
Abstract
Epilepsy has been characterized a disease whose social and occupational behavioural has had devastating economical consequences and is associated with great cumulative brain damage and neurological deficits. From different forms of epilepsy, the most frequent type is temporal lobe epilepsy (TLE), being the most common form of drug refractory epilepsy. Although there are a great amount of studies about the mechanisms involved in neuronal damage and death during critical phases of epileptogenesis, it is crucial to construct strategies for neuroprotection that may prevent the development of epilepsy. In this chapter, some molecular mechanisms involved in the neuronal death, which are induced by excitotoxicity phenomena following the signalling pathways activation and studied in animal models under seizure conditions or expressed in the epilepsy are discussed, mainly those as the mitogen-activated protein kinases, Jak/Stat, and Pi3k/Akt pathways those genes responsible to participate in the apoptosis and cell cycle regulation are also analysed.
In summary, the structural and molecular changes at cellular level are believed to play a key role in the generation of convulsive seizures and its possible identification should facilitate the develop of potential therapeutic targets heading towards of specific genes, proteins, and signalling pathways altered during the different stages of epileptogenesis process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
Epilepsy has devastating behavioural, social, and occupational consequences and is associated with cumulative brain damage and neurological deficits. In addition, it is characterized by the occurrence of repeated and sudden transitory episodes of motor, sensory, autonomic, and physical origin known as seizures, which at the cellular level are characterized by synchronized discharges of large groups of neurons that interfere their functions. Temporal lobe epilepsy (TLE) is the most common form of partial epilepsy and affects 40% of the patients. Seizures arising from the mesial temporal lobe structures (i.e., amygdala and hippocampus) can be progressive and often becomes refractory to drug treatment. It is characterized by the presence of complex partial seizures and generalized tendency to produce multiple epileptic foci. One of the most common histologic abnormalities observed in approximately 66% of patients with TLE is hippocampal sclerosis or mesial temporal sclerosis, characterized by a remarkable loss of neurons in the hippocampus leading to excessive glial proliferation, particularly in the hilar region of the dentate gyrus and the CA1 and CA3 regions (Thom et al. 2005). The majority of the patients with TLE suffer from symptomatic focal epilepsies, which are frequently a consequence of brain trauma, complicated febrile convulsions, prolonged seizures (status epilepticus—SE), ischemic lesions and brain tumours, encephalitis or childhood febrile seizures (Cendes 2002; Engel 2001; French et al. 2004). It has been established that each of these initial events leads to the activation of molecular signalling cascades, which in turn induce selective cell death that is directly related to the epileptogenic process, although even now it is not well known if cell death is the cause or effect of the establishment of the phenomenon of epilepsy. Hippocampal sclerosis (HS), also known as Ammon’s horn sclerosis, is characterized by the loss of pyramidal cells and gliosis in CA1 (Ammon’s horn) and end folium, dispersion of the granule cell layer of the dentate gyrus (DG), neurogenesis of granule cells, axonal sprouting, and synaptic reorganization of the mossy fibres (Wieser 2004; Thom et al. 2005). Cell loss is typically asymmetric between the hippocampus; the most affected regions are the CA1 and CA3 subfields and hilar region of the DG, while the CA2 subfield and granule cells of the DG usually show much less cell loss (Mathern et al. 1997). In spite of damage to other limbic regions, the cerebellum and cerebral cortex are also commonly affected.
TLE represents the final stage of a long and complex process of cellular and molecular events that are determined by the initial stimulus that triggers the process. There is usually a latent period of several years between this injury and the emergence of the chronic TLE characterized by spontaneous recurrent seizures originating from the temporal lobe, as well as learning and memory impairments (Bartolomei et al. 2005; Detour et al. 2005; Devinski 2004).
The TLE can be reproduced in laboratory animals (typically rodents) by the systemic or intracerebral administration of powerful convulsant agents such as glutamatergic (kainic acid) or cholinergic (pilocarpine) agonists (Pitkänen et al. 2005; Covolan et al. 2000). Over the last few decades, there has been considerable progress in the pharmacotherapy of epilepsy, including the introduction of several new antiepileptic drugs (AEDs) (McCabe 2000). The mechanisms of action of most clinically used drugs in human epilepsies are based upon the synchronized neuronal activity and imbalance between inhibitory and excitatory neurotransmission, events commonly linked to the pathogenesis of epilepsy (Dalby and Mody 2001). However, approximately 30–40% of all patients with TLE are estimated to be drug resistant, therefore identification of specific biological processes and biochemical pathways that trigger cell death during critical phases of epileptogenesis is crucial to design strategies for neuroprotection that may prevent epileptogenesis process.
6.2 Epileptogenesis and Animal Models
The term epileptogenesis refers to the transformation through the normal process of the plastic neuronal network into a chronically hyperexcitable state. The epileptogenic processes emerge after precipitating insults (i.e., local infections, SE, ischemia, or trauma) in concert with genetic susceptibility factors, which they are possible to trigger such persistent pathophysiological changes (Coulter and DeLorenzo 1999). However, the study of human epileptic hippocampus does not allow revealing the sequence of events leading to neuronal loss and the regulation of plastic events. For this reason, different animal models have been designed using electrical or drug stimulation, among which include systemic administration of kainic acid, an analogue of glutamate, or cholinergic agonist-pilocarpine (Pitkänen et al. 2005). Both experimental models and postmortem human studies support the idea that cell death is a common pathological feature of insult to the brain, which triggers a chronic epileptic condition (Sutula 2004). Briefly, such injuries invariably set in signal various cell and molecular processes including gliosis, inflammation and vascular changes, neurogenesis and rewiring, axonal reorganization, dispersion of granule cells, and changes in expression of ion channels and signalling molecules including neuronal death. Collectively, this process is identified as epileptogenesis (Blumcke et al. 1999; Mathern et al. 1993; Sloviter 1999).
In the animal models of TLE, the damage within the hippocampus precedes the appearance of spontaneous seizures. Moreover SE induced by systemic injection of pilocarpine or kainic acid or by repeated electrical stimulation-caused structural brain damage in rats (Sloviter 2008). Cell loss has been observed in these models in the hilus and CA3 regions, as well as amygdala and entorhinal cortex (Turski et al. 1989; Bartolomei et al. 2005). Moreover, prominent mossy fibre sprouting occurred. The primary cause of neuronal death following seizures is probably over-activation of ion channels gated by glutamate, the principal excitatory neurotransmitter in the brain (Meldrum 1991; Fujikawa 2006). In these conditions, biological processes, including activation of signalling pathways related to stress response, ion transport, signal transduction, and synaptic transmission are triggered (Aronica and Gorter 2007).
6.3 Mechanisms of Cell Death
6.3.1 Excitotoxicity
The excitotoxicity is a pathological process into the cells. The pioneering work of Meldrum (1993) provided evidence that seizure-induced cell death and other events that induce neurodegeneration result from over-activation of ionotropic glutamate receptors which leads to increased intracellular levels of Ca2+ and Na+ and causes swelling and cell lysis. There is also energy failure, production of free radicals, activation of enzymatic complex, and cell death (Smolders et al. 2009). The death of cells has been classified generally into two distinct types: apoptosis and necrosis. Both forms of cell death can be induced by excitotoxicity. It has been shown that the excitotoxic damage induced by seizures activates programmed cell death pathways through changes in the expression of specific genes (Engel and Henshall 2009).
6.3.2 Apoptotic Pathways
Apoptotic signals are given through a highly ordered molecular cascade that is energy dependent. Several different stimuli can initiate the apoptotic death of neurons. Complex intracellular and intercellular cell-death-regulatory pathways are increasingly recognized as important contributors to seizure-induced neuronal death; however, apoptotic pathways converge on a restricted number of common effector (Sastry and Rao 2000; Engel and Henshall 2009). Two principal pathways have been described as apoptotic death. These two signalling pathways and the final caspase executor activation pathway are also regulated by several proteins such as glycogen synthase kinase (GSK3), ataxia-telangiectasia-mutated protein (ATM)/p53, Bcl-2, cyclin-dependent kinases (CDKs), and mitogen-activated protein kinases (MAPKs), which act on both pathways (Wang et al. 2007; Kroemer et al. 2007).
There are two main cell-signalling pathways that have been identified in the control of apoptosis: the intrinsic pathway, or core pathway, and the extrinsic pathway.
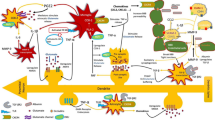
The extrinsic pathway mediates cell death in response to extracellular stimuli and is initiated by cell-surface receptors called death receptors (DR) of the tumour necrosis factor (TNF) superfamily (Wajant 2002). Ligands for these receptors include TNFα, Fas ligand, and TNF receptor apoptosis-inducing ligand (TRAIL). Activation follows binding of the ligand to its receptor and oligomerization of the receptor. In the case of Fas, the cell death signal is propagated inside the cell by recruitment of Fas-Associated protein with Death Domain (FADD) and an initiator caspase (e.g., 8 or 10 caspases) to the intracellular side of the plasma membrane, resulting in formation of a death-inducing signalling complex (DISC). In contrast, activation of TNFR1 leads to direct association with TNF receptor-associated death domain (TRADD); the recruited to this complex can then modulate the nuclear factor-κB pathway (Fig. 6.1) (Wajant 2002; Strasser et al. 2000).

Major apoptosis pathways activated by seizures. Both the excitotoxicity which may directly trigger mitochondrial dysfunction or activate enzymatic pathways (e.g., mitogen activated protein kinases-MAPK) and the pathway activated by death receptors (DR) converge in the activation of modulators (e.g., inhibitor of caspase-activated DNase-ICAD) and executors proteins of death process (e.g., caspases). Other pathways may compensate these processes (e.g., pathway mediated by Growth-factor receptor or cytokines) promoting activity or expression (e.g., inhibitors of apoptosis proteins-IAP) of anti-apoptotic proteins or decreasing their activity (e.g., Bad and glycogen synthase kinase-3β (GSK-3β))
Extrinsic pathway activation after seizures is documented in various seizure models where the presence or activity for 2 and 8 caspases has been reported (Henshall et al. 2001a, b). While death receptors are constitutively expressed in the brain, their participation has been linked to the activity of caspase 8 observed after seizures through scaffolding proteins and receptors for TNFα and FasL, both present in the adult rat hippocampus before and after seizures (Shinoda et al. 2003; Henshall et al. 2001a, b).
On the other hand, an intrinsic pathway that is associated to apoptosis is regulated by mitochondria, which integrates a lethal or pro-survival signal that eventually determines the cell density. It is initiated after cell stressors that perturb intracellular organelle function (Danial and Korsmeyer 2004; Xu et al. 2005) and include raised intracellular Ca2+ and reactive oxygen species (ROS) (Orrenius et al. 2003). Mitochondrial apoptosis in neurons can be triggered by a variety of structurally related agents (Sastry and Rao 2000). A key event in this pathway is the release of apoptogenic molecules from mitochondria, which is caused by a change in permeability of the outer mitochondrial membrane and the release of molecules from mitochondria, and in particular cytochrome c (cyt c), binds the apoptotic protease, activating factor 1 (Apaf1) and recruiting caspase 9. This forms the so-called apoptosome, which processes downstream effector caspases such as caspase 3, culminating in cleavage of various structural and other proteins (Fig. 6.1) (Bratton and Salvesen 2010).
The permeability of the mitochondrial outer membrane (MOMP) is regulated by the activity of several proteins that belong to the Bcl2 family. The Bcl-2 gene family comprises more than 20 different members that either positively or negatively regulate apoptosis primarily by affecting the mitochondria (Cory and Adams 2002; Liou et al. 2003; Kroemer et al 2007). The proteins of this family with anti-apoptotic function include Bcl-2, Bcl-XL, and Mcl-1, which preserve the integrity of the outer mitochondrial membrane. The major pro-apoptotic proteins here include Bad, Bid, Bik, Bim, Noxa, p53-upregulated mediator of apoptosis (PUMA), Bax, and Bak. Interestingly, Bid protein constitutes one link between the extrinsic and intrinsic pathways through the cleavage of caspase-8, which further amplifies the apoptotic death signal. Bid interacts with Bax–Bak, which forms pores that allow the release of cyt c (Cory and Adams 2002). The anti-apoptotic Bcl-2 and Bcl-XL proteins can prevent Bax translocation towards the mitochondria, but additionally Bcl-XL may bind to Apaf-1 and in doing so suppresses caspase-9 activation (Fig. 6.1) (Kroemer et al. 2007; Stavrovskaya and Kristal 2005).
As previously mentioned, ROS production is another common stressor factor that is triggered by excitotoxicity. One target of ROS is DNA, which is extremely sensitive to oxidative stress. One of the sensors of DNA damage is the ATM, which belongs to the family of phosphatidylinositol-3 kinases (PI3K) (Roos and Kaina 2006; Chipuk and Green 2009). Once activated, ATM stimulates p53 (a nuclear transcription factor). Thus, DNA damage and the subsequent p53 activation; both contribute to other apoptotic signals that the mitochondria receive through the intrinsic pathway. In fact, if the neuronal DNA damage cannot be repaired, over-activation of p53 triggers the neuronal apoptotic process. In a second step, p53 mainly induces the expression of the pro-apoptotic Bcl-2 family members PUMA (Roos and Kaina 2006; Chipuk and Green 2009). Noxa is another p53-activated mediator that can contribute to apoptosis. Interestingly, PUMA activates the intrinsic apoptotic pathway by binding to Bax, which acts directly on the mitochondria (Chipuk and Green 2009). In addition, PUMA can bind with and consequently inhibit anti-apoptotic Bcl-2 family members, including Bcl-2, Bcl-XL, Bcl-w, and Mcl-1 (Fig. 6.1) (Roos and Kaina 2006). A tight balance exists between the activities of pro- and anti-apoptotic Bcl-2 family members in resting conditions. The cell’s fate will progress to apoptosis only when this equilibrium is altered toward enhanced activity of pro-apoptotic proteins.
While both extrinsic and intrinsic pathways have different beginnings, they eventually converge in the massive activation of catabolic enzymes (including a class proteases known as caspases, no caspases proteases, lipases, and endonucleases); at present there are 14 known mammalian caspases (named from cysteinyl-aspartate-specific proteases) that are enzymes that cleave other proteins next to an aspartate residue. The apoptosis regulatory caspases are divided into initiators of apoptosis that include caspases 8, 9, and 10, and the apoptotic executioners are caspases 3, 6, and 7 (Schindler et al. 2006; Bozzi et al. 2011). Each caspase is initially synthesized as a zymogen and requires processing at specific cleavage sites to generate the active enzymes. The caspases that are the first to be activated trigger downstream other caspases giving rise to a proteolytic cascade that culminates in the execution of apoptosis. Different subsets of caspases are activated depending on the pro-apoptotic stimulus (Salvesen and Riedl 2007). For example, caspases 3, 6, and 8 are part of the Fas/TNFα-mediated death pathway, while caspases 3 and 9 together with apoptosis protease-activated factor 1 (Apaf1) and cyt c participate in mitochondria-associated cell death (see Fig. 6.1) (Bratton and Salvesen 2010).
Activation of intrinsic, mitochondria-dependent cell death pathways after seizures would be predicted based on the assumed significance of glutamate excitotoxicity and mitochondrial dysfunction due to both calcium (Ca2+) and ROS loading (Orrenius et al. 2003). Several authors have observed that neuroprotection is also less pronounced when mitochondrial-activated caspase-9 is blocked after seizures, and other data suggests the extrinsic cell death pathway-associated caspase 8 is activated following seizures in vitro (Henshall et al. 2001b, c; Meller et al. 2006).
6.3.3 Apoptosis and Cell Cycle Regulation
The biochemical mechanisms of the different phases of the cell cycle are highly regulated by intracellular signalling elements such as protein kinases (e.g., MAPKs) as well as their target substrates in particular cell cycle regulators. A family of cyclins act as regulatory subunits for CDKs, and thus regulate passage through the four phases of the cell cycle. The activities of the various cyclin/CDK complexes regulate the progression through G1/S/G2/M phases of the cell cycle (Nigg 1995). MAPKs are involved in regulating the protein expression of cell cycle regulators; in particular those that regulate passage of cells of phase G0 to G1 (Yeste-Velasco et al. 2009).
In particular, the differentiated neurons are post-mitotic cells and completely lacking in replicative capability. These cells enter a phase of mitotic quiescence commonly referred to as the Go phase, and as such were believed to be unable to re-enter the cell cycle. Postmortem studies have revealed pathological evidence of aberrant cell cycle re-entry occurring in neurons of patients with Alzheimer’s disease (Yang et al. 2001, 2003), epilepsy (Nagy and Esiri 1998), and Parkinson’s disease (Jordan-Sciutto et al. 2003). Moreover, it has been observed experimentally that cell cycle regulators such as CDKs are produced and abnormally activated in different models of induction of cell damage (e.g., ischemia, epilepsy, excitotoxicity, and trauma) (Timsit and Menn 2007; Sutula 2004). The activation of these events leads to cell death. Various markers of this event have been detected before neuronal death occurs suggesting its participation as an initiator of the execution of the cell death program (Katchanov et al. 2001; Timsit and Menn 2007). The atypical expression of mitogenic genes may promote entry and progression of neurons into the cell cycle through an increase in the expression level of cyclin D and phosphorylation of the retinoblastome protein (Rb), regulating the E2F activity which induces modifications to the transcription of pro-apoptotic molecules as caspases 3, 8, and 9, as well as Apaf-1 or members of the Bcl-2 family (Greene et al. 2004).
Although few studies have evaluated the role of cell cycle regulators in epilepsy, there is enough evidence to link changes in the expression and activity of these molecules in epileptogenesis. A study following kainate-induced seizures showed that the cyclin D1 mRNA was induced in the vulnerable CA3 region, and to a lesser extent, in non-vulnerable regions, while that the expression of CDK4 and cyclin D1 was upregulated in neurons of the rat piriform cortex and amygdala 1–3 days after KA administration in vivo. CDK4 and cyclin D1 proteins were induced in the cytoplasm and nuclei of neurons, with a concomitant increase of CDK4- and cyclin D1-positive microglia in the affected areas; these results suggest that CDK4 and cyclin D1 are essential for KA-induced neuronal apoptosis in vivo (Timsit and Menn 2007; Ino and Chiba 2001).
6.4 Signal Pathways in Survival or Cell Damage
Epilepsy activates several signalling cascades that are essential to regulate the survival or cell damage, which are evoked by multiple stimuli, including excitotoxicity, oxidative stress, and inflammation processes (Henshall and Murphy 2008; Okamoto et al. 2010). Particularly the inflammatory processes, including activation microglia and astrocytes and production of proinflammatory cytokines and related molecules, have been described in human epilepsy patients as well as in experimental models of epilepsy (Vezzani et al. 2008). A number of proinflammatory mediators, thus initiating a cascade of processes in brain tissue, alter neuronal excitability and affect the physiological functions of glia by paracrine or autocrine actions, thus interfering with the neuronal communications and may compromise neuronal survival (Riazi et al. 2010; Vezzani et al. 2008). Chronic brain inflammation may also contribute to susceptibility to seizures and comorbidity in chronic epilepsy patients. Prototypical inflammatory cytokines such as interleukine-1β (IL-1β), TNF-alpha, and interleukine-6 (IL-6) are over-expressed in experimental models of seizures in brain areas of seizure generation and propagation, and are prominent in glia, and to a lesser extent by neurons. Cytokines receptors are also upregulated, and the related intracellular signalling is activated in both cell populations highlighting autocrine and paracrine actions of cytokines in the brain (Riazi et al. 2010; Vezzani et al 2008). The recent demonstration of functional interactions between cytokines and classical neurotransmitters such as glutamate and gamma amino butyric acid (GABA), as well as intracellular signalling mechanisms, suggest the possibility that these interactions underlie the cytokine-mediated changes in neuronal excitability, thus promoting seizure phenomena and the associated neuropathology (Balosso et al. 2008, 2009; Stellwagen et al. 2005; Pickering et al. 2005).
6.4.1 Protein Kinases Activated by Mitogen
Extracellular stimuli evoked by neurotransmitters, neurotrophins, and growth factors in the brain regulate critical cellular events, including synaptic transmission, neuronal plasticity, morphological differentiation, and survival. A pathway known to influence seizure-induced neuronal damage and epileptogenesis includes the MAPK cascades (Liou et al. 2003; Shinoda et al. 2003). The MAPK pathways are used by eukaryotic cells for the transduction of extracellular signals to the nucleus and other intracellular targets (Chang and Karin 2001).
There are two known major pathways of MAPK including the extracellular signal-regulated kinases (ERK) and stress-activated protein kinases (SAPK). The latter is divided into the kinases c-Jun NH2-terminal (JNK/SAPK) and p38 kinase pathway (p38/SAPK) (Pearson et al. 2001; Okuno et al. 2004). The MAPKs are also involved in apoptosis and may, therefore, play a role in neurodegeneration (Borsello and Forloni 2007; Guan et al. 2006; Kyosseva 2004). These kinases are activated by phosphorylation on threonine and tyrosine residues. Subsequently, phosphorylated and other intracellular enzymes or transcription factors regulate the expression of genes involved in cellular response (Kyosseva 2004). The substrates that are identified are phosphorylated; for MAPKs in the nucleus they include some hormone receptors, as well as transcription factors such as the activator protein-1 (AP-1), the family of Jun factors (c-Jun, Jun-B, and Jun-D), Elk-1, p53, transcription factor-2 (ATF-2), JDP2, c-Myc, the NAFT family, the STAT family, and the PAX family (Chen et al. 2001; Kyosseva 2004).
Commonly the activation of signalling pathways JNK/SAPK and p38/SAPK has been associated with the promotion of cell damage (Borsello and Forloni 2007; Guan et al. 2006; Kyosseva 2004). Nevertheless, extracellular signal-regulated kinase1/2 (ERK1/2) has been implicated in several cellular functions including regulation of cell proliferation, differentiation, survival, and apoptosis in response to a wide variety of external stimuli (Cheung and Slack 2004; Miller and Gauthier 2007; Yoon and Seger 2006).
ERK pathway exhibits dynamic changes following several types of seizure activity and may function in the regulation of neuronal excitability (Dudek and Fields 2001; Houser et al. 2008). This signalling pathway is strongly activated in neurons following severe, chemically induced seizures. In an initial SE episode-increased ERK activation may be neuroprotective and limit the damage of some neurons, such as dentate granule cells (Choi et al. 2008). Conversely, a lack of ERK activation in other neurons may contribute to their vulnerability to excitotoxic damage (Choi et al. 2007). At later stages, ERK phosphorylation may decrease as a compensatory mechanism to control increased network excitability (Dudek and Fields 2001).
Certain evidence has shown that neuronal activity-dependent modulation of the ERK signalling pathway plays an important role in synaptic plasticity (Yoon and Seger 2006).
Moreover, in vivo studies have implicated that the SAPKs play an important role in mediating glutamate receptor (GluR) responses, possibly involving the normal physiology of glutamate and associated pathophysiology. For example, the activation of the N-methyl-d-aspartate (NMDA) receptor stimulates JNK and p38 MAPK in cultured CGCs (Kawasaki et al. 1997); and in hippocampal neurons α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainite (KA) receptors stimulate ERKs, JNK, and p38 kinase (Mukherjee et al. 1999).
The JNK pathway has a central position in cellular damage particularly in apoptosis and participates in the death cell program through regulation of the function of pro-apoptotic activators members of bcl-2 family (BH3-only) or phosphorylates Bim- and Bcl2-associated agonist of cell death (Bad) at distinct serine residues (Donovan et al. 2002; Putcha et al. 2003). Moreover, JNK enhances bim-gene expression through activation of the transcription factor c-Jun. Therefore, the deletion or inhibition of JNKs components substantially limits the cellular potential to undergo death in neuronal and non-neuronal cells, principally the caspases dependent. The most convincing evidence to suggest that JNK is implicated in excitotoxic neuronal death has come from studies using JNK3 knockout mice, where KA-mediated seizures in vivo failed to cause apoptosis in hippocampal neurons, coincident with the reduction of c-Jun phosphorylation (Yang et al. 1997). The principal substrate for JNK is c-Jun; however, it is not known which isoform is responsible for its phosphorylation. High expression of both the gene and protein of c-Jun precedes or coincides with periods of cell death, such as that occurring during embryonic development (Sun et al. 2005), after trauma (Raivich et al. 2004), cerebral ischemia (Wessel et al. 1991), and seizures (Morgan and Curran 1991).
This dual role of MAPKs may make it possible to design alternative and/or synergistic approaches to the management of degenerative diseases, either by using specific inhibitors of the MAPKs involved in apoptosis or by increasing the activation of the MAPKs involved in neuronal survival and differentiation.
6.4.2 JAK/STAT and PI3K/AKT Pathways
The Janus kinases (JAKs) are a family of non-receptor protein tyrosine kinases. They are activated in a variety of different ways. In the canonical pathway, two JAK molecules bind to two receptors that dimerized in response to ligand binding and the juxtaposed JAKs trans and/or autophosphorylate resulting in their activation (Yamaoka et al. 2004). This mode of activation applies, for example, to cytokine receptors, growth hormone-like receptors, and the leptin receptor. Alternatively, JAKs may be activated following stimulation of G protein-coupled receptors and/or via intracellular Ca2+ changes. Once activated, JAKs phosphorylate and activate downstream targets. For instance, the recruitment of JAK2 mediates the activation of several signalling pathways, including STAT5, ERK/MAPK, and PI3K/Akt (Silva et al. 1999; Kretz et al. 2005). The best-established downstream effector of JAK is the signal transducer and activator of transcription (STAT) family. Seven STAT isoforms, named STAT1 to STAT4, STAT5A, STAT5B, and STAT6, have been identified (Battle and Frank 2002). Once phosphorylated by JAK, STATs dimerize and are translocated to the nucleus where they regulate the expression of numerous genes (Aaronson and Horvath 2002). The JAK/STAT pathway is involved in many physiological processes including those governing cell survival, proliferation, differentiation, development, and inflammation. There is increasing evidence that this pathway also has neuronal specific functions in the central nervous system (Yadav et al. 2005). The cellular and molecular mechanism by which the JAK/STAT pathway is involved in neuronal function is unknown. However, it has been shown that STAT can regulate the expression or function of several neurotransmitter receptors, including GABA (Lund et al. 2008), muscarinic acetylcholine (Chiba et al. 2009), and NMDA and AMPA receptors. Particularly, STAT5 is a predominantly pro-survival signal (Debierre-Grockiego 2004).
A role for Akt in mediating neuronal survival was first demonstrated by Datta and colleagues (Datta et al. 1997) in a primary postnatal cerebellar granule cell culture model, in which apoptosis is induced by either low potassium or growth factor withdrawal (D’Mello et al. 1993). Moreover, Akt is a serine/threonine kinase with diverse roles related to the regulation of cell growth, proliferation, migration, glucose metabolism, transcription, protein synthesis, and angiogenesis (Bazil et al. 2002). Activation of Akt occurs following the binding of a protein growth factor to its receptor on the cell surface. Ligand binding induces autophosphorylation of tyrosine residues in the cytoplasmic portion of the receptor, resulting in the recruitment and activation of phosphatidylinositol 3-kinase (PI3K).
One best-characterized signalling survival mediated by Akt is activated by NMDA receptors (Datta et al. 1997); the inhibition of this kinase activity contributes to NMDA receptor-mediated apoptosis. Moreover the protein kinase serine/threonine (Akt), also known as protein kinase B (PKB) has two sites of phosphorylation that determine the regulation of Akt activity: threonine 308 (Thr308), located in the kinase domain, and serine 473 (Ser473), which is in the regulatory domain (Coffer and Woodgett 1991; Song et al. 2005).
The activation of Akt by trophic factors depends on PI3-K (Burgering and Coffer 1995). When the trophic factor specifically binds to its receptor, PI3-K is recruited by activating Akt modulating, and an anti-apoptotic effect may be through:
-
1.
Direct regulation of the apoptotic pathway
-
2.
Transcriptional control of molecules that promote cell survival, and
-
3.
Regulation of cellular metabolism (Song et al. 2005)
With respect to direct regulation, Akt can phosphorylate different members of the pro-apoptotic Bcl-2 family such as Bad (Datta et al. 1997) and Bim. Once phosphorylated bind to proteins called chaperones 14-3-3 in the cytoplasm; they are thereby inactive in a pro-apoptotic function. Other direct effects involve the inactivation of caspase-9 by phosphorylation or the negative regulation of JNK/SAPK. Regarding the transcriptional control, Akt can phosphorylate different transcription factors indirectly by modulating its activity. Phosphorylation of the family of FoxO transcription factors, whose function includes the induction of apoptosis through the redistribution of these factors from the nucleus to the cytoplasm, prevents its activity (Huang and Tindal 2007).
6.5 Mechanisms of Neuronal Death in Both Experimental Models and Patients with Intractable TLE
6.5.1 Experimental Models of TLE and Cell Signalling
Molecular analyses of epilepsy-induced hippocampal plasticity have largely focused on individual candidate genes, with particular emphasis on genes with known functions for specific pathogenetic aspects (Aronica and Gorter 2007; Mefford et al. 2010). Various studies have reported changes in gene expression in the SE induced by kainic acid (Hunsberger et al. 2005) and pilocarpine (Becker et al. 2003), as well as by electric stimulation (Gorter et al. 2006; Engel and Henshall 2009). Moreover, the analyses of these studies have shown an overlap in gene expression profiling in epileptogenesis revealing that the biological process emerges as the most frequently encountered in this context and is related to glial activation, immune response (e.g., inflammation), signal transduction, synaptic transmission (e.g., dopaminergic, glutamatergic, and GABAergic), and the induction of immediate early genes (IEGs) (De Lanerolle and Lee 2005; Aronica and Gorter 2007; Okamoto et al. 2010).
In the work of Okamoto et al. (2010), all possible changes in the rat transcriptome were monitored at distinct time points corresponding from the latent to chronic phase of the pilocarpine model of epilepsy, one the most extensively studied models of TLE. Genes identified as being differentially expressed were classified based on their respective biological functions to envisage processes and pathways likely implicated in epileptogenesis. The hyper-expression of 128 genes was described in this model, indicating stable modulation of the p38/MAPK, JAK/STAT, and PI3K signalling pathways (Okamoto et al. 2010), some of which displaying a parallel expression pattern in humans with epilepsy.
The involvement of caspases in SE-induced neurodegeneration has also been studied after systemic injection of kainic acid or lithium-pilocarpine, both of which produce vast and severe neuronal damage (Fujikawa et al. 1999, 2000). Henshall et al (2001b) and Li et al (2006) reported that caspases-8 and -9 are activated in the hippocampus after focal SE was induced by kainic acid. The expression of activated caspase-3 in hippocampal neurons and astrocytes have been also detected after pilocarpine-induced SE (Narkilahti et al. 2003; Weise et al. 2005). The different location of caspase after SE suggests different functions in the brain.
Additionally, López–Meraz et al. (2010) using the lithium–pilocarpine model of SE in 2-week-old rat pups showed that dying neurons in the DG and CA1-subiculum area do not share the same mechanism of death. In CA1-subiculum, caspase-8 upregulation preceded caspase-3 activation in morphologically necrotic neurons, while in the DG dying neurons were caspases-9 and -3 immunoreactive and morphologically apoptotic. SE-induced neuronal necrosis can be an active mechanism involving the activation of a caspase cascade (Niquet et al. 2007; Lopez-Meraz et al. 2010).
Weak evidence for apoptotic mitochondrial pathways has been described after lithium–pilocarpine-induced SE in degenerating neuronal populations (Fujikawa et al. 2002). Some works have shown that SE triggered by intra-amygdala kainic acid in mice causes rapid p53 accumulation and subsequent hippocampal damage. Expression of PUMA, a pro-apoptotic protein under p53 control, was increased within a few hours of SE. Induction of PUMA was blocked by pharmacologic inhibition of p53, and hippocampal damage was also reduced. Compared to PUMA-expressing mice, PUMA-deficient mice had significantly smaller hippocampal lesions after SE. Moreover, PUMA-deficient mice were found to develop fewer epileptic seizures than wild-type animals after SE (Engel et al. 2010). Nevertheless, functional-proteomics studies are needed to determine which molecules are active during the process of epileptogenesis or after SE (Engel and Henshall 2009).
On the other hand, the neuronal stem cells in the hippocampus appear to be sensitive to a prolonged seizure resulting in an increase in stem or progenitor cell numbers (Walker et al. 2008). In agreement, a quantitative real-time PCR analysis of cell cycle genes confirmed hyper-expression of Cdk1, a gene regulating the G1 to S and G2 to M transition of the cell cycle, and Nestin, a marker of neural stem cells and neural progenitor cells. However, expression of the cell cycle inhibitor p18(INK4c) was paradoxically enhanced after SE induced by pilocarpine and coincided with the peak of Cdk1 and Nestin expression at day 3 post-SE (Okamoto et al. 2010). These findings suggest that the proliferative stage may be inhibited by such activation p18 in pilocarpine model.
Cells born after seizure have altered synaptic inputs and neurotransmitter expression (Jessberger et al. 2007; Parent et al. 2006; Jakubs et al. 2006). These alterations have also been shown in neurogenesis in pilocarpine-induced SE (Radley and Jacobs 2003). Experimental TLE is associated with an increase in neurogenesis following amygdala kindling (Parent et al. 1998, 2006; Scott et al. 1998); since many of the newborn neurons eventually integrate into hippocampal circuitry and they may either contribute to the hippocampal network plasticity associated with epilepsy or, possibly, limit seizure activity (Jakubs et al. 2008; Overstreet-Wadiche et al. 2006).
Moreover, phosphorylated ERK (pERK) is increased in many hippocampal neurons following recurrent spontaneous seizures in pilocarpine-treated mice (Houser et al. 2008). Its activity appears to be involved in regulation of seizure-induced neurogenesis during the first few days after SE, since ERK activation returns to control levels within 1 week (Choi et al. 2008).
Experimental evidence suggests that seizures evoked by microinjection of kainic acid into the amygdala of the rat activate multiple cell death pathways involving Bcl-2 and caspase family proteins in brain regions destined to die, whereas survival promoting responses predominate in cortical populations that survive (Henshall 2001a). Pro-apoptotic BAD and the counteractive effects of Akt-pathway may underlie in part, the cell death outcome after seizures, providing a more complete understanding of the mechanisms by which seizures damage brain and highlighting novel targets for treatment of brain injury associated with seizure disorders (Henshall 2001a, b). Moreover, the end effector of the signalling pathway regulated by STAT5 proteins includes Bcl-xL and XIAP. Both have shown anti-apoptotic effects in diverse damage animal models (Okamoto et al. 2010).
In summary, although there are several models for the study of epileptogenesis, SE, and convulsive seizures, it is important to continue with additional studies for search potential molecular elements that can participate in the process of neuroprotection and/or as therapeutic targets for the treatment of epilepsy.
6.5.2 Studies of Signalling in Patients with Pharmacoresistant TLE
Studies have shown that both pro- and anti-apoptotic proteins, as well as death receptors are found in surgically removed brain samples from patients with pharmacoresistant TLE (Nagy and Esiri 1998; Henshall et al. 2000a, b; Dorr et al 2002).
Analysis of hippocampus from patients with intractable TLE from several groups has confirmed altered expression of Bcl-2 and caspase family genes. In particular, it has been observed that the Bcl-2 and bax immunoreactivity increases predominantly in cells with the morphologic appearance of neurons, whereas bcl-xL immunoreactivity augments in cells with the appearance of glia (for review Engel and Henshall 2009). In another studies, the expression of anti-apoptotic proteins Bcl-2, Bcl-x, and Bcl-w has been reported to be higher in brain tissue obtained from patients with intractable seizures; however, some pro-apoptotic changes are also seen in this gene family. It may suggest the possible predominance of apoptotic protective pathway-Bcl2 in human epileptic brain (Henshall et al. 2000a, b; Shinoda et al. 2004). DNA fragmentation was also detected in some but not all brain sections from patients undergoing temporal lobectomy for intractable seizures (Henshall et al. 2000a, b).
Nagy and Esiri (1998) described cell cycle disturbances and a possible apoptotic mechanism of hippocampal neuronal cell death in hippocampus obtained from patients with pharmacoresistant epilepsy, suggesting that neurons have re-entered the cell division cycle and reached the G2 phase. Another interaction was described between the cell cycle machinery and the intrinsic processes in apoptotic neurons, with evidence that Cdk1 activates pro-apoptotic bad protein. Moreover, that interaction has also been associated with different pathological conditions such as stress, depression, and epilepsy (Becker and Bonni 2004).
Other studies trying to prove a causal relationship between changes in neurogenesis and the disease state (i.e., depression and epilepsy) have been controversial (Jung et al. 2006; Malberg et al. 2000; Scharfman et al. 2000).
6.6 Conclusions and Perspectives
In TLE, structural changes in the hippocampus are believed to play a key role in the generation of epileptic seizures. However, given the complexity of hippocampal circuitry and cell damage in case of hippocampal sclerosis, structural repair of epileptic hippocampal networks will require complex strategies in which proper integration and rewiring of the implanted neurons will be of crucial importance. The activation of cell-signalling pathways in response to acute seizures has dramatic consequences such as neuronal loss and irreversible loss of function. In the past 10 years, researchers have found molecular “signatures” of gene-directed cell death signalling strategy linked to apoptosis in brain samples from a subpopulation of patients with pharmacoresistant epilepsy who experience frequent seizures. Using animal models, researchers have shown that evoked seizures or epilepsy often activates the same signalling pathways, and drugs or genetic modulation of these cascades can reduce brain injury.
The analysis of molecular responses to seizure is not simple because biological processes are not uniform and experimental protocols differ. The identification of potential therapeutic targets should be facilitated by the knowledge of genes, proteins, and altered signalling pathways during the different stages of epilepsy development.
It is important to consider that most of the functional studies reviewed here support targeting apoptosis signalling pathways to prevent seizure-induced neuronal death. However, not all groups detect an apoptotic signature (Henshal and Murphy 2008). Also, protection of some cell populations may be more critical than others. Translating short-term to long-term neuroprotection will also be challenging. Apoptosis-regulatory genes with neuromodulatory properties may be particularly promising but, of course, raises concerns of its effects on brain function that targeting apoptosis pathways was originally expected to avoid.
A deep understanding of signalling pathways involved in both acute and long-term responses to seizures continues to be crucial to unravel the origins of epileptic behaviours. Therefore, additional studies would be necessary to identify those genes related to neuroprotection and/or those involved in neuronal activities related to epileptogenesis and could potentially represent target genes in design new preventive drugs for epilepsy.
References
Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–5.
Aronica E, Gorter JA. Gene expression profile in temporal lobe epilepsy. Neuroscientist. 2007;151:272–92.
Balosso S, Maroso M, Sanchez-Alavez M, Ravizza T, Frasca A, Bartfai T, et al. A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1beta. Brain. 2008;131:3256–65.
Balosso S, Ravizza T, Pierucci M, Calcagno E, Invernizzi R, Di Giovanni G, et al. Molecular and functional interactions between tumor necrosis factor-alpha receptors and the glutamatergic system in the mouse hippocampus: implications for seizure susceptibility. Neuroscience. 2009;161:293–300.
Bartolomei F, Khalil M, Wendling F, Sontheimer A, Regis J, Ranjeva JP. Entorhinal cortex involvement in human mesial temporal lobe epilepsy: an electrophysiologic and volumetric study. Epilepsia. 2005;46:677–87.
Battle TE, Frank DA. The role of STATs in apoptosis. Curr Mol Med. 2002;2:381–92.
Bazil DP, Park J, Hemmings BA. PKB binding proteins: getting in on the Akt. Cell. 2002;111:293–303.
Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25.
Becker AJ, Chen J, Zien A, Sochivko D, Normann S, Schramm J, et al. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2003;18:2792–802.
Blumcke I, Beck H, Lie AA, Wiestler OD. Molecular neuropathology of human mesial temporal lobe epilepsy. Epilepsy Res. 1999;36:205–23.
Borsello T, Forloni G. JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des. 2007;13:1875–86.
Bozzi Y, Dunleavy M, Henshall DC. Cell signaling underlying epileptic behavior. Front Behav Neurosci. 2011;5:1–11.
Bratton SB, Salvesen GS. Regulation of the Apaf-1-caspase-9 apoptosome. J Cell Sci. 2010;123:3209–14.
Burgering PJC, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602.
Cendes F. Febrile seizures and mesial temporal sclerosis. Curr Opin Neurol. 2002;17:161–4.
Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40.
Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, et al. MAP kinases. Chem Rev. 2001;101:2449–76.
Cheung EC, Slack RS. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci STKE. 2004;2004(251):pe45.
Chiba T, Yamada M, Aiso S. Targeting the JAK2/STAT3 axis in Alzheimer’s disease. Expert Opin Ther Targets. 2009;13:1155–67.
Chipuk JE, Green DR. PUMA cooperates with direct activator proteins to promote mitochondrial outer membrane permeabilization and apoptosis. Cell Cycle. 2009;8:2692–6.
Choi YS, Lin SL, Lee B, Kurup P, Cho HY, Naegele JR, et al. Status epilepticus induced somatostatinergic hilar interneuron degeneration is regulated by striatal enriched protein tyrosine phosphatase. J Neurosci. 2007;27:2999–3009.
Choi YS, Cho HY, Hoyt KR, Naegele JR, Obrietan K. IGF-1 receptor-mediated ERK/MAPK signaling couples status epilepticus to progenitor cell proliferation in the subgranular layer of the dentate gyrus. Glia. 2008;56:791–800.
Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991;13:1401–9.
Cory S, Adams JM. The bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56.
Coulter DA, DeLorenzo RJ. Basic mechanisms of status epilepticus. Adv Neurol. 1999;79:725–33.
Covolan L, Ribeiro LT, Longo BM, Mello LE. Cell damage and neurogenesis in the dentate granule cell layer of adult rats after pilocarpine- or kainate-induced status epilepticus. Hippocampus. 2000;10:169–80.
D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;3(171):1–9.
Dalby NO, Mody I. The process of epileptogenesis: a pathophysiological approach. Curr Opin Neurol. 2001;14:187–92.
Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41.
De Lanerolle NC, Lee TS. New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav. 2005;7:190–203.
Debierre-Grockiego F. Anti-apoptotic role of STAT5 in haematopoietic cells and in the pathogenesis of malignancies. Apoptosis. 2004;9:717–28.
Detour J, Schroeder H, Desor D, Nehlig A. A 5-month period of epilepsy impairs spatial memory, decreases anxiety, but spares object recognition in the lithium-pilocarpine model in adult rats. Epilepsia. 2005;46:499–508.
Devinski O. Diagnosis and treatment of temporal lobe epilepsy. Rev Neurol Dis. 2004;1:2–9. http://ukpmc.ac.uk/abstract/MED/16397445.
Donovan N, Becker EB, Konishi Y, Bonni A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem. 2002;277:40944–9.
Dorr J, Bechmann I, Waiczies S, Aktas O, Walckzac H, Krammer PH, et al. Lack of tumor necrosis factor-related apoptosis-inducing ligand but presence of its receptors in human brain. J Neurosci. 2002;22:RC209.
Dudek SM, Fields RD. Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and mode of calcium entry. J Neurosci. 2001;21:RC122.
Engel J. Mesial temporal lobe epilepsy: what have we learned? Neuroscientist. 2001;7:340–52.
Engel T, Henshall DC. Apoptosis, Bcl-2 family proteins and caspases: the ABCs of seizure-damage and epileptogenesis? Int J Physiol Pathophysiol Pharmacol. 2009;8:267–71.
Engel T, Murphy BM, Hatazaki S, Jimenez-Mateos EM, Concannon CG, Woods I, et al. Reduced hippocampal damage and epileptic seizures after status epilepticus in mice lacking proapoptotic Puma. FASEB J. 2010;24:853–61.
French JA, Kanner AM, Bautista J, About- Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs I: Treatment of new onset epilepsy. Neurology. 2004; 62(8):1252–60.
Fujikawa DG. Neuroprotective strategies in status epilepticus. In: Wasterlain CG, Treiman DM, editors. Status epilepticus: mechanisms and management. Cambridge: MIT Press; 2006. p. 463–80.
Fujikawa DG, Shinmei SS, Cai B. Lithium-pilocarpine-induced status epilepticus produces necrotic neurons with internucleosomal DNA fragmentation in adults rats. Eur J Neurosci. 1999;11:1605–14.
Fujikawa DG, Shinmei SS, Cai B. Kainic acid-induced seizures produce necrotic, not apoptotic neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience. 2000;98:41–53.
Fujikawa DG, Ke X, Trinidad RB, Shinmei SS, Wu A. Caspase-3 is not activated in seizure-induced neuronal necrosis with internucleosomal DNA cleavage. J Neurochem. 2002;83:229–40.
Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, Lopes da Silva FH, et al. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26:11083–110.
Greene LA, Biswas SC, Liu DX. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 2004;11:49–60.
Guan QH, Pei DS, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139:609–27.
Henshall DC, Murphy BM. Modulators of neuronal cell death in epilepsy. Curr Opin Pharmacol. 2008;8:75–81.
Henshall DC, Araki T, Schindler CK, Lan J-Q, Tiekoter KL, Taki W, et al. Activation of Bcl-2-associated death protein and counter-response of Akt within cell populations during seizure-induced neuronal death. Neurology. 2000a;55:250–7.
Henshall DC, Clark RS, Adelson PD, Chen M, Simon RP, Watkins SC. Alterations in bcl2 and caspases gene family protein expression in human temporal lobe epilepsy. Neurology. 2000b;55:250–7.
Henshall DC, Bonislawski DP, Skradski SL, Meller R, Lan J-Q, Simon RP. Cleavage of Bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol Dis. 2001a;8:568–80.
Henshall DC, Bonislawski DP, Skradski SL, Araki T, Lan J-Q, Schindler CK, et al. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure induced neuronal death. Cell Death Differ. 2001b;8:1169–81.
Henshall DC, Skradski SL, Bonislawski DP, Lan JQ. Simon RP (2001c) Caspase-2 activation is redundant during seizure-induced neuronal death. J Neurochem. 2001c;77:886–95.
Houser CR, Huang CS, Peng Z. Dynamic seizure-related changes in extracellular signal-regulated kinase activation in a mouse model of temporal lobe epilepsy. Neuroscience. 2008;156:222–37.
Huang H, Tindal DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–87.
Hunsberger JG, Bennett AH, Selvanayagam E, Duman RS, Newton SS. Gene profiling the response to kainic acid induced seizures. Brain Res Mol Brain Res. 2005;141:95–112.
Ino H, Chiba T. Cyclin-dependent kinase 4 and Cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J Neurosci. 2001;21:6086–94. http://www.jneurosci.org/content/21/16/6086.short.
Jakubs K, Nanobashvili A, Bonde S, Ekdahl CT, Kokaia Z, Kokaia M, et al. Environment matters: synaptic properties of neurons born in the epileptic adult brain develop to reduce excitability. Neuron. 2006;52:1047–59.
Jakubs K, Bonde S, Iosif RE, Ekdahl CT, Kokaia Z, Kokaia M, et al. Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 2008;28:12477–88.
Jessberger S, Zhao C, Toni N, Clemenson Jr GD, Li Y, Gage FH. Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J Neurosci. 2007;27:9400–7.
Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, Achim CL. Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol. 2003;62:68–74.
Jung KH, Chu K, Lee ST, Kim J, Sinn DI, Kim JM, et al. Cyclooxygenase-2 inhibitor, celecoxib, inhibits the altered hippocampal neurogenesis with attenuation of spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neurobiol Dis. 2006;23:237–46.
Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, et al. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–53.
Kawasaki H, Morooka T, Shimohama S, Kimura J, Hirano T, Gotoh Y, Nishida E. Activation and involvement of p38 mitogen-activated protein kinase in glutamate-induced apoptosis in rat cerebelar granulle cells. J Biol Chem. 1997;272:18518–521.
Kretz A, Happold CJ, Marticke JK, Isenmann S. Erythropoietin promotes regeneration of adult CNS neurons via Jak2/Stat3 and PI3K/AKT pathway activation. Mol Cell Neurosci. 2005;29:569–79.
Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163.
Kyosseva SV. Mitogen-activated protein kinase signaling. Int Rev Neurobiol. 2004;59:201–20.
Li T, Lu C, Xia Z, Xiao B, Luo Y. Inhibition of caspase-8 attenuates neuronal death induced by limbic seizures in a cytochrome c-dependent and Smac/DIABLOindependent way. Brain Res. 2006;1098:204–11.
Liou AKF, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103–42.
Lopez-Meraz ML, Niquet J, Wasterlain CG. Distinct caspase pathways mediate necrosis and apoptosis in subpopulations of hippocampal neurons after status epilepticus. Epilepsia. 2010;51:56–60.
Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, et al. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. 2008;1:ra9.
Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–10.
Mathern GW, Cifuentes F, Leite JP, Pretorius JK, Babb TL. Hippocampal EEG excitability and chronic spontaneous seizures are associated with aberrant synaptic reorganization in the rat intrahippocampal kainate model. Electroencephalogr Clin Neurophysiol. 1993;87:326–39.
Mathern GW, Babb TL, Armstrong DL. Hippocampal sclerosis. In: Engel JJ, Pedley TA, editors. Epilepsy: a comprehensive textbook. Philadelphia: Lippincott-Raven Publishers; 1997. p. 133–55.
McCabe PH. New anti-epileptic drugs for the 21st century. Expert Opin Pharmacother. 2000;1:633–74.
Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, et al. Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010;6:e1000962.
Meldrum B. Excitotoxicity and epileptic brain damage. Epilepsy Res. 1991;10:55–61.
Meldrum B. Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 1993;3:405–12.
Meller R, Clayton C, Torrey DJ, Schindler CK, Lan JQ, Cameron JA, et al. Activation of the caspase 8 pathway mediates seizure-induced cell death in cultured hippocampal neurons. Epilepsy Res. 2006;70:3–14.
Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–69.
Morgan JI, Curran T. Proto-oncogene transcription factors and epilepsy. Trends Pharmacol Sci. 1991;12:459–62.
Mukherjee PK, Decoster M, Campbell FZ, Davis RJ, Bazan NG. Glutamate receptor signaling interplay modulates stress-sensitive mitogen-activated protein kinases and neuronal cell death. J Biol Chem. 1999;274:6493–8.
Nagy Z, Esiri MM. Neuronal cyclin expression in the hippocampus in temporal lobe epilepsy. Exp Neurol. 1998;150:240–7.
Narkilahti S, Pirttila TJ, Lukasiuk K, Tuunanen J, Pitkanen A. Expression and activation of caspase 3 following status epilepticus in the rat. Eur J Neurosci. 2003;18:1486–96.
Nigg A. Cyclin-dependent protein kinases: key regulation of the eukaryotic cell cycle. Bioessays. 1995;17:471–80.
Niquet J, Auvin S, Archie M, Seo DW, Allen S, Sankar R, et al. Status epilepticus triggers caspase-3 activation and necrosis in the immature rat brain. Epilepsia. 2007;48:1203–6.
Okamoto OK, Janjoppi L, Bonone FM, Pansani AP, Da Silva AV, Scorza FA, et al. Whole transcriptome analysis of the hippocampus: toward a molecular portrait of epileptogenesis. BMC Genomics. 2010;11:230.
Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24(36):7879–87.
Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–65.
Overstreet-Wadiche LS, Bromberg DA, Bensen AL, Westbrook GL. Seizures accelerate functional integration of adult-generated granule cells. J Neurosci. 2006;26:4095–103.
Parent JM, Janumpalli S, McNamara JO, Lowenstein DH. Increased dentate granule cell neurogenesis following amygdala kindling in the adult rat. Neurosci Lett. 1998;247:9–12.
Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, Lowenstein DH. Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann Neurol. 2006;59:81–91.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22(2):153–83.
Pickering M, Cumiskey D, O’Connor JJ. Actions of TNFalpha on glutamatergic synaptic transmission in the central nervous system. Exp Physiol. 2005;90:663–70.
Pitkänen A, Schwartzkroin P, Moshé S. Models of seizures and epilepsy. 1st ed. Burlington, MA: Academic; 2005.
Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914.
Radley JJ, Jacobs BL. Pilocarpine-induced status epilepticus increases cell proliferation in the dentate gyrus of adult rats via a 5-HT1A receptor-dependent mechanism. Brain Res. 2003;966:1–12.
Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, et al. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:7–67.
Riazi K, Galic MA, Pittman QJ. Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res. 2010;89:34–42.
Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–50.
Salvesen GS, Riedl SJ. Programmed cell death in cancer progression and therapy. Adv Exp Med Biol. 2007;615:13–23.
Sastry PS, Rao KS. Apoptosis and the nervous system. J Neurochem. 2000;74(1):1–20.
Scharfman HE, Goodman JH, Sollas AL. Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure induced neurogenesis. J Neurosci. 2000;20:6144–58.
Schindler CK, Pearson EG, Bonner HP, So NK, Simon RP, Prehn JH, et al. Caspase-3 cleavage and nuclear localization of caspase-activated DNase in human temporal lobe epilepsy. J Cereb Blood Flow Metab. 2006;26:583–9.
Scott BW, Wang S, Burnham WM, De BU, Wojtowicz JM. Kindling-induced neurogenesis in the dentate gyrus of the rat. Neurosci Lett. 1998;248:73–6.
Shinoda S, Skradski SL, Araki T, Schindler CK, Meller R, Lan J-Q. Formation of a tumour necrosis factor receptor 1 molecular scaffolding complex and activation of apoptosis signal-regulating kinase 1 during seizure-induced neuronal death. Eur J Neurosci. 2003;17:2065–76.
Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, et al. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J Clin Invest. 2004;113:1059–68.
Silva M, Benito A, Sanz C. Erythropoietin can induce the expression of bcl-x(L) through Stat5 in erythropoietin-dependent progenitor cell lines. J Biol Chem. 1999;274:22165–9.
Sloviter RS. Status epilepticus-induced neuronal injury and network reorganization. Epilepsia. 1999;40:s34–9.
Sloviter RS. Hippocampal epileptogenesis in animal models of mesial temporal lobe epilepsy with hippocampal sclerosis: the importance of the “latent period” and other concepts. Epilepsia. 2008;49:85–92.
Smolders I, Khan GM, Manil J, Ebinger G, Michotte Y. NMDA receptor-mediated pilocarpine-induced seizures: characterization in freely moving rats by microdialysis. Br J Pharmacol. 2009;121:1171–9.
Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9:59–71.
Stavrovskaya IG, Kristal BS. The powerhouse takes control of the cell: is the mitochondrial permeability transition a viable therapeutic target against neuronal dysfunction and death? Free Radic Biol Med. 2005;38:687–97.
Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci. 2005;25(12):3219–28.
Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–45.
Sun W, Gould TW, Newbern J, Milligan C, Choi SY, Kim H, et al. Phosphorylation of c-Jun in avian and mammalian motoneurons in vivo during programmed cell death: an early reversible event in the apoptotic cascade. J Neurosci. 2005;25:5595–603.
Sutula TP. Mechanisms of epilepsy progression: current theories and perspectives from neuroplasticity in adulthood and development. Epilepsy Res. 2004;60:161–71.
Thom M, Martinian L, Williams G, Stoeber K, Sisodiya SM. Cell proliferation and granule cell dispersion in human hippocampal sclerosis. J Neuropathol Exp Neurol. 2005;64:194–201.
Timsit S, Menn B. Cerebral ischemia, cell cycle elements and Cdk5. Biotechnol J. 2007;2:958–66.
Turski L, Ikonomidou CH, Turski WA, Bortolotto ZA, Cavalheiro EA. Cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse. 1989;3:154–71.
Vezzani A, Balosso S, Ravizza T. The role of cytokines in the pathophysiology of epilepsy. Brain Behav Immun. 2008;22:797–803.
Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296(5573):1635–6.
Walker TL, White A, Black DM, Wallace RH, Sah P, Bartlett PF. Latent stem and progenitor cells in the hippocampus are activated by neural excitation. J Neurosci. 2008;28:5240–7.
Wang W, Yang Y, Ying C, Li W, Ruan H, Zhu X, et al. Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology. 2007;52:1678–84.
Weise J, Engelhorn T, Dorfler A, Aker S, Bahr M, Hufnagel A. Expression time course and spatial distribution of activated caspase-3 after experimental status epilepticus: contribution of delayed neuronal cell death to seizure-induced neuronal injury. Neurobiol Dis. 2005;18:582–90.
Wessel TC, Joh TH, Volpe BT. In situ hybridization analysis of c-fos and c-jun expression in the rat brain following transient forebrain ischemia. Brain Res. 1991;567:231–40.
Wieser HG. ILAE Commission report. Mesial temporal lobe epilepsy with hippocampal sclerosis. Epilepsia. 2004;45:695–714.
Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115(10):2656–64.
Yadav A, Kalita A, Dhillon S, Banerjee K. JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem. 2005;280:31830–40.
Yamaoka K, Saharinen P, Pesu M, Holt V, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5:253.
Yang DD, Kuan CY, Withmarsh AJ, Rincon M, Zheng TS, Davis RJ, et al. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the jnk3 gene. Nature. 1997;389:865–70.
Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21:2661–8.
Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurosci. 2003;23:2557–63.
Yeste-Velasco M, Folch J, Pallàs M, Camins A. The p38(MAPK) signaling pathway regulates neuronal apoptosis through the phosphorylation of the retinoblastoma protein. Neurochem Int. 2009;54:99–105.
Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse celular functions. Growth Factors. 2006;24:21–44.
Acknowledgements
This work was partially supported by grants: CYTED (http://www.cyted.org/) through Group study of neuroscience Iberoamerican Network and CONACyT 00177594 to CBZ.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Rivera-Cervantes, M., Feria-Velasco, A.I., Junyent, F., Espuny, A.C., Beas-Zárate, C. (2013). Intracellular Pathways Associated with Neuronal Survival and Death in Epilepsy. In: Rocha, L., Cavalheiro, E. (eds) Pharmacoresistance in Epilepsy. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6464-8_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6464-8_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6463-1
Online ISBN: 978-1-4614-6464-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)