
Abstract
Early epidemiological studies on sporadic Creutzfeldt–Jakob disease did not identify blood transfusion as a risk factor for the disease. However, the emergence of variant Creutzfeldt–Jakob disease (vCJD) in 1996 and the identification of PrPSc in lymphoid tissues in this novel disorder led to concerns that transmission of infectivity by blood transfusion might be a possibility. These concerns were fully realised in 2004, when the first case of vCJD associated with transmission by blood transfusion was identified in a recipient who was a methionine homozygote at codon 129 in the prion protein gene, as in all other vCJD patients. Other similar cases have subsequently emerged, along with cases of asymptomatic vCJD infection in a blood transfusion recipient and a plasma product recipient, both of whom were heterozygous at codon 129 of the prion protein gene. This chapter reviews the experimental evidence for the transmission of prion infectivity by blood transfusion in a range of experimental models, discusses the evidence for the transmission of vCJD by blood transfusion and plasma products and considers the future possibilities for the development and potential uses of blood-based screening tests for human prion diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
8.1 Introduction
Despite several decades of research in many different countries, the cause of the commonest form of human prion disease, sporadic Creutzfeldt–Jakob disease (sCJD), remains unclear. sCJD appears to have been transmitted as an iatrogenic infection following a variety of medical and surgical procedures, but evidence to support infection via blood transfusion appears lacking to date. The emergence of variant Creutzfeldt–Jakob disease (vCJD) in the UK 15 years ago and subsequent evidence for the transmission of vCJD infectivity by the transfusion of non-leucodepleted red cell concentrates from donors who were asymptomatic at the time of donation, but who subsequently died from vCJD, have focused attention on the potential for transmission of other forms of CJD by this route (Puopolo et al. 2011).
In this chapter, we review the evidence for the transmission of prions by blood transfusion in experimental models of prion disease and in sCJD and vCJD in humans, describe recent and developing methods to detect prions in blood and discuss the prospects of a blood screening test for prions and the issues surrounding the implementation of such a test.
8.2 Experimental Evidence for Prion Disease Transmission by Blood Transfusion
8.2.1 Cellular Prion Protein in Blood
Expression of the cellular prion protein (PrPC) is thought to be an absolute requirement for the development of prion infection. PrPC is widely expressed in different tissues and cell types, including neurones in the central nervous system and follicular dendritic cells in lymphoreticular tissues. It is also present in blood, in which the distribution and cellular physiology of PrPC has been intensively studied. PrPC is present in plasma and is also found to be cell associated in human blood (MacGregor et al. 1999). Platelets contribute the greatest amount of cell-associated PrPC to blood with lesser amounts contributed by white blood cells (WBC) and lower levels still by red blood cells (RBC) (MacGregor et al. 1999; Choi et al. 2009). The highest levels of PrPC (on a per cell basis) in normal human blood are in specific WBC subpopulations (MacGregor et al. 1999; Durig et al. 2000; Choi et al. 2009). Platelets act as a dynamic reservoir for PrPC in that it is stored in their α-granules, being recruited to the cell surface or released during platelet activation and storage (Perini et al. 1996; MacGregor et al. 1999; Bessos et al. 2001; Holada et al. 2002a). The activation-dependent upregulation of expression in, or release of PrPC from leucocytes, dendritic cells, and mast cells, has been interpreted as indicative of the normal cellular functions for PrPC in blood and suggestive of a role for these cells in prion disease pathogenesis (Durig et al. 2000; Burthem et al. 2001; Lee et al. 2001; Haddon et al. 2009). In so far as PrPC expression and function in blood might relate to prion disease pathogenesis, it should be noted that clear differences in PrPC expression between different blood components are evident when human blood is compared with blood of species that are commonly used as models of prion disease, such as rodents and sheep (Barclay et al. 2002).
8.2.2 Animal Models
The study of human prion diseases, such as CJD, continues to be informed by analogous diseases of animals, specifically sheep scrapie and bovine spongiform encephalopathy (BSE) and the establishment of experimental animal models of those animal diseases and of the human diseases themselves. The adaptation of sheep scrapie isolates to rodents has been of fundamental significance to the field, providing a series of well-characterised meta-stable strains in both hamsters and mice, but the modelling of blood transfusion has recently been particularly well served by the development of an experimental blood transfusion paradigm using the BSE agent experimentally transmitted to sheep.
8.2.3 Rodent Models
Reports of the existence of a “viraemia” associated with prion disease have a long history and quite naturally these observations raised fears of transfusion-related transmission of CJD. Guinea pig-adapted CJD, serially transmitted by intracerebral (i.c.) inoculation was reported to have infectivity detectable throughout the incubation period in buffy coat samples, as determined by further i.c. challenge (Manuelidis et al. 1978). This finding was supported by a study using a different human prion disease, a mouse-adapted Gerstmann–Straussler–Scheinker disease isolate, termed Fukuoka-1. When challenged with Fukuoka-1 by the i.c route, mice showed detectable infectivity in circulating whole blood from around half way through the incubation period onwards, as determined by intraperitoneal (i.p.) challenge of further susceptible mice (Kuroda et al. 1983). Direct (but limited and poorly documented) testing of blood and buffy coat specimens from CJD patients also indicated the presence of infectivity in human blood during the clinical illness, when inoculated into guinea pigs and hamsters (Manuelidis et al. 1985; Deslys et al. 1994).
The further development of high titre, well-characterised rodent scrapie models has provided more consistent, reliable and perhaps more relevant data. A sustained low level of infectivity was found to characterise blood throughout the incubation period in the 263K hamster scrapie model, following i.p. inoculation (Diringer 1984; Casaccia et al. 1989). At the clinical stage, the infectivity was reported to be associated with the mononuclear leucocyte fraction and not with platelets (Holada et al. 2002b). The hamster 263K model has been used extensively in the development and evaluation of prion reduction filters (Gregori et al. 2004a, 2006a, b; Sowemimo-Coker et al. 2005, 2010) and to investigate partitioning during plasma protein manufacture (Lee et al. 2000; Foster et al. 2000; Li et al. 2001; Gregori et al. 2004b; Hartwell et al. 2005; Burdick et al. 2006).
Similar results to those obtained with the 263K scrapie strain hamster model have also been obtained using the Fukuoka-1 mouse model. Following i.c. inoculation, blood was found to contain ∼10 infectious units per ml (IU/ml) during the pre-clinical phase, rising to ∼100 IU/ml during the clinical phase and largely associated with the buffy coat fraction, as measured by bioassay using the same (i.c.) route (Brown et al. 1998, 1999). Infectivity levels in plasma were found to be low and further reduced by plasma processing (Brown et al. 1998, 1999). When comparisons were made between the blood-borne infectivity levels in the Fukuoka-1 GSS model and RIII mouse-adapted vCJD, the latter was found to contain 20–30 ID/ml at both the pre-clinical and clinical phase, primarily in buffy coat and plasma, with lower levels in platelets and no infectivity detectable in red blood cells (Cervenakova et al. 2003).
These experiments demonstrated clear proof of principle of blood-borne prion infectivity and they also provided information on infectivity levels, on which risk assessments could be based. However, direct extrapolation to blood transfusion and the risk posed by vCJD is difficult due to the possible effects of route and agent/host interaction. Consequently, the use of large animal models offers distinct advantages over rodents where blood transfusion is concerned.
8.2.4 Primate Models
Early attempts to transmit human spongiform encephalopathy by transfusion of unit quantities of blood to chimpanzees were reported to be negative (Brown et al. 1994). Nevertheless, non-human primates experimentally infected with the BSE/vCJD agent have been used to model vCJD (Lasmezas et al. 2001, 2005 Herzog et al. 2005; Williams et al. 2007). Both brain and buffy coat from a clinically affected lemur (previously exposed by the i.c. route) were found to transmit disease when inoculated i.c. into naive lemurs (Bons et al. 2002). Conversely, brain tissue from clinically affected macaques (previously exposed by the i.c. route) was shown to transmit disease when further macaques were exposed orally or intravenously (Herzog et al. 2004). Reported use of primate models to directly mimic transfusion practice has been surprisingly limited in scope and has recently been complicated by the finding of a novel myelopathic syndrome in macaques exposed intravenously (i.v.) to blood components from vCJD infected macaques and from a vCJD patient (Comoy et al. 2012; Lescoutra-Etchegaray et al. 2012).
8.2.5 Sheep Models
To date, only in sheep models have relevant agents (principally BSE) been used to infect animals using the relevant route (orally, to model zoonotic transmission to humans) to produce donors of blood (at clinical and pre-clinical time points) that can be used to transfuse recipients, using protocols that closely mimic human transfusion practice. The report of one successful transmission by intravenous administration of a unit of whole blood from a pre-clinical BSE orally exposed donor sheep to a naive recipient (Houston et al. 2000, and see an accompanying commentary by Brown 2000) was confirmed and has been fully justified by subsequent publications describing the whole study (Hunter et al. 2002; Siso et al. 2006; Houston et al. 2008; McCutcheon et al. 2011). The overall BSE transfusion transmission rate was 36% and included blood from donors throughout the second half of the (asymptomatic) incubation period, suggesting that either the titre of the infectious agent in blood is higher than anticipated or that transfusion of blood is a very efficient mode of transmission (Houston et al. 2008). Using the same experimental paradigm, components separated from orally exposed pre-clinical BSE sheep blood have shown infectivity to be present in red cell concentrates, plasma and platelet units, even when the blood has first been leucoreduced (McCutcheon et al. 2011). Interestingly, efficient transfusion transmission is not a property restricted to the BSE agent. Similar transmission rates (43%) were seen in parallel experiments conducted using clinical and pre-clinical sheep scrapie (Houston et al. 2008). The neuropathological phenotype of experimental ovine BSE is largely unaffected by route (Siso et al. 2006), whereas that of scrapie appears to differ between natural infection and transfusion transmission (Siso et al. 2009). The efficiency of transfusion mediated transmission has been further explored using a different sheep scrapie model system in which transfusion-mediated transmission rates approach 100% (Andreoletti et al. 2012; Lacroux et al. 2012). The results using this model system demonstrate a marked discrepancy between prion titres in sheep blood as defined by i.c. challenge of susceptible (ovinised) transgenic mice and the efficiency of disease transmission following intravenous transfusion of viable cells between sheep. This may not be surprising from a biological perspective, but it does provide an important caveat for calculations previously based on blood infectivity measurements obtained by i.c. inoculation of rodents (Andreoletti et al. 2012).
Each of the above rodent, primate and sheep experimental systems is at one or more removes from the events they seek to model, and not all of the evidence accumulated to date, such as the kinetics of accumulation or cell types involved, is entirely consistent. Titre is a key case in point. Rodent studies have previously supported an estimate of 10IU/ml of blood, whereas this has now been revised downwards to less than 1IU/unit of blood (∼400ml) based on examination of existing ovine and human data (Gregori et al. 2011). However, when taken together three conclusions can be drawn: first that low levels of infectivity in blood occur during the pre-clinical phase in these acquired prion diseases. Second, that some of this infectivity is cell associated, and third, that intravenous delivery, especially the transfusion of fresh blood and its components is an efficient mode of prion disease transmission.
8.3 Evidence for vCJD Transmission by Blood Transfusion and Plasma
8.3.1 Secondary Transmission of vCJD by Blood Transfusion
There have been four known cases of vCJD in recipients of blood components from asymptomatic donors who subsequently developed vCJD (Llewelyn et al. 2004; Peden et al. 2004; Wroe et al. 2006; Health Protection Agency 2007) and a fifth case in which only circumstantial evidence implicates blood transfusion as the cause (Chohan et al. 2010). These individuals were all members of a cohort identified by the Transfusion Medicine Epidemiology Review (TMER), a collaboration of the National CJD Research & Surveillance Unit (NCJDRSU) and the UK Blood Services (Hewitt et al. 2006; http://www.cjd.ed.ac.uk/TMER/TMER.htm). Figure 8.1 summarises information on the time of the relevant transfusions and the deaths or onsets of vCJD in both the donors and the recipients. In all four cases, secondary vCJD infection in the recipient appears to have resulted from the transfusion of a single unit of non-leucodepleted red cells from a pre-clinical vCJD donor. These transfusions occurred prior to the phasing in of leucodepletion of all blood for transfusion in the UK during 1998–1999; to date there have been no secondary vCJD cases in patients receiving leucodepleted blood.

Time lines for the donors and recipients of blood in the four known cases of blood transfusion-associated vCJD infection. The interval between donation/transfusion (circles) and death (square) or vCJD disease onset (triangles) are represented by lines drawn to a scale indicated at the top and bottom of the figure. The donations/transfusions are indicated by open symbols for the blood donors and filled symbols for the recipients. The recipients’ ages at the time of transfusion (where published) are shown. The asymptomatic haemophiliac patient that showed evidence of vCJD infection in the spleen had been a recipient of two identified vCJD-implicated batches of Factor VIII (indicated by hexagons). Key references are shown on the right; the data are also reviewed in Hewitt et al. (2006). Recipient 2 and the haemophiliac patient died of non-neurological disorders and recipients 3 and 4 shared a common donor
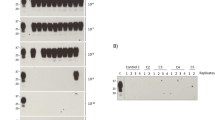
The clinical reports of recipients 1, 3 and 4 were typical for vCJD and genotype analysis showed they were all methionine homozygous (M/M) at codon 129 of the prion protein gene PRNP. All neuropathologically confirmed clinical cases of vCJD to date have also been homozygous for methionine. The neuropathological findings for recipients 1 and 3 were typical for vCJD (Head et al. 2009; Wroe et al. 2006) (Fig. 8.2a–d). In both of these recipients, Western blotting analysis of brain homogenate following treatment with proteinase K revealed the presence of disease-associated protease-resistant PrP (PrPres) with a banding pattern of type 2B, characteristic of vCJD.

Pathology and biochemistry of autopsy tissues from symptomatic and asymptomatic vCJD infected individuals following blood transfusion and plasma product administration. Haematoxylin and eosin stained sections of the cerebellum from blood donor 1 (A) and the corresponding blood transfusion recipient 1 (C) show spongiform change and florid plaques. Corresponding immunohistochemistry for PrP in sections of cerebral cortex (B for donor 1 and D for recipient 1) shows florid plaques, cluster plaques and other deposits of disease-associated PrP. E and F show PrP-labelling of germinal centres of the spleen (E) and the cervical lymph node (F) from the asymptomatic blood transfusion recipient (recipient 2). Panels G and H show the presence of protease resistant PrP by NaPTA/WB analysis in spleen from blood transfusion recipient 2 (marked r2) and the case of asymptomatic vCJD infection in a plasma product recipient with haemophilia (marked h). These samples have been run alongside spleen from a clinical case of vCJD (‘v’), non-CJD control spleen (‘c’) and vCJD brain homogenate alone (‘b’) or spiked into control spleen (‘c + b’) for comparison
Mice inoculated with cerebral frontal cortex samples from recipient 1 became infected with incubation times and brain lesion profiles that were consistent with previous transmissions of vCJD to mice of the same lines, suggesting that there had been no alteration of agent strain (Bishop et al. 2008). Therefore, in PRNP codon 129M/M individuals, the strain properties and clinicopathological features of secondary vCJD following blood transfusion are currently indistinguishable from those in patients with vCJD resulting from exposure to BSE.
The second case of blood transfusion-associated vCJD infection differed from the other three in that the transfused recipient (recipient 2) died 5 years after transfusion from a non-neurological disorder and was methionine/valine (M/V) heterozygous at PRNP codon 129 (Fig. 8.1) (Peden et al. 2004). Evidence for vCJD infection in this recipient was obtained when autopsy tissues were examined for the presence of PrPSc by sodium phosphotungstate precipitation/Western blotting (NaPTA/WB) (Fig. 8.2g), paraffin embedded tissue blotting (PET) and immunohistochemistry (IHC). PrPres was found to be restricted to the spleen (NaPTA/WB, PET and IHC) and a cervical lymph node (IHC). The PrPres banding pattern in spleen was type 2B. No pathological signs of vCJD were detected in the central nervous system. Recipient 2 thus provided the first evidence that PRNP codon 129M/V individuals might be either susceptible to vCJD or capable of incubating this disease.
PrPres was not detected in tonsil tissue from recipient 2 (Peden et al. 2004). This finding highlights a potential caveat in the use of resected tonsil for estimating the prevalence of vCJD in the population and the use of tonsil biopsies for the pre-mortem diagnosis of secondary vCJD. Interestingly, PrPres was detected in tonsil tissue taken at autopsy from recipient 3, but a pre-mortem tonsil biopsy had not been performed on this recipient (Wroe et al. 2006).
8.3.2 Evidence for vCJD Transmission by Plasma Products
There has been one case of vCJD infection detected at autopsy in a patient who had been treated with large doses of UK-produced Factor VIII (Peden et al. 2010). The patient was a haemophiliac who died of a non-neurological disorder in 2008, aged 73. This patient was heterozygous (M/V) at PRNP codon 129. PrPres was detected by NaPTA/WB in only one sample of spleen with a banding pattern of type 2B (Fig. 8.2h). All other tissues tested from this patient, including brain and tonsil, were negative.
This case of vCJD infection was identified through a United Kingdom (UK) Department of Health funded study to undertake active surveillance of haemophiliac patients for vCJD infection. All haemophiliacs undergoing surgery on tissues from the central nervous system and lymphoid tissues were invited to participate and give consent for analysis of tissue samples at NCJDRSU for PrPres. In addition, consent was sought for the analysis of samples from autopsy tissues from relatives of patients who died during this study. A variable range of biopsy and autopsy specimens from 17 patients have been analysed by NaPTA/WB, PET and IHC. All tissues tested negative for PrPres apart from one spleen sample from the patient described above (Peden et al. 2010).
A number of possibilities have been considered to explain how this haemophiliac patient became infected with vCJD. Prior to 1998 in the UK, blood products such as Factor VIII and Factor IX were manufactured from blood plasma sourced in the UK. Units of blood from asymptomatic donors, who went onto develop vCJD, contributed to pooled plasma for the manufacture of batches of clotting factor concentrates (Hewitt et al. 2006). The patient described above had been treated with two of these “vCJD-implicated” batches of Factor VIII, totalling 9,025 units, in 1994 and 1996. However, this person’s medical history also included treatment with approximately 400,000 units of non-implicated Factor VIII between 1980 and 2001, four blood transfusions and multiple endoscopic procedures. An assessment of all risk factors, including dietary exposure to BSE, concluded the most likely route of exposure for this patient was non-implicated batches of Factor VIII (Bennett and Ball 2009). This conclusion was based on (1) the large number of units of Factor VIII received by this patient, (2) an estimated prevalence of vCJD in the UK population of 1/10,000 (Spongiform Encephalopathy Advisory Committee 2008) and (3) the routine pooling of around 20,000 units of plasma to make a single batch of clotting factor concentrate (Clarke and Ghani 2005; Clewley et al. 2009; Hilton et al. 2004).
8.4 Methods to Detect Prions in Blood and the Prospect of Implementation of a Blood Screening Test for vCJD
8.4.1 The Challenge
The development of a workable blood screening test for vCJD faces a series of formidable obstacles. Some of these are biochemical in nature: if prions are equated with abnormal forms of the prion protein (PrPSc), then a prospective blood test must be able to detect extremely low levels of PrPSc in the analyte (whole blood, plasma or buffy coat), in which the normal precursor protein, PrPC is more abundant by orders of magnitude. The property of PrPSc being measured must be unique to the disease-associated or infected state. Whilst brain PrPC and PrPSc are well characterised, both PrPC and PrPSc are now recognised as being biochemically heterogeneous with protease-resistant forms of PrPC being found in normal brain and protease-sensitive forms of PrPSc being found in CJD brain (Safar et al. 2005; Yuan et al. 2006). Moreover, the exact biochemical form of PrPSc in blood is unknown. This may result in a practical problem for test development, in that an assay developed with, and optimised for brain PrPSc, even if spiked into blood or plasma at high dilution, may not be applicable for the detection of endogenous blood PrPSc. Blood from analogous animal diseases or animal models may therefore appear an attractive option, especially since blood from pre-clinical stages can be taken to mimic screening for asymptomatic vCJD infection, but translation may be complicated by differences in the prion strain and host species involved. Given all of these difficulties, a framework for CJD blood test evaluation has been developed by the UK National Institute for Biological Standards and Control (http://www.nibsc.ac.uk/spotlight/cjd_resource_centre/cjd_tests.aspx).
Implementation presents a further series of challenges: the actual prevalence of vCJD infection in the UK population can only be estimated with very wide confidence intervals (Hilton et al. 2004; Clewley et al. 2009; de Marco et al. 2010; Garske and Ghaini 2010) but the most recent prevalence estimate is of 1:2,000 based on retrospective screening of archived tonsil specimens in England (Health Protection Agency 2012). A routine blood screening test with an exceptionally high specificity, if applied routinely to all blood donations, would still generate significant numbers of false positives (Turner 2006; Ludlam and Turner 2006; Peden et al. 2008). One way to mitigate the effects of these unavoidable false positive screening test results (for donors and for the transfusion services alike) would be to implement a second (confirmatory) assay in parallel with a screening assay. Therefore two assays are actually being sought. Ideally the screening assay and confirmatory test would work by different principles, and only one (the screening assay) would need to be high throughput and rapid.
8.4.2 Approaches to Sensitive Detection of PrPSc
A wide variety of approaches have been taken to the development of blood tests for vCJD. We have reviewed these recently (Peden et al. 2008) and a detailed description is beyond the scope of this chapter. In general, they involve a step that distinguishes PrPC and PrPSc, followed by a sensitive end detection method. Despite considerable scientific and commercial interest none of these conventional approaches have, as yet, delivered a prototype assay for the detection of PrPSc in human blood.
8.4.3 PrPSc Amplification and Current Blood Test Development
Prion disease pathogenesis is thought to depend on the autocatalytic conversion of PrPC by PrPSc. Using an in vitro cell-free system to model this process could effectively amplify PrPSc from sub-detectable levels to levels readily detectable by conventional means. Capitalising on earlier work by Byron Caughey and co-workers (Kocisko et al. 1994; Caughey et al. 1999), Claudio Soto and colleagues developed a method termed protein misfolding cyclic amplification (PMCA) in which a “seed” of PrPSc promotes the conversion of PrPC “substrate” supplied by an appropriate (usually brain) tissue homogenate. Accelerated by cycles of sonication and incubation, the amplified PrPSc product is then detected by protease digestion and Western blotting (Saborio et al. 2001). The sensitivity of detection can be further enhanced by using the product from one PMCA reaction to seed further rounds in a process termed serial PMCA or sPMCA (Bieschke et al. 2004; Castilla et al. 2005). Working with the experimental hamster 263K scrapie model, serial PMCA has been able to distinguish between bloods from infected and uninfected hamsters at the clinical phase (Castilla et al. 2005) and during the asymptomatic pre-clinical phase (Saa et al. 2006). This general PMCA methodology has been adopted by numerous researchers and has been further developed towards basic science (Deleault et al. 2007), medical (Jones et al. 2007) and veterinary (Thorne and Terry 2008) applications. Blood or plasma appears to require the introduction of additional preparative steps in part to avoid inhibition of the amplification reaction by plasma constituents (Castilla et al. 2005; Saa et al. 2006; Thorne and Terry 2008). Our own approach (in collaboration with the Scottish National Blood Transfusion Service) has been to collect PrPSc from plasma, perform serial PMCA using out-dated human platelet extracts as substrate, followed by detection using conformation-dependent immunoassay (Jones et al. 2009). Other configurations of the PMCA methodology aimed at human blood testing also appear promising (Tattum et al. 2010).
A third generation amplification method termed QuIC has been described in which recombinant PrP replaces natural PrPC substrates, periodic shaking replaces sonication, and (in the real-time variant, RT-QuIC), amyloid formation is monitored in real time by thioflavin T fluorescence (Atarashi et al. 2007; Atarashi et al. 2008). RT-QuIC is already under evaluation as a clinical diagnostic using cerebrospinal fluid from suspected cases of sporadic CJD (Atarashi et al. 2011; McGuire et al. 2012). Problems with relatively inefficient detection of vCJD brain and CSF samples (Peden et al. 2012) and with inhibitors of QuIC in plasma appear to have been overcome by a further modification of the methodology (termed e-QuIC) that incorporates PrPSc-specific antibody immunoprecipitation, a chimeric recombinant PrP substrate and a reaction buffer replacement step (Orru et al. 2011). e-QuIC is reported to be able to detect a 1014-fold dilution of vCJD brain or 2 ag/ml of vCJD PrPres making it the most sensitive assay yet reported as judged by limit of detection (LoD) of human CJD brain. However, the method has not yet been tested on clinical vCJD blood specimens and relevant controls. A prospective blood test with a somewhat higher LoD has been tested using whole blood from clinical vCJD patients (n = 21) against 142 blood specimens from donors (n = 100) and neurological controls (n = 42) giving sensitivity and specificities of 71.4% and 100%, respectively (Edgeworth et al. 2011). The novelty and biochemical point of interest of this assay is the use of stainless steel particles to concentrate, modify or present PrP in advance of a sensitive immunoassay. The assay failed to detect PrPSc in blood taken from sCJD patients (n = 27).
8.4.4 Future Perspectives
The above assays, in addition to a further blood test under development by Prionics AG, all have a considerable distance to go before they could be considered validated as vCJD blood screening tests. Moreover, none of these tests currently meet the assay time requirements demanded by blood donation testing. At present, e-QuIC (Orru et al. 2011) and the assay of Edgeworth et al. (2011) appear most promising. The serial format of PMCA involved in achieving the appropriate analytical sensitivity makes assays such as sPMCA/CDI better suited to development as a confirmatory blood test. It is tempting to speculate that these technologies could be in implementable forms within the next 2 or 3 years. However, the track record in this area indicates that this is far from certain and the potential benefits of implementation of any test will need to be weighed carefully against the costs and potential consequences.
8.5 Conclusions
The emergence of vCJD has had a major impact on blood transfusion in the UK and other affected countries. It is greatly to the credit of the UK transfusion services that several precautionary measures to protect the blood supply were put into place even before the first cases of transfusion-associated vCJD were identified. The measures taken to reduce the risks of vCJD transmission by blood and blood products in the UK are summarised in Table 8.1. It remains to be seen whether any further measures will be implemented, for example the introduction of “prion filters”. The cases of transfusion-associated vCJD infection all occurred prior to the full introduction of leucodepletion in the UK. The most recent data from sheep models indicate that whilest leucodepletion alone does not prevent disease transmission completely (McCutcheon et al. 2011), it does have a pronounced effect, and that it is the leucoreduction component of combined leucodepletion/prion reduction filters that is responsible the prion removal (Lacroux et al. 2012). Both of these sheep studies also show that all blood components may be considered potential vectors for prion transmission (McCutcheon et al. 2011; Lacroux et al. 2012). These findings reinforce the need for multiple control measures to reduce the risk of vCJD transmission by blood and blood products and raise the possibility that future cases of transfusion-associated vCJD may be identified in recipients of blood products other than non-leucodepleted red blood cell concentrates. In view of the uncertainties over the prevalence of asymptomatic vCJD infection in the UK, it seems likely that these control measures will continue to be required, perhaps until an effective test for asymptomatic vCJD infection is available.
The evidence for the transmission of other forms of CJD by blood transfusion is far less clear-cut: although data from experimental models indicate that different strains of prions can be transmitted by blood, the epidemiological evidence in humans to support these findings is largely absent. The recent contention that blood transfusion may be a risk factor for sCJD (Puopolo et al. 2011) has renewed interest in this field of research and will hopefully generate new research that is better designed to answer this problem than many of the previously published studies. Until these new studies are concluded, continued surveillance and analysis of risk factors for all forms of human prion disease is required.
References
Andreoletti O, Litaise C, Simmons H, Corbiere F, Lugan S, Costes P, Schelcher F, Vilette D, Grassi J, Lacroux (2012) Highly efficient prion transmission by blood transfusion. PLoS Pathog 8:e1002782
Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B (2007) Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods 4:645–650
Atarashi R, Wilham JM, Christensen L, Hughson AG, Moore RA, Johnson LM, Onwubiko HA, Priola SA, Caughey B (2008) Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 5:211–212
Atarashi R, Satoh K, Sano K, Fuse T, Yamaguchi N, Ishibashi D, Matsubara T, Nakagaki T, Yamanaka H, Shirabe S, Yamada M, Mizusawa H, Kitamoto T, Klug G, McGlade A, Collins SJ, Nishida N (2011) Utrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 17:175–178
Barclay GR, Houston EF, Halliday SI, Farquhar CF, Turner ML (2002) Comparative analysis of normal prion protein expression on human, rodent, and ruminant blood cells by a panel of prion antibodies. Transfusion 42:517–526
Bennett PG, Ball J (2009) vCJD Risk Assessment Calculations for a Patient With Multiple Routes of Exposure. http://www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/documents/digitalasset/dh_100337.pdf. Accessed 15 September 2011
Bessos H, Drummond O, Prowse C, Turner M, MacGregor I (2001) The release of prion protein from platelets during storage of apheresis platelets. Transfusion 41:61–66
Bieschke J, Weber P, Sarafoff N, Beekes M, Giese A, Kretzschmar H (2004) Autocatalytic self-propagation of misfolded prion protein. Proc Natl Acad Sci USA 101:12207–12211
Bishop MT, Ritchie DL, Will RG, Ironside JW, Head MW, Thomson V, Bruce M, Manson JC (2008) No major change in vCJD agent strain after secondary transmission via blood transfusion. Plos One 3:e2878
Bons N, Lehmann S, Mestre-France N, Dormont D, Brown P (2002) Brain and buffy coat transmission of bovine spongiform encephalopathy to the primate Microcebus murinus. Transfusion 42:513–516
Brown P, Gibbs CJ, Rodgers-Johnson P, Asher DM, Sulima PM, Bacote A, Goldfarb LG, Gajdusek DC (1994) Human spongiform encephalopathy: The National Institute of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 35:513–529
Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, Drohan WN (1998) The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 38:810–816
Brown P, Cervenakova L, McShane LM, Barber P, Rubenstein R, Drohan WN (1999) Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion 39:1169–1179
Brown P (2000) BSE and transmission through blood. Lancet 356:955–956
Burdick MD, Pifat DY, Petteway SR, Cai K (2006) Clearance of prions during plasma protein manufacture. Transfus Med Rev 20:57–62
Burthem J, Urban B, Pain A, Roberts DJ (2001) The normal cellular prion protein is strongly expressed in myeloid dendritic cells. Immunobiology 98:3733–3738
Casaccia P, Ladogana A, Xi YG, Pocchiari M (1989) Levels of infectivity in the blood throughout the incubation period of hamsters peripherally injected with scrapie. Arch Virol 108:146–149
Castilla J, Saa P, Soto C (2005) Detection of prions in blood. Nat Med 11:982–985
Caughey B, Horiuchi M, Demaimay Raymond GJ (1999) Assays of protease-resistant prion protein and its formation. Methods Enzymol 309:122–133
Cervenakova L, Yakovleva O, McKenzie C, Kolchinsky S, McShane L, Drohlan WN, Brown P (2003) Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43:1687–1694
Chohan G, LLewelyn C, Mackenzie J, Cousens S, Kennedy A, Will R, Hewitt P (2010) Variant Creutzfeldt-Jakob disease in a transfusion recipient: coincidence or cause? Transfusion 50:1003–1006
Choi EM, Geschwind MD, Deering C, Pomeroy K, Kuo A, Miller BL, Safar JG, Prusiner SB (2009) Prion proteins in subpopulations of white blood cells from patients with sporadic Creutzfeldt-Jakob disease. Lab Invest 89:624–635
Clarke P, Ghani AC (2005) Projections of the future course of the primary vCJD epidemic in the UK: inclusion of subclinical infection and the possibility of wider genetic susceptibility. J R Soc Interface 2:19–31
Clewley JP, Kelly CM, Andrews N, Vogliqi K, Mallinson G, Kaisar M, Hilton DA, Ironside JW, Edwards P, McCardle M, Ritchie DL, Dabagian R, Ambrose HE, Gill ON (2009) Prevalence of disease related prion protein in anonymous tonsil specimens in Britain: a cross sectional opportunistic survey. Br Med J 338:b1442
Comoy E, Jaffre N, Mikol J, Durand V, Jas-Duval C, Lebon V, Cheval J, Quadrio I, Lescoutra-Etchegaray N, Streichenberger, Haik S, Sumian C, Perret-Liaudet A, Eloit M, Hantraye P, Brown P, Deslys JP (2012) A new neurological disease in primates inoculated with prion-infected blood or blood components. Prion 6S:19–20
De Marco MF, Linehan J, Gill ON, Clewley JP, Brandner S (2010) Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J Pathol 222:380–387
Deleault NR, Harris BT, Rees JR, Supattapone S (2007) Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA 104:9741–9746
Deslys JP, Lasmezas C, Dormont D (1994) Selection of specific strains in iatrogenic Creutzfeldt-Jakob disease. Lancet 343:848–849
Diringer H (1984) Sustained viremia in experimental hamster scrapie. Arch Virol 82:105–109
Durig J, Giese A, Schulz-Schaeffer W, Rosenthal C, Schmucker U, Bieschke J, Duhrsen U, Kretzschmar HA (2000) Differential constitutive and activation-dependent expression of prion protein in human peripheral blood leucocytes. Br J Haematol 108:488–495
Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, Jackson GS (2011) Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377:487–493
Foster PR, Welch AG, McLean C, Griffin BD, Hardy JC, Bartley A, MacDonald S, Bailey AC (2000) Studies on the removal of abnormal prion protein by processes used in the manufacture of human plasma products. Vox Sang 78:86–95
Garske T, Ghaini AC (2010) Uncertainty in the tail of the variant Creutzfeldt-Jakob disease epidemic in the UK. PLoS One 5:e15626
Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, Rohwer RG (2004a) Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet 364:529–531
Gregori L, Maring JA, MacAuley C, Dunston B, Rentsch M, Kempf C, Rohwer RG (2004b) Partitioning of TSE infectivity during ethanol fractionation of human plasma. Biologicals 32:1–10
Gregori L, Lambert BC, Gurgel PV, Gheorghiu L, Edwardson P, Lathrop JT, MacAuley C, Carbonell RG, Burton SJ, Hammond D, Rohwer RG (2006a) Reduction of transmissible spongiform encephalopathy infectivity from red blood cells with prion protein affinity ligands. Transfusion 46:1152–1161
Gregori L, Gurgel PV, Lathrop JT, Edwardson P, Lambert BC, Carbonell RG, Burton SJ, Hammond DJ, Rohwer RG (2006b) Reduction of infectivity of endogenous transmissible spongiform encephalopathies present in blood by adsorption to selective affinity resins. Lancet 368:2226–2230
Gregori L, Yang H, Anderson S (2011) Estimation of variant Creutzfeldt-Jakob disease infectivity titres in human blood. Transfusion 51:2596–2603
Haddon DJ, Hughes MR, Antignano F, Westaway D, Cashman NR, McNagny KM (2009) Prion protein expression and release by mast cells after activation. J Infect Dis 200:827–831
Hartwell RC, Nelson MS, Kislan MM, Stenland CJ, Miller JLC, Pifat DY, Petteway SR, Cai K (2005) An improved Western blot assay to assess the clearance of prion protein from plasma-derived therapeutic proteins. J Virol Methods 125:187–193
Head MW, Yull HM, Ritchie DL, Bishop MT, Ironside JW (2009) Pathological investigation of the first blood donor and recipient pair linked by transfusion-associated variant Creutzfeldt-Jakob disease transmission. Neuropathol Appl Neurobiol 35:433–436
Health Protection Agency (2007) Fourth case of transfusion-associated variant-CJD infection. Health Protect Report 2007; 1: 2–3. http://www.hpa.org.uk/hpr/archives/2007/hpr0307.pdf. Accessed September 15, 2011
Health Protection Agency (2012) Summary results of the second national survey of abnormal prion protein prevalence in archived appendix specimens. Health Protection Report 6: 3–4. http://www.hpa.org/hpr/archives/2012/news3212.htm#bnrmlprn. Accessed September 11, 2012
Herzog C, Sales N, Etchegaray N, Charbonnier A, Freire S, Dormant D, Deslys JP, Lasmezas CI (2004) Tissue distribution of bovine spondiform encephalopathy agent in primates after intravenous or oral infection. Lancet 363:422–428
Herzog C, Riviere J, Lescoutra-Eschegaray N, Charbonnier A, Leblanc V, Sales N, Deslys JP, Lasmezas CI (2005) PrPTSE distribution in a primate model of variant, sporadic and iatrogenic Creutzfeldt-Jakob disease. J Virol 70:14339–14345
Hewitt PE, Llewelyn CA, Mackenzie J, Will RG (2006) Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang 91:221–230
Hilton DA, Ghani AC, Conyers L, Edwards P, McCardle L, Ritchie D, Penney M, Hegazy D, Ironside JW (2004) Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol 203:733–739
Holada K, Simak J, Risitano AM, Maciejewski J, Young NS, Vostal JG (2002a) Activated platelets of patients with paroxysmal nocturnal hemoglobinuria express cellular prion protein. Blood 100:341–343
Holada K, Vostal JG, Theisen PW, MacAuley C, Gregori L, Rohwer RG (2002b) Scrapie infectivity in hamster blood is not associated with platelets. J Virol 76:4649–4650
Houston F, Foster JD, Chong A, Hunter N, Bostock CJ (2000) Transmission of BSE by blood transfusion in sheep. Lancet 356:999–1000
Houston F, McCutcheon S, Goldman W, Chong A, Foster J, Siso S, Gonzalez L, Jeffrey M, Hunter N (2008) Prion diseases are efficiently transmitted by transfusion in sheep. Blood 112:4739–4745
Hunter N, Foster J, Chong A, McCutcheon S, Parnham D, Eaton S, MacKenzie C, Houston F (2002) Transmission of prion diseases by blood transfusion. J Gen Virol 83:2897–2905
Jones M, Peden AH, Prowse CV, Groener A, Manson JC, Turner ML, Ironside JW, MacGregor IR, Head MW (2007) In vitro amplification and detection of variant Creutzfeldt-Jakob disease PrPSc. J Pathol 213:21–26
Jones M, Peden AH, Yull H, Wight D, Bishop MT, Prowse CV, Turner ML, Ironside JW, MacGregor IR, Head MW (2009) Human platelets as a substrate source for the in vitro amplification of the abnormal prion protein (PrP) associated with variant Creutzfeldt-Jakob disease. Transfusion 49:376–384
Kocisko DA, Come JH, Priola S, Chesebro B, Raymond GJ, Lansbury PT, Caughey B (1994) Cell-free formation of protease-resistant prion protein. Nature 370:471–474
Kuroda Y, Gibbs CJ, Amyx HL, Gajdusek DC (1983) Creutzfeldt-Jakob disease in mice: persistent viremia and preferential replication of virus in low-density lymphocytes. Infect Immun 41:154–161
Lacroux C, Bougard D, Litaise C, Simmons H, Corbiere F, Dernis D, Tardivel R, Morel N, Simon S, Lugan S, Costes P, Weisbecker JL, Schlcher F, Grassi J, Coste J Andeoletti O (2012) Impact of leucocyte depletion and prion reduction filters on TSE blood borne transmission. PLoS One 7:e42019
Lasmezas CI, Fournier JG, Nouvel V, Boe H, Marce D, Lamoury F, Kopp N, Hauw JJ, Ironside JW, Bruce M, Dormont D, Deslys JP (2001) Adaptation of the bovine spongiform encephalopathy agent to primates and comparison with Creutzfeldt-Jakob disease: Implications for human health. Proc Natl Acad Sci USA 98:4142–4147.
Lasmezas CI, Comoy E, Hawkins S, Herzog C, Mouthon F, Timm K, Auvre F, Corriea E, Lescoutra-Etchagaray N, Sales N, Wells G, Brown P, Deslys JP (2005) Risk of oral infection with bovine spongiform encephalopathy agent in primates. Lancet 365:781–783
Lee DC, Stenland CJ, Hartwell RC, Ford EK, Cai K, Miller JLC, Gilligan KJ, Rubenstein R, Fournel M, Petteway SR (2000) Monitoring plasma processing steps with a sensitive Western blot assay for the detection of the prion protein. J Virol Methods 84:77–89
Lee DC, Stenland CJ, Miller JL, Cai K, Ford EK, Gilligan KJ, Hartwell RC, Terry JC, Rubenstein R, Fournel M, Petteway SR (2001) Direct relationship between the partitioning of pathogenic prion protein and transmissible spongiform encephalopathy infectivity during the purification of plasma proteins. Transfusion 41:449–455
Lescoutra-Etchegaray N, Jaffre N, Culeux A, Sumian C, Durand V, Deslys JP, Comoy E (2012) Prion removal PCapt device delays onset of atypical neurological disease observed in primates exposed to BSE-infected blood products. Prion 6S:141
Li R, Liu D, Zanusso G, Liu T, Fayen JD, Huang JH, Petersen RB, Gambetti P, Sy MS (2001) The expression and potential function of cellular prion protein in human lymphocytes. Cell Immunol 207:49–58
Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG (2004) Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363:417–421
MacGregor I, Hope J, Barnard G, Kirby L, Drummond O, Pepper D, Hornsey V, Barclay R, Bessos H, Turner M, Prowse C (1999) Application of time-resolved fluoroimmunoassay for the analysis of normal human prion protein in human blood and its components. Vox Sang 77:88–96
Manuelidis EE, Gorgacz EJ, Manuelidis L (1978) Viremia in experimental Creutzfeldt-Jakob disease. Science 200:1069–1071
Manuelidis EE, Kim JH, Mericangas JR, Manuelidis L (1985) Transmission to animals of Creutzfeldt-Jakob disease from human blood. Lancet 2:896–897
McCutcheon S, Blanco ARA, Houston EF, de Wolf C, Tan BC, Smith A, Groschup MH, Hunter N, Hornsey VS, MacGregor IR, Prowse CV, Turner M, Manson JC (2011) All clinically-relevant blood components transmit prion disease following a single blood transfusion: A sheep model of vCJD. PLoS One 6:e23169
McGuire LI, Peden AH, Orru CD, Wilham JM, Appleford NE, Mallinson G, Andrews M, Head MW, Caughey B, Will RG, Knight RSG, Green AJE (2012) Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol 72:278–285
Orru CD, Wilham JM, Raymond LD, Kuhn F, Schroeder B, Raeber AJ, Caughey B (2011) Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. M Biol 2:e00078–11
Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW (2004) Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364:527–529
Peden AH, Head MW, Jones M, MacGregor I, Turner M, Ironside J (2008) Advances in the development of a screening test for variant Creutzfeldt-Jakob disease. Expert Opin Med Diag 2:207–219
Peden A, McCardle L, Head MW, Love S, Ward HJT, Cousens SN, Keeling DM, Millar CM, Hill FGH, Ironside JW (2010) Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16:296–304
Peden AH, McGuire LI, Appleford NEJ, Mallinson G, Wilham JM, Orru CD, Caughey B, Ironside J, Knight RS, Will RG, Green AJE, Head MW (2012) Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J Gen Virol 93:438–449
Perini F, Vidal R, Ghetti B, Tagliavini F, Frangione B, Prelli F (1996) PRP27-30 is a normal soluble protein fragment released by human platelets. Biochem Biophys Res Commun 223:572–577
Puopolo M, Ladogana A, Vetrugno V, Pocchiari M (2011) Transfusion of sporadic Creutzfeldt-Jakob disease by blood transfusion: risk factor or possible biases. Transfusion 51:1556–1566
Saa P, Castilla J, Soto C (2006) Presymptomatic detection of prions in blood. Science 313:92–94
Saborio GP, Permanne B, Soto C (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813
Safar J, Geschwind MD, Deering C, Didorenko S, Sattavat M, Sanchez H, Serban A, Vey M, Baron H, Giles K, Miller BL, DeArmond SJ, Prusiner SB (2005) Diagnosis of human prion protein. Proc Natl Acad Sci USA 102:3501–3506
Siso S, Gonzalez L, Houston F, Hunter N, Martin S, Jeffrey M (2006) The neuropathological phenotype of experimental ovine BSE is maintained after blood transfusion. Blood 108:745–748
Siso S, Jeffrey M, Houston F, Hunter N, Martin S, Gonzalez L (2009) Pathological phenotype of sheep scrapie after blood transfusion. J Comp Pathol 142:27–35
Sowemimo-Coker S, Kascsak R, Kim A, Andrade F, Pesci S, Kascsak R, Meeker C, Carp R, Brown P (2005) removal of exogenous (spiked) and endogenous prion infectivity from red cells with a new prototype of leucoreduction filter. Transfusion 45:1839–1844
Sowemimo-Coker SO, Demczyk CA, Andrade F, Baker CA (2010) Evaluation of prion infectivity from red blood cells with prion reduction filters using a new rapid and highly sensitive cell culture-based infectivity assay. Transfusion 50:980–988
Spongiform Encephalopathy Advisory Committee (2008) Position Statement: Prevalence of Subclinical Variant Creutzfeldt-Jakob Disease Infections. http://webarchive.nationalarchives.gov.uk/20110316162913/http://www.seac.gov.uk/statements/state-cjd-infections.pdf. Accessed September 15, 2011
Tattum MH, Jones S, Pal S, Collinge J, Jackson GS (2010) Discrimination between prion-infected and normal blood samples by protein misfolding cyclic amplification. Transfusion 50:996–1002
Thorne L, Terry LA (2008) In vitro amplification of PrPSc derived from the brain and blood of sheep infected with scrapie. J Gen Virol 89:3177–3184
Turner M (2006) Transfusion safety with regards to prions: ethical, legal and societal considerations. Transfus Clin Biol 13:317–319
Williams L, Brown P, Ironside J, Gibson S, Will R, Ritchie D, Kreil TR, Abee C (2007) Clinical, neuropathological and immunocytochemical features of sporadic and variant forms of Creutzfeldt-Jakob disease in the squirrel monkey (Saimiri sciureus). J Gen Virol 88:688–695
Wroe SJ, Pal S, Siddique D, Hyare H, Macfarlane R, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Hewitt P, Collinge J (2006) Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: a case report. Lancet 368:2061–2067
Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H, Kong Q, Kneale G, Gambetti P, Zou WQ (2006) Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem 281:34848–34858
Acknowledgements
The work of the National CJD Research & Surveillance Unit is supported by the Department of Health, UK, and the Scottish Government. Dr. Alexander Peden has been supported by grants funding from the Scottish Government Health Directorate Chief Scientists Office. The views expressed in the publication are those of the authors and not necessarily those of the Department of Health, UK. The authors would like to thank relatives of patients for the opportunity to conduct research on tissue specimens at the National CJD Research & Surveillance Unit that contributed to the identification of blood transfusion-related transmission of variant CJD.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Peden, A.H., Head, M.W., Ironside, J.W. (2013). Risk of Transmission of Creutzfeldt–Jakob Disease by Blood Transfusion. In: Zou, WQ., Gambetti, P. (eds) Prions and Diseases. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-5338-3_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5338-3_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-5337-6
Online ISBN: 978-1-4614-5338-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)