
Abstract
Twenty years ago marks a fascinating beginning of the new fledgling life of a novel signaling cascade—the Hippo Pathway. Today we are still grasping to understand its biological context and scrabbling to find new modulators of the pathway. Research is in full swing, and does not show any signs of slowing down in the foreseeable future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Twenty years ago marks a fascinating beginning of new fledgling life of a novel signaling cascade—the Hippo Pathway. Today we are still grasping to understand its biological context and scrabbling to find new modulators of the pathway. Research is in full swing, and does not show signs of slowing down in the foreseeable future.
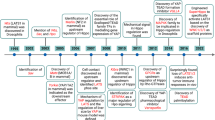
The founding member of the Hippo pathway was Yap (YES-associated protein), first described in 1994 (Sudol 1994). As its name implies, Yap cDNA was isolated from a lambda phage expression library in a screen for proteins that bind to the Yes kinase (Sudol 1994). Sequence comparison between Yap proteins of different organisms revealed a new protein module; the WW domain. Subsequently, using a cDNA expression library, the WW domain of Yap was found to bind proline-rich peptides (Sudol et al. 1995). These motifs were to become linchpins of Hippo interactions.
key to the ensuing discovery of additional Hippo components has been the rewarding exchange between mammalian and fly researchers (fruitful fruit flies!). Mosaic screens in Drosophila have facilitated the isolation of hyperproliferation mutations that are lethal at earlier developmental stages, since clusters of somatic cells mutated in genes that encode negative regulators of cell proliferation were easily detected. By 1995, more than 22 putative fly “tumor suppressor” genes had been cloned and characterized at the molecular level (Watson et al. 1994), four of which functioned in imaginal discs. These four were homologous to human genes: fat, a gene encoding a large cadherin-like transmembrane molecule involved in cell adhesion (Mahoney et al. 1991); discs-large (dlg), encoding an SH3-containing kinase localized to cell junctions (Woods and Bryant 1991); lethal2giant larvae (lgl), localized either in the cytoplasm or in association with cell membranes at sites of cell–cell contact (Strand et al. 1994); and expanded (exp), the homolog of the mammalian NF2 tumor suppressor gene (Boedigheimer et al. 1993), which encodes a membrane-cytoskeleton linker protein. This is interesting not only because loss of apical–basal polarity and cell contact inhibition are critical attributes driving epithelial tumor progression but also because each of the proteins encoded by these genes was ultimately implicated in signaling to the Hippo pathway (Grusche et al. 2010).
dLats (Large tumor suppressor or warts, wts) is an archetypal example of a hippo-related tumor suppressor isolated in a Drosophila screen. dLats was identified in 1995 in two independent screens for recessive hyperproliferation mutations (Justice et al. 1995; Xu et al. 1995). It was recognized as a member of the NDR family of kinases and was subsequently the first ser/thr kinase demonstrated to negatively regulate cell cycle (Xu et al. 1995). Loss of dLats caused a massive hyperproliferation phenotype with enlarged adult somatic structures (Xu et al. 1995). Mutant flies also exhibited apical hypertrophy of epithelial cells, leading to abnormal deposition of extracellular matrix during adult development (“warts”) (Justice et al. 1995). Proline-rich regions were identified in the N-terminus of the protein. In retrospect, this is evocative of the PP domain that had been highlighted previously by Sudol within the Yap-binding context (Sudol 1994).
Mammals harbor two dLats orthologs, Lats1 and Lats2. Mice lacking Lats1 develop soft-tissue sarcomas and ovarian stromal tumors with 100% penetrance, and are highly sensitive to exposure to carcinogens (St John et al. 1999). This was an important milestone, since it indisputably pegged Lats as a tumor suppressor.
In 2000, Taz (Transcriptional co-activator with PDZ-binding motif) was isolated in a cDNA screen for novel 14-3-3-binding proteins (Kanai et al. 2000). BLAST analysis revealed significant sequence homology and domain conservation with the Hippo component Yap (Both Yap and Taz contain WW domains, although Yap does not harbor a PDZ domain and Taz does not harbor a Yes-binding SH3 domain). The transcriptional co-activation function of Taz was dependent on its C-terminal PDZ domain. This was intriguing, since PDZ domains historically have been found in membrane-associated signal transduction molecules, such as the tight junction protein ZO1. Consistent with this, Taz could be found at the plasma membrane, as well as in punctate nuclear foci. The authors foresaw that competition between PDZ domain-mediated membrane and nuclear targeting, along with phosphorylation-dependent 14-3-3 binding and cytoplasmic sequestration, might provide a mechanism for spatial control of Taz function (Kanai et al. 2000).
Meanwhile, work on dissecting the transcriptional function of Yap bustled on. Yap was shown to function as a coactivator for a number of transcription factors, such as the Runx family member PEBP2a (Yagi et al. 1999) and p73 (Strano et al. 2001). Runx family members play an important role in regulating mesenchymal stem cell differentiation during bone formation (Lian et al. 2004). Yap and p73, a well-known member of the p53 family, act together in a feed-forward circuit to drive apoptosis (Basu et al. 2003; Lapi et al. 2008; Levy et al. 2007). The above interactions were mediated by the WW domains of Yap and the PPxY motifs of PEBP2a and p73. more recent work has highlighted the direct interaction of Yap and Taz with the four TEAD/TEF family transcription factors, which mediate Yap- and Taz-dependent tissue growth and progenitor cell expansion (Vassilev et al. 2001). Interestingly, this binding was independent of the Yap and Taz WW domains (Chen et al. 2010; Li et al. 2010; Tian et al. 2010).
In another Drosophila screen, akin to that of Xu et al. described above, Tapon (2002) identified mutations in Salvador (sav, named after the surrealist painter Salvador Dali who, while alive, claimed to be immortal). Concurrently, in a genetic screen to identify mutations that affected Drosophila eye size, the group of Georg Halder isolated the same gene and called it shar-pei (Kango-Singh et al. 2002). Sav protein contained two WW domains that were necessary for its interaction with dLats (Tapon et al. 2002). Similar to dLats, sav mutant cells proliferated more than their wild-type counterparts. Although tissue patterning appeared unaffected, an excess of a subtype of cells whose number was normally “pruned” by apoptosis was suggestive of a defect in cell death. These similarities, as well as a physical interaction, led the authors to postulate that sav and dLats may work epistatically. Complicating this possibility, however, was the fact that double mutants exhibited more severe phenotypes than either single mutant, suggesting that the two genes did not work in a simple linear manner.
In 2003, hippo (hpo), a ser/thr kinase and ortholog of Mst1/2, was identified by no less than five independent groups: four by genetic mosaic screens (similar to those described above) for mutants exhibiting hyperproliferation (Udan et al. 2003; Wu et al. 2003; Harvey et al. 2003; Jia et al. 2003) and one in a yeast two hybrid screen using sav as bait (Pantalacci et al. 2003). Reminiscent of dLats and sav, hpo mutants displayed high levels of cyclin E which drives cell proliferation, as well as increased dIAP1, an inhibitor of apoptosis. Hpo physically bound sav, which in turn interacted with dLats, suggesting that the three proteins functioned as a complex to negatively regulate cell proliferation. The trio was postulated to act via transcriptional repression of cyclin E and dIAP1, by unknown mechanisms. This new “Hippo” complex had only a handful of identified kinase substrates; a destabilizing phosphorylation of dIAP1 (Tapon et al. 2002; Harvey et al. 2003; Pantalacci et al. 2003), the G2/M regulator cdc2 (Tapon et al. 2002; Tao et al. 1999) and actin regulators zyxin and LIMK1 (Hirota et al. 2000; Yang et al. 2004). Furthermore, sav was shown to be a target of hpo kinase, and sav and hpo, jointly, promoted phosphorylation of dLats (Wu et al. 2003; Pantalacci et al. 2003; Chan et al. 2005). although the “Hippo” complex clearly affected transcriptional levels of cyclin E and dIAP1, nothing was known about how the pertinent signals were transduced into the nucleus and/or integrated with other transcriptional programs.
In 2005 the pieces started to come together. Implementing a yeast two hybrid screen using the N-terminus of dLats as bait, Duojia Pan’s group (Huang et al. 2005) identified yorkie (yki), the fly ortholog of Yap, as a critical target of the Lats kinase. Accordingly, yki was required for dIAP1 transcription, whereas overexpression of yki phenocopied loss-of-function mutations of hpo, sav, and dLats (Huang et al. 2005). Thus, yki was the first substrate identified for the Hippo pathway, and, more broadly, for any of the NDR kinases. The authors also noted that NDR kinases are often regulated by a family of proteins called Mob. Congruently, in Drosophila, the Mob family protein Mats was identified as a tumor suppressor putatively regulating Lats in the Hippo signaling pathway (Lai et al. 2005).
Subsequent work reinforced the notion that the “canonical” mechanism of Hippo regulation is via cell–cell contact. In tissue culture, high cell density induced phosphorylation, cytoplasmic translocation, (Zhao et al. 2007) and rapid degradation (Zhao et al. 2010) of Yap. Accordingly, disruption of cell junctions in epithelial cells resulted in the nuclear localization of both Yap and Taz (Varelas et al. 2010).
Thus, the Hippo signaling pathway was born—but where are we now?
The study of Hippo function continues to be enormously exciting and persistently surprising. The authors of the chapters in this book are at the cutting edge of the Hippo field. We will allow their chapters to speak for themselves.
Lest we be lulled into the complacent opinion that the Hippo pathway has been “deciphered,” let us remember that while a coherent conception of Hippo functioning is now emerging, additional evidence of more complex networks of interactions is also discernible. Illustrating just one of many examples, even at the time of placing Mst as the central Hippo kinase, data had already accumulated of seemingly Hippo-unrelated functions of Mst kinases. Mst1 and 2 had been described as MAPKKK kinases that incite c-Jun, p38 and caspase activation (Graves et al. 1998). Once activated, caspase 6/7 cleaves Mst1 (whereas caspase 3 cleaves Mst2) (Feig and Buchsbaum 2002), creating a constitutively activated kinase that is transported into the nucleus to phosphorylate histone H2B and potentiate apoptotic chromatin condensation (Cheung et al. 2003). The new Hippo pathway added another level of complexity to this preexisting story. The cleaved portion of Mst harbors a SARAH (SAlvador-RAssf-Hpo binding) motif, which keeps the Mst pro-apoptotic function in check. In response to oncogene activation, for instance, the Hippo components sav and Rassf1a displace inhibitory Raf1, thereby activating an apoptotic Mst-Lats kinase signaling cascade (O’Neill and Kolch 2005).
Evidence for the involvement of subpopulations of Hippo components in non-Yap/Taz effector outcomes continues to crop up. Most of these processes have been less “neatly” resolved than the above Mst story. In fact, the first description of an in vivo upstream activating signal (in this case, DNA damage) of the Hippo pathway actually involved dmp53, the fly ortholog of the p53 tumor suppressor (Colombani et al. 2006). Similarly to its mammalian counterpart, dmp53 mediates the DNA damage response in the fly. Importantly, Hpo signaling is required for a maximal dmp53 response. In turn, Hpo kinase activity is activated in a dmp53-dependent manner (Colombani et al. 2006). Concurrently, our laboratory uncovered a somewhat analogous feedback circuit in mammalian cells (Aylon et al. 2006). In the mammalian system, mitotic or oncogenic stress causes Lats2 to translocate from the centrosome to the nucleus. In the nucleus, Lats2 binds the negative regulator of p53, Mdm2, leading to inhibition of p53 degradation and induction of a p53-driven transcriptional response. Since the Lats2 gene itself is directly transcriptionally activated by p53, this leads to a gradual and continuous increase in Lats2 protein levels.
The mention of p53 is not coincidental. p53 is historically one of the most studied tumor suppressor genes, making it the prototypic tumor suppressor. p53 was identified in 1979 by four independent laboratories (Lane and Crawford 1979; Linzer and Levine 1979; DeLeo et al. 1979; Kress et al. 1979). Following its discovery, p53 has evolved from an obscure molecule to a key tumor suppressor gene with potentially great clinical impact. In many ways, the p53 pathway is long considered to have come of age. In this analogy, the Hippo pathway is still a toddler, but perhaps we can learn from the flip-flopped evolution of our concept of p53 tumor suppressor function and apply similar principles to the burgeoning Hippo pathway. Three major factors have contributed to the overwhelming success of p53 research; (1) reliable working “tools” and infrastructure; (2) recognition of cross-talk with other pathways and (3) clinical relevance.
How do these attributes apply to the Hippo pathway? For tools, we need a battery of reliable and sensitive measuring and detection methods; good antibodies, strong mouse models, identification of a robust list of target genes; good database infrastructure to make information accessible and interchangeable to all researchers. We need to continue to meet at conferences, talk, discuss, exchange reagents, and ideas.
As to recognition of cross-talk with other pathways, our understanding of the intricacies of cell signaling begins on the single molecule level. Genetic and physical interactions develop into pathways, which subsequently evolve into cellular networks. But even networks do not function in a vacuum. Cell fate decisions are the sum total of innumerous signaling inputs and outputs, the weight of each signal being determined (among many other factors) by cell density, cell type, developmental stage, neighboring cells, and whether those cells are normal or transformed. Complicating the “untangling” of distinct networks is the fact that adult organisms often reuse signaling cassettes that were previously used for different purposes earlier in development. Furthermore, miswiring or hijacking of pathway members from diverse networks is often associated with severe diseases, such as cancer. From the Hippo perspective, different cells have distinct modifications of hippo function and output. One of the most glaring examples of this is the ability of Yap to promote tremendously diverse cellular outcomes such as apoptosis, cell growth, or “stemness.” Whereas, on a broad level, this is suggestive of fail-safe mechanisms to check and limit the oncogenic potential of Yap-TEAD, it also implies a complex interaction between cellular signaling pathways.
As for clinical relevance, let us keep in mind that model organisms are just, well, model organisms. The exchange of information between Drosophila and mammalian systems has been very rewarding. However, care needs to be exercised against hasty analogies, since mammals are not merely wingless flies. Human genomics, such as identifying single nucleotide polymorphisms, copy number variations, and somatic mutations, are becoming more and more mainstream. Although confronting the genetic variations among humans is more “inconvenient” than working with inbred strains of model organisms, an immense advantage of humans is the detailed phenotypes that can be followed in clinical records. Comprehensive records of patient outcome, together with detailed genetic information, are rapidly being assembled in central facilities. Accessible and user-friendly databases will be critical for human-as-an-ultimate-model-organism Hippo researchers.
With this high-throughput vision in place, as well as the functions of the Hippo pathway expanding, and considering its central role in tumorigenesis and development, the opportunities for drug development increase. Drugs that disrupt Yap-TEAD binding (Verteporfin), Taz-TEAD binding (TM-25659), or Yap nuclear translocation (Dobutamine) are already available (Jang et al. 2012; Liu-Chittenden et al. 2012; Bao et al. 2011). Conceivably, negative modulators of Hippo function are also potential drug targets. For instance, the PP2A phosphatase complex, an antagonist of Hpo (Ribeiro et al. 2010), is targeted by Fostriecin, which entered phase I clinical studies in 2002 as a cancer-killing agent (Lewy et al. 2002). Another exciting approach will be the search for targets in pathways that show synthetic lethality with either loss of Hippo function or excessive Yap/Taz function. Clinical applications are important not only because they save lives but also because they provide glimpses of the complex modes of action of molecules and pathways within a holistic human context.
We hope that by bringing together contributions from many leading experts, this volume will provide a great introduction to the field for newcomers to the Hippo pathway, as well as a starting point for vigorous debate among the already converted. The many unknowns in this system, detailed and discussed exquisitely in this volume, provide us all with inspiration for future work.
We are enormously indebted to the team of authors who took a timeout from their ongoing investigations to consider their work in a broader context and share it with us all, in true Hippo spirit!
References
Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006;20(19):2687–700.
Bao Y, Nakagawa K, Yang Z, Ikeda M, Withanage K, Ishigami-Yuasa M, et al. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J Biochem. 2011;150(2):199–208.
Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell. 2003;11(1):11–23.
Boedigheimer M, Bryant P, Laughon A. Expanded, a negative regulator of cell proliferation in Drosophila, shows homology to the NF2 tumor suppressor. Mech Dev. 1993;44(2–3):83–4.
Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24(12):2076–86.
Chen L, Chan SW, Zhang X, Walsh M, Lim CJ, Hong W, et al. Structural basis of YAP recognition by TEAD4 in the hippo pathway. Genes Dev. 2010;24(3):290–300.
Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell. 2003;113(4):507–17.
Colombani J, Polesello C, Josue F, Tapon N. Dmp53 activates the Hippo pathway to promote cell death in response to DNA damage. Curr Biol. 2006;16(14):1453–8.
DeLeo AB, Jay G, Appella E, Dubois GC, Law LW, Old LJ. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proc Natl Acad Sci U S A. 1979;76(5):2420–4.
Feig LA, Buchsbaum RJ. Cell signaling: life or death decisions of ras proteins. Curr Biol. 2002;12(7):R259–61.
Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, et al. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 1998;17(8):2224–34.
Grusche FA, Richardson HE, Harvey KF. Upstream regulation of the hippo size control pathway. Curr Biol. 2010;20(13):R574–82.
Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell. 2003;114(4):457–67.
Hirota T, Morisaki T, Nishiyama Y, Marumoto T, Tada K, Hara T, et al. Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor suppressor. J Cell Biol. 2000;149(5):1073–86.
Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122(3):421–34.
Jang EJ, Jeong H, Kang JO, Kim NJ, Kim MS, Choi SH, et al. TM-25659 enhances osteogenic differentiation and suppresses adipogenic differentiation by modulating the transcriptional co-activator TAZ. Br J Pharmacol. 2012;165(5):1584–94.
Jia J, Zhang W, Wang B, Trinko R, Jiang J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 2003;17(20):2514–9.
Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995;9(5):534–46.
Kanai F, Marignani PA, Sarbassova D, Yagi R, Hall RA, Donowitz M, et al. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 2000;19(24):6778–91.
Kango-Singh M, Nolo R, Tao C, Verstreken P, Hiesinger PR, Bellen HJ, et al. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development. 2002;129(24):5719–30.
Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol. 1979;31(2):472–83.
Lai ZC, Wei X, Shimizu T, Ramos E, Rohrbaugh M, Nikolaidis N, et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120(5):675–85.
Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278(5701):261–3.
Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell. 2008;32(6):803–14.
Levy D, Adamovich Y, Reuven N, Shaul Y. The Yes-associated protein 1 stabilizes p73 by preventing Itch-mediated ubiquitination of p73. Cell Death Differ. 2007;14(4):743–51.
Lewy DS, Gauss CM, Soenen DR, Boger DL. Fostriecin: chemistry and biology. Curr Med Chem. 2002;9(22):2005–32.
Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, et al. Structural insights into the YAP and TEAD complex. Genes Dev. 2010;24(3):235–40.
Lian JB, Javed A, Zaidi SK, Lengner C, Montecino M, van Wijnen AJ, et al. Regulatory controls for osteoblast growth and differentiation: role of Runx/Cbfa/AML factors. Crit Rev Eukaryot Gene Expr. 2004;14(1–2):1–41.
Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17(1):43–52.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300–5.
Mahoney PA, Weber U, Onofrechuk P, Biessmann H, Bryant PJ, Goodman CS. The fat tumor suppressor gene in Drosophila encodes a novel member of the cadherin gene superfamily. Cell. 1991;67(5):853–68.
O’Neill E, Kolch W. Taming the Hippo: Raf-1 controls apoptosis by suppressing MST2/Hippo. Cell Cycle. 2005;4(3):365–7.
Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol. 2003;5(10):921–7.
Ribeiro PS, Josue F, Wepf A, Wehr MC, Rinner O, Kelly G, et al. Combined functional genomic and proteomic approaches identify a PP2A complex as a negative regulator of Hippo signaling. Mol Cell. 2010;39(4):521–34.
St John MA, Tao W, Fei X, Fukumoto R, Carcangiu ML, Brownstein DG, et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet. 1999;21(2):182–6.
Strand D, Raska I, Mechler BM. The Drosophila lethal(2) giant larvae tumor suppressor protein is a component of the cytoskeleton. J Cell Biol. 1994;127(5):1345–60.
Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276(18):15164–73.
Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9(8):2145–52.
Sudol M, Bork P, Einbond A, Kastury K, Druck T, Negrini M, et al. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J Biol Chem. 1995;270(24):14733–41.
Tao W, Zhang S, Turenchalk GS, Stewart RA, St John MA, Chen W, et al. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet. 1999;21(2):177–81.
Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber DA, et al. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell. 2002;110(4):467–78.
Tian W, Yu J, Tomchick DR, Pan D, Luo X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc Natl Acad Sci U S A. 2010;107(16):7293–8.
Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol. 2003;5(10):914–20.
Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell. 2010;19(6):831–44.
Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15(10):1229–41.
Watson KL, Justice RW, Bryant PJ. Drosophila in cancer research: the first fifty tumor suppressor genes. J Cell Sci Suppl. 1994;18:19–33.
Woods DF, Bryant PJ. The discs-large tumor suppressor gene of Drosophila encodes a guanylate kinase homolog localized at septate junctions. Cell. 1991;66(3):451–64.
Wu S, Huang J, Dong J, Pan D. Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114(4):445–56.
Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development. 1995;121(4):1053–63.
Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18(9):2551–62.
Yang X, Yu K, Hao Y, Li DM, Stewart R, Insogna KL, et al. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6(7):609–17.
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61.
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24(1):72–85.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Oren, M., Aylon, Y. (2013). Introduction. In: Oren, M., Aylon, Y. (eds) The Hippo Signaling Pathway and Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6220-0_1
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6220-0_1
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6219-4
Online ISBN: 978-1-4614-6220-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)