
Abstract
Autophagy is a highly conserved lysosomal degradation pathway that is important for maintaining cellular homeostasis by degrading bulk cytoplasm and superfluous or damaged organelles. Autophagy plays a dual role in cancer because it suppresses tumorigenesis and also promotes cancer cell survival for existing tumors. Therefore, targeting autophagy has become a promising therapeutic approach for preventing or treating cancers. With the rapid progression of autophagy research and our expanding knowledge on autophagy machinery and regulation pathways, many high-throughput screening assays have been established and conducted. Here, we summarize potential autophagy proteins and signaling pathways that could be drug targets for modulating autophagy. We also summarize novel compounds that have been discovered from high-throughput screening, which can either inhibit or promote autophagy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
A fundamental goal in cancer research is to develop therapeutic drugs that selectively kill cancer cells, but have minimal cytotoxicity to the surrounding normal tissues. Over the past two decades, induction of apoptosis by therapeutically targeting the apoptotic pathway has become a major strategy to kill cancer cells because of its well-known genetically controlled pathways (Letai 2008). With the expanding understanding of molecular mechanisms of apoptosis, many chemotherapy drugs have been developed to treat cancer patients by inducing apoptosis. However, some tumors have developed resistance to traditional chemotherapeutic drugs that induce apoptosis. Therefore, an emerging and novel strategy for cancer therapy is to target not only the cell death pathway, but also the cell survival pathway.
Macroautophagy (referred to as autophagy hereafter) is a major intracellular degradation system that is mainly responsible for the degradation of long-lived proteins and other cellular contents (Levine and Klionsky 2004; Lum et al. 2005b). Autophagy is a bulk degradation system that is usually activated in response to an adverse environment, such as the deprivation of nutrients or growth factors (Kamada et al. 2004). Autophagy also plays a role in development (Levine and Klionsky 2004), aging, defense against microbial infections (Kirkegaard et al. 2004), and pathogenesis of many diseases, such as neurodegenerative diseases (Shintani and Klionsky 2004). As an important cellular homeostasis regulator, it is not surprising that autophagy also plays an important role in cancer. However, accumulating evidence suggests that autophagy plays differential roles in tumorigenesis and existing cancer.
2 Role of Autophagy in Cancer
2.1 Autophagy Is a Tumor Suppressor During Tumorigenesis
Accumulating evidence supports that autophagy functions as a tumor suppressor. The first evidence comes from the findings that there are increased spontaneous tumors in Beclin 1 monoallelically deleted mice, a gene involved in the autophagy process in mammals (Huang and Tindall 2011; Tzivion and Hay 2011). Later, it was also found that knockout of Bif-1, another gene involved in autophagy, also leads to increased tumorigenesis in multiple organs (Huang and Tindall 2011; Takahashi et al. 2007). Because both Beclin 1 and Bif-1 also play a role in apoptosis by interacting with Bcl-2 family proteins, it is suspected that the tumor suppressor role of Beclin 1 and Bif-1 could be due to their multiple functions in addition to autophagy. However, the finding that liver-specific knockout of Atg5 or Atg7 leads to increased liver tumors clearly supports the notion that autophagy is a bona fide tumor suppressor (Takamura et al. 2011). More recently, it was found that liver-specific TSC1 knockout mice, which have increased mTOR activation and decreased autophagy, also had increased liver tumors (Tzivion et al. 2011). Furthermore, various other tumor suppressors such as p53, DAPK, p19ARF, LKB, and PTEN all have shown to be able to induce autophagy, whereas oncogenes such as Bcl-2 and AKT suppress autophagy. Although the exact mechanisms by which autophagy suppress tumorigenesis are not completely understood, it is suggested that autophagy may help to remove damaged mitochondria, which in turn reduces mitochondrial mediated free radicals and, subsequently, genome instability (Mathew et al. 2009). In addition, increased inflammation has been consistently observed in the mouse liver with allelic loss of Beclin 1 (Mathew et al. 2009), in autophagy-deficient tumor allografts (Degenhardt et al. 2006), and in Crohn’s disease with the expression of a hypomorphic allele Atg16L1 (Cadwell et al. 2008). These results suggest that autophagy can limit inflammation. Furthermore, p62/SQSTM1, a multifunctional adapter protein, is usually accumulated in autophagy-deficient tissues (Komatsu et al. 2007; Ni et al. 2012). p62/SQSTM1 has been found to be a potential molecular link between autophagy inhibition and tumor development. Indeed, p62/SQSTM1 knockout mice were protected from Ras-induced lung carcinomas compared to wild-type mice. p62/SQSTM1 and Atg7 double-knockout mice also have few liver tumors compared to Atg7 liver-specific knockout mice (Takamura et al. 2011). p62/SQSTM1 may promote liver tumorigenesis through multiple mechanisms. First, p62/SQSTM1 interacts with Keap1, which is an inhibitor of the Nrf2 transcription factor. Nrf2 is important for regulation of cellular redox homeostasis. Interaction of p62/SQSTM1 with keap1 releases its inhibitory effect on Nrf2 and causes persistent Nrf2 activation (Komatsu et al. 2010). Activation of Nrf2 has also been found in liver tumor cells but not in adjacent normal tissues (Inami et al. 2011). Recent evidence indicates that expression of oncogenes such as Kras, Braf, and Myc can increase Nrf2 activation, which increases expression of antioxidant genes and leads to the detoxification of reactive oxygen species to promote cell survival and oncogene-driven tumorigenesis (DeNicola et al. 2011). Moreover, Nrf2 activation can lead to up-regulation of Bcl-2, which inhibits apoptosis and increases cancer cell survival (Niture and Jaiswal 2012). Furthermore, Nrf2 activation can also redirect glucose and glutamine into anabolic pathways to promote cell proliferation (Mitsuishi et al. 2012). In addition to regulating Nrf2 activation, p62/SQSTM1 interacts with mTOR, raptor, and RAG to promote mTORC1 activation and cell proliferation (Duran et al. 2011). Finally, p62/SQSTM1 could also serve as a modulator of mitotic transit and genomic stability as well as the activation of nuclear factor-kappaB (NF-kappaB) (Duran et al. 2008; Mathew et al. 2009). Together, these findings indicate that autophagy suppresses tumorigenesis by multiple mechanisms, and autophagy inducers might be effective in preventing tumorigenesis.
2.2 Autophagy Acts as a Cell Survival Mechanism in Cancer Cells
While autophagy serves as a tumor suppressor mechanism during tumorigenesis, autophagy is activated in existing tumors to maintain tumor cell survival. Many conditions can activate autophagy in cancer cells such as starvation, growth factor deprivation, and hypoxia. Autophagy may also help recycle amino acids to maintain cellular biosynthesis and ATP levels for cell survival. It has been found that the number of autophagosomes is significantly increased in tumor cells that are located in hypoxic tumor regions, and cells with the deletion of autophagy genes in these regions are prone to undergo cell death. The induction of autophagy in hypoxic tumor regions can be hypoxia-inducing factor 1 α dependent or independent. Furthermore, pharmacological inhibition of autophagy or genetic knockdown of autophagy genes renders cancer cells as more sensitive to various chemotherapies both in vitro and in vivo, further supporting the notion that autophagy promotes cancer cell survival (Amaravadi et al. 2011; Ding et al. 2007a; Lum et al. 2005a). Oncogenic transformation by activation of RAS, which is important to promote tumor growth, also activates autophagy. Interestingly, we found that the combination of autophagy suppression and proteasome inhibition killed more RAS-transformed tumor cells than non-transformed normal cells (Ding et al. 2009). This indicates that RAS-transformed tumor cells seem to rely more on autophagy for survival than normal non-transformed cells. Thus, this finding supports the current idea for cancer treatment, which involves a combination of autophagy inhibition with traditional chemotherapy treatments to more selectively kill cancer cells with less cytotoxic effects on normal cells. Indeed, inhibition of autophagy using the antimalarial agent hydroxychloroquine (HCQ), a lysosomal inhibitor that inhibits autophagy by increasing lysosomal pH, together with other chemotherapy drugs is being actively assessed in the clinic (Amaravadi et al. 2011). Thus, identification of more novel autophagy inhibitors in addition to HCQ will help for development of more efficient cancer treatments that involve inhibition of autophagy in cancer cells.
2.3 Paradoxical Role of Autophagy in Cell Death
As discussed above, cancer cells use autophagy as a cell survival pathway when facing a variety of stresses including hypoxia, growth factor deprivation, starvation, endoplasmic reticulum (ER) stress, or damaging stimuli as well as proteasome inhibition (Ding et al. 2007a, b; Hu et al. 2012; Lum et al. 2005a). By doing so, autophagy helps to maintain mitochondrial quality control and genome stability and also provides more fuel for supporting mitochondrial metabolism and energy homeostasis. In addition to cancer cells, genetic deletion of Atg5 or Atg7 from mouse livers leads to increased cell death and liver injury, suggesting that even basal autophagy is an important cell survival pathway for normal liver cells (Komatsu et al. 2007; Ni et al. 2012). Together, this evidence strongly supports the notion that autophagy is a cellular protective mechanism. Indeed, accumulating evidence has demonstrated that inhibition of autophagy can lead to more cell death in cancer cells or make cancer cells more susceptible to chemotherapy (Degenhardt et al. 2006; Ding et al. 2007b; White 2012). These findings are very significant for cancer therapy because many cancer cells eventually become resistant to chemotherapeutic drugs, and inhibition of autophagy can make these drug-resistant cancer cells more susceptible for drug-induced cell death. In line with this, discovery of novel autophagy inhibitors by biomedical research would be beneficial for cancer therapy. While most evidence supports that autophagy is a cell survival mechanism for cancer cells, it has also been hotly debated whether autophagy could be a cell death mechanism, which is referred to as “autophagic cell death.” Many early studies only employed morphological approaches when defining autophagic cell death. Therefore, criteria used in these studies for autophagic cell death may not be appropriate for definition of autophagic cell death because coexistence of autophagy with cell death does not guarantee that autophagy contributes to cell death. In this case, autophagy could be detrimental, protective, or just a bystander. However, with better understanding of autophagy machinery and molecular pathways, it is now relatively easier to address this issue. It seems that under certain circumstances or in some particular cell types, autophagy may contribute to cell death. During the developmental stage of Drosophila, programmed cell death is required for the degradation of the salivary glands. Interestingly, both autophagy and apoptosis are induced during this process. Using Atg8 and Atg18 mutants, autophagy was found to contribute to the cell death (Berry and Baehrecke 2007). In mammalian cells, autophagy has also been found to contribute to cell death when cells are exposed to certain chemotherapeutic drugs (Kanzawa et al. 2005; Shimizu et al. 2004), radiation(Moretti et al. 2007), hypoxia (Azad et al. 2008), and cytokines such as INF-γ (Pyo et al. 2005). In all these cases, siRNA knockdown or genetic deletion of key autophagy genes suppresses cell death, while overexpression of these genes promotes cell death. Exactly how autophagy induces cell death is not clear, although it is generally thought that excessive autophagy may non-selectively degrade essential cell components. Moreover, whether there is a real “autophagic cell death” is also debatable, because the presence of autophagy may actually just promote either apoptosis or necrosis (Shen et al. 2012). Nevertheless, in certain cancer cells such as murine fibrosarcoma L929 cells (Yu et al. 2004), breast cancer MCF7 cells (Akar et al. 2008), or glioblastoma cells (Kanzawa et al. 2005; Takeuchi et al. 2005), induction of autophagy seems to promote cell death. In this case, autophagy inducers would be beneficial for the treatment of these cancers. Therefore, usefulness of autophagy inhibitors or inducers for cancer treatment may depend on the specific cancer type. More work is definitely needed to further clarify why certain cancers behave differently in response to the modulation of autophagy. However, it should be noted that in most cases, inhibition of autophagy can further promote cell death. In line with this notion, the approach to use autophagy inhibitors alone or in combination with a chemotherapy drug for clinical trials is currently under investigation.
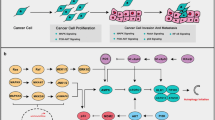
3 Targeting Different Autophagy Pathways for Cancer Therapy
More than 30 autophagy-related (Atg) genes have been identified in mammalian cells, and many of them play a critical role in regulating the formation of double-membrane autophagosomes, which is described in Chap. 2 in detail. Many signal transduction pathways that regulate autophagy have also been revealed and described in detail in Chap. 4. Here we focus on these pathways and autophagy molecular machinery that have been shown to be potential drug targets for modulating autophagy.
3.1 Drug Development for Targeting Signaling Pathways Regulating Mammalian Autophagy
3.1.1 Targeting mTOR
Among the signaling pathways that regulate autophagy, the inhibition of the mammalian target of rapamycin (mTOR) has been placed as a central key signaling pathway for regulation of autophagy induction (Jung et al. 2010; Yang and Klionsky 2010). mTOR exists in two heteromeric complexes, mTORC1 and mTORC2. However, it seems that rapamycin-sensitive mTORC1 plays a major role in the regulation of autophagy and cell growth, although there is evidence that rapamycin-insensitive mTORC2 may also regulate autophagy in some specific tissues.
The first generation of mTOR inhibitors is rapamycin and its analogs. Rapamycin (also known as Sirolimus) was first discovered as a product of the bacterium Streptomyces hygroscopicus in a soil sample and has an antifungal function (Sehgal et al. 1975; Vezina et al. 1975). Later it was found that it also has potent immunosuppressive and anti-proliferative effects. In mammalian cells, rapamycin binds with an intracellular protein FKBP12, and the rapamycin–FKBP12 complex binds to mTOR in a region adjacent to the mTOR kinase domain to inhibit mTORC1 but not mTORC2. However, chronic treatment with rapamycin can also block mTORC2 function in some cells. Many rapamycin analogs have also been discovered, and they all inhibit mTOR activity by binding with FKBP12. All of these rapamycin analogs contain only minor structure modifications to increase their solubility and stability. Currently, rapamycin and its analogs CCI-779, RAD-001, and AP23573 are in clinical trials for treating various cancer patients. As we discussed above, although rapamycin and its analogs have showed inhibitory effects on cell growth, they also simultaneously induce the cell survival autophagy process in cancer cells. This may help to explain why the therapeutic effects of rapamycin for cancer are far more satisfactory. However, in various cell culture and animal models, the combination of autophagy inhibitors, such as chloroquine with rapamycin or other mTOR inhibitors, has shown increased tumor cell death and tumor regression (Huang et al. 2011; Loehberg et al. 2012), although the beneficial effects of this approach on human cancer therapy are currently not clear.
In addition to rapamycin and its analogs, the second generation of mTOR inhibitors has been developed, and these are small-molecule mTOR kinase inhibitors that inhibit mTOR by targeting its ATP-binding site pocket. By binding the catalytic site of mTOR, these mTOR kinase inhibitors can inhibit both mTORC1 and mTORC2, and therefore have broad applications. These inhibitors include Torin1, PP242, and PP30. It has been shown that both Torin1 and PP242 could fully suppress 4EBP-1 phosphorylation, whereas rapamycin has only mild inhibitory effects on 4EBP-1 phosphorylation. Thus, these mTOR kinase inhibitors have been shown to have greater efficacy for inhibiting cell proliferation and inducing autophagy than rapamycin (Guertin and Sabatini 2009). The mTOR inhibitors that have been shown to induce autophagy in cancer cells are listed in Table 12.1 including rapamycin and its analogs, ATP-binding competitive inhibitors, and inhibitors with unknown mechanisms.
3.1.2 Targeting PI3K and AKT
Many PI3K and AKT inhibitors have been, and are being, developed. For the PI3K inhibitors, there are adenosine triphosphate (ATP)-competitive inhibitors of PI3K, which can target to all class I PI3K such as XL147 (Exelixis), BKM120 (Novartis), and GDC0941 (Genetech). The isoform-specific PI3K inhibitors include BYL719 (Novartis) and CAL-101 (Calistoga) (Zhang et al. 2011). The advantage of the isoform-specific inhibitors of PI3K is that they have increased potency, fewer off-target effects, and decreased side effects. For the AKT inhibitors, both ATP-competitive and allosteric AKT inhibitors are being developed. AZD5363, GDC-0068, GSK2141795, and GSK690693 are ATP-competitive inhibitors which target to three different isoforms of AKT. Allosteric AKT inhibitors, such as MK-2206, bind to the AKT PH domain to promote an inactive conformation of the AKT protein, which is unable to bind to the plasma membrane (Zhang et al. 2011). All of these PI3K and AKT inhibitors have either been in phase I or II clinical trials and have shown some antitumor activities. Because all these inhibitors inhibit AKT, which ultimately leads to the suppression of mTOR, it is suggested that these inhibitors will activate autophagy, which will limit their antitumor effects. Indeed, the allosteric AKT inhibitor MK-2206 has been shown to increase autophagy in leukemia cells (Simioni et al. 2012). However, because of the structure similarity among class I, II, and III PI3Ks, the specificities of these PI3Ks will affect their effects on autophagy. For example, both LY294002 and wortmannin are ATP-competitive PI3K inhibitors which have similar inhibition potency on class I, II, and III PI3K in vitro (Vanhaesebroeck et al. 2001). Indeed both the inhibition and induction of autophagy by LY294002 have been reported in many cell culture models (Blommaart et al. 1997; Takeuchi et al. 2005). 3-Methyladenine (3-MA), another widely used autophagy inhibitor, which was thought to inhibit class III PI3K to suppress autophagosome formation (Seglen and Gordon 1982), actually shows a dual-effect on autophagy depending on the duration of its treatment. At a shorter time point (less than 9 h), 3-MA inhibits autophagic flux but it increases autophagic flux after prolonged treatment (more than 9 h) in cultured cells. The dual-effect of 3-MA on autophagy regulation is due to its differential temporal effects on class I and III PI3K in which 3-MA blocks class I PI3K persistently, but only suppresses class III PI3K transiently (Wu et al. 2010). It is possible that the effects of PI3K inhibitors on autophagy would depend on different cell types (the basal expression level and activity of class I, II, and III PI3K), dose of PI3K inhibitors, and duration of treatment. Nevertheless, it does not matter if PI3K and AKT inhibitors induce or inhibit autophagy because, when combined with other autophagy inhibitors that target downstream lysosomes, such as chloroquine or bafilomycin A1, they should further suppress tumor cell proliferation and increase cell death. Not all known PI3K and AKT inhibitors [such as CH5132799, AS-252424, CAY10505, A66, and BKM120 (NVP-BKM120)] have been tested for their effects on autophagy (Maira et al. 2012; Pomel et al. 2006; Tanaka et al. 2011; Tyagi et al. 2012). The PI3K and AKT inhibitors that have been shown to inhibit or activate autophagy in cancer cells are listed in Table 12.2.
3.1.3 Targeting PI3K and mTOR
Activation of mTOR is the most downstream target of the PI3K signaling pathway, and thus inhibition of mTOR has been a critical strategy for cancer therapy. As discussed above, because of the negative-feedback loop inhibition on AKT by mTOR, inhibition of mTOR leads to activation of PI3K and AKT. To circumvent this problem, dual inhibitors that target both PI3K and mTOR have been developed. Dual PI3K–mTOR inhibitors such as NVP-BEZ235, NVP-BGT226, XL765, and SF-1126 have been extensively studied in preclinical animal models for cancer treatment (Amaravadi et al. 2011; Zhang et al. 2011). However, results from these mouse models reveal that the dual PI3K–mTOR inhibitors induce tumor stasis rather than tumor regression. It was further revealed that the dual PI3K–mTOR inhibitors activate autophagy, and in combination with the autophagy inhibitor (chloroquine) can promote tumor cell death and cause tumor regression in a preclinical glioblastoma model. More importantly, this combination approach also increased cell death in glioma cells mutant for PTEN. Interestingly, combination of inhibitors for mTORC1 only with autophagy inhibitors did not promote cell death (Fan et al. 2010). These results suggest that blockade of three targets, PI3K, mTOR, and autophagy, may be required for inducing efficient cell death in PTEN mutant glioma. More studies are needed to determine whether simultaneous inhibition of PI3K–mTOR and autophagy would be a general effective approach for treating cancers with activated PI3K/AKT/mTOR signaling. The dual PI3K–mTOR inhibitors that have been shown to induce autophagy in cancers are listed in Table 12.3.
3.1.4 Targeting AMPK
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase which acts as a sensor of cellular energy levels (Hardie et al. 2012). AMPK is a heterotrimeric complex which is highly conserved through evolution. The AMPK complex contains a catalytic subunit (AMPK-α) and two regulatory subunits (AMPK-β and AMPK-γ). A high AMP/ATP ratio reflects low-energy status, which can directly activate AMPK through AMP binding. AMP binds to the CBS domain in the γ subunit resulting in conformational changes in the heterotrimeric complex. This conformational change then promotes the phosphorylation of Thr172 in the α subunit by several upstream kinases which include the tumor-suppressor liver kinase B1 (LKB1), calmodulin-dependent protein kinase kinases (CaMKKs), and transforming growth factor-β-activated kinase-1 (TAK1) (Alexander and Walker 2011; Hurley et al. 2005; Xie et al. 2006). AMPK is a master regulator of glucose, cholesterol, and lipid metabolism in various organs such as liver, skeletal muscle, and adipose tissue. AMPK stimulates glucose transport and fatty acid oxidation in skeletal muscle while it increases fatty acid oxidation and decreases cholesterol and triglyceride synthesis as well as glucose output in the liver. Thus, AMPK plays a critical role in lowering blood glucose levels in hyperglycemic individuals and is a key therapeutic target for diabetes. In addition to the regulation of glucose and lipid metabolism, AMPK can also regulate mTOR activity. Once activated, AMPK phosphorylates TSC2 and stimulates TSC2 GAP activity toward Rheb, which results in the inhibition of mTOR. AMPK-induced inhibition of mTOR leads to the inhibition of protein translation and activation of autophagy (Inoki et al. 2003). In addition, AMPK can also directly phosphorylate UNC-51-like kinase 1 (ULK1), the mammalian homologue of yeast Atg1, leading to ULK1 activation and induction of autophagy (Egan et al. 2011; Kim et al. 2011; Zhao and Klionsky 2011). Thus, AMPK agonists seem to behave similarly to mTOR inhibitors that can simultaneously inhibit cancer cell growth and induce autophagy.
Indeed, epidemiology studies indicate that there is strong evidence that AMPK agonists have suppressive inhibitory effects on tumorigenesis and cancer growth. In an observational cohort study, it was found that people with type 2 diabetes who used metformin had a reduced risk of cancer (Libby et al. 2009). Several other epidemiology studies have also found that oral short-term low-dose metformin treatment suppressed colorectal cancer (Higurashi et al. 2012), liver cancer (Zhang et al. 2012), pancreatic cancer (Bodmer et al. 2012), breast cancer (Col et al. 2012), and lung cancer (Lai et al. 2012). Metformin was also associated with a survival benefit for patients with various solid tumors (Currie et al. 2012; Romero et al. 2012). Experimentally, administration of metformin alone reduced human gastric cancer cell proliferation in vitro and in vivo (Kato et al. 2012). However, because AMPK agonists can induce autophagy, it remains to be studied whether the combination of AMPK agonists (such as metformin) with autophagy inhibitors (such as chloroquine) would further enhance the anticancer effects beyond each agent alone.
3.1.5 Targeting Bcl-2 Family Proteins
Bcl-2 family proteins play an important role in regulating cell death. However, recent evidence suggests that Bcl-2 can also regulate autophagy. By directly binding to Beclin 1, anti-apoptotic proteins such as Bcl-2, Bcl-xL, and Mcl-1 can suppress autophagy (Liang et al. 1999; Pattingre et al. 2005). The binding of Bcl-2 to Beclin 1 dissociates Beclin 1 from Vps34, resulting in decreased Vps34 kinase activity and inhibition of autophagy. In contrast to anti-apoptotic Bcl-2 family proteins, pro-apoptotic BH3-only proteins such as Bnip3, Bnip3L/Nix, Bad, Bik, Noxa, Puma, and Bim induce autophagy by displacing the inhibitory Bcl-2 from the Beclin 1 complex. In general, it seems that anti-apoptotic Bcl-2 family proteins inhibit, but pro-apoptotic Bcl-2 family proteins promote, autophagy. Thus, the small-molecule BH3 mimetics, which are pharmacological ligands of the BH3-binding domain of Bcl-2, would induce autophagy by disrupting Bcl-2–Beclin 1 interaction. Indeed, several BH3 mimetics such as ABT737, HA14-1, and (−)-gossypol can induce autophagy in various types of cancer cells (Lian et al. 2011; Maiuri et al. 2007).
BH3-only proteins (or BH3 mimetics) have been shown to induce mitochondria-mediated apoptosis by direct or indirect activation of pro-apoptotic multidomain proteins from the Bcl-2 family such as Bax and Bak (Labi et al. 2008). Thus, similar to mTOR inhibitors and AMPK agonists, BH3 mimetics also have dual roles on cell death and autophagy. Because of their abilities to induce apoptosis alone or augment the anticancer effects of other conventional chemotherapy drugs, some of the BH3 mimetics are currently being evaluated in clinical trials. Although it is promising to use the combination of BH3 mimetics and autophagy inhibitors strategy in treating cancers, the results are not yet clear.
3.2 Targeting Transcription Factors
Autophagy involves the formation of double-membrane autophagosomes, which is a dynamic process requiring multiple protein complexes. Although it has been suggested that some of the existing membranes such as ER and mitochondrial membranes could be used, it is not surprising that transcriptional regulation of many autophagy genes can play an important role in autophagy. Three decades ago, it was shown that administration of cycloheximide, a general inhibitor of protein synthesis, could block glucagon-induced autophagy as demonstrated in rat hepatocytes (Papadopoulos and Pfeifer 1986). In yeast, it was also found that in the presence of cycloheximide, autophagosomes are significantly smaller than normal, indicating that de novo protein synthesis could play a role in the regulation of autophagosome expansion (Abeliovich et al. 2000). Indeed, recent evidence has supported that autophagy genes are regulated at a transcriptional level in response to stress. Below, we discuss several key transcription factors that regulate gene expression for autophagy and lysosome biogenesis.
3.2.1 FoxO3
FoxO (forkhead box transcription factor class O) transcription factors regulate diverse cellular functions including metabolism, oxidative stress, differentiation, cell cycle, and cell death. It has been shown that FoxO is the first transcription factor that is necessary and sufficient to induce autophagy in the Drosophila larval fat body (Dobson et al. 2011). Over-expression of FoxO3 induces the transcription of multiple autophagy genes including LC3B, Gabarapl1, atg12, atg4B, vps34, ulk2, beclin 1, Bnip3, and Bnip3l in mouse skeletal muscle (Zhao et al. 2007). It has been demonstrated that FoxO3 directly binds to the promoters of LC3B, Gabarapl1, atg12, Bnip3l, and Bnip3 to activate their gene transcription (Mammucari et al. 2007). As a result, constitutively active FoxO3 promotes lysosomal proteolysis and leads to muscle wasting by activating autophagy in mouse skeletal muscle.
The activity of FoxO is inhibited by growth factors and PI3K/AKT pathways. AKT phosphorylates FoxO and negatively regulates its transcriptional activity by promoting its nuclear extrusion into the cytosol. In addition to AKT, FoxO can also be phosphorylated on different sites by other kinases including AMPK and JNK, which promote its transcription activity, and ERK and IKKb, which inhibit its transcription activity (Tzivion et al. 2011). FoxO protein levels are also regulated by ubiquitin-dependent protein degradation. The E3 ligase SKP2 binds to AKT-phosphorylated FoxO1 at Ser 256 whereas the E3 ligase MDM2 binds to ERK-phosphorylated FoxOs. Interestingly, MDM2 can induce both FoxO mono-ubiquitination and poly-ubiquitination. Mono-ubiquitination promotes FoxO translocation to the nucleus and increased transcriptional activity whereas poly-ubiquitination targets FoxO for degradation (Huang and Tindall 2011). Sirt1, a mammalian NAD+-dependent protein deacetylase, deacetylates FoxO to increase a subset of its target genes such as antioxidant genes. In addition to regulating expression of autophagy genes, it was recently found that activation of FoxO increased levels of glutamine production (van der Vos et al. 2012). This resulted in mTOR inhibition by preventing the translocation of mTOR to lysosomal membranes, which in turn activated autophagy (Fu and Tindall 2008).
Although the small molecules that directly activate FoxO remain to be discovered, the activity of FoxO could be modulated by indirectly targeting its posttranslational modifications. For example, the PI3K/AKT, Sirt1, and JNK inhibitors can all modulate FoxO activity and thus modulate the expression of autophagy genes and autophagy. Because these agents are not specific for FoxO, future work is definitely needed to identify direct specific FoxO agonists.
3.2.2 TFEB
Completion of the autophagic process relies on the fusion of autophagosomes with lysosomes to form autolysosomes. Within autolysosomes, the autophagic contents are then broken down by lysosomal enzymes. Thus, it is important that lysosomal activities are coordinated to respond to cellular stress and needs. Recently, it was found that the transcriptional factor EB (TFEB), a basic helix-loop-helix leucine zipper transcription factor of the Myc family, is a master regulator for controlling the expression of lysosomal genes (Settembre et al. 2011, 2012). Over-expression of TFEB in cultured cells results in the biogenesis of lysosome and enhanced lysosomal degradation capacity (Settembre et al. 2012). Indeed, over-expression of TFEB can reduce mutant huntingtin levels in cultured cells (Settembre et al. 2012). In addition to regulating lysosome biogenesis, TFEB can also directly regulate autophagy. Over-expression of TFEB increases autophagy flux in HeLa cells by increasing the expression of autophagy genes. Under starvation conditions in both cultured cells and in mice in vivo, it was found that TFEB translocated from cytosol into the nuclei, and this was associated with increased transcription of TFEB target genes both for autophagy and lysosomal biogenesis (Settembre et al. 2011). Mechanistically, it was found that TFEB can be phosphorylated by the extracellular signal-regulated kinase (ERK) and mTOR, which retains TFEB in the cytoplasm under normal nutrient conditions (Martina et al. 2012; Settembre et al. 2011, 2012). Moreover, TFEB colocalizes with mTORC1 on the lysosomal membrane and serves as the sensor for cellular nutrients. When levels of cellular nutrients are high, mTORC1 phosphorylates TFEB and inhibits its activity. Conversely, starvation as well as pharmacological inhibition of mTORC1 activate TFEB by promoting its nuclear translocation and in turn promoting autophagy (Martina et al. 2012; Settembre et al. 2012). Therefore, inhibition of mTOR can activate autophagy not only through the modulation of the ULK1 complex but also through the increased expression of autophagy and lysosomal genes by activating TFEB.
However, it should be noted that TFEB has been suggested to function as an oncogene in the kidney. In a subset of renal tumors, both mRNA and protein levels of TFEB are dramatically increased in tumor cells compared to normal tissues (Davis et al. 2003; Kuiper et al. 2003). Translocation carcinoma, which has aberrant expression of melanocytic markers, is thought to also be driven by enhanced TFEB nuclear translocation and activation (Srigley and Delahunt 2009). Moreover, genetic deletion of the tumor-suppressor gene tsc2 leads to persistent activation of mTORC1 and in turn phosphorylates TFEB (Pena-Llopis et al. 2011). Contradictory to other studies, this results in TFEB nuclear translocation and activates expression of a subset of genes including V-ATPase, an important component of the late endosomes/lysosome which regulates their acidification. The reasons behind these contradictory data are not clear, but it could be possible that the amino acid-mediated mTOR activation (through the regulator) and growth factor-mediated mTOR activation (through PI3K-tsc1/tsc2) are different. More studies are needed to clarify these conflicting findings in the future. Nevertheless, although reagents that directly activate TFEB have not been reported, modulation of mTOR may serve as an indirect approach for TFEB activation.
3.2.3 C/EBPβ
Cellular metabolic states are dynamically changed with circadian oscillation, which is a well-regulated process in response to limited nutrients for maintaining energy homeostasis in mammals. As an important catabolic pathway, it was recently found that autophagy activation fluctuates to coordinate circadian rhythm in mouse liver (Ma et al. 2011). There is a cyclic induction of autophagy gene expression to meet the needs for autophagy induction during circadian rhythm changes. C/EBPβ was identified as a potent activator of autophagy in the mouse liver during circadian changes through functional analysis of various transcription factors and cofactors in the liver (Ma et al. 2011). C/EBPβ is a basic leucine zipper transcription factor transcribed from an intronless gene. There are three protein isoforms from this single mRNA due to alternative translation initiation at three in-frame methionine initiator codons. C/EBPβ is an important regulator for a variety of physiological processes including metabolism, cellular differentiation, and stress response. In the liver, C/EBPβ is rhythmically expressed in response to both circadian and nutritional signals. In primary cultured hepatocytes, starvation-increased expression of C/EBPβ stimulates the expression of autophagy genes and is sufficient to trigger autophagy induction. siRNA knockdown of C/EBPβ in mouse liver in vivo abolishes diurnal hepatic autophagy rhythm (Ma et al. 2011). All these results support that C/EBPβ is a key integrator of nutritional and circadian signals that regulates autophagy status in the liver.
Interestingly, over-expression of C/EBPβ leads to increased autophagy resulting in non-apoptotic cell death in various human breast cancer cell lines (Abreu and Sealy 2010). Although the expression of autophagy genes was not determined in the C/EBPβ over-expressed breast cancer cells, over-expression of C/EBPβ resulted in an increased number of acidic vesicles (Abreu and Sealy 2010). Moreover, cells with over-expressed C/EBPβ can engulf neighboring cells in culture. However, whether autophagy is involved in this process is not clear (Abreu and Sealy 2012). Interestingly, C/EBPβ over-expression stimulates the engulfment of live cells but not dead apoptotic cells. It is suggested that C/EBPβ-mediated engulfment may serve as a tumor suppressor by removing unwanted cells that may have DNA mutations. However, more studies are needed to further expand our knowledge on how C/EBPβ affects autophagy in cancer cells, and more importantly, to identify pharmacological approaches to modulate C/EBPβ.
3.3 Protein Posttranslational Modifications
Autophagy is regulated by various posttranslational modification processes. These modifications include ubiquitination, phosphorylation, lipidation, and acetylation. The autophagy core machinery proteins such as Atg5, Atg7, Atg12, and LC3 undergo ubiquitin-like conjugation to form complexes that regulate the formation of double-membrane autophagosomes. Autophagy cargos and damaged mitochondria are often ubiquitinated, which seems to be important for their selective removal by autophagy (Ding et al. 2010; Johansen and Lamark 2011; Karbowski and Youle 2011). Phosphorylation of ULK1 and dephosphorylation of Atg13 are required for autophagy induction (Egan et al. 2011; Kim et al. 2011). LC3 needs to be lipidated by conjugation with phosphatidylethanolamine (PE) to ensure the formation of autophagosomes. Accumulating evidence now suggests that protein acetylation is an evolutionarily conserved mechanism regulating autophagy. In yeast, Atg3 is acetylated during starvation by Esa1p, a histone acetylase. The acetylation of Atg3 is necessary for Atg8 lipidation and autophagy activation (Yi et al. 2012). In mammals, TIP60, a yeast Esa1p homologue, mediates the acetylation of ULK1 to promote autophagy induction. In response to starvation, glycogen synthase kinase-3 (GSK3) phosphorylates TIP60, which enhances its affinity to bind with ULK1 resulting in ULK1 acetylation. Acetylation increases ULK1 kinase activity and thus promotes autophagy induction in response to starvation (Lin et al. 2012). Moreover, other autophagy core machinery proteins have also been shown to be regulated by acetylation. Under nutrient-rich conditions, the acetyltransferase p300 directly acetylates Atg5, Atg7, LC3, and Atg12 to inhibit autophagy (Lee et al. 2008; Lee and Finkel 2009). In contrast, Sirt1, an NAD-dependent deacetylase, deacetylates Atg5, Atg7, LC3, and Atg12 to promote autophagy when p300 is dissociated from these Atg proteins under starvation conditions (Lee et al. 2008). Sirt1 knockout mice die during the neonatal stage, which resembles the phenotype of Atg5 or Atg7 knockout mice (Lee et al. 2008). Activation of Sirt1 by resveratrol or inhibition of histone acetylases by spermidine induces autophagy and increases longevity in yeast, nematodes, and flies (Eisenberg et al. 2009; Marino et al. 2011; Morselli et al. 2011). Although it seems that acetylation of different autophagy proteins may differentially regulate autophagic response, targeting acetylation modification may be a useful approach for modulating autophagy.
3.4 mTOR-Independent Autophagy
Autophagy can also be activated independent of mTOR. Drugs such as lithium, carbamazepine, and valproic acid are used to treat a range of neurological and psychiatric diseases. These drugs lower intracellular inositol and inositol 1,4,5-trisphosphate (IP3) levels and induce autophagy independent of mTOR activity (Sarkar et al. 2005). Mechanistically, it was found that these drugs can affect the intracellular levels of Ca2+ and cyclic AMP (cAMP). Indeed, calpain inhibitors, l-type Ca2+ channel agonists, and chemicals which decrease intracellular cAMP can all induce autophagy independent of mTOR (Ravikumar et al. 2010). In a GFP-LC3 image-based high-throughput screen, eight compounds were identified that induce autophagy in an mTOR-independent manner (see below Sect. 4.1 and Table 12.4), although the exact mechanisms by which these compounds induce autophagy are not well understood (Zhang et al. 2007).
4 Methods to Discover Drugs That Modulate Autophagy
With the rapidly expanding understanding of autophagic machinery and the molecular signaling pathways that regulate autophagy, many high-throughput autophagy screening assays have been developed, and have led to identification of various small molecules that either activate or inhibit autophagy. Here, we discuss several reported assays for autophagy screening.
4.1 GFP-LC3 High-Content and High-Throughput Assay
One high-throughput image-based assay to screen small molecules that modulate autophagy is to use a cell line that stably expresses GFP-LC3. This assay takes advantage of the distinctive pattern of cellular GFP-LC3 upon autophagy induction. Under nutrient-rich conditions, the level of autophagy is low and GFP-LC3 displays a diffuse pattern. However, during autophagy induction, GFP-LC3 translocates to the autophagosomal membrane and displays a punctate pattern. The number of GFP-LC3 puncta per cell can be quantified and represent the number of autophagosomes in each cell.
We recently developed an image-based assay using mouse embryonic fibroblasts (MEF) stably expressing GFP-LC3. Briefly, MEF-GFP-LC3 cells were dispensed at 2,000 cells/well/25 μl in a 384-well black wall/clear bottom, collagen-coated plate (BD Bioscience, San Jose, CA) for 5 h to allow cell attachment to the plate followed by addition of various concentrations of chloroquine (0.31–20 μM) or DMSO as a vehicle control. After 16-h incubation, the cells were fixed with 8 % paraformaldehyde and nuclei were stained with Hoechst 33342. After washing twice with PBS, the assay plate was subjected to quantitative image analysis by an ArrayScan VTI HCS Reader (Thermo Scientific, Pittsburgh, PA) with a 20× objective and an Omega XF100 dual-bandpass filter set at excitation/emission wavelengths of 385 nm/461 nm (Hoechst) and 485 nm/515 nm (GFP), respectively. Typically <200 cells per well were examined using the Compartmental Analysis BioApplication. These results suggest that the GFP-LC3 puncta formation assay is robust and suitable for high-throughput screening.
Because autophagy is a dynamic process, autophagosomes need to fuse with lysosomes to form autolysosomes where the enveloped contents are degraded. However, not only the autophagic cargos but also GFP-LC3 itself are degraded in the autolysosomes. Therefore, inhibition of lysosomal functions, such as with bafilomycin A1 and CQ, can also increase the number of GFP-LC3 puncta. Thus the increased number of GFP-LC3 puncta does not necessarily reflect autophagic degradation activity. Therefore, identified compounds that increase GFP-LC3 puncta need to be further examined by other biochemical assays, such as by the long-lived protein degradation assay.
Based on the high-content GFP-LC3 imaging screening followed by functional biochemical characterization, various compounds have been identified that can modulate autophagy (see Table 12.4). For example, in one screening using H4 human neuroglioma GFP-LC3 cells, eight compounds were identified to induce autophagy and seven of them are FDA-approved drugs. These drugs are able to degrade misfolded proteins with low cytotoxicity and may be used to treat expanded polyglutamine diseases such as Huntington’s disease (Zhang et al. 2007). In another high-content GFP-LC3 imaging screening, four chemicals (perhexiline, niclosamide, amiodarone, and rottlerin) were identified from a collection of 3,584 drugs and pharmacologically active chemicals that can induce autophagy through inhibition of mTOR (Balgi et al. 2009) (see Table 12.4). This image-based autophagy screen has also led to the discovery of a novel autophagy inhibitor, specific and potent autophagy inhibitor 1 (Spautin-1). Spautin-1 inhibits autophagy by inhibiting ubiquitin-specific peptidase 10 (USP10) and USP13 which usually target to the Beclin 1–Vps34 complex resulting in enhanced degradation of the Beclin 1–Vps34 complex (Liu et al. 2011). Interestingly, the Beclin 1–Vps34 complex also regulates the stability of USP10 and USP13 which can control the levels of p53. This provided the molecular basis for why the levels of p53 are reduced in the tissues of beclin1 ± mice, which have increased tumorigenesis.
In addition, the high-throughput GFP-LC3 cell-based assay has been modified and used to identify Atg genes. The library of human cDNA clones (Castrillon et al. 2003) and genome-wide siRNA have both been used for this GFP-LC3 image-based screening (Tothova et al. 2007). Three genes (TM9SF1, TMEM166, and TMEM74) were identified that induce high levels of autophagosome formation when they are over-expressed from a library of 1,050 human cDNA clones. The genome-wide siRNA screen identified nine novel autophagy regulators including short coiled-coil protein (SCOC) and elongation protein zeta 1 (FEZ1). SCOC may form a complex with UVRAG and FEZ1 and may regulate ULK1 and Beclin 1 complex activities to regulate autophagy (Tothova et al. 2007).
4.2 BiFC–FRET Screening
Bimolecular fluorescence complementation–fluorescence resonance energy transfer (BiFC–FRET) is used to determine the interaction of three proteins in live cells whereas BiFC can only determine the interactions of two proteins. Autophagy is a highly regulated process which involves several stages including initiation, nucleation, elongation, docking, maturation, and degradation. Each step is regulated by complicated multiple protein complexes. For example, initiation is regulated by mTORC1 via the ULK1/2–Atg13–FIP200–Atg101 complex. Autophagosomal membrane nucleation is regulated by the Beclin 1–Atg14–Vps34–Vps15–UVRAG complex, and the elongation step is mediated by the Atg12–Atg5–Atg16 complex. These multiple protein complexes make them good candidates for BiFC–FRET screen. Dai et al. (2012) have elegantly established this screen by targeting the Atg12–Atg5–Atg16 complex. Briefly, Atg5 and Atg12 genes were fused with the N′- and C′-fragments of a red fluorescence protein (RFP), and Atg16 was cloned into a pEGFP-C1 plasmid. After co-transfection with these three plasmids, the interaction between Atg5 and Atg12 yielded an intact RFP signal, and this process was called BiFC. In contrast, FRET would occur due to the interaction between the Atg12–Atg5 heterodimer and Atg16. The interaction between Atg5, Atg12, and Atg16 can be determined using a microplate reader at 610 and 509 nm after excitation at 488 nm.
Using this screening method, 15 medicinal plants were identified that could inhibit the Atg12–Atg5–Atg16 complex from 83 types of traditional Chinese medicines. Furthermore, one compound evodiamine, which is the major active component from one of the 15 identified medicinal plants, was confirmed to inhibit the formation of the Atg12–Atg5–Atg16 heterotrimer resulting in autophagy inhibition. Because influenza A virus (IAV) requires the autophagy process to replicate, it was found that IAV replication was significantly repressed by evodiamine (Dai et al. 2012).
4.3 High-Throughput Assay for Small-Molecule Inhibitors of Atg4
Among all the autophagy core proteins that have been identified, Atg4 is the only protein that has protease activity, which makes it an ideal target for developing high-throughput assays to measure protease activity. During autophagosome biogenesis, Atg4 cleaves Atg8 to allow its conjugation with phosphatidylethanolamine, which is a key step for the formation of autophagosomes. Furthermore, Atg4 can also regulate autophagy by deconjugating Atg8 from autophagosomal membranes. Therefore, compounds that affect the activity of Atg4 could be a target for therapeutic intervention by modulating autophagy.
A couple of high-throughput assays have been developed for measuring Atg4 activity. One assay uses LC3B fused to an assayable enzyme phospholipase A2 (PLA2) as an Atg4B substrate. PLA2 is inactive when it is expressed as a fusion protein when LC3B is fused on its N-terminus. When LC3B-PLA2 is cleaved by Atg4B, the released PLA2 then cleaves a fluorescence NBD-C6-HPC substrate to generate NBD fluorescence in a concentration-dependent manner, which can be measured using a plate reader. Four compounds were identified from a library of 3,282 bioactive molecules showing inhibitory effects against Atg4B; however, these compounds were not further characterized by other functional autophagy assays (Shu et al. 2011).
Another assay to determine Atg4 activity was developed using a FRET-based approach (Li et al. 2012). LC3B was fused with CFP and YFP at its N- and C-terminus, respectively, allowing FRET to occur. When the LC3B fusion was cleaved by Atg4, which separates the two fluorescence proteins, the FRET signals decreased when measured using a fluorescence spectrometer. Although this FRET-based assay to measure Atg4-specific activity is simple and easy to use, high-throughput screening has not been done using this assay.
4.4 Other Potential Methods for Autophagy Screening
In addition to the above assays, other assays for monitoring autophagy, which may be developed into high-throughput screening, have also been reported. One assay is to use the fluorescence-activated cell sorter (FACS) to quantify the turnover of GFP-LC3 in living mammalian cells (Shvets et al. 2008). The principle of this assay is based on the fact that during autophagy induction, the GFP-LC3 fluorescence intensity is decreased. One advantage of this assay is that FACS analysis can be used to evaluate more samples with much larger cell sample sizes compared to the image-based assays. Moreover, this assay can directly measure autophagy activity. Thus, this method may also be used to perform large-scale screens for identification of autophagy modulators.
To directly measure autophagic flux in real time in living cells, a luciferase-based reporter assay has also been established (Farkas et al. 2009). This assay is also based on the concept that LC3 fluorescent fusion partners are degraded within lysosomes. To quantitatively measure the dynamic change of LC3 fusion protein, LC3 is fused with the Renilla Reinformis luciferase (RLuc). The autophagy-dependent turnover of RLuc-LC3 can be measured using cell lysates or living cells for monitoring autophagic flux. Using this assay, a screen using a small-molecule kinase inhibitor library containing 80 compounds has led to the identification of 12 compounds as inducers of autophagic flux. In addition, three potent autophagy inhibitors were also identified from this screen (Farkas et al. 2011). KU559339 [a specific inhibitor of ataxia telangiectasia-mutated (ATM)], Gö6976 (a broad-spectrum protein kinase C inhibitor), and Janus 3 kinase (Jak3) inhibitor VI could all effectively inhibit rapamcyin-induced autophagy. Interestingly, subsequent studies revealed that KU55933 and Gö6976 directly and effectively inhibited PtdIns3K, suggesting that their effects on autophagy are independent of their known target kinases. Given the rapid progress of autophagy research, there is no doubt that more assays for monitoring autophagy will be, or are being, developed. This will definitely lead to discovery of more agents that effectively modulate autophagy, and could be potentially used for treating cancer patients in combination with other chemotherapy drugs.
5 Concluding Remarks
It is now well known that autophagy plays a critical role in both tumorigenesis and growth of existing cancers. With the expanding understanding of molecular machinery and regulating signaling pathways for autophagy, more efforts have been put into the area of drug development for targeting autophagy. With many high-throughput screening assays available, more efficient and specific autophagy inducers or inhibitors will be identified. These agents will definitely provide a useful tool for treating cancer patients in combination with other chemotherapy drugs, in particular for cancers that are resistant to conventional chemotherapy drugs. However, many fundamental questions still remain to be answered before the autophagy modulation approach can be efficiently used for cancer prevention and therapy. How does autophagy mitigate the efficacy of mTOR inhibitor-induced inhibition on tumor cell growth? How does autophagy affect the immune response to cancer, and what would be the response to autophagy inhibition among different cancer cells that have different basal levels of autophagy? Nevertheless, identifying more agents that target different aspects of the autophagy pathway will definitely improve basic and translational research on autophagy and may also help to answer the above questions.
Abbreviations
- 3-MA:
-
3-Methyladenine
- 4EBP1:
-
4E-binding protein 1
- AMP:
-
Adenosine monophosphate
- AMPK:
-
5′ Adenosine monophosphate-activated protein kinase
- Atg:
-
Autophagy-related
- ATM:
-
Ataxia telangiectasia mutated
- ATP:
-
Adenosine-5′-triphosphate
- BiFC–FRET:
-
Bimolecular fluorescence complementation–fluorescence resonance energy transfer
- CaMKKs:
-
Calmodulin-dependent protein kinase kinases
- DAPK1:
-
Death-associated protein kinase 1
- eIF4E:
-
Eukaryotic translation initiation factor 4E
- FACS:
-
Fluorescence-activated cell sorter
- FEZ1:
-
Fasciculation and elongation protein zeta-1
- FoxO:
-
Forkhead box transcription factor class O
- FRET:
-
Fluorescence resonance energy transfer
- GFP:
-
Green fluorescent protein
- GPCRs:
-
G-protein-coupled receptors
- HCQ:
-
Hydroxychloroquine
- IAV:
-
Influenza A virus
- IRS:
-
Insulin receptor substrate
- Jak3:
-
Janus 3 kinase
- LKB1:
-
Liver kinase B1
- MEF:
-
Mouse embryonic fibroblasts
- mLST8:
-
Target of rapamycin complex subunit LST8
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
mTOR complex 1
- mTORC2:
-
mTOR complex 2
- NF-kappaB:
-
Nuclear factor-kappaB
- Nrf2:
-
Nuclear factor (erythroid-derived 2)-like 2
- p19ARF:
-
p19 Alternative reading frame
- p70S6K1:
-
p70 Ribosomal protein S6 kinase 1
- PDK:
-
Phosphoinositide-dependent protein kinase
- PE:
-
Phosphatidylethanolamine
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIP2:
-
Phosphatidylinositol 4,5-bisphosphate
- PIP3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- PKB:
-
Protein kinase B
- PKC:
-
Protein kinase C
- PLA2:
-
Phospholipase A2
- PPAR-γ:
-
Peroxisome proliferator-activated receptor γ
- PTEN:
-
Phosphatase and tensin homolog
- Raptor:
-
Regulatory associated protein of mTOR
- RFP:
-
Red fluorescence protein
- Rheb:
-
Ras homolog enriched in brain
- Rictor:
-
Rapamycin-insensitive companion of mTOR
- RLuc:
-
Renilla Reinformis luciferase
- RTK:
-
Receptor tyrosine kinase
- SCOC:
-
Short coiled-coil protein
- SGK:
-
Serum- and glucocorticoid-induced protein kinase
- SREBP1/2:
-
Sterol regulatory element-binding protein1/2
- TAK1:
-
Transforming growth factor-β-activated kinase-1
- TFEB:
-
Transcriptional factor EB
- TSC:
-
Tuberous sclerosis complex
References
Abeliovich H, Dunn WA Jr, Kim J, Klionsky DJ (2000) Dissection of autophagosome biogenesis into distinct nucleation and expansion steps. J Cell Biol 151:1025–1034
Abreu MM, Sealy L (2010) The C/EBPbeta isoform, liver-inhibitory protein (LIP), induces autophagy in breast cancer cell lines. Exp Cell Res 316:3227–3238
Abreu M, Sealy L (2012) Cells expressing the C/EBPbeta isoform, LIP, engulf their neighbors. PLoS One 7:e41807
Akar U, Chaves-Reyez A, Barria M, Tari A, Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G, Ozpolat B (2008) Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 4:669–679
Alexander A, Walker CL (2011) The role of LKB1 and AMPK in cellular responses to stress and damage. FEBS Lett 585:952–957
Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17:654–666
Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, Gibson SB (2008) Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 4:195–204
Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M (2009) Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 4:e7124
Berry DL, Baehrecke EH (2007) Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131:1137–1148
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ (1997) The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem 243:240–246
Bodmer M, Becker C, Meier C, Jick SS, Meier CR (2012) Use of metformin is not associated with a decreased risk of colorectal cancer: a case-control analysis. Cancer Epidemiol Biomarkers Prev 21:280–286
Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, Virgin HW IV (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal paneth cells. Nature 456:259–263
Carayol N, Vakana E, Sassano A, Kaur S, Goussetis DJ, Glaser H, Druker BJ, Donato NJ, Altman JK, Barr S, Platanias LC (2010) Critical roles for mTORC2- and rapamycin-insensitive mTORC1-complexes in growth and survival of BCR-ABL-expressing leukemic cells. Proc Natl Acad Sci USA 107:12469–12474
Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA (2003) Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 301:215–218
Chang KY, Tsai SY, Wu CM, Yen CJ, Chuang BF, Chang JY (2011) Novel phosphoinositide 3-kinase/mTOR dual inhibitor, NVP-BGT226, displays potent growth-inhibitory activity against human head and neck cancer cells in vitro and in vivo. Clin Cancer Res 17: 7116–7126
Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X, Zhou L, Liao J, Keihack H, Yan L, Rubin E, Yang JM (2012) MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 11:154–164
Col NF, Ochs L, Springmann V, Aragaki AK, Chlebowski RT (2012) Metformin and breast cancer risk: a meta-analysis and critical literature review. Breast Cancer Res Treat 135:639–646
Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL (2012) Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes Care 35:299–304
Dai JP, Li WZ, Zhao XF, Wang GF, Yang JC, Zhang L, Chen XX, Xu YX, Li KS (2012) A drug screening method based on the autophagy pathway and studies of the mechanism of evodiamine against influenza A virus. PLoS One 7:e42706
Davis IJ, Hsi BL, Arroyo JD, Vargas SO, Yeh YA, Motyckova G, Valencia P, Perez-Atayde AR, Argani P, Ladanyi M, Fletcher JA, Fisher DE (2003) Cloning of an Alpha-TFEB fusion in renal tumors harboring the t(6;11)(p21;q13) chromosome translocation. Proc Natl Acad Sci USA 100:6051–6056
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10:51–64
DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475:106–109
Dickstein RJ, Nitti G, Dinney CP, Davies BR, Kamat AM, McConkey DJ (2012) Autophagy limits the cytotoxic effects of the AKT inhibitor AZ7328 in human bladder cancer cells. Cancer Biol Ther 13:1325–1338
Ding WX, Ni HM, Gao W, Hou YF, Melan MA, Chen X, Stolz DB, Shao ZM, Yin XM (2007a) Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J Biol Chem 282:4702–4710
Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM (2007b) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol 171:513–524
Ding WX, Ni HM, Gao W, Chen X, Kang JH, Stolz DB, Liu J, Yin XM (2009) Oncogenic transformation confers a selective susceptibility to the combined suppression of the proteasome and autophagy. Mol Cancer Ther 8:2036–2045
Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW II, Yin XM (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285:27879–27890
Dobson M, Ramakrishnan G, Ma S, Kaplun L, Balan V, Fridman R, Tzivion G (2011) Bimodal regulation of FoxO3 by AKT and 14-3-3. Biochim Biophys Acta 1813:1453–1464
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J (2008) The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 13:343–354
Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT (2011) p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell 44:134–146
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331:456–461
Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Frohlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F (2009) Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11:1305–1314
Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA, Debnath J, Shokat KM, Weiss WA (2010) Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal 3:ra81
Farkas T, Hoyer-Hansen M, Jaattela M (2009) Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 5:1018–1025
Farkas T, Daugaard M, Jaattela M (2011) Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem 286:38904–38912
Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, Khuri FR, Sun SY (2009) Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res 69:8967–8976
Fu Z, Tindall DJ (2008) FOXOs, cancer and regulation of apoptosis. Oncogene 27:2312–2319
Guertin DA, Sabatini DM (2009) The pharmacology of mTOR inhibition. Sci Signal 2(67):pe 24
Hao J, Pei Y, Ji G, Li W, Feng S, Qiu S (2011) Autophagy is induced by 3beta-O-succinyl-lupeol (LD9-4) in A549 cells via up-regulation of Beclin 1 and down-regulation mTOR pathway. Eur J Pharmacol 670:29–38 Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
Higurashi T, Takahashi H, Endo H, Hosono K, Yamada E, Ohkubo H, Sakai E, Uchiyama T, Hata Y, Fujisawa N, Uchiyama S, Ezuka A, Nagase H, Kessoku T, Matsuhashi N, Yamanaka S, Inayama Y, Morita S, Nakajima A (2012) Metformin efficacy and safety for colorectal polyps: a double-blind randomized controlled trial. BMC Cancer 12:118
Hu C, Zou MJ, Zhao L, Lu N, Sun YJ, Gou SH, Xi T, Guo QL (2012) E Platinum, a newly synthesized platinum compound, induces autophagy via inhibiting phosphorylation of mTOR in gastric carcinoma BGC-823 cells. Toxicol Lett 210:78–86 Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, Aghi MK (2012) Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 72:1773–1783
Huang H, Tindall DJ (2011) Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta 1813:1961–1964
Huang S, Yang ZJ, Yu C, Sinicrope FA (2011) Inhibition of mTOR kinase by AZD8055 can antagonize chemotherapy-induced cell death through autophagy induction and down-regulation of p62/sequestosome 1. J Biol Chem 286:40002–40012
Hurley RL, Anderson KA, Franzone JM, Kemp BE, Means AR, Witters LA (2005) The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280:29060–29066
Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee MS, Tanaka K, Komatsu M (2011) Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193:275–284
Inoki K, Zhu T, Guan KL (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115:577–590
Ito S, Koshikawa N, Mochizuki S, Takenaga K (2007) 3-Methyladenine suppresses cell migration and invasion of HT1080 fibrosarcoma cells through inhibiting phosphoinositide 3-kinases independently of autophagy inhibition. Int J Oncol 31:261–268
Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, Konopleva M, Martin MB, Ren P, Liu Y, Rommel C, Fruman DA (2010) Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med 16:205–213
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7:279–296
Jung CH, Ro SH, Cao J, Otto NM, Kim DH (2010) mTOR regulation of autophagy. FEBS Lett 584:1287–1295
Kamada Y, Sekito T, Ohsumi Y (2004) Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol 279:73–84
Kanzawa T, Zhang L, Xiao L, Germano IM, Kondo Y, Kondo S (2005) Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 24:980–991
Karbowski M, Youle RJ (2011) Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23:476–482
Kato K, Gong J, Iwama H, Kitanaka A, Tani J, Miyoshi H, Nomura K, Mimura S, Kobayashi M, Aritomo Y, Kobara H, Mori H, Himoto T, Okano K, Suzuki Y, Murao K, Masaki T (2012) The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol Cancer Ther 11:549–560
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141
Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B (2008) Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 4:659–668 Kirkegaard K, Taylor MP, Jackson WT (2004) Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat Rev Microbiol 2:301–314
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223
Koul D, Shen R, Kim YW, Kondo Y, Lu Y, Bankson J, Ronen SM, Kirkpatrick DL, Powis G, Yung WK (2010) Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro Oncol 12:559–569
Kuiper RP, Schepens M, Thijssen J, van Asseldonk M, van den Berg E, Bridge J, Schuuring E, Schoenmakers EF, van Kessel AG (2003) Upregulation of the transcription factor TFEB in t(6;11)(p21;q13)-positive renal cell carcinomas due to promoter substitution. Hum Mol Genet 12:1661–1669 Labi V, Grespi F, Baumgartner F, Villunger A (2008) Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ 15:977–987
Lai SW, Liao KF, Chen PC, Tsai PY, Hsieh DP, Chen CC (2012) Antidiabetes drugs correlate with decreased risk of lung cancer: a population-based observation in Taiwan. Clin Lung Cancer 13:143–148
Lee IH, Finkel T (2009) Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 284:6322–6328
Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T (2008) A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci USA 105:3374–3379
Letai AG (2008) Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer 8:121–132
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Li M, Chen X, Ye QZ, Vogt A, Yin XM (2012) A high-throughput FRET-based assay for determination of Atg4 activity. Autophagy 8(3):401–412
Lian J, Wu X, He F, Karnak D, Tang W, Meng Y, Xiang D, Ji M, Lawrence TS, Xu L (2011) A natural BH3 mimetic induces autophagy in apoptosis-resistant prostate cancer via modulating Bcl-2-Beclin1 interaction at endoplasmic reticulum. Cell Death Differ 18:60–71
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM (2009) New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 32:1620–1625
Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Ruan K, Wang Z, Zhang CS, Chien KY, Wu J, Li Q, Han J, Lin SC (2012) GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science 336:477–481
Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Jin M, Zhu Z, Wang H, Yu J, Hao Y, Choi A, Ke H, Ma D, Yuan J (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147:223–234
Loehberg CR, Strissel PL, Dittrich R, Strick R, Dittmer J, Dittmer A, Fabry B, Kalender WA, Koch T, Wachter DL, Groh N, Polier A, Brandt I, Lotz L, Hoffmann I, Koppitz F, Oeser S, Mueller A, Fasching PA, Lux MP, Beckmann MW, Schrauder MG (2012) Akt and p53 are potential mediators of reduced mammary tumor growth by cloroquine and the mTOR inhibitor RAD001. Biochem Pharmacol 83:480–488
Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB (2005a) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120:237–248
Lum JJ, DeBerardinis RJ, Thompson CB (2005b) Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 6:439–448
Ma D, Panda S, Lin JD (2011) Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J 30:4642–4651
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF (2012) Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 11:317–328
Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G (2007) BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3:374–376
Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M (2007) FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6:458–471
Mahoney E, Lucas DM, Gupta SV, Wagner AJ, Herman SE, Smith LL, Yeh YY, Andritsos L, Jones JA, Flynn JM, Blum KA, Zhang X, Lehman A, Kong H, Gurcan M, Grever MR, Johnson AJ, Byrd JC (2012) ER stress and autophagy: new discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood 120:1262–1273 Marino G, Morselli E, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Benit P, Rustin P, Criollo A, Kepp O, Galluzzi L, Shen S, Malik SA, Maiuri MC, Horio Y, Lopez-Otin C, Andersen JS, Tavernarakis N, Madeo F, Kroemer G (2011) Longevity-relevant regulation of autophagy at the level of the acetylproteome. Autophagy 7:647–649
Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8(6):903–914
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137:1062–1075
Mirzoeva OK, Hann B, Hom YK, Debnath J, Aftab D, Shokat K, Korn WM (2011) Autophagy suppression promotes apoptotic cell death in response to inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J Mol Med 89:877–889 Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22:66–79
Moretti L, Attia A, Kim KW, Lu B (2007) Crosstalk between Bak/Bax and mTOR signaling regulates radiation-induced autophagy. Autophagy 3:142–144
Morselli E, Marino G, Bennetzen MV, Eisenberg T, Megalou E, Schroeder S, Cabrera S, Benit P, Rustin P, Criollo A, Kepp O, Galluzzi L, Shen S, Malik SA, Maiuri MC, Horio Y, Lopez-Otin C, Andersen JS, Tavernarakis N, Madeo F, Kroemer G (2011) Spermidine and resveratrol induce autophagy by distinct pathways converging on the acetylproteome. J Cell Biol 192:615–629
Ni HM, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, Ding WX (2012) Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci 127:438–450
Niture SK, Jaiswal AK (2012) Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J Biol Chem 287:9873–9886
Nyfeler B, Bergman P, Triantafellow E, Wilson CJ, Zhu Y, Radetich B, Finan PM, Klionsky DJ, Murphy LO (2011) Relieving autophagy and 4EBP1 from rapamycin resistance. Mol Cell Biol 31:2867–2876 Papadopoulos T, Pfeifer U (1986) Regression of rat liver autophagic vacuoles by locally applied cycloheximide. Lab Invest 54:100–107
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J (2011) Regulation of TFEB and V-ATPases by mTORC1. EMBO J 30:3242–3258
Pomel V, Klicic J, Covini D, Church DD, Shaw JP, Roulin K, Burgat-Charvillon F, Valognes D, Camps M, Chabert C, Gillieron C, Francon B, Perrin D, Leroy D, Gretener D, Nichols A, Vitte PA, Carboni S, Rommel C, Schwarz MK, Ruckle T (2006) Furan-2-ylmethylene thiazolidinediones as novel, potent, and selective inhibitors of phosphoinositide 3-kinase gamma. J Med Chem 49:3857–3871
Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280:20722–20729
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435
Romero IL, McCormick A, McEwen KA, Park S, Karrison T, Yamada SD, Pannain S, Lengyel E (2012) Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet Gynecol 119:61–67
Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, Lin L, Ellis MJ (2011) Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res 13:R21 Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596–603 Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170:1101–1111
Seglen PO, Gordon PB (1982) 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 79:1889–1892
Sehgal SN, Baker H, Vezina C (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 28:727–732
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332:1429–1433
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31:1095–1108
Shen S, Kepp O, Kroemer G (2012) The end of autophagic cell death? Autophagy 8:1–3
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol 6:1221–1228
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Shu CW, Madiraju C, Zhai D, Welsh K, Diaz P, Sergienko E, Sano R, Reed JC (2011) High-throughput fluorescence assay for small-molecule inhibitors of autophagins/Atg4. J Biomol Screen 16:174–182
Shvets E, Fass E, Elazar Z (2008) Utilizing flow cytometry to monitor autophagy in living mammalian cells. Autophagy 4:621–628
Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL, Melchionda F, Pagliaro P, Pession A, McCubrey JA, Capitani S, Martelli AM (2012) Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 26:2336–2342
Srigley JR, Delahunt B (2009) Uncommon and recently described renal carcinomas. Mod Pathol 22(Suppl 2):S2–S23
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9:1142–1151
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N (2011) Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25:795–800
Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S (2005) Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res 65:3336–3346
Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y (2011) The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res 17:3272–3281
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS (2009) An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 284:8023–8032 Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128:325–339
Tyagi S, Sharma S, Budhiraja RD (2012) Effect of phosphatidylinositol 3-kinase-gamma inhibitor CAY10505 in hypertension, and its associated vascular endothelium dysfunction in rats. Can J Physiol Pharmacol 90:881–885
Tzivion G, Hay N (2011) PI3K–AKT–FoxO axis in cancer and aging. Biochim Biophys Acta 1813:1925
Tzivion G, Dobson M, Ramakrishnan G (2011) FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta 1813:1938–1945
van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, Verhagen LP, Groot Koerkamp MJ, Braat AK, Dansen TB, Holstege FC, Gebhardt R, Burgering BM, Coffer PJ (2012) Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 14:829–837
Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70:535–602
Vezina C, Kudelski A, Sehgal SN (1975) Rapamycin (AY-22,989), a new antifungal antibiotic I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo) 28:721–726
Viola G, Bortolozzi R, Hamel E, Moro S, Brun P, Castagliuolo I, Ferlin MG, Basso G (2012) MG-2477, a new tubulin inhibitor, induces autophagy through inhibition of the Akt/mTOR pathway and delayed apoptosis in A549 cells. Biochem Pharmacol 83:16–26 White E (2012) Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 12:401–410
Wu YT, Tan HL, Huang Q, Ong CN, Shen HM (2009) Activation of the PI3K-Akt-mTOR signaling pathway promotes necrotic cell death via suppression of autophagy. Autophagy 5: 824–834 Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM (2010) Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem 285:10850–10861
Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, Mann DL, Taffet GE, Baldini A, Khoury DS, Schneider MD (2006) A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci USA 103:17378–17383
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22:124–131
Yazbeck VY, Buglio D, Georgakis GV, Li Y, Iwado E, Romaguera JE, Kondo S, Younes A (2008) Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol 36:443–450 Yi C, Ma M, Ran L, Zheng J, Tong J, Zhu J, Ma C, Sun Y, Zhang S, Feng W, Zhu L, Le Y, Gong X, Yan X, Hong B, Jiang FJ, Xie Z, Miao D, Deng H, Yu L (2012) Function and molecular mechanism of acetylation in autophagy regulation. Science 336:474–477
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304:1500–1502
Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, Zhu H, Yu AD, Xie X, Ma D, Yuan J (2007) Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA 104:19023–19028
Zhang J, Roberts TM, Shivdasani RA (2011) Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology 141:50–61
Zhang ZJ, Zheng ZJ, Shi R, Su Q, Jiang Q, Kip KE (2012) Metformin for liver cancer prevention in patients with type 2 diabetes: a systematic review and meta-analysis. J Clin Endocrinol Metab 97:2347–2353
Zhao M, Klionsky DJ (2011) AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab 13:119–120
Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL (2007) FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6:472–483
Acknowledgements
The research work in W.X. Ding’s lab was supported in part by the NIAAA funds R01 AA020518-01, R21 AA017421, National Center for Research Resources (5P20RR021940-07), and P20 RR016475 from the IDeA Networks of Biomedical Research Excellence (INBRE) program of the National Center for Research Resources. J.A. Williams was supported by the “Training Program in Environmental Toxicology” (grant 5 T32 ES007079) from the National Institute of Environmental Health Sciences. Y.F. Hou was supported by National Natural Science Foundation of China (Contract grant numbers: 81072165), and the Shanghai Science and Technology Committee (Contract grant numbers: 09PJ1402700). The authors would like to thank Bonnie Goodwin for technical support of ArrayScan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Yang, H. et al. (2013). Autophagy and Cancer Drug Discovery. In: Wang, HG. (eds) Autophagy and Cancer. Current Cancer Research, vol 8. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6561-4_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6561-4_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6560-7
Online ISBN: 978-1-4614-6561-4
eBook Packages: MedicineMedicine (R0)