, 140–147.
Street, S. F. (1983). Lateral transmission of tension in frog myofibers: A myofibrillar network and transverse cytoskeletal connections are possible transmitters. Journal of Cellular Physiology, 114, 346–364.
Bloch, R. J., & Gonzalez-Serratos, H. (2003). Lateral force transmission across costameres in skeletal muscle. Exercise and Sport Sciences Reviews, 31, 73–78.
Collinsworth, A. M., Zhang, S., Kraus, W. E., & Truskey, G. A. (2002). Apparent elastic modulus and hysteresis of skeletal muscle cells throughout differentiation. American Journal of Physiology. Cell Physiology, 283, C1219–C1227.
Goldstein, M. A., Schroeter, J. P., & Michael, L. H. (1991). Role of the Z band in the mechanical properties of the heart. The FASEB Journal, 5, 2167–2174.
Kumar, A., Chaudhry, I., Reid, M. B., & Boriek, A. M. (2002). Distinct signaling pathways are activated in response to mechanical stress applied axially and transversely to skeletal muscle fibers. The Journal of Biological Chemistry, 277, 46493–46503.
Senyo, S. E., Koshman, Y. E., & Russell, B. (2007). Stimulus interval, rate and direction differentially regulate phosphorylation for mechanotransduction in neonatal cardiac myocytes. FEBS Letters, 581, 4241–4247.
Pardo, J. V., Siliciano, J. D., & Craig, S. W. (1983). Vinculin is a component of an extensive network of myofibril-sarcolemma attachment regions in cardiac muscle fibers. The Journal of Cell Biology, 97, 1081–1088.
Danowski, B. A., Imanaka-Yoshida, K., Sanger, J. M., & Sanger, J. W. (1992). Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. The Journal of Cell Biology, 118, 1411–1420.
Mansour, H., de Tombe, P. P., Samarel, A. M., & Russell, B. (2004). Restoration of resting sarcomere length after uniaxial static strain is regulated by protein kinase Cε and focal adhesion kinase. Circulation Research, 94, 642–649.
Michele, D. E., Kabaeva, Z., Davis, S. L., Weiss, R. M., & Campbell, K. P. (2009). Dystroglycan matrix receptor function in cardiac myocytes is important for limiting activity-induced myocardial damage. Circulation Research, 105, 984–993.
Li, R., Wu, Y., Manso, A. M., Gu, Y., Liao, P., Israeli, S., et al. (2012). β1 integrin gene excision in the adult murine cardiac myocyte causes defective mechanical and signaling responses. The American Journal of Pathology, 180, 952–962.
Ervasti, J. M. (2003). Costameres: The Achilles’ heel of Herculean muscle. The Journal of Biological Chemistry, 278, 13591–13594.
Elsherif, L., Huang, M. S., Shai, S. Y., Yang, Y., Li, R. Y., Chun, J., et al. (2008). Combined deficiency of dystrophin and β1 integrin in the cardiac myocyte causes myocardial dysfunction, fibrosis and calcification. Circulation Research, 102, 1109–1117.
Sussman, M. A., McCulloch, A., & Borg, T. K. (2002). Dance band on the Titanic: Biomechanical signaling in cardiac hypertrophy. Circulation Research, 91, 888–898.
Bershadsky, A. D., Balaban, N. Q., & Geiger, B. (2003). Adhesion-dependent cell mechanosensitivity. Annual Review of Cell and Developmental Biology, 19, 677–695.
Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signaling machines. Cell, 110, 673–687.
Keller, R. S., Shai, S. Y., Babbitt, C. J., Pham, C. G., Solaro, R. J., Valencik, M. L., et al. (2001). Disruption of integrin function in the murine myocardium leads to perinatal lethality, fibrosis, and abnormal cardiac performance. The American Journal of Pathology, 158, 1079–1090.
Terracio, L., Rubin, K., Gullberg, D., Balog, E., Carver, W., Jyring, R., et al. (1991). Expression of collagen binding integrins during cardiac development and hypertrophy. Circulation Research, 68, 734–744.
Ross, R. S., & Borg, T. K. (2001). Integrins and the myocardium. Circulation Research, 88, 1112–1119.
Schwartz, M. A. (2009). Cell biology. The force is with us. Science, 323, 588–589.
Campbell, I. D., & Ginsberg, M. H. (2004). The talin-tail interaction places integrin activation on FERM ground. Trends in Biochemical Sciences, 29, 429–435.
Anthis, N. J., Wegener, K. L., Ye, F., Kim, C., Goult, B. T., Lowe, E. D., et al. (2009). The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. The EMBO Journal, 28, 3623–3632.
Zemljic-Harpf, A., Manso, A. M., & Ross, R. S. (2009). Vinculin and talin: Focus on the myocardium. Journal of Investigative Medicine, 57, 849–855.
Senetar, M. A., Moncman, C. L., & McCann, R. O. (2007). Talin2 is induced during striated muscle differentiation and is targeted to stable adhesion complexes in mature muscle. Cell Motility and the Cytoskeleton, 64, 157–173.
Conti, F. J., Monkley, S. J., Wood, M. R., Critchley, D. R., & Muller, U. (2009). Talin 1 and 2 are required for myoblast fusion, sarcomere assembly and the maintenance of myotendinous junctions. Development (Cambridge, England), 136, 3597–3606.
Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., de Pereda, J. M., et al. (2003). Talin binding to integrin β tails: A final common step in integrin activation. Science, 302, 103–106.
Ye, F., Hu, G., Taylor, D., Ratnikov, B., Bobkov, A. A., McLean, M. A., et al. (2010). Recreation of the terminal events in physiological integrin activation. The Journal of Cell Biology, 188, 157–175.
Anthis, N. J., Haling, J. R., Oxley, C. L., Memo, M., Wegener, K. L., Lim, C. J., et al. (2009). β-integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. The Journal of Biological Chemistry, 284, 36700–36710.
Dabiri, G. A., Turnacioglu, K. K., Sanger, J. M., & Sanger, J. W. (1997). Myofibrillogenesis visualized in living embryonic cardiomyocytes. Proceedings of the National Academy of Sciences of the United States of America, 94, 9493–9498.
Stout, A. L., Wang, J., Sanger, J. M., & Sanger, J. W. (2008). Tracking changes in Z-band organization during myofibrillogenesis with FRET imaging. Cell Motility and the Cytoskeleton, 65, 353–367.
Dumbauld, D. W., Michael, K. E., Hanks, S. K., & Garcia, A. J. (2011). Focal adhesion kinase-dependent regulation of adhesive forces involves vinculin recruitment to focal adhesions. Biology of the Cell, 102, 203–213.
Humphries, J. D., Wang, P., Streuli, C., Geiger, B., Humphries, M. J., & Ballestrem, C. (2007). Vinculin controls focal adhesion formation by direct interactions with talin and actin. The Journal of Cell Biology, 179, 1043–1057.
Brown, M. C., Perrotta, J. A., & Turner, C. E. (1996). Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. The Journal of Cell Biology, 135, 1109–1123.
Hayashi, I., Vuori, K., & Liddington, R. C. (2002). The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nature Structural Biology, 9, 101–106.
Kanchanawong, P., Shtengel, G., Pasapera, A. M., Ramko, E. B., Davidson, M. W., Hess, H. F., et al. (2010). Nanoscale architecture of integrin-based cell adhesions. Nature, 468, 580–584.
Kuppuswamy, D., Kerr, C., Narishige, T., Kasi, V. S., Menick, D. R., & Cooper, G. T. (1997). Association of tyrosine-phosphorylated c-Src with the cytoskeleton of hypertrophying myocardium. The Journal of Biological Chemistry, 272, 4500–4508.
Laser, M., Willey, C. D., Jiang, W., Cooper, G., Menick, D. R., Zile, M. R., et al. (2000). Integrin activation and focal complex formation in cardiac hypertrophy. The Journal of Biological Chemistry, 275, 35624–35630.
Domingos, P. P., Fonseca, P. M., Nadruz, W., Jr., & Franchini, K. G. (2002). Load-induced focal adhesion kinase activation in the myocardium: Role of stretch and contractile activity. American Journal of Physiology. Heart and Circulatory Physiology, 282, H556–H564.
Bayer, A. L., Heidkamp, M. C., Patel, N., Porter, M. J., Engman, S. J., & Samarel, A. M. (2002). PYK2 expression and phosphorylation increases in pressure overload-induced left ventricular hypertrophy. American Journal of Physiology. Heart and Circulatory Physiology, 283, H695–H706.
Torsoni, A. S., Fonseca, P. M., Crosara-Alberto, D. P., & Franchini, K. G. (2003). Early activation of p160ROCK by pressure overload in rat heart. American Journal of Physiology. Cell Physiology, 284, C1411–C1419.
Ross, R. S., Pham, C., Shai, S. Y., Goldhaber, J. I., Fenczik, C., Glembotski, C. C., et al. (1998). β1 Integrins participate in the hypertrophic response of rat ventricular myocytes. Circulation Research, 82, 1160–1172.
Eble, D. M., Strait, J. B., Govindarajan, G., Lou, J., Byron, K. L., & Samarel, A. M. (2000). Endothelin-induced cardiac myocyte hypertrophy: Role for focal adhesion kinase. American Journal of Physiology. Heart and Circulatory Physiology, 278, H1695–H1707.
Taylor, J. M., Rovin, J. D., & Parsons, J. T. (2000). A role for focal adhesion kinase in phenylephrine-induced hypertrophy of rat ventricular cardiomyocytes. The Journal of Biological Chemistry, 275, 19250–19257.
Pham, C. G., Harpf, A. E., Keller, R. S., Vu, H. T., Shai, S. Y., Loftus, J. C., et al. (2000). Striated muscle-specific β1D-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. American Journal of Physiology. Heart and Circulatory Physiology, 279, H2916–H2926.
Kovacic-Milivojevic, B., Roediger, F., Almeida, E. A., Damsky, C. H., Gardner, D. G., & Ilic, D. (2001). Focal adhesion kinase and p130Cas mediate both sarcomeric organization and activation of genes associated with cardiac myocyte hypertrophy. Molecular Biology of the Cell, 12, 2290–2307.
Heidkamp, M. C., Bayer, A. L., Kalina, J. A., Eble, D. M., & Samarel, A. M. (2002). GFP-FRNK disrupts focal adhesions and induces anoikis in neonatal rat ventricular myocytes. Circulation Research, 90, 1282–1289.
Heidkamp, M. C., Bayer, A. L., Scully, B. T., Eble, D. M., & Samarel, A. M. (2003). Activation of focal adhesion kinase by protein kinase Cε in neonatal rat ventricular myocytes. American Journal of Physiology. Heart and Circulatory Physiology, 285, H1684–H1696.
Wang, N., Butler, J. P., & Ingber, D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science, 260, 1124–1127.
Schlaepfer, D. D., Mitra, S. K., & Ilic, D. (2004). Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochimica et Biophysica Acta, 1692, 77–102.
Schaller, M. D., Hildebrand, J. D., Shannon, J. D., Fox, J. W., Vines, R. R., & Parsons, J. T. (1994). Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Molecular and Cellular Biology, 14, 1680–1688.
Frame, M. C., Patel, H., Serrels, B., Lietha, D., & Eck, M. J. (2010). The FERM domain: Organizing the structure and function of FAK. Nature Reviews. Molecular Cell Biology, 11, 802–814.
Lim, Y., Han, I., Jeon, J., Park, H., Bahk, Y. Y., & Oh, E. S. (2004). Phosphorylation of focal adhesion kinase at tyrosine 861 is crucial for Ras transformation of fibroblasts. The Journal of Biological Chemistry, 279, 29060–29065.
Yi, X. P., Zhou, J., Huber, L., Qu, J., Wang, X., Gerdes, A. M., et al. (2006). Nuclear compartmentalization of FAK and FRNK in cardiac myocytes. American Journal of Physiology. Heart and Circulatory Physiology, 290, H2509–H2515.
Chu, M., Iyengar, R., Koshman, Y. E., Kim, T., Russell, B., Martin, J. L., et al. (2011). Serine-910 phosphorylation of focal adhesion kinase is critical for sarcomere reorganization in cardiomyocyte hypertrophy. Cardiovascular Research, 92, 409–419.
Seko, Y., Takahashi, N., Tobe, K., Kadowaki, T., & Yazaki, Y. (1999). Pulsatile stretch activates mitogen-activated protein kinase (MAPK) family members and focal adhesion kinase (p125FAK) in cultured rat cardiac myocytes. Biochemical and Biophysical Research Communications, 259, 8–14.
Eble, D. M., Qi, M., Strait, J., & Samarel, A. M. (2000). Contraction-dependent hypertrophy of neonatal rat ventricular myocytes: Potential role for focal adhesion kinase. In N. Takeda & N. S. Dhalla (Eds.), The hypertrophied heart (pp. 91–107). Boston: Kluwer.
Bayer, A. L., Ferguson, A. G., Lucchesi, P. A., & Samarel, A. M. (2001). Pyk2 expression and phosphorylation in neonatal and adult cardiomyocytes. Journal of Molecular and Cellular Cardiology, 33, 1017–1030.
Ilic, D., Furuta, Y., Kanazawa, S., Takeda, N., Sobue, K., Nakatsuji, N., et al. (1995). Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature, 377, 539–544.
Ilic, D., Kovacic, B., McDonagh, S., Jin, F., Baumbusch, C., Gardner, D. G., et al. (2003). Focal adhesion kinase is required for blood vessel morphogenesis. Circulation Research, 92, 300–307.
Hakim, Z. S., DiMichele, L. A., Doherty, J. T., Homeister, J. W., Beggs, H. E., Reichardt, L. F., et al. (2007). Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Molecular and Cellular Biology, 27, 5352–5364.
Peng, X., Kraus, M. S., Wei, H., Shen, T. L., Pariaut, R., Alcaraz, A., et al. (2006). Inactivation of focal adhesion kinase in cardiomyocytes promotes eccentric cardiac hypertrophy and fibrosis in mice. The Journal of Clinical Investigation, 116, 217–227.
DiMichele, L. A., Doherty, J. T., Rojas, M., Beggs, H. E., Reichardt, L. F., Mack, C. P., et al. (2006). Myocyte-restricted focal adhesion kinase deletion attenuates pressure overload-induced hypertrophy. Circulation Research, 99, 636–645.
Nadruz, W., Jr., Corat, M. A., Marin, T. M., Guimaraes Pereira, G. A., & Franchini, K. G. (2005). Focal adhesion kinase mediates MEF2 and c-Jun activation by stretch: Role in the activation of the cardiac hypertrophic genetic program. Cardiovascular Research, 68, 87–97.
Avraham, H., Park, S. Y., Schinkmann, K., & Avraham, S. (2000). RAFTK/Pyk2-mediated cellular signalling. Cellular Signalling, 12, 123–133.
Park, S. Y., Avraham, H. K., & Avraham, S. (2004). RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. The Journal of Biological Chemistry, 279, 33315–33322.
Melendez, J., Welch, S., Schaefer, E., Moravec, C. S., Avraham, S., Avraham, H., et al. (2002). Activation of Pyk2/related focal adhesion tyrosine kinase and focal adhesion kinase in cardiac remodeling. The Journal of Biological Chemistry, 277, 45203–45210.
Kodama, H., Fukuda, K., Takahashi, T., Sano, M., Kato, T., Tahara, S., et al. (2002). Role of EGF receptor and Pyk2 in endothelin-1-induced ERK activation in rat cardiomyocytes. Journal of Molecular and Cellular Cardiology, 34, 139–150.
Bayer, A. L., Heidkamp, M. C., Howes, A. L., Heller Brown, J., Byron, K. L., & Samarel, A. M. (2003). Protein kinase Cε-dependent activation of proline-rich tyrosine kinase 2 in neonatal rat ventricular myocytes. Journal of Molecular and Cellular Cardiology, 35, 1121–1133.
Melendez, J., Turner, C., Avraham, H., Steinberg, S. F., Schaefer, E., & Sussman, M. A. (2004). Cardiomyocyte apoptosis triggered by RAFTK/pyk2 via Src kinase is antagonized by paxillin. The Journal of Biological Chemistry, 279, 53516–53523.
Hirotani, S., Higuchi, Y., Nishida, K., Nakayama, H., Yamaguchi, O., Hikoso, S., et al. (2004). Ca(2+)-sensitive tyrosine kinase Pyk2/CAKβ-dependent signaling is essential for G-protein-coupled receptor agonist-induced hypertrophy. Journal of Molecular and Cellular Cardiology, 36, 799–807.
Heidkamp, M. C., Scully, B. T., Vijayan, K., Engman, S. J., Szotek, E. L., & Samarel, A. M. (2005). PYK2 regulates SERCA2 gene expression in neonatal rat ventricular myocytes. American Journal of Physiology. Cell Physiology, 289, C471–C482.
Blaukat, A., Ivankovic-Dikic, I., Gronroos, E., Dolfi, F., Tokiwa, G., Vuori, K., et al. (1999). Adaptor proteins Grb2 and Crk couple Pyk2 with activation of specific mitogen-activated protein kinase cascades. The Journal of Biological Chemistry, 274, 14893–14901.
Pandey, P., Avraham, S., Kumar, S., Nakazawa, A., Place, A., Ghanem, L., et al. (1999). Activation of p38 mitogen-activated protein kinase by PYK2/related adhesion focal tyrosine kinase-dependent mechanism. The Journal of Biological Chemistry, 274, 10140–10144.
Tokiwa, G., Dikic, I., Lev, S., & Schlessinger, J. (1996). Activation of Pyk2 by stress signals and coupling with JNK signaling pathway. Science, 273, 792–794.
Hart, D. L., Heidkamp, M. C., Iyengar, R., Vijayan, K., Szotek, E. L., Barakat, J. A., et al. (2008). CRNK gene transfer improves function and reverses the myosin heavy chain isoenzyme switch during post-myocardial infarction left ventricular remodeling. Journal of Molecular and Cellular Cardiology, 45, 93–105.
Fukuda, K., Gupta, S., Chen, K., Wu, C., & Qin, J. (2009). The pseudoactive site of ILK is essential for its binding to α-parvin and localization to focal adhesions. Molecular Cell, 36, 819–830.
Wickstrom, S. A., Lange, A., Montanez, E., & Fassler, R. (2010). The ILK/PINCH/parvin complex: The kinase is dead, long live the pseudokinase! The EMBO Journal, 29, 281–291.
Hannigan, G. E., Leung-Hagesteijn, C., Fitz-Gibbon, L., Coppolino, M. G., Radeva, G., Filmus, J., et al. (1996). Regulation of cell adhesion and anchorage-dependent growth by a new β1-integrin-linked protein kinase. Nature, 379, 91–96.
Wu, C., & Dedhar, S. (2001). Integrin-linked kinase (ILK) and its interactors: A new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. The Journal of Cell Biology, 155, 505–510.
Liang, X., Zhou, Q., Li, X., Sun, Y., Lu, M., Dalton, N., et al. (2005). PINCH1 plays an essential role in early murine embryonic development but is dispensable in ventricular cardiomyocytes. Molecular and Cellular Biology, 25, 3056–3062.
White, D. E., Coutu, P., Shi, Y. F., Tardif, J. C., Nattel, S., St Arnaud, R., et al. (2006). Targeted ablation of ILK from the murine heart results in dilated cardiomyopathy and spontaneous heart failure. Genes & Development, 20, 2355–2360.
Persad, S., Attwell, S., Gray, V., Mawji, N., Deng, J. T., Leung, D., et al. (2001). Regulation of protein kinase B/Akt-serine 473 phosphorylation by integrin-linked kinase: Critical roles for kinase activity and amino acids arginine 211 and serine 343. The Journal of Biological Chemistry, 276, 27462–27469.
Attwell, S., Roskelley, C., & Dedhar, S. (2000). The integrin-linked kinase (ILK) suppresses anoikis. Oncogene, 19, 3811–3815.
Bock-Marquette, I., Saxena, A., White, M. D., Dimaio, J. M., & Srivastava, D. (2004). Thymosin β4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature, 432, 466–472.
Disatnik, M. H., Buraggi, G., & Mochly-Rosen, D. (1994). Localization of protein kinase C isozymes in cardiac myocytes. Experimental Cell Research, 210, 287–297.
Huang, X. P., Pi, Y., Lokuta, A. J., Greaser, M. L., & Walker, J. W. (1997). Arachidonic acid stimulates protein kinase C-ε redistribution in heart cells. Journal of Cell Science, 110(Pt 14), 1625–1634.
Borg, T. K., Goldsmith, E. C., Price, R., Carver, W., Terracio, L., & Samarel, A. M. (2000). Specialization at the Z line of cardiac myocytes. Cardiovascular Research, 46, 277–285.
Ping, P., Zhang, J., Pierce, W. M., Jr., & Bolli, R. (2001). Functional proteomic analysis of protein kinase Cε signaling complexes in the normal heart and during cardioprotection. Circulation Research, 88, 59–62.
Vondriska, T. M., Zhang, J., Song, C., Tang, X. L., Cao, X., Baines, C. P., et al. (2001). Protein kinase Cε-Src modules direct signal transduction in nitric oxide-induced cardioprotection: Complex formation as a means for cardioprotective signaling. Circulation Research, 88, 1306–1313.
Ping, P., Song, C., Zhang, J., Guo, Y., Cao, X., Li, R. C., et al. (2002). Formation of protein kinase Cε-Lck signaling modules confers cardioprotection. The Journal of Clinical Investigation, 109, 499–507.
Song, C., Vondriska, T. M., Wang, G. W., Klein, J. B., Cao, X., Zhang, J., et al. (2002). Molecular conformation dictates signaling module formation: Example of PKCε and Src tyrosine kinase. American Journal of Physiology. Heart and Circulatory Physiology, 282, H1166–H1171.
Heidkamp, M. C., Iyengar, R., Szotek, E. L., Cribbs, L. L., & Samarel, A. M. (2007). Protein kinase Cε-dependent MARCKS phosphorylation in neonatal and adult rat ventricular myocytes. Journal of Molecular and Cellular Cardiology, 42, 422–431.
Strait, J. B., Martin, J. L., Bayer, A., Mestril, R., Eble, D. M., & Samarel, A. M. (2001). Role of protein kinase Cε in hypertrophy of cultured neonatal rat ventricular myocytes. American Journal of Physiology. Heart and Circulatory Physiology, 280, H756–H766.
Liliental, J., & Chang, D. D. (1998). RACK1, a receptor for activated protein kinase C, interacts with integrin β subunit. The Journal of Biological Chemistry, 273, 2379–2383.
Mochly-Rosen, D., Khaner, H., & Lopez, J. (1991). Identification of intracellular receptor proteins for activated protein kinase C. Proceedings of the National Academy of Sciences of the United States of America, 88, 3997–4000.
Chang, B. Y., Conroy, K. B., Machleder, E. M., & Cartwright, C. A. (1998). RACK1, a receptor for activated C kinase and a homolog of the β subunit of G proteins, inhibits activity of Src tyrosine kinases and growth of NIH 3T3 cells. Molecular and Cellular Biology, 18, 3245–3256.
Schechtman, D., & Mochly-Rosen, D. (2001). Adaptor proteins in protein kinase C-mediated signal transduction. Oncogene, 20, 6339–6347.
Besson, A., Wilson, T. L., & Yong, V. W. (2002). The anchoring protein RACK1 links protein kinase Cε to integrin β chains. Requirements for adhesion and motility. The Journal of Biological Chemistry, 277, 22073–22084.
Berrier, A. L., Mastrangelo, A. M., Downward, J., Ginsberg, M., & LaFlamme, S. E. (2000). Activated R-Ras, Rac1, PI 3-kinase and PKCε can each restore cell spreading inhibited by isolated integrin β1 cytoplasmic domains. The Journal of Cell Biology, 151, 1549–1560.
Robia, S. L., Ghanta, J., Robu, V. G., & Walker, J. W. (2001). Localization and kinetics of protein kinase Cε anchoring in cardiac myocytes. Biophysical Journal, 80, 2140–2151.
Huang, X., & Walker, J. W. (2004). Myofilament anchoring of protein kinase C-ε in cardiac myocytes. Journal of Cell Science, 117, 1971–1978.
Vuori, K., & Ruoslahti, E. (1993). Activation of protein kinase C precedes α5β1 integrin-mediated cell spreading on fibronectin. The Journal of Biological Chemistry, 268, 21459–21462.
Parsons, J. T. (2003). Focal adhesion kinase: The first ten years. Journal of Cell Science, 116, 1409–1416.
Ruwhof, C., van Wamel, J. T., Noordzij, L. A., Aydin, S., Harper, J. C., & van der Laarse, A. (2001). Mechanical stress stimulates phospholipase C activity and intracellular calcium ion levels in neonatal rat cardiomyocytes. Cell Calcium, 29, 73–83.
Decker, M. L., Simpson, D. G., Behnke, M., Cook, M. G., & Decker, R. S. (1990). Morphological analysis of contracting and quiescent adult rabbit cardiac myocytes in long-term culture. The Anatomical Record, 227, 285–299.
Sharp, W. W., Simpson, D. G., Borg, T. K., Samarel, A. M., & Terracio, L. (1997). Mechanical forces regulate focal adhesion and costamere assembly in cardiac myocytes. The American Journal of Physiology, 273, H546–H556.
Peng, X., Wu, X., Druso, J. E., Wei, H., Park, A. Y., Kraus, M. S., et al. (2008). Cardiac developmental defects and eccentric right ventricular hypertrophy in cardiomyocyte focal adhesion kinase (FAK) conditional knockout mice. Proceedings of the National Academy of Sciences of the United States of America, 105, 6638–6643.
DiMichele, L. A., Hakim, Z. S., Sayers, R. L., Rojas, M., Schwartz, R. J., Mack, C. P., et al. (2009). Transient expression of FRNK reveals stage-specific requirement for focal adhesion kinase activity in cardiac growth. Circulation Research, 104, 1201–1208.
Quach, N. L., & Rando, T. A. (2006). Focal adhesion kinase is essential for costamerogenesis in cultured skeletal muscle cells. Developmental Biology, 293, 38–52.
Hunger-Glaser, I., Salazar, E. P., Sinnett-Smith, J., & Rozengurt, E. (2003). Bombesin, lysophosphatidic acid, and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: Requirement for ERK activation. The Journal of Biological Chemistry, 278, 22631–22643.
Subauste, M. C., Pertz, O., Adamson, E. D., Turner, C. E., Junger, S., & Hahn, K. M. (2004). Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. The Journal of Cell Biology, 165, 371–381.
Zemljic-Harpf, A. E., Miller, J. C., Henderson, S. A., Wright, A. T., Manso, A. M., Elsherif, L., et al. (2007). Cardiac-myocyte-specific excision of the vinculin gene disrupts cellular junctions, causing sudden death or dilated cardiomyopathy. Molecular and Cellular Biology, 27, 7522–7537.
Hagel, M., George, E. L., Kim, A., Tamimi, R., Opitz, S. L., Turner, C. E., et al. (2002). The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Molecular and Cellular Biology, 22, 901–915.
Honda, H., Oda, H., Nakamoto, T., Honda, Z., Sakai, R., Suzuki, T., et al. (1998). Cardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nature Genetics, 19, 361–365.
Goldstein, M. A., Michael, L. H., Schroeter, J. P., & Sass, R. L. (1988). Structural states in the Z band of skeletal muscle correlate with states of active and passive tension. The Journal of General Physiology, 92, 113–119.
Knoll, R., Hoshijima, M., & Chien, K. (2003). Cardiac mechanotransduction and implications for heart disease. Journal of Molecular Medicine, 81, 750–756.
Pyle, W. G., & Solaro, R. J. (2004). At the crossroads of myocardial signaling: The role of Z-discs in intracellular signaling and cardiac function. Circulation Research, 94, 296–305.
Knoll, R., Buyandelger, B., & Lab, M. (2011). The sarcomeric Z-disc and Z-discopathies. Journal of Biomedicine & Biotechnology, 2011, 569628.
Buyandelger, B., Ng, K. E., Miocic, S., Piotrowska, I., Gunkel, S., Ku, C. H., et al. (2011). MLP (muscle LIM protein) as a stress sensor in the heart. Pflügers Archiv, 462, 135–142.
Boateng, S. Y., Senyo, S. E., Qi, L., Goldspink, P. H., & Russell, B. (2009). Myocyte remodeling in response to hypertrophic stimuli requires nucleocytoplasmic shuttling of muscle LIM protein. Journal of Molecular and Cellular Cardiology, 47, 426–435.
Sanger, J. W., Wang, J., Fan, Y., White, J., & Sanger, J. M. (2010). Assembly and dynamics of myofibrils. Journal of Biomedicine & Biotechnology, 2010, 858606.
Zhu, W., Zou, Y., Shiojima, I., Kudoh, S., Aikawa, R., Hayashi, D., et al. (2000). Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. The Journal of Biological Chemistry, 275, 15239–15245.
Pyle, W. G., Hart, M. C., Cooper, J. A., Sumandea, M. P., de Tombe, P. P., & Solaro, R. J. (2002). Actin capping protein: An essential element in protein kinase signaling to the myofilaments. Circulation Research, 90, 1299–1306.
Yu, J. G., & Russell, B. (2005). Cardiomyocyte remodeling and sarcomere addition after uniaxial static strain in vitro. The Journal of Histochemistry and Cytochemistry, 53, 839–844.
Hartman, T. J., Martin, J. L., Solaro, R. J., Samarel, A. M., & Russell, B. (2009). CapZ dynamics are altered by endothelin-1 and phenylephrine via PIP2- and PKC-dependent mechanisms. American Journal of Physiology. Cell Physiology, 296, C1034–C1039.
Maruyama, K., & Obinata, T. (1965). Presence of β-actinin in the soluble fraction of the muscle cells of the chick embryo. Journal of Biochemistry, 57, 575–577.
Maruyama, K. (1966). Effect of β-actinin on the particle length of F-actin. Biochimica et Biophysica Acta, 126, 389–398.
Maruyama, K., Kimura, S., Ishi, T., Kuroda, M., & Ohashi, K. (1977). β-actinin, a regulatory protein of muscle. Purification, characterization and function. Journal of Biochemistry, 81, 215–232.
Asakura, S. (1961). F-actin adenosine triphosphatase activated under sonic vibration. Biochimica et Biophysica Acta, 52, 65–75.
Isenberg, G., Aebi, U., & Pollard, T. D. (1980). An actin-binding protein from Acanthamoeba regulates actin filament polymerization and interactions. Nature, 288, 455–459.
Hart, M. C., Korshunova, Y. O., & Cooper, J. A. (1997). Vertebrates have conserved capping protein α isoforms with specific expression patterns. Cell Motility and the Cytoskeleton, 38, 120–132.
Hug, C., Miller, T. M., Torres, M. A., Casella, J. F., & Cooper, J. A. (1992). Identification and characterization of an actin-binding site of CapZ. The Journal of Cell Biology, 116, 923–931.
Schafer, D. A., Korshunova, Y. O., Schroer, T. A., & Cooper, J. A. (1994). Differential localization and sequence analysis of capping protein β-subunit isoforms of vertebrates. The Journal of Cell Biology, 127, 453–465.
von Bulow, M., Rackwitz, H. R., Zimbelmann, R., & Franke, W. W. (1997). CP β3, a novel isoform of an actin-binding protein, is a component of the cytoskeletal calyx of the mammalian sperm head. Experimental Cell Research, 233, 216–224.
Hart, M. C., & Cooper, J. A. (1999). Vertebrate isoforms of actin capping protein β have distinct functions in vivo. The Journal of Cell Biology, 147, 1287–1298.
Yamashita, A., Maeda, K., & Maeda, Y. (2003). Crystal structure of CapZ: Structural basis for actin filament barbed end capping. The EMBO Journal, 22, 1529–1538.
Barron-Casella, E. A., Torres, M. A., Scherer, S. W., Heng, H. H., Tsui, L. C., & Casella, J. F. (1995). Sequence analysis and chromosomal localization of human Cap Z. Conserved residues within the actin-binding domain may link Cap Z to gelsolin/severin and profilin protein families. The Journal of Biological Chemistry, 270, 21472–21479.
Wear, M. A., Yamashita, A., Kim, K., Maeda, Y., & Cooper, J. A. (2003). How capping protein binds the barbed end of the actin filament. Current Biology, 13, 1531–1537.
Kim, K., McCully, M. E., Bhattacharya, N., Butler, B., Sept, D., & Cooper, J. A. (2007). Structure/function analysis of the interaction of phosphatidylinositol 4,5-bisphosphate with actin-capping protein: Implications for how capping protein binds the actin filament. The Journal of Biological Chemistry, 282, 5871–5879.
Wear, M. A., & Cooper, J. A. (2004). Capping protein binding to S100B: Implications for the tentacle model for capping the actin filament barbed end. The Journal of Biological Chemistry, 279, 14382–14390.
Takeda, S., Minakata, S., Koike, R., Kawahata, I., Narita, A., Kitazawa, M., et al. (2010). Two distinct mechanisms for actin capping protein regulation-steric and allosteric inhibition. PLoS Biology, 8, e1000416.
Kim, T., Cooper, J. A., & Sept, D. (2010). The interaction of capping protein with the barbed end of the actin filament. Journal of Molecular Biology, 404, 794–802.
Narita, A., Takeda, S., Yamashita, A., & Maeda, Y. (2006). Structural basis of actin filament capping at the barbed-end: A cryo-electron microscopy study. The EMBO Journal, 25, 5626–5633.
Dorn, G. W., Souroujon, M. C., Liron, T., Chen, C. H., Gray, M. O., Zhou, H. Z., et al. (1999). Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proceedings of the National Academy of Sciences of the United States of America, 96, 12798–12803.
Swanson, E. R., Warren, C. M., Solaro, R. J., Samarel, A. M., & Russell, B. (2011). Cyclic mechanical strain alters CapZ β1 but not CapZ β2 dynamics and phosphorylation via PKCε-dependent mechanisms. Journal of Molecular and Cellular Cardiology, 51, S23 (abstract).
Rybin, V. O., & Steinberg, S. F. (1994). Protein kinase C isoform expression and regulation in the developing rat heart. Circulation Research, 74, 299–309.
Prekeris, R., Mayhew, M. W., Cooper, J. B., & Terrian, D. M. (1996). Identification and localization of an actin-binding motif that is unique to the epsilon isoform of protein kinase C and participates in the regulation of synaptic function. The Journal of Cell Biology, 132, 77–90.
Mochly-Rosen, D., Wu, G., Hahn, H., Osinska, H., Liron, T., Lorenz, J. N., et al. (2000). Cardiotrophic effects of protein kinase Cε: Analysis by in vivo modulation of PKCε translocation. Circulation Research, 86, 1173–1179.
Gupta, M. P., Samant, S. A., Smith, S. H., & Shroff, S. G. (2008). HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. The Journal of Biological Chemistry, 283, 10135–10146.
Takeishi, Y., Ping, P., Bolli, R., Kirkpatrick, D. L., Hoit, B. D., & Walsh, R. A. (2000). Transgenic overexpression of constitutively active protein kinase Cε causes concentric cardiac hypertrophy. Circulation Research, 86, 1218–1223.
Sanger, J. M., & Sanger, J. W. (2008). The dynamic Z bands of striated muscle cells. Science Signaling, 1, pe37.
Lemmon, M. A. (2008). Membrane recognition by phospholipid-binding domains. Nature Reviews. Molecular Cell Biology, 9, 99–111.
Huang, S., Gao, L., Blanchoin, L., & Staiger, C. J. (2006). Heterodimeric capping protein from Arabidopsis is regulated by phosphatidic acid. Molecular Biology of the Cell, 17, 1946–1958.
Kilimann, M. W., & Isenberg, G. (1982). Actin filament capping protein from bovine brain. The EMBO Journal, 1, 889–894.
Hartmann, H., Noegel, A. A., Eckerskorn, C., Rapp, S., & Schleicher, M. (1989). Ca2+-independent F-actin capping proteins. Cap 32/34, a capping protein from Dictyostelium discoideum, does not share sequence homologies with known actin-binding proteins. The Journal of Biological Chemistry, 264, 12639–12647.
Nachmias, V. T., Golla, R., Casella, J. F., & Barron-Casella, E. (1996). Cap Z, a calcium insensitive capping protein in resting and activated platelets. FEBS Letters, 378, 258–262.
Janmey, P. A., Iida, K., Yin, H. L., & Stossel, T. P. (1987). Polyphosphoinositide micelles and polyphosphoinositide-containing vesicles dissociate endogenous gelsolin-actin complexes and promote actin assembly from the fast-growing end of actin filaments blocked by gelsolin. The Journal of Biological Chemistry, 262, 12228–12236.
Janmey, P. A., & Stossel, T. P. (1987). Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature, 325, 362–364.
Papa, I., Astier, C., Kwiatek, O., Raynaud, F., Bonnal, C., Lebart, M. C., et al. (1999). α-Actinin-CapZ, an anchoring complex for thin filaments in Z-line. Journal of Muscle Research and Cell Motility, 20, 187–197.
Schafer, D. A., Jennings, P. B., & Cooper, J. A. (1996). Dynamics of capping protein and actin assembly in vitro: Uncapping barbed ends by polyphosphoinositides. The Journal of Cell Biology, 135, 169–179.
Visser, M. B., Koh, A., Glogauer, M., & Ellen, R. P. (2011). Treponema denticola major outer sheath protein induces actin assembly at free barbed ends by a PIP2-dependent uncapping mechanism in fibroblasts. PLoS One, 6, e23736.
Smith, J., Diez, G., Klemm, A. H., Schewkunow, V., & Goldmann, W. H. (2006). CapZ-lipid membrane interactions: A computer analysis. Theoretical Biology & Medical Modelling, 3, 30.
Pyle, W. G., La Rotta, G., de Tombe, P. P., Sumandea, M. P., & Solaro, R. J. (2006). Control of cardiac myofilament activation and PKC-βII signaling through the actin capping protein, CapZ. Journal of Molecular and Cellular Cardiology, 41, 537–543.
Dellefave, L., & McNally, E. M. (2010). The genetics of dilated cardiomyopathy. Current Opinion in Cardiology, 25, 198–204.
Seidman, C. E., & Seidman, J. G. (2011). Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circulation Research, 108, 743–750.
Moore, J. R., Leinwand, L., & Warshaw, D. M. (2012). Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circulation Research, 111, 375–385.
Dorn, G. W. (2012). Decoding the cardiac message: The 2011 Thomas W. Smith memorial lecture. Circulation Research, 110, 755–763.
Arimura, T., Hayashi, T., Terada, H., Lee, S. Y., Zhou, Q., Takahashi, M., et al. (2004). A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C. The Journal of Biological Chemistry, 279, 6746–6752.
Arimura, T., Inagaki, N., Hayashi, T., Shichi, D., Sato, A., Hinohara, K., et al. (2009). Impaired binding of ZASP/Cypher with phosphoglucomutase 1 is associated with dilated cardiomyopathy. Cardiovascular Research, 83, 80–88.
Moric-Janiszewska, E., & Markiewicz-Loskot, G. (2008). Genetic heterogeneity of left-ventricular noncompaction cardiomyopathy. Clinical Cardiology, 31, 201–204.
Zhou, Q., Chu, P. H., Huang, C., Cheng, C. F., Martone, M. E., Knoll, G., et al. (2001). Ablation of Cypher, a PDZ-LIM domain Z-line protein, causes a severe form of congenital myopathy. The Journal of Cell Biology, 155, 605–612.
Zheng, M., Cheng, H., Li, X., Zhang, J., Cui, L., Ouyang, K., et al. (2009). Cardiac-specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Human Molecular Genetics, 18, 701–713.
Mochly-Rosen, D., Smith, B. L., Chen, C. H., Disatnik, M. H., & Ron, D. (1995). Interaction of protein kinase C with RACK1, a receptor for activated C-kinase: A role in β protein kinase C mediated signal transduction. Biochemical Society Transactions, 23, 596–600.
Zhou, Q., Ruiz-Lozano, P., Martone, M. E., & Chen, J. (1999). Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to α-actinin-2 and protein kinase C. The Journal of Biological Chemistry, 274, 19807–19813.
Nakagawa, N., Hoshijima, M., Oyasu, M., Saito, N., Tanizawa, K., & Kuroda, S. (2000). ENH, containing PDZ and LIM domains, heart/skeletal muscle-specific protein, associates with cytoskeletal proteins through the PDZ domain. Biochemical and Biophysical Research Communications, 272, 505–512.
Taniguchi, K., Takeya, R., Suetsugu, S., Kan, O. M., Narusawa, M., Shiose, A., et al. (2009). Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. The Journal of Biological Chemistry, 284, 29873–29881.
Iskratsch, T., Lange, S., Dwyer, J., Kho, A. L., dos Remedios, C., & Ehler, E. (2010). Formin follows function: A muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. The Journal of Cell Biology, 191, 1159–1172.
Iskratsch, T., & Ehler, E. (2011). Formin-g muscle cytoarchitecture. BioArchitecture, 1, 66–68.
Kan-o, M., Takeya, R., Taniguchi, K., Tanoue, Y., Tominaga, R., & Sumimoto, H. (2012). Expression and subcellular localization of mammalian formin Fhod3 in the embryonic and adult heart. PLoS One, 7, e34765.
Bai, J., Hartwig, J. H., & Perrimon, N. (2007). SALS, a WH2-domain-containing protein, promotes sarcomeric actin filament elongation from pointed ends during Drosophila muscle growth. Developmental Cell, 13, 828–842.
Pruyne, D., Evangelista, M., Yang, C., Bi, E., Zigmond, S., Bretscher, A., et al. (2002). Role of formins in actin assembly: Nucleation and barbed-end association. Science, 297, 612–615.
Kovar, D. R., Bestul, A. J., Li, Y. J., & Scott, B. J. (2010). Formin-mediated actin assembly. In M.-F. Carlier (Ed.), Actin-based motility: Cellular, molecular and physical aspects (pp. 279–316). New York: Springer.
Li, F., & Higgs, H. N. (2005). Dissecting requirements for auto-inhibition of actin nucleation by the formin, mDia1. The Journal of Biological Chemistry, 280, 6986–6992.
Westendorf, J. J. (2001). The formin/diaphanous-related protein, FHOS, interacts with Rac1 and activates transcription from the serum response element. The Journal of Biological Chemistry, 276, 46453–46459.
Katoh, M., & Katoh, M. (2004). Identification and characterization of human FHOD3 gene in silico. International Journal of Molecular Medicine, 13, 615–620.
Kanaya, H., Takeya, R., Takeuchi, K., Watanabe, N., Jing, N., & Sumimoto, H. (2005). Fhos2, a novel formin-related actin-organizing protein, probably associates with the nestin intermediate filament. Genes to Cells, 10, 665–678.
Takeya, R., Taniguchi, K., Narumiya, S., & Sumimoto, H. (2008). The mammalian formin FHOD1 is activated through phosphorylation by ROCK and mediates thrombin-induced stress fibre formation in endothelial cells. The EMBO Journal, 27, 618–628.
Zigmond, S. H., Evangelista, M., Boone, C., Yang, C., Dar, A. C., Sicheri, F., et al. (2003). Formin leaky cap allows elongation in the presence of tight capping proteins. Current Biology, 13, 1820–1823.
Cooper, J. A., & Sept, D. (2008). New insights into mechanism and regulation of actin capping protein. International Review of Cell and Molecular Biology, 267, 183–206.
Acknowledgments
Supported in part by NIH P01 HL62426, NIH 1F32 HL096143, and a grant from the Dr. Ralph and Marian Falk Medical Research Trust.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Samarel, A.M., Koshman, Y., Swanson, E.R., Russell, B. (2013). Biophysical Forces Modulate the Costamere and Z-Disc for Sarcomere Remodeling in Heart Failure. In: Solaro, R., Tardiff, J. (eds) Biophysics of the Failing Heart. Biological and Medical Physics, Biomedical Engineering. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7678-8_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7678-8_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7677-1
Online ISBN: 978-1-4614-7678-8
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)